
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalyst-dependent selectivity in sulfonium ylide cycloisomerization reactions†
Rik
Oost
a,
James D.
Neuhaus
a,
Antonio
Misale
a,
Ricardo
Meyrelles
 b,
Luís F.
Veiros
b and
Nuno
Maulide
*a
b,
Luís F.
Veiros
b and
Nuno
Maulide
*a
aUniversity of Vienna, Institute of Organic Chemistry, Währinger Straße 38, 1090 Vienna, Austria. E-mail: Nuno.Maulide@univie.ac.at
bUniversidade de Lisboa, Centro de Química Estrutural, Instituto Superior Técnico, Av. Rovisco Pais 1, 1049-001 Lisbon, Portugal
First published on 25th July 2018
Abstract
Divergent catalysis is an emerging field whereby access to structurally diverse compounds from a common precursor is achieved through controlled reaction pathways. Herein we present an unusual example of π-acid catalyst dependent selectivity in the cycloisomerization of alkene-tethered sulfonium ylides. Computational mechanistic studies revealed how the ability of palladium to cycle through oxidation states largely controls the selectivity.
Introduction
Divergent catalysis provides a quick access to structurally different compounds from a common precursor through controlled reaction pathways. It is considered a highly attractive tool for the discovery of functional materials and novel drugs.1 However, the conceptual development of such processes is still rare in the literature.A distinctive reactivity feature of Au(I), Pt(II) and Pd(II) complexes is their propensity towards electrophilic activation of unsaturated C–C bonds.2 Although these metals are intrinsically different from one another, it is not unusual that their catalysis of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C or C
C or C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C activation often leads to similar products (a notable exception being ene-yne cyclisations3). Nonetheless, Au(I) catalysts only have one free coordination site, where Pd(II) and Pt(II) salts are square planar and can have two coordination sites. Moreover, Pd(II) and Pt(II) salts often behave as bifunctional Lewis acids and even form σ-complexes with heteroatoms.4 Last but not least, Pd and Pt complexes can undergo oxidation state changes far more readily than gold.
C activation often leads to similar products (a notable exception being ene-yne cyclisations3). Nonetheless, Au(I) catalysts only have one free coordination site, where Pd(II) and Pt(II) salts are square planar and can have two coordination sites. Moreover, Pd(II) and Pt(II) salts often behave as bifunctional Lewis acids and even form σ-complexes with heteroatoms.4 Last but not least, Pd and Pt complexes can undergo oxidation state changes far more readily than gold.
Sulfonium ylides have long occupied a privileged position in organic synthesis.5 In recent years, stabilised sulfonium ylides have seen their reactivity greatly expanded through transition-metal-mediated activation.6 We and others have shown that electrophilic activation of alkene- and alkyne-tethered sulfur ylides with gold catalysts affords cyclopropanes7 and furans.8 Nonetheless, cyclopropanation is still dominated by the chemistry of diazo compounds using transition metal catalysis.9 The resulting metallocarbenes are mild and selective cyclopropanating agents. As a notable exception, the cyclopropanation of olefins containing a sulfur atom does not lead to the desired cyclopropane product. Instead, the sulfonium ylide is formed with complete chemoselectivity (Scheme 1).10 In face of this limitation of metal carbenoid strategies, we became interested in the possibility of achieving cycloisomerization of the corresponding sulfonium ylides through π-acid catalysis. Herein we report on an intriguing reactivity switch that allows the divergent catalytic synthesis of cyclopropanes or dihydrofurans at will from the same precursor as well as a mechanistic study of these transformations.
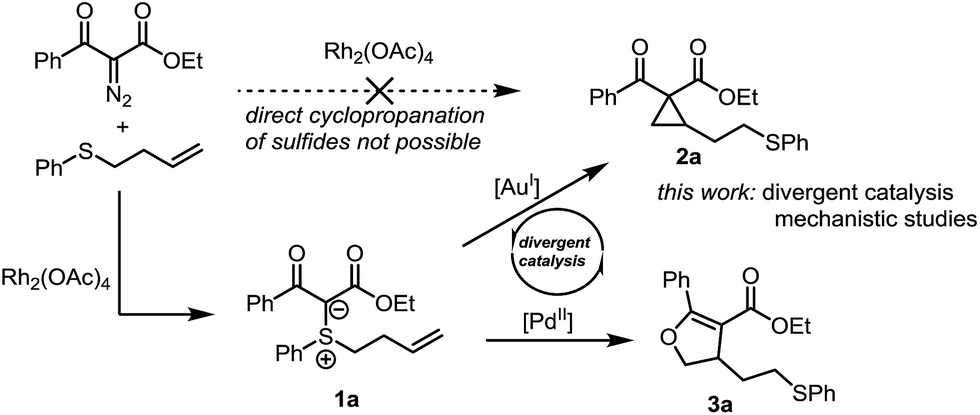 | ||
| Scheme 1 Synthesis of cyclopropanes on homoallyl sulfides. | ||
Results and discussion
Based on our prior success using gold catalysis for the activation of sulfonium ylides, we started our investigations with sulfonium ylide 1a (Scheme 1) and screened several gold catalysts.11 Using Echavarren's catalyst in toluene at 75 °C, the reaction proceeded smoothly to the desired cyclopropane in excellent 95% yield (for other gold catalysts, see the ESI† for details).Different π-acid catalysts were screened in attempts to optimize the reaction further. While AuCl3 and Hg(OTf)2 only gave low yields of the cyclopropane, Pd(II) and Pt(II) salts surprisingly led to formation of a different product 3a (Scheme 1). Using PdCl2(MeCN)2 in 1,4-dioxane enabled full conversion and an isolated yield of 82%.12
From the outset, the catalyst-controlled divergence was intriguing and we performed experiments aimed at elucidating the mechanism of these reactions. Critically, conversion of the cyclopropane product 2a to the dihydrofuran 3a is not possible under Pd(II) catalysis under the reaction conditions, nor in the presence of sulfonium ylide 1b. This suggests that neither the palladium catalyst nor an active species formed in the reaction, is responsible for cyclopropane isomerization to the corresponding dihydrofuran and such an isomerization can therefore be excluded as a possible reaction pathway (Scheme 2a).13
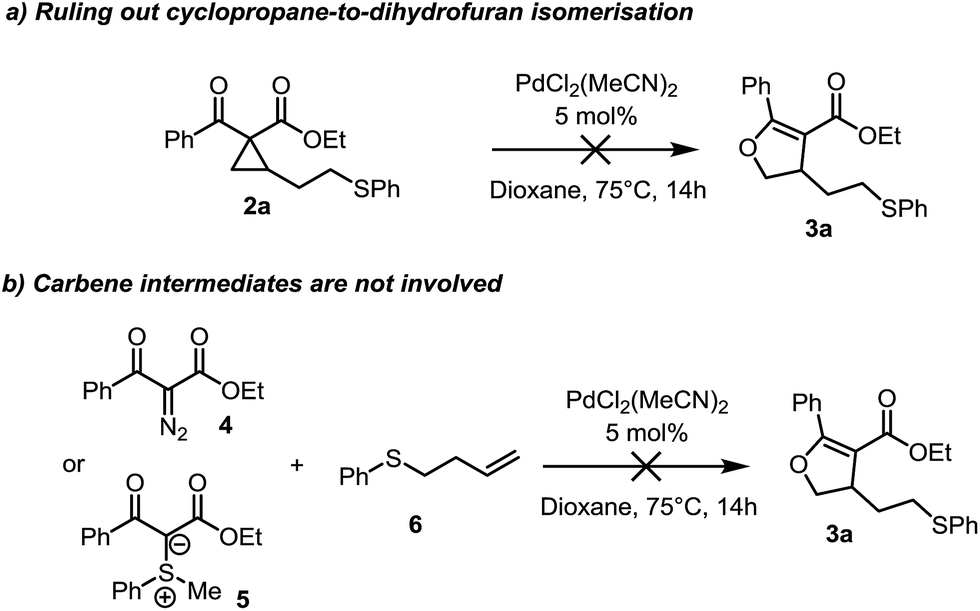 | ||
| Scheme 2 Mechanistic experiments. | ||
Suspicious of the involvement of carbene intermediates,14,15 we probed the reaction of diazo compound 4 in the presence of the homoallyl sulfide 6 under our reaction conditions (Scheme 2b). Full consumption of the diazo compound was observed, however, no trace of the dihydrofuran 3a, nor sulfonium ylide 1a was obtained. Similarly, no dihydrofuran was obtained from the combination of (phenylmethyl)sulfonium ylide 5 and homoallyl sulfide 6, suggesting that carbene intermediates are not involved.16
Selected isotope labelling experiments are depicted in Scheme 3. Substrate 1a–d, bearing a deuterium label on the terminal position of the double bond, was synthesized and exposed to the two sets of conditions. In both cases, the deuterium label ended up at the expected position. Interestingly, the high 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E/Z ratio of the substrate resulted in cyclopropane and dihydrofuran products with a similar 3:1 trans/cis ratio. This suggests that, while the reaction is not fully stereospecific, both pathways might share a common cyclization step.
:1 E/Z ratio of the substrate resulted in cyclopropane and dihydrofuran products with a similar 3:1 trans/cis ratio. This suggests that, while the reaction is not fully stereospecific, both pathways might share a common cyclization step.
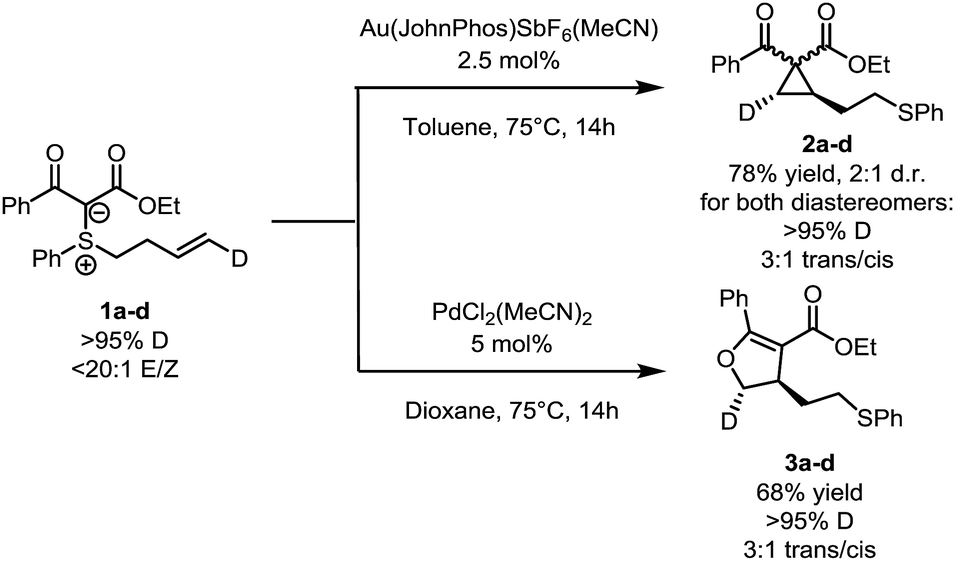 | ||
| Scheme 3 Deuterium labelling studies. | ||
The mechanism of both reactions was further studied by DFT calculations17 (see ESI† for a full account of the Computational Details and references), employing a ketoester substrate (methyl substituents on both the ketone and the ester group) as model and replacing JohnPhos by PPhMe2, for computational expediency.18 The free energy profile obtained for cyclopropanation with the Au catalyst is represented in Fig. 1.
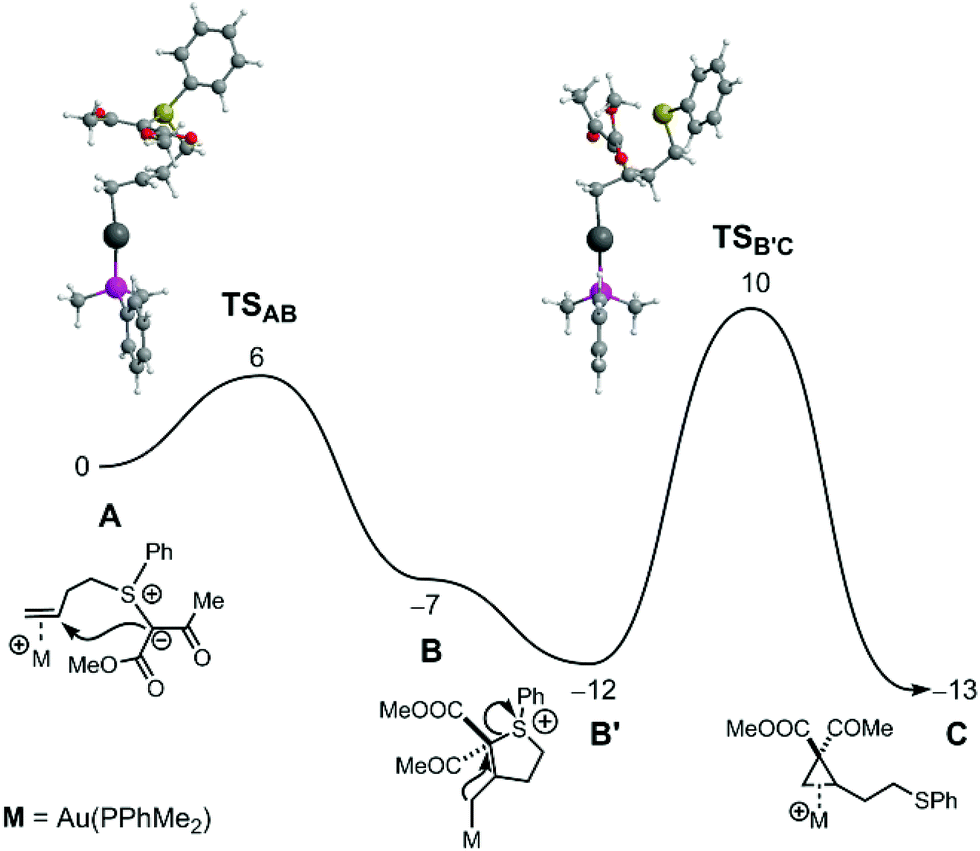 | ||
| Fig. 1 Free energy profile (kcal mol−1) for the cyclopropanation reaction with the Au catalyst. | ||
As shown, following π-coordination of Au to the CC double bond, an attack of the ylide C-atom to the internal olefinic carbon forms intermediate B, an Au-alkyl complex with a 5-member tetrahydrothiophenium (SC4) ring. After a conformational rearrangement (B→B′), nucleophilic attack of the coordinated C-atom to the formerly ylidic carbon results in cleavage of the C–S bond and formation of the cyclopropane product. This second step has a barrier of 22 kcal mol−1 and represents the rate-determining event of the process. The overall reaction A→C is exergonic with ΔGR = −13 kcal mol−1.19
A pathway for dihydrofuran formation under Au catalysis was also calculated.11 However, comparison of the two mechanisms shows a clear preference for the cyclopropanation reaction, due to a 21 kcal mol−1 lower overall barrier. Aiming to elucidate the differences between Au and Pd, the corresponding mechanisms with Pd catalysis were also addressed by means of DFT calculations. The free energy profiles obtained are represented in Fig. 2 and 3.
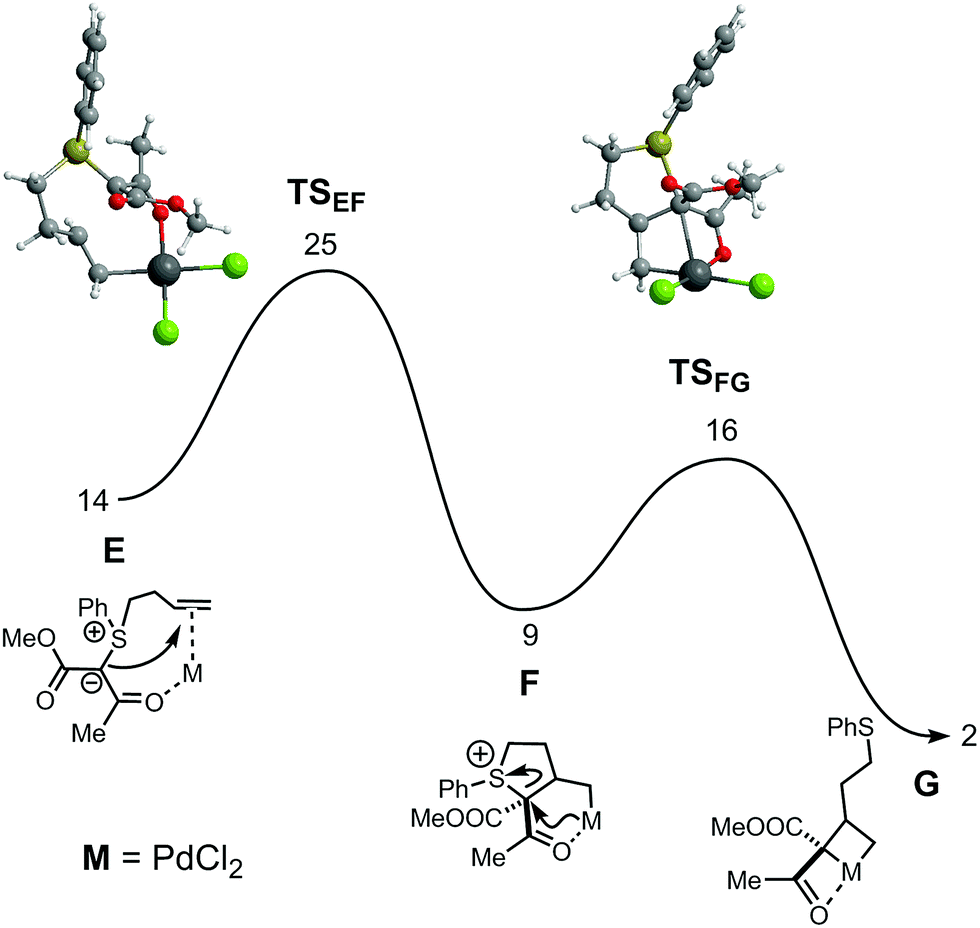 | ||
| Fig. 2 Free energy profile (kcal mol−1) for the formation of the PdIV intermediate common to both reactions. Free energy values relative to the separated reactants: [PdCl2(MeCN)2] and substrate. | ||
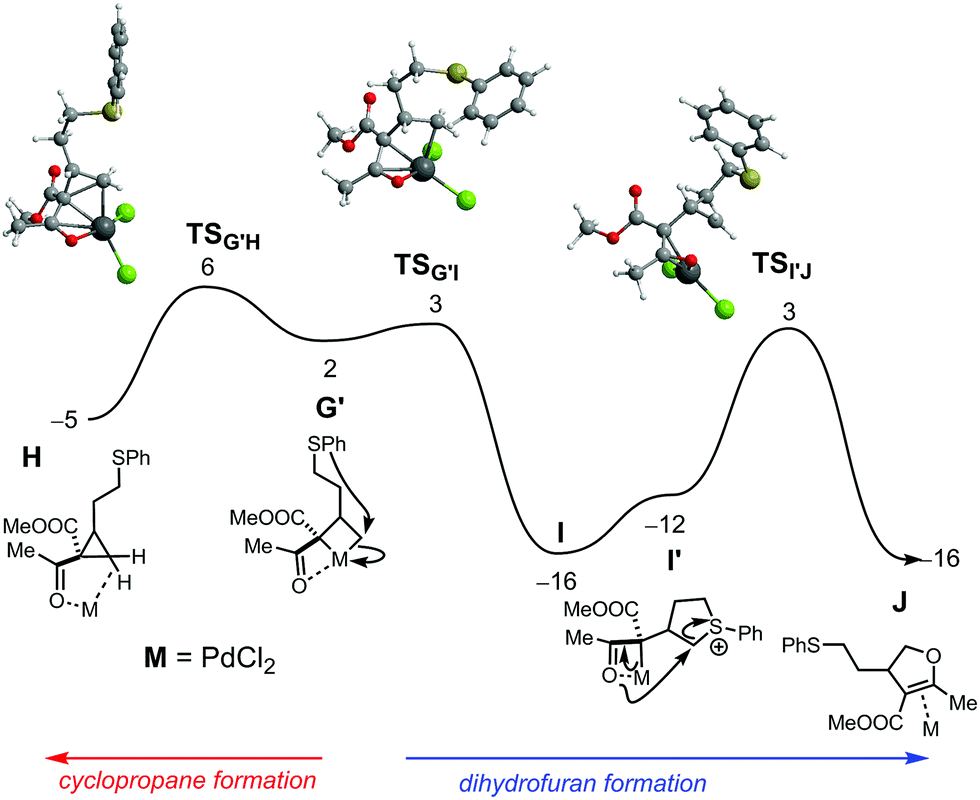 | ||
| Fig. 3 Free energy profile (kcal mol−1) for the formation of dihydrofuran (right side) and for cyclopropanation (left side) from intermediate G. Free energy values relative to the separated reactants: [PdCl2(MeCN)2] and substrate. | ||
After substitution of two acetonitrile ligands in [PdCl2(MeCN)2] by the chelating substrate, the first step of the mechanism parallels that found for Au catalysis – formation of the SC4 ring after nucleophilic attack of the ylide C-atom to the internal olefinic carbon. The associated barrier is significant (25 kcal mol−1, relative to the separated reactants) but the process is exergonic, with F more stable than E by 5 kcal mol−1. In the following step there is C–S bond breaking in the substrate with concomitant coordination of the ylide C-atom, forming a metallacyclobutane G. That formally corresponds to an oxidative addition process, with a change from PdII (in F) to PdIV (in G). In the corresponding transition state, TSFG, both S–C bond breaking as well as Pd–C coordination are rather incipient. In fact, the S–C bond distance in TSFG, is only 0.34 Å longer than in F, and the Pd–C separation is 0.46 Å longer than it will become in intermediate G. The process involves a significant rearrangement of the ligand. In F, there is a square planar geometry around the metal whereas in G there is a square pyramidal geometry with the two chlorides, the O-atom and the former ylidic C-atom occupying the four base positions and the olefin terminal C-atom in the apical position. The barrier for this second step is modest, TSFG being 16 kcal mol−1 less stable than the initial reactants, and it corresponds to an exergonic process with ΔG = −7 kcal mol−1.
The free energy profile for the formation of both the cyclopropane and dihydrofuran products from common PdIV intermediate G′ (a conformer of G) is represented in Fig. 3. Pd-catalysed cyclopropanation (Fig. 3, left) occurs in a single step, from G′ to H, in a formal reductive elimination process that brings the metal back to PdII. In H, the product is coordinated by both a C–H bond (σ-complex) and the ketone O-atom. In the corresponding transition state, TSG′H, the formation of the new C–C bond is well advanced, with a distance of 1.94 Å, only 0.41 Å longer than the final one, in H.
Formation of the dihydrofuran (Fig. 3, right) starts with attack of the S-atom onto the coordinated carbon and formation of a stabilized Pd enolate (G′→I). This is formally a reductive step, as the metal changes from PdIV to PdII – regenerating the characteristic square planar environment. The associated barrier is negligible (1 kcal mol−1) and the step is significantly exergonic. Following conformational change at S (I→I′), attack from the ketone oxygen with concomitant C–S bond breaking, from I′ to J, yields the dihydrofuran product (as a π-complex). Both TSG′I and TSI′J are 3 kcal mol−1 more stable than TSG′H. This indicates that dihydrofuran formation is slightly more favourable than cyclopropanation, for the Pd catalyst. Overall, the free energy balance indicates an exergonic process (ΔGR = −16 kcal mol−1).
Taken together, the mechanisms found for the Pd catalyst involve oxidation of the metal from PdII to PdIV in an intermediate step. This is accomplished with C–S bond breaking prior to C–C coupling and formation of a metallacyclobutane (in intermediates G/G′). As a result, the sulfur atom is freed for formation of a tetrahydrothiophenium intermediate, effectively providing a natural leaving group for the final attack from the ketone oxygen that produces the dihydrofuran. On the other hand, gold oxidation from AuI to AuIII is much less favourable and therefore the favourable route becomes cyclopropanation.20
At this juncture, we decided to investigate the scope of both transformations. As shown in Scheme 4, the cyclopropanation reaction is quite general and works well for a wide variety of substrates, tolerating electron-rich or electron-poor arene moieties or a hindered ketone. An electronically diverse range of substituents (R3) was tolerated on sulfur. Even with an extra carbon in the tether, cyclization could still be achieved, albeit only in 25% yield. Unfortunately, di- and trisubstituted alkenes did not lead to the desired product in synthetically useful yields.11
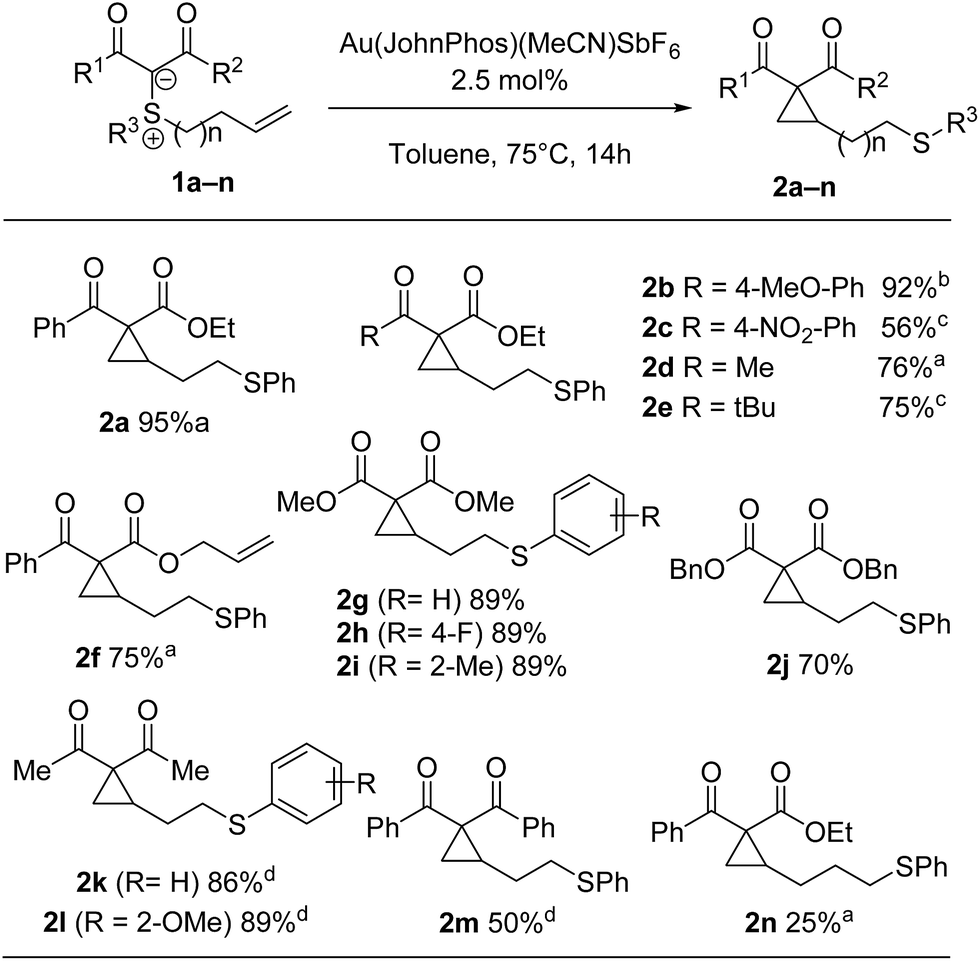 | ||
| Scheme 4 Scope of Au-catalyzed cyclopropanation. a Isolated yield of combined diastereomers (2:1 d.r.). b Isolated yield of combined diastereomers (1.8:1 d.r.). c Isolated yield of combined diastereomers (1.5:1 d.r.). d Reaction was performed at 75 °C for 24 h. | ||
Interestingly, exquisite selectivity was observed in the formation of cyclopropane 2f (Scheme 4). Noteworthy, the corresponding diphenylsulfonium ylide was an excellent substrate for cyclopropanation of tethered allyl esters in our prior work.7 The observed reactivity could be explained by the difference in calculated activation energy (17.8 vs. 6 kcal mol−1). This is also qualitatively apparent from a cursory analysis of reaction temperature. Indeed, while the reactions in the present report are performed at 75 °C, intramolecular cyclopropanation of the allyl ester requires 100 °C.7
Next, we surveyed the palladium(II)-catalysed cyclization leading to dihydrofurans (Scheme 5). A qualitative difference in reactivity between electron-poor and electron-rich substrates was observed, with electron-donating substituents requiring a decrease in temperature to avoid side product formation. Selectivity analogous to that of the cyclopropanation process was observed for 3f, for which the substrate carries two double bonds susceptible of activation. Malonate derivatives (cf.3g) gave a complex mixture of products with no dihydrofuran detected. Interestingly, bicyclic product 3o was formed in low yield, while cyclopropanation of the same substrate didn't take place.
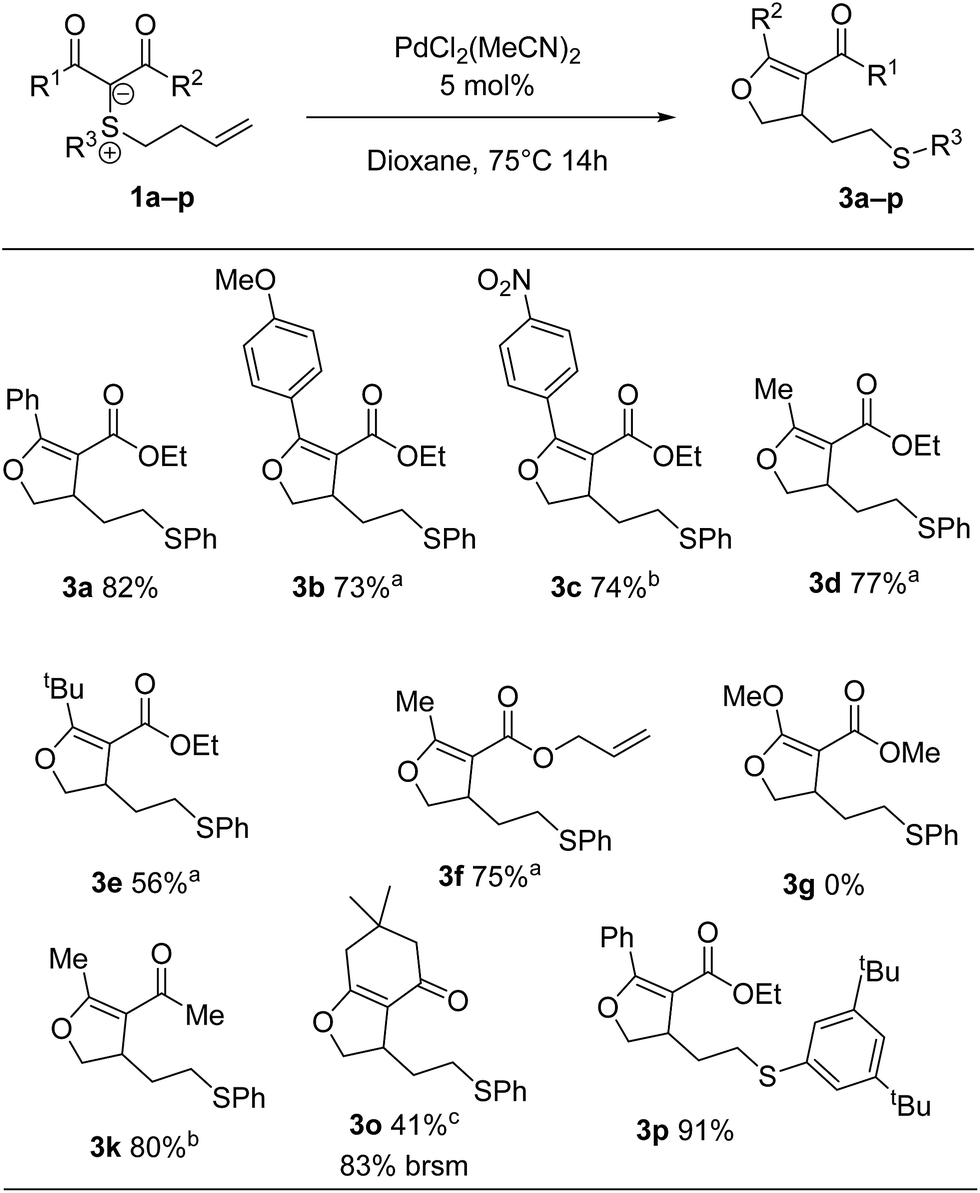 | ||
| Scheme 5 Scope of dihydrofurans. a Reactions were carried out at 55 °C for 14 h. b Reactions were carried out at 75 °C for 72 h. c Reactions were carried out at 55 °C for 72 h. | ||
Conclusions
In summary, we have presented an unusual example of π-acid catalyst-dependent selectivity in the cycloisomerization of alkene-tethered sulfonium ylides. While gold catalysis leads to cyclopropane products, palladium(II) leads to a unique dihydrofuran synthesis. Gold effectively enables the formal cyclopropanation of sulfur-containing olefins, a reaction currently beyond the realm of diazo chemistry; palladium unlocks an oxidative addition/reductive elimination catalytic cycle rarely seen in sulfonium ylide chemistry. Computational studies clarified the mechanism and shed light on the factors responsible for the selectivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Generous support of this research by the DFG (Grants MA 4861/4-1 and 4-2 to N. M.) is acknowledged. L. F. V. thanks Fundação para a Ciência e Tecnologia, UID/QUI/00100/2013. We are very grateful to the University of Vienna for continued support of our research programs.Notes and references
- (a) J. Mahatthananchai, A. M. Dumas and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 10954 CrossRef PubMed; (b) L.-W. Xu, L. Li and C.-Q. Lai, Mini-Reviews Org. Chem., 2007, 4, 217 CrossRef.
- (a) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef PubMed; (b) Z. G. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239 CrossRef PubMed; (c) H. Huang, Y. Zhou and H. Liu, Beilstein J. Org. Chem., 2011, 7, 897 CrossRef PubMed; (d) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028 CrossRef PubMed.
- E. Jiménez-Núñez and A. M. Echavarren, Chem. Commun., 2007, 4, 333 RSC.
- Y. Yamamoto, J. Org. Chem., 2007, 72, 7817 CrossRef PubMed.
- (a) X.-L. Sun and Y. Tang, Acc. Chem. Rev., 2008, 41, 937 CrossRef PubMed; (b) E. M. McGarrigle, E. L. Myers, O. Illa, M. A. Shaw, S. L. Riches and V. K. Aggarwal, Chem. Rev., 2007, 107, 5841 CrossRef PubMed; (c) L.-Q. Lu, T.-R. Li, Q. Wang and W.-J. Xiao, Chem. Soc. Rev., 2017, 46, 4135 RSC.
- R. Oost, R. J. D. Neuhaus, J. Merad and N. Maulide, Sulfur Ylides in Organic Synthesis and Transition Metal Catalysis, in Structure and Bonding, Springer, Berlin, Heidelberg, 2017 Search PubMed.
- (a) X. Huang, S. Klimczyk, L. F. Veiros and N. Maulide, Chem. Sci., 2013, 4, 1105 RSC; (b) S. Klimczyk, X. Huang, H. Kählig, L. F. Veiros and N. Maulide, J. Org. Chem., 2015, 80, 5719 CrossRef PubMed; (c) S. Klimczyk, A. Misale, X. Huang and N. Maulide, Angew. Chem., Int. Ed., 2015, 54, 10365 CrossRef PubMed.
- X. Huang, B. Peng, M. Luparia, L. F. R. Gomes, L. F. Veiros and N. Maulide, Angew. Chem., Int. Ed., 2012, 51, 8886 CrossRef PubMed.
- (a) M. P. Doyle, M. A. McKervey and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, John Wiley & Sons, New York, NY, 1998 Search PubMed; (b) M. P. Doyle, Chem. Rev., 1986, 86, 919 CrossRef; (c) H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977 CrossRef PubMed; (d) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861 CrossRef PubMed.
- W. Ando, Acc. Chem. Res., 1976, 10, 179 CrossRef.
- See ESI† for further details..
- PtCl2/PtCl4 were selective for dihydrofuran formation in lower yields. Rh, Ir, Cu or Ag displayed virtually no reactivity. See ESI† for details..
- Classical Lewis acids (e.g. FeCl3 or Sc(OTf)3) could convert 2a into dihydrofuran 3a in moderate yield. See ESI† for details..
- For reactions of palladium carbenoids see: O. Reiser, Cyclopropanation and Other Reactions of Palladium–Carbene (and Carbyne) Complexes, in Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi, John Wiley & Sons, New York, 2002, vol. 2, pp. 1561–1577 Search PubMed.
- For metal carbenoid reactions leading to dihydrofurans see: (a) L. Xia and Y. R. Lee, Adv. Synth. Catal., 2013, 355, 2361 CrossRef; (b) P. Müller and S. Chappellet, Helv. Chim. Acta, 2005, 88, 1010 CrossRef.
- In the case of Pd(OAc)2, decomposition of the diazo-compound was observed, but no trace of dihydrofuran, nor sulfonium ylide was obtained. See ESI† for details..
- (a) R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed; (b) Calculations performed at the PBE0/(SDD*, 6-311++G**, PCM)//PBE0/(SDD*, 6-31G**) level using the Gaussian 09 package. A full account of the computational details and a complete list of references are provided as ESI.†.
- The simplicity of the model used in the calculations prevents the discussion of the reaction stereochemistry..
- Closure of the catalytic cycle is thermodynamically favorable with a free energy balance of ΔGR = −16 kcal mol−1. See the ESI† for further computational details..
- Transition states for gold oxidation, i.e., the equivalents of TSFG for the Au system, have associated barriers above 40 kcal mol−1..
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, full characterization for new compounds, spectral data and computational details. See DOI: 10.1039/c8sc02815j |
| This journal is © The Royal Society of Chemistry 2018 |