DOI:
10.1039/C8SC02739K
(Edge Article)
Chem. Sci., 2018,
9, 6853-6859
Conjugated double helices via self-dimerization of α,α′-dianilinotripyrrins
Received
22nd June 2018
, Accepted 16th July 2018
First published on 17th July 2018
Abstract
A new motif for artificial double helices was developed on the basis of α,α′-disubstituted tripyrrin. α,α′-Dibromotripyrrin 3 was prepared by gentle bromination at the pyrrolic α-positions of 5,10-diphenyltripyrrane followed by oxidation with DDQ. Nucleophilic substitution reactions of 3 with anilines proceeded efficiently to furnish a series of α,α′-dianilinotripyrrins 4–11, which displayed monomeric and dimeric forms depending upon the solvent used for crystallization and the structures of the substituted anilines. Dimeric forms show double helical structures with smooth π-conjugation as indicated by their absorption spectra. van't-Hoff plot analyses revealed that the dimerizations in CDCl3 are enthalpy-driven. Larger association constants of the dimerization are attained for 3,5-di-t-butylanilino- and 3,5-bis(trifluoromethyl)anilino-substituted tripyrrins (7 and 8) via additional multiple intermolecular interactions. In a nonpolar and aprotic solvent, tripyrrins (9 and 10) bearing bulkier 1-naphthylamino and mesitylamino groups do not dimerize but undergo unique tautomerization.
Introduction
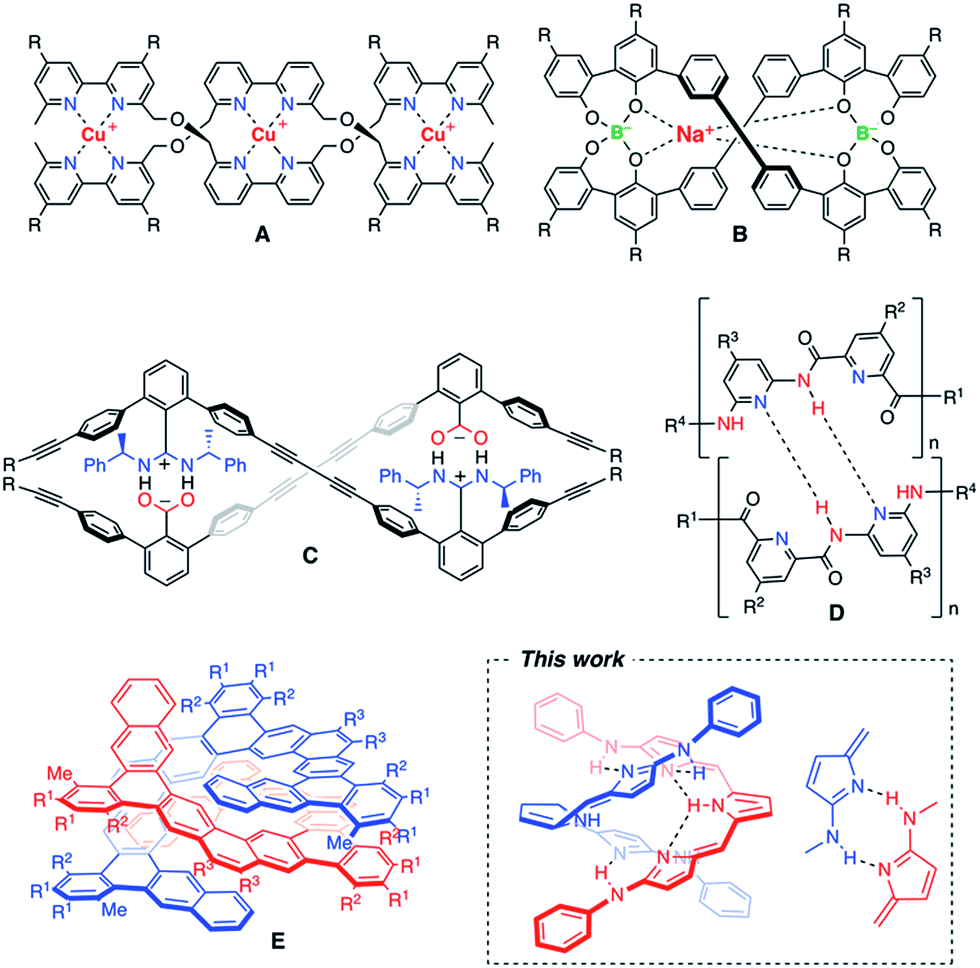
An artificial double helix motif has stimulated chemists' interest not only to mimic the biological functions as seen in DNAs, but also to develop new functional devices for organic electronics and photonics.1 Significant efforts have been made for synthetic oligomers that can form bimolecular double helices through different types of non-covalent interactions.2–5 One of the most useful non-covalent interactions is metal–ligand coordination. Metal ion-directed self-assemblies in which the organic ligands form double-helical complexes are called double helicates.2 As a pioneering study, J. M. Lehn et al. employed copper(I)-chelation by bipyridyl ligands to obtain double helicates (A).2a A variety of metallohelicates utilizing pyridyl coordinations have been extensively studied later.2b–d Not only transition metal complexes, but also spiroborates (B) were found to form double helicates, in which coordination of the central sodium ion plays an important role in shielding the electrostatic repulsion between the two borate groups.3 As for purely organic approaches, effective salt-bridge formation was utilized for m-terphenylene-based double helical oligomers (C).4 Yashima and co-workers have beautifully extended this motif to well-defined double helical polymers.4 On the other hand, Huc and co-workers synthesized double-stranded helices of aromatic oligoamides (D) in which the NH site of the amide moiety and the N site of the pyridine moiety serve as the hydrogen bonding donor and acceptor, respectively.5 As such, hydrogen bonding is a useful intermolecular interaction to hold the supramolecular arrangement, and a variety of hydrogen bonding donor and acceptor sites have been properly allocated to form double helical structures.6 However, there are only a few examples of fully π-conjugated double helices that may be promising for visible-light harvesting or effective career transporting along the helix axis.7 Recently, a large polycyclic helical molecule (E), so called expanded helicene, was reported to display a homochiral double helical structure in solution and in the solid-state due to the favourable π-stacking interaction.8 A new motif of artificial double helices having smooth π-conjugation still remains highly desired (Chart 1).
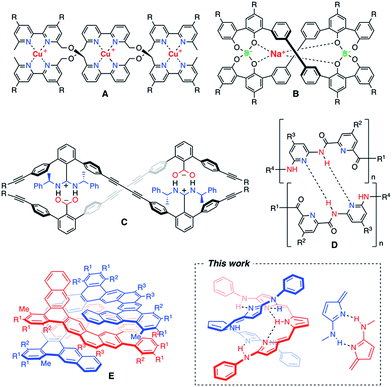 |
| | Chart 1 Examples of artificial double helix molecules.2–5 | |
Here, we report a new motif of a fully conjugated double helix based on tripyrrin derivatives. Tripyrrin is a conjugated tripyrrolic chain consisting of three pyrrolic moieties and two bridging methine carbons.9–11 Since the tripyrrin itself is generally not so stable, the isolation of tripyrrin derivatives has been limited to its protonated salts,9a metal complexes,9b–f oxygenated derivatives (tripyrrinones)10 or those with steric protections at the labile α-positions.11 We developed α,α′-dianilinotripyrrins as stable entities exhibiting a large absorption band reaching the NIR region. Surprisingly, some of these molecules spontaneously form doubly helical structures via effective intramolecular and intermolecular hydrogen bonding interactions.
Results and discussion
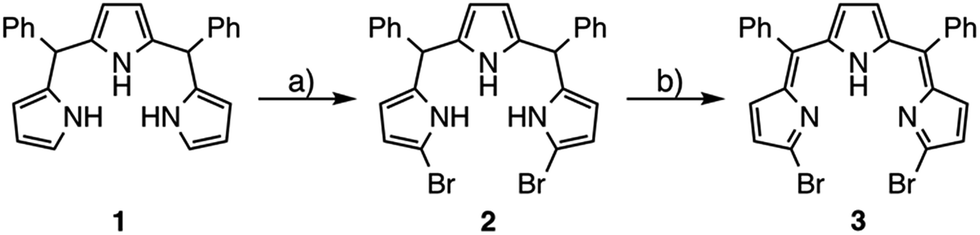
Synthesis of α,α′-dibromotripyrrin
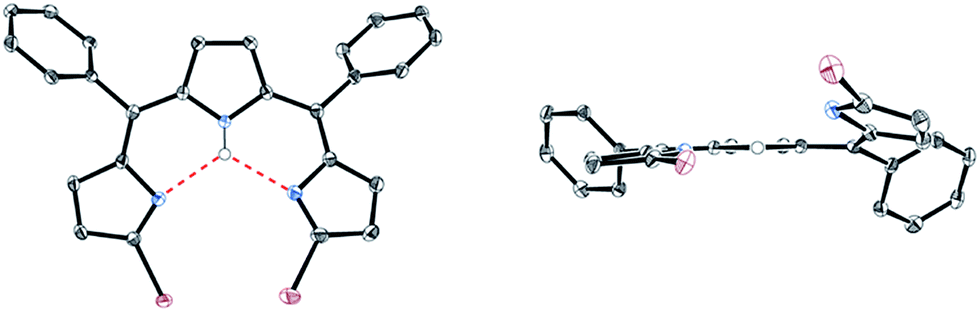
5,10-Diphenyltripyrrane 1 was easily prepared according to Gryko's report.12 Bromination of 1 with 2.1 equivalents of N-bromosuccinimide at −78 °C afforded α,α′-dibrominated tripyrrane 2 in 72% yield (Scheme 1).12 Subsequently, oxidation of 2 with 2.1 equivalents of DDQ gave a key compound, α,α′-dibromotripyrrin 3, in 87% yield. Fortunately, 3 is fairly stable under aerobic conditions and can be purified by silica-gel column chromatography. Indeed, recrystallization from CH2Cl2/MeOH gave crystals suitable for X-ray diffraction analysis (Fig. 1). Due to the intramolecular hydrogen bonding networks among the two imine-like pyrroles and one pyrrole-NH, 3 takes a cis-conformation with a slight deformation from planarity due to the steric repulsion between the two bromine atoms. The 1H NMR spectrum of 3 is consistent with the structure, featuring two doublets at 6.75 and 6.54 ppm due to the β-protons of imine-like pyrroles, a singlet at 6.31 ppm due to the β-protons of the central pyrrole and the NH proton signal at 13.44 ppm.
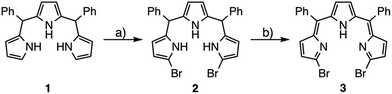 |
| | Scheme 1 Synthesis of α,α′-dibromotripyrrin 3: (a) NBS, THF, −78 °C, 2.5 h, 72%; (b) DDQ, CH2Cl2, 0 °C, 10 min, 87%. | |
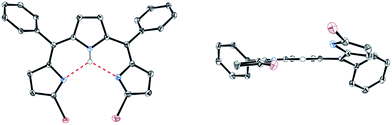 |
| | Fig. 1 X-ray crystal structure of 3. Solvent molecules and hydrogen atoms except for NH are omitted for clarity. The thermal ellipsoids are scaled to 50% probability. | |
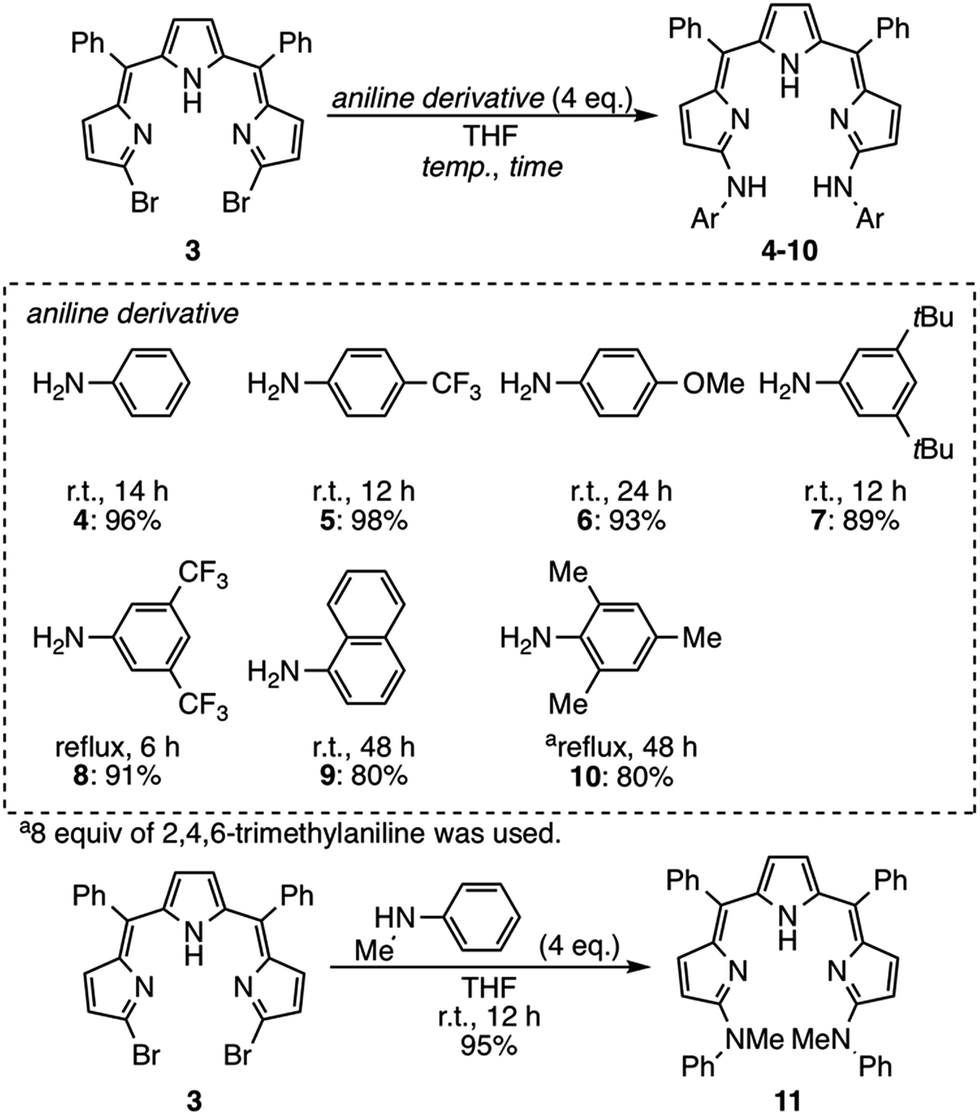
Nucleophilic substitution reactions of 3 with anilines
Next, nucleophilic substitution reactions of 3 with anilines were examined (Scheme 2). With 4 equivalents of aniline in dry THF at room temperature, the substitution reaction of 3 proceeded very smoothly to give α,α′-dianilinotripyrrin 4 in 96% yield.13 Treatment of 3 with an equimolar amount of aniline in THF/acetonitrile at room temperature afforded a mixture, from which α-anilino-α′-bromotripyrrin S1† was isolated in 90% yield.14 The substitution reactions of 3 were also examined with different types of aniline derivatives. p-Trifluoromethylaniline and p-methoxyaniline were reacted to give the corresponding anilinotripyrrin derivatives 5 and 6 in 98% and 93% yields, respectively. Sterically encumbering 3,5-di-t-butylaniline, 3,5-bis(trifluoromethyl)aniline and 1-naphthylaniline also reacted to afford 7, 8 and 9 in 89%, 91% and 80% yields, respectively. Sterically more hindered 2,4,6-trimethylaniline did not react at room temperature, but upon refluxing in THF for 48 h, 10 was obtained in 80% yield. Secondary N-methylaniline also reacted smoothly to afford N,N′-dimethylanilinoltripyrrin 11 in 95% yield.
 |
| | Scheme 2 Substitution reactions of 3 with aniline derivatives. | |
X-ray crystallographic analysis
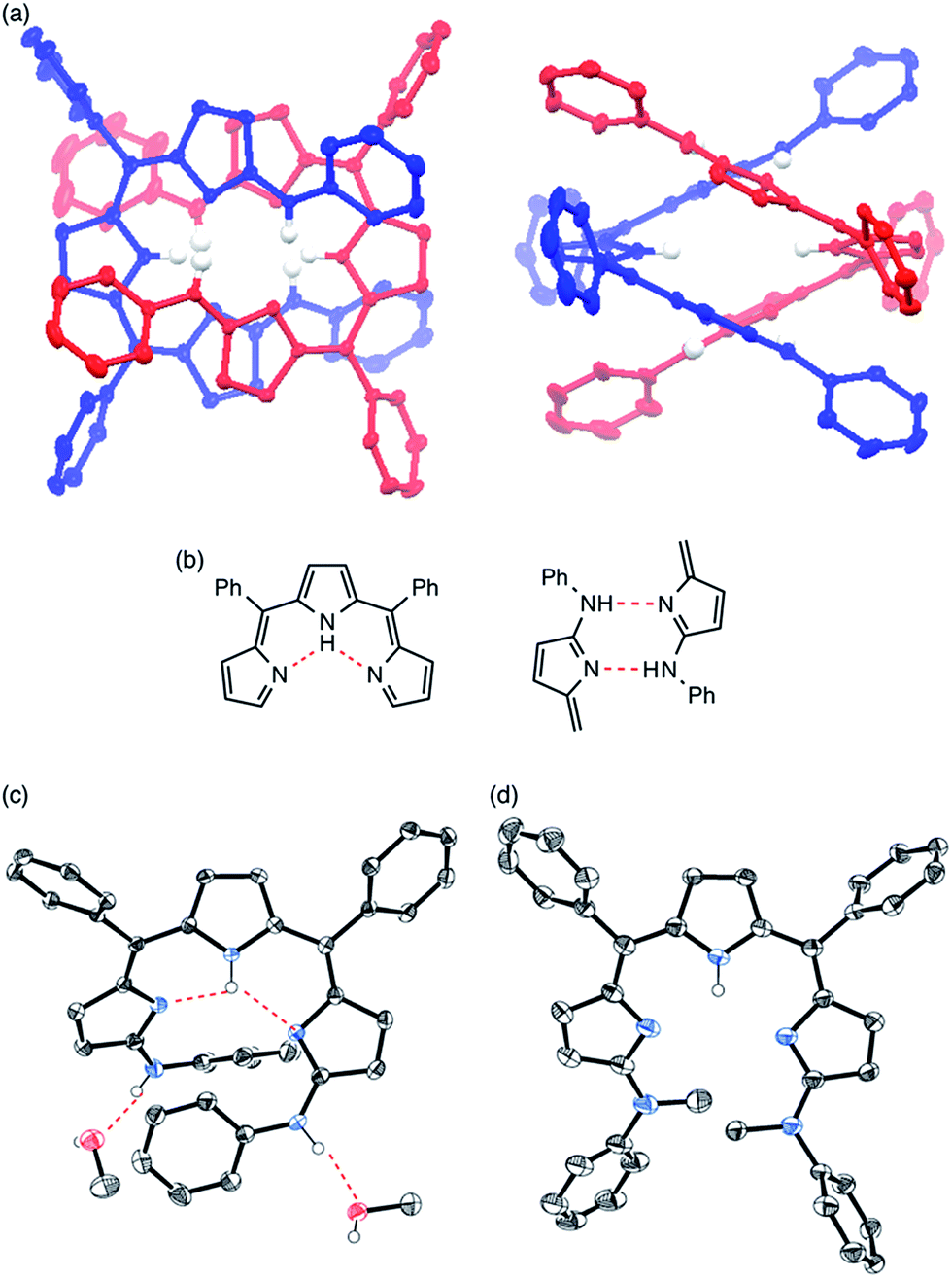
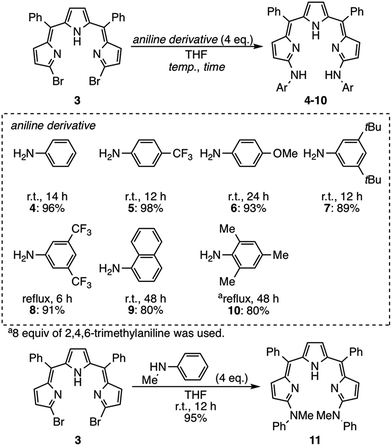
Single crystals of 4 were obtained from its solution in CHCl3 and n-hexane (Fig. 2a). The X-ray diffraction analysis revealed that 4 formed a double helix in the solid state due to the interstrand hydrogen bonding interaction between the aniline-NH and the imine-type pyrrole (Fig. 2b, right) as well as the intramolecular hydrogen bonding interaction among pyrrole-NH and imine-type pyrroles (Fig. 2b, left). When grown from a protic solvent system such as a mixture of methanol and H2O, single crystals of 4 showed a monomeric helical structure featuring intermolecular hydrogen bonding interactions between the aniline-NH and methanol (Fig. 2c). While doubly helical structures were also observed in crystals of substituted anilinotripyrrins 5, 6, 7 and 8 (Fig. S5-4–S5-8; ESI†),15N,N′-dimethyanilinoltripyrrin 11 showed a monomeric structure without any intermolecular hydrogen bonding with solvent molecules (Fig. 2d). Therefore, it is obvious that the two aniline NH segments and the tripyrrin segment are both crucial to form the double stranded helix. The helical pitches are calculated to be 5.44–5.88 Å.16 The pyrrole planes are smoothly conjugated with torsion angles along the helical twist of 3.92–45.29°. It should be noted that tripyrrins 9 and 10 could not form double helical structures probably due to serious steric hindrance around the aniline NH segments. Detailed solid-state structures of 9 and 10 will be discussed later.
 |
| | Fig. 2 (a) X-ray crystal structure of 4 obtained from CHCl3/n-hexane. Representations for intramolecular hydrogen bonding and interstrand hydrogen bonding networks are shown in (b). (c) X-ray crystal structure of 4 obtained from MeOH/H2O. (d) X-ray crystal structure of 11. Solvent molecules and hydrogen atoms except for NHs and OHs are omitted for clarity. The thermal ellipsoids are scaled to 50% probability. | |
NMR analysis
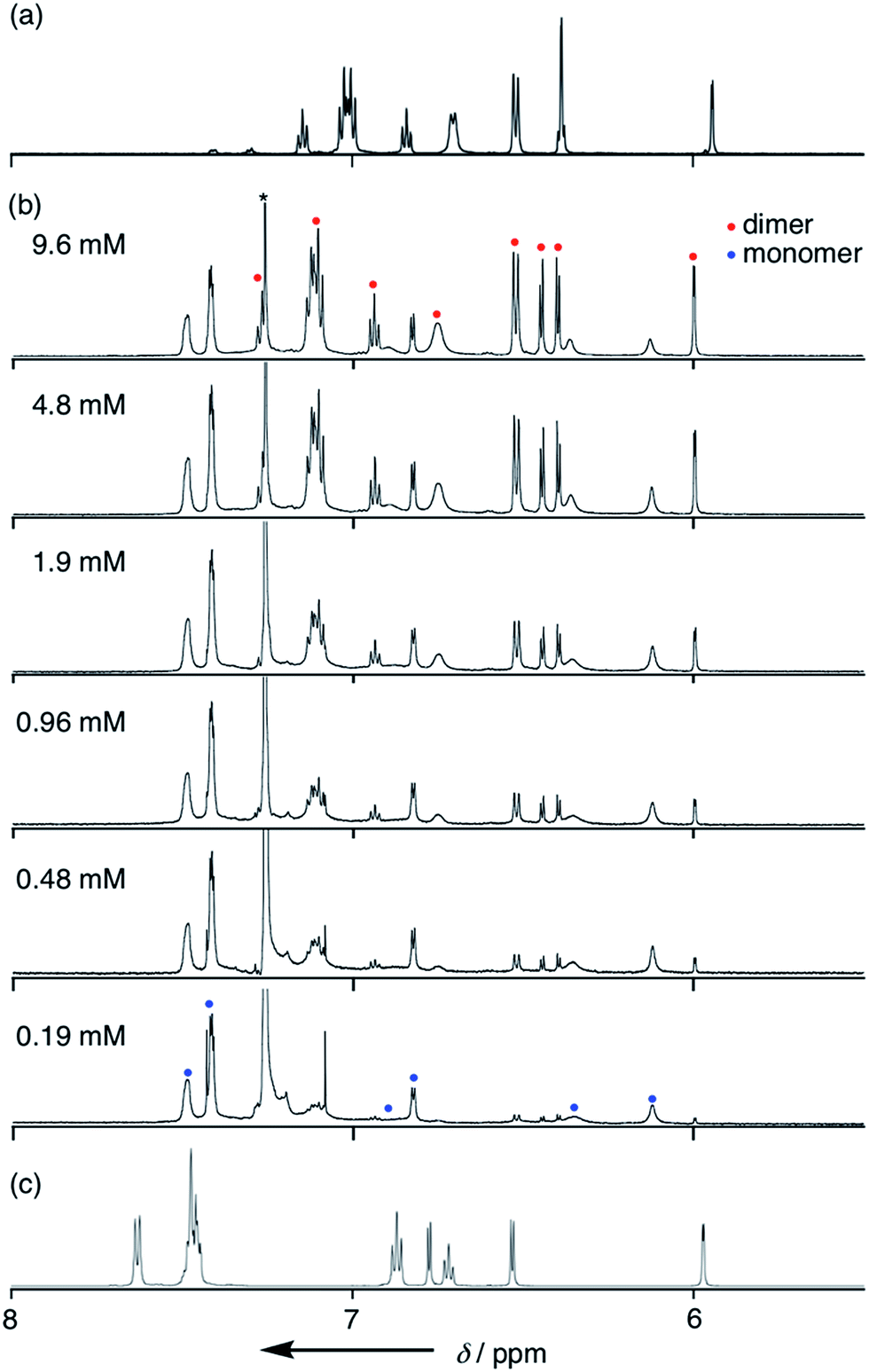
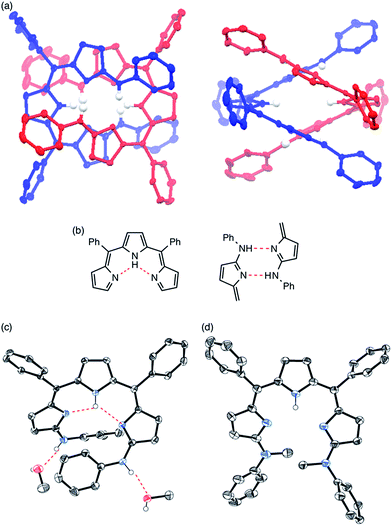
Solvent-dependent structural changes were also evident in the 1H NMR analysis. The 1H NMR spectrum of 4 in CDCl3 at room temperature showed two sets of signals, namely monomer and dimer, whose ratio was concentration dependent (Fig. 3b). The 1H NMR spectrum of 4 in a hydrogen-bonding-accepting polar solvent, DMSO-d6, showed clear signals featuring three doublets at 6.77, 6.53 and 5.97 ppm due to the pyrrolic β-protons and two singlets at 13.06 and 9.55 ppm due to the tripyrrin-NH and aniline-NH protons, respectively (Fig. 3c), which can be assigned as a monomeric species. On the other hand, the 1H NMR spectrum of 4 in a nonpolar and aprotic solvent, cyclohexane-d12, exhibited sharp peaks but different spectral features: a quartet at 6.39 ppm and a doublet at 5.94 ppm due to the β-protons and two singlets at 12.41 and 11.95 ppm due to the tripyrrin-NH and aniline-NH protons, respectively (Fig. 3a), which can be assigned as a dimer. Therefore, collectively, the 1H NMR spectra of 4 in CDCl3 indicate a slow equilibrium between the monomeric and dimeric forms of 4. Diffusion ordered spectroscopy (DOSY) in CDCl3 supports this assignment (Fig. S3-35; ESI†). The association constant Kdim for dimerization of 4 at 25 °C in CDCl3 was determined to be 2.7 × 102 M−1 by 1H NMR spectral analysis. In contrast, the 1H NMR spectra of 11 were not significantly changed by solvents. The 1H NMR spectrum of 11 in CDCl3 showed only a set of signals: a broad singlet peak at 6.4 ppm, doublet peaks at 6.21 and 6.68 ppm due to the β-protons and a singlet at 13.22 ppm due to the inner NH protons, indicating the predominant existence of the monomer in solution.
 |
| | Fig. 3
1H NMR spectra of 4 (a) in cyclohexane-d12, (b) in CDCl3 with various concentrations and (c) in DMSO-d6. | |
The dimerization equilibrium was also affected by temperature (Fig. S3-6; ESI†). At low temperature, the amount of monomeric 4 decreased and the amount of dimeric 4 increased, leading to a larger Kdim value. A van't Hoff plot of Kdim gave thermodynamic parameters, ΔH = −53.6 kJ mol−1 and ΔS = −134 J mol−1 K−1, indicating that the dimerization is mostly enthalpy-driven.
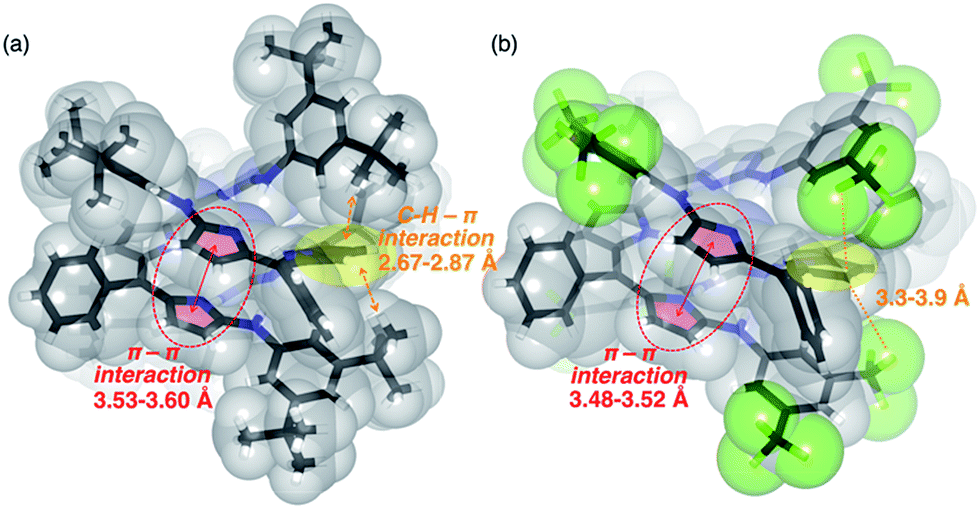
Similar to 4, the 1H NMR spectra of 5, 6, 7 and 8 in CDCl3 displayed concentration- and temperature-dependent equilibriums between monomeric and dimeric species (Fig. S3-10, S3-11, S3-14, S3-15, S3-19, S3-20, S3-24 and S3-25; ESI†). The Kdim values of 5, 6, 7 and 8 at 25 °C in CDCl3 were estimated to be 52, 58, 1100 and 4000 M−1, respectively. The thermodynamic parameters of 5, 6, 7 and 8 were also determined and are summarized in Table 1. Among these, meta-substituted dianilinotripyrrins 7 and 8 exhibited larger Kdim values and large negative ΔG values, while para-substituted dianilinotripyrrins 5 and 6 were less amenable to form the double helices, as compared with non-substituted 4. Fig. 4 displays the space-filling models of the solid-state structures of 7 and 8. In both cases, aromatic stacking interactions are observed in the imine-like pyrrole moieties with average stacking distances of 3.53–3.60 Å for 7 and 3.48–3.52 Å for 8. Additionally, the meta-substituents on the aniline moiety are in close proximity to the central pyrrolic plane to gain a certain thermodynamic stabilization. In the case of 7, however, the 3,5-di-t-butylphenyl groups are slightly tilted in order to have an appropriate arrangement to gain such stabilization, that likely resulted in some entropic losses.
Table 1 Summary of the thermodynamic parameters for 4–8 in CDCl3
|
|
K
dim
|
ΔH kJ mol−1 |
ΔS J mol−1 K−1 |
ΔG298 kJ mol−1 |
|
T = 298 K.
|
|
4
|
2.7 × 102 |
−53.6 |
−134 |
−13.7 |
|
5
|
5.2 × 10 |
−49.1 |
−131 |
−10.0 |
|
6
|
5.8 × 10 |
−47.1 |
−125 |
−9.85 |
|
7
|
1.1 × 103 |
−57.3 |
−133 |
−17.7 |
|
8
|
4.0 × 103 |
−56.1 |
−119 |
−20.6 |
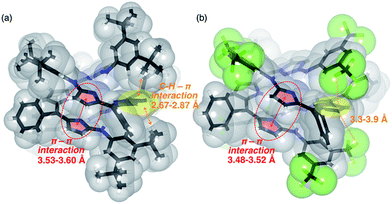 |
| | Fig. 4 Space-filling and stick representations of the solid-state structures of (a) 7 and (b) 8. Solvent molecules included in the unit cell are omitted. Selected intermolecular short-contacts are shown. | |
Different conformations
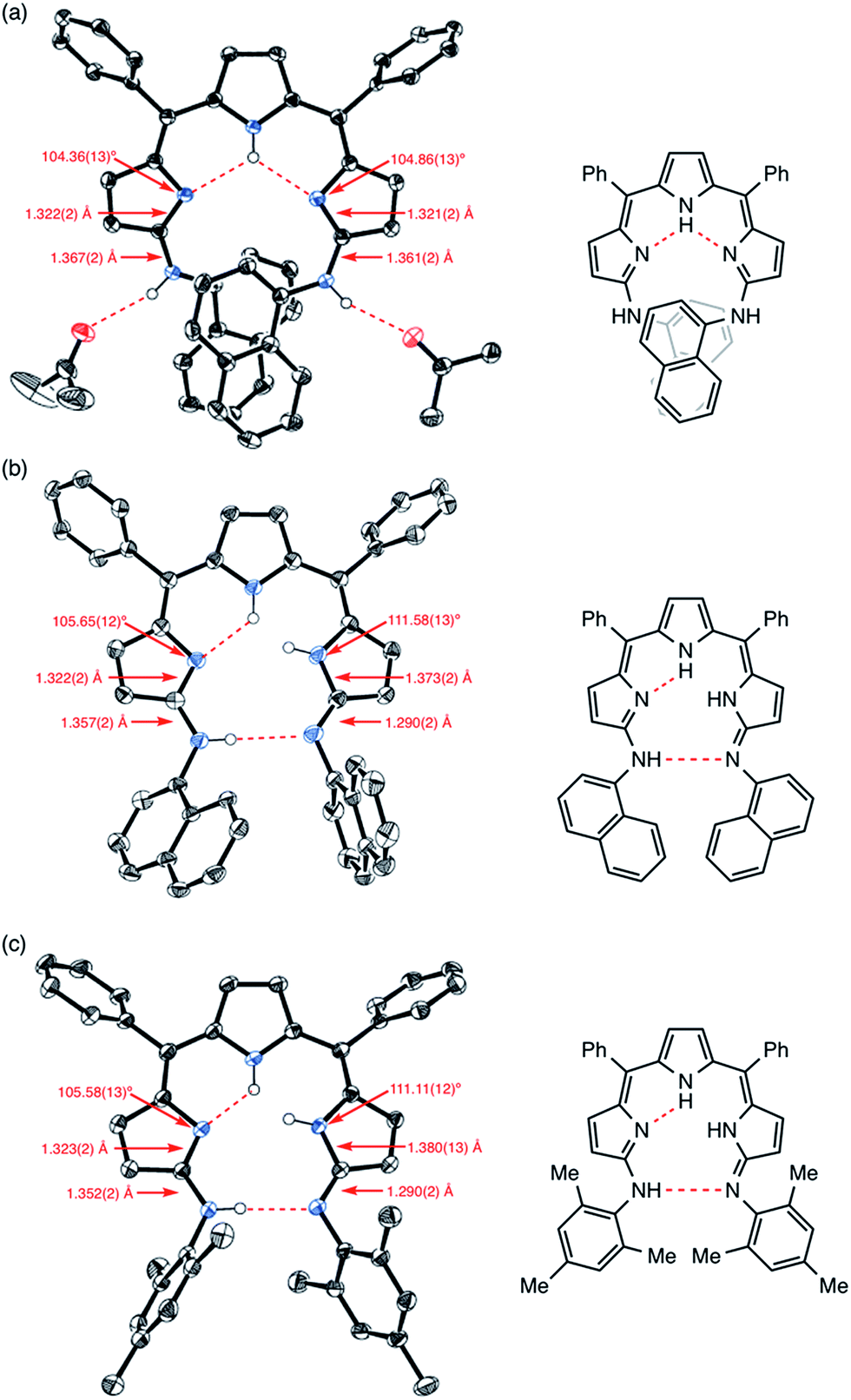
Similar to 4, the crystal structure of 9 obtained from acetone/H2O showed intramolecular and intermolecular hydrogen bonding interactions as shown in Fig. 5a. The intramolecular hydrogen bonding interactions are between the pyrrolic NH and the pyrrolic imine nitrogen atoms, and the intermolecular hydrogen bonding interactions are between the aniline-NH and solvent molecules (acetone). Curiously, the crystal structure of 9 obtained from an aprotic solvent such as a mixture of CH2Cl2 and n-hexane did not show a dimeric structure but displayed a tautomeric structure with different hydrogen-bonding interactions. Judging from the bond lengths and angles, one of the arylamino-substituted imine-type pyrroles was assigned as a different arylimino-substituted amino-type pyrrole tautomer (Fig. 5b). Thus, the overall structure is asymmetric, and two sets of intramolecular hydrogen bonding networks are formed. The same structure was observed in the crystal structure of 10 (Fig. 5c and S5-13–S5-16†). Presumably due to the steric hindrance of the 1-naphthyl and mesityl groups around the aniline-NHs, 9 and 10 were not able to form dimeric structures. In the case of 10, even intermolecular hydrogen bonding with solvent molecules seemed to be prevented in the solid state (Fig. S5-15; ESI†).
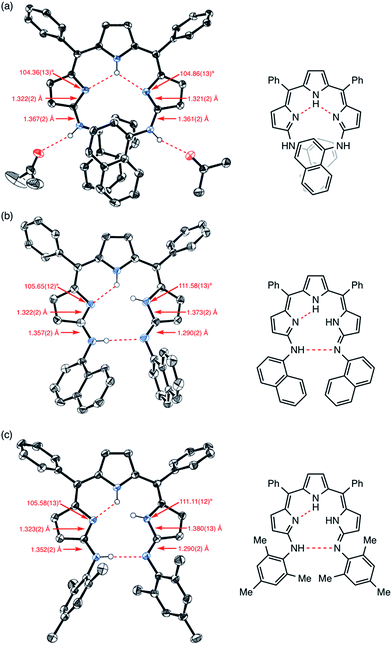 |
| | Fig. 5 X-ray crystal structures of (a) 9 obtained from acetone/H2O, (b) 9 obtained from CH2Cl2/n-hexane and (c) 10 obtained from CH2Cl2/n-hexane. Solvent molecules except for acetone and hydrogen atoms except for NHs are omitted for clarity. The thermal ellipsoids are scaled to 50% probability. | |
The 1H NMR spectrum of 9 in CDCl3 showed broad peaks probably due to the slower tautomerism with regard to the 1H NMR timescale. In contrast, the 1H NMR spectrum of 9 in DMSO-d6 showed sharp peaks featuring three doublets at 6.86, 6.82, and 5.98 ppm due to the β-protons and two singlets at 13.31 and 9.27 ppm due to the tripyrrin-NH and amine-NH, respectively. This signal pattern is similar to that of 4 measured in DMSO-d6 although signals assignable to the naphthyl protons were largely shifted.17
Mixing of the two different strands in solution may give homo-sorted or hetero-sorted dimers. To test this sorting behaviour, to a CDCl3 solution of 8 was added an equimolar amount of 7 or 10. In the case of 7 and 8, a new signal that can be assigned to the hetero-strand [7 + 8], was observed in the 1H NMR spectrum (Fig. S3-40†). Interestingly, the peak intensity of [7 + 8] is larger than that of 7 or 8, indicating a larger association constant of the hetero-strand.18 In contrast, a mixture of 10 and 8 only showed the superposition of the signal sets (Fig. S3-41†).
UV/Vis absorption spectra
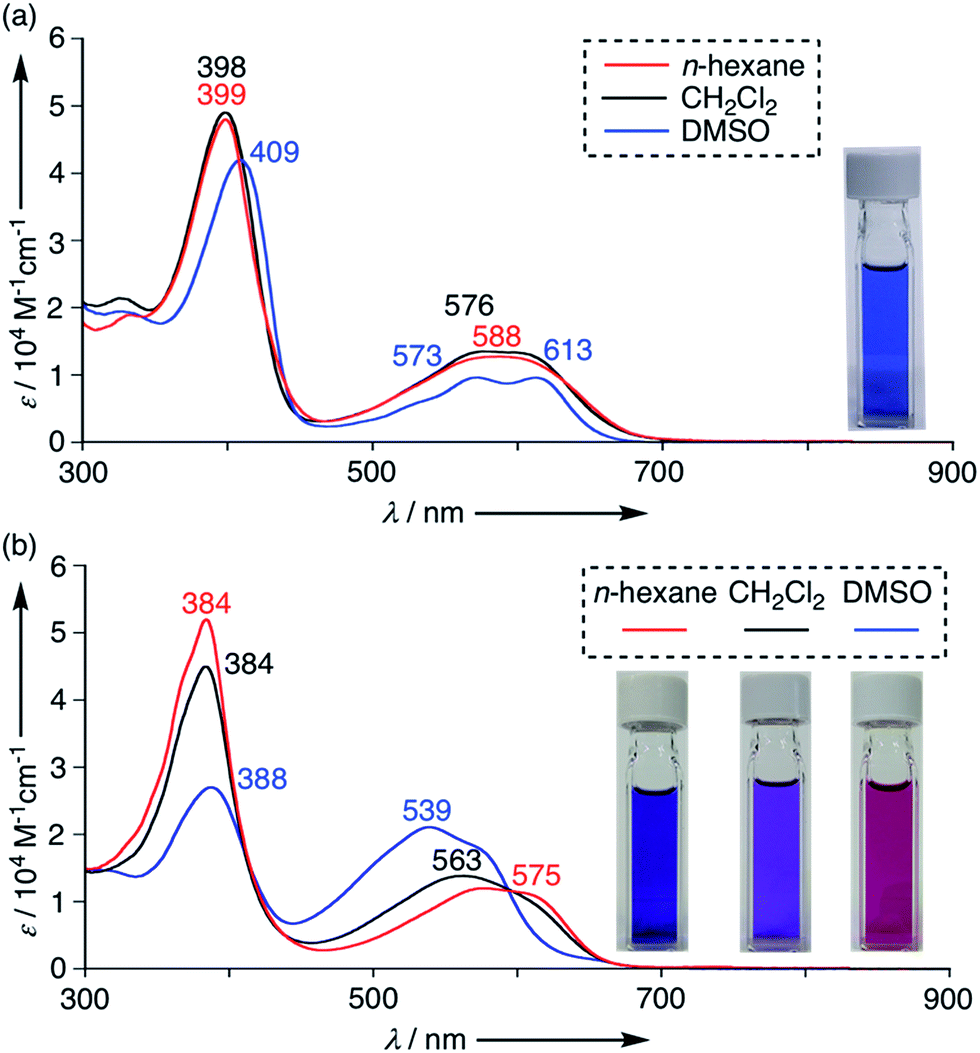
The UV/Vis absorption spectra of 4–11 were measured in various solvents. These tripyrrin derivatives exhibit mainly two absorption bands around 400 nm and 570 nm and the absorption tails reach a deep visible region (∼700 nm) due to the extended π-conjugation. These absorption features resemble those of porphyrinoids and natural chlorophyll analogs,20 thus demonstrating promising applications as functional dyes. Indeed, the solution colour of 4 is vivid blue in CH2Cl2 owing to the large extinction coefficient (ca. 49![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 at 398 nm and 14000 M−1 cm−1 at 576 nm in CH2Cl2). In addition, the absorption spectra of 4 showed a slight shift in polar solvents (Fig. 6a and S6-2; ESI†). Tripyrrin derivatives 5–11 also display similar spectral features in some solvents, while the UV/Vis absorption spectra of 10 are more dramatically changed by solvent effects. In DMSO, 10 displays blue-shifted absorption bands at 539 nm. The degree of such shifts is likely solvent-polarity dependent, thus demonstrating a vivid colour change from blue to purple as shown in Fig. 6b.
000 M−1 cm−1 at 398 nm and 14000 M−1 cm−1 at 576 nm in CH2Cl2). In addition, the absorption spectra of 4 showed a slight shift in polar solvents (Fig. 6a and S6-2; ESI†). Tripyrrin derivatives 5–11 also display similar spectral features in some solvents, while the UV/Vis absorption spectra of 10 are more dramatically changed by solvent effects. In DMSO, 10 displays blue-shifted absorption bands at 539 nm. The degree of such shifts is likely solvent-polarity dependent, thus demonstrating a vivid colour change from blue to purple as shown in Fig. 6b.
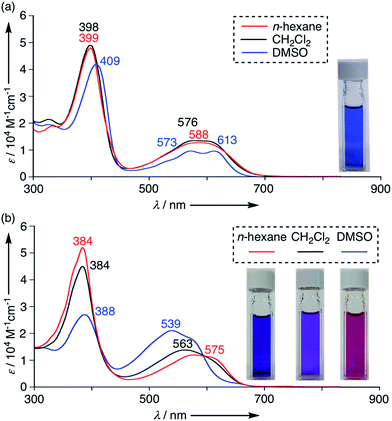 |
| | Fig. 6 UV/Vis absorption spectra of (a) 4 and (b) 10 in n-hexane (red), CH2Cl2 (black) and DMSO (blue). Concentrations of 4 are set at 1.0 × 10−5 M.19 | |
Molecular orbital calculations have revealed that the effects of substituted aniline moieties are more significant in the HOMOs rather than the LUMOs (Fig. S7-1–S7-7; ESI†). Strong CT-like contributions are not evident in these systems. In 9 and 10, the NH tautomers are proved to be the lowest-energy conformers when solvent molecules are not considered (Fig. S7-8 and S7-9; ESI†). Variable probabilities of this form in solution may result in the observed solvent-dependent spectral shifts.
Conclusions
In summary, the nucleophilic substitution reactions of α,α′-dibromotripyrrin 3 with anilines gave dianilinotripyrrins 4–11 in high yields. Dianilinotripyrrins 4–8 form double helix structures in the solid states and in nonpolar and aprotic solvents. The dimerization constants are larger for 7 and 8 bearing 3,5-di-t-butylanilino- and 3,5-bis(trifluoromethyl)anilino-substituents, respectively, in which the dimeric structures are stabilized via the close contacts between the meta-substituents and the central pyrrolic π-plane as well as aromatic stacking interactions. In nonpolar and aprotic solvents, 9 and 10 carrying bulky substituents at the aniline moiety do not form dimers but form different tautomeric structures. These compounds retain π-conjugated networks as evinced by their absorption spectra and π-orbital calculations. Collectively, these α,α′-dianilinotripyrrins may offer a unique platform for novel artificial double helix molecules. Further structural modifications and elongation of the helical structure are actively ongoing in our laboratory.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers (JP26810021, JP18H03910 and JP18K14199). T. T. acknowledges the TOBEMAKI Scholarship Foundation, Murata Science Foundation and Izumi Science and Technology Foundation. M. U. acknowledges a JSPS Fellowship for Young Scientists.
Notes and references
-
(a) M. Albrecht, Chem. Rev., 2001, 101, 3457 CrossRef PubMed;
(b) M. Albrecht, Angew. Chem., Int. Ed., 2005, 44, 6448 CrossRef PubMed;
(c) E. Yashima, K. Maeda and Y. Furusho, Acc. Chem. Res., 2008, 41, 1166 CrossRef PubMed;
(d) D. Haldar and C. Schmuck, Chem. Soc. Rev., 2009, 38, 363 RSC;
(e) Y. Furusho and E. Yashima, Macromol. Rapid Commun., 2011, 32, 136 CrossRef PubMed;
(f) G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933 RSC;
(g) P. Prabhakaran, G. Priya and G. J. Sanjayan, Angew. Chem., Int. Ed., 2012, 51, 4006 CrossRef PubMed;
(h) M. Yamaguchi, M. Shigeno, N. Saito and K. Yamamoto, Chem. Rec., 2014, 14, 15 CrossRef PubMed;
(i) M. Boiocchi and L. Fabbrizzi, Chem. Soc. Rev., 2014, 43, 1835 RSC;
(j) E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752 CrossRef PubMed.
-
(a) J.-M. Lehn, A. Rigault, J. Siegel, J. Harrowfield, B. Chevrier and D. Moras, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2565 CrossRef PubMed;
(b) M. J. Hannon and L. J. Childs, Supramol. Chem., 2004, 16, 7 CrossRef;
(c) M. Albrecht, Top. Curr. Chem., 2005, 248, 105 CrossRef;
(d) H. Miyake and H. Tsukube, Supramol. Chem., 2005, 17, 53 CrossRef.
-
(a) H. Katagiri, T. Miyagawa, Y. Furusho and E. Yashima, Angew. Chem., Int. Ed., 2006, 45, 1741 CrossRef PubMed;
(b) K. Miwa, Y. Furusho and E. Yashima, Nat. Chem., 2010, 2, 444 CrossRef PubMed;
(c) Y. Furusho, K. Miwa, R. Asai and E. Yashima, Chem.–Eur. J., 2011, 17, 13954 CrossRef PubMed;
(d) S. Yamamoto, H. Iida and E. Yashima, Angew. Chem., Int. Ed., 2013, 52, 6849 CrossRef PubMed;
(e) H. Iida, K. Ohmura, R. Noda, S. Iwahana, H. Katagiri, N. Ousaka, T. Hayashi, Y. Hijikata, S. Irle and E. Yashima, Chem.–Asian J., 2017, 12, 927 CrossRef PubMed.
-
(a) Y. Tanaka, H. Katagiri, Y. Furusho and E. Yashima, Angew. Chem., Int. Ed., 2005, 44, 3867 CrossRef PubMed;
(b) M. Ikeda, Y. Tanaka, T. Hasegawa, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2006, 128, 6806 CrossRef PubMed;
(c) T. Maeda, Y. Furusho, S.-i. Sakurai, J. Kumaki, K. Okoshi and E. Yashima, J. Am. Chem. Soc., 2008, 130, 7938 CrossRef PubMed;
(d) H. Ito, Y. Furusho, T. Hasegawa and E. Yashima, J. Am. Chem. Soc., 2008, 130, 14008 CrossRef PubMed;
(e) H. Yamada, K. Maeda and E. Yashima, Chem.–Eur. J., 2009, 15, 6794 CrossRef PubMed;
(f) H. Yamada, Z.-Q. Wu, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2012, 134, 9506 CrossRef PubMed;
(g) W. Makiguchi, J. Tanabe, H. Yamada, H. Iida, D. Taura, N. Ousaka and E. Yashima, Nat. Commun., 2015, 6, 7236 CrossRef PubMed.
-
(a) V. Berl, I. Huc, R. G. Khoury, M. J. Krische and J.-M. Lehn, Nature, 2000, 407, 720 CrossRef PubMed;
(b) V. Berl, I. Huc, R. G. Khoury and J.-M. Lehn, Chem.–Eur. J., 2001, 7, 2810 CrossRef PubMed;
(c) H. Jiang, V. Maurizot and I. Huc, Tetrahedron, 2004, 60, 10029 CrossRef;
(d) C. Dolain, C. Zhan, J.-M. Léger, L. Daniels and I. Huc, J. Am. Chem. Soc., 2005, 127, 2400 CrossRef PubMed;
(e) C. Zhan, J.-M. Léger and I. Huc, Angew. Chem., Int. Ed., 2006, 45, 4625 CrossRef PubMed;
(f) D. Haldar, H. Jiang, J.-M. Léger and I. Huc, Angew. Chem., Int. Ed., 2006, 45, 5483 CrossRef PubMed;
(g) E. Berni, B. Kauffmann, C. Bao, J. Lefeuvre, D. M. Bassani and I. Huc, Chem.–Eur. J., 2007, 13, 8463 CrossRef PubMed;
(h) D. Haldar, H. Jiang, J.-M. Léger and I. Huc, Tetrahedron, 2007, 63, 6322 CrossRef;
(i) Q. Gan, C. Bao, B. Kauffmann, A. Grélard, J. Xiang, S. Liu, I. Huc and H. Jiang, Angew. Chem., Int. Ed., 2008, 47, 1715 CrossRef PubMed;
(j) B. Baptiste, J. Zhu, D. Haldar, B. Kauffmann, J.-M. Léger and I. Huc, Chem.–Asian J., 2010, 5, 1364 Search PubMed;
(k) M. L. Singleton, G. Pirotte, B. Kauffmann, Y. Ferrand and I. Huc, Angew. Chem., Int. Ed., 2014, 53, 13140 CrossRef PubMed;
(l) J. Shang, Q. Gan, S. J. Dawson, F. Rosu, H. Jiang, Y. Ferrand and I. Huc, Org. Lett., 2014, 16, 4992 CrossRef PubMed.
-
(a) J. Li, J. A. Wisner and M. C. Jennings, Org. Lett., 2007, 9, 3267 CrossRef PubMed;
(b) H. Abe, H. Machiguchi, S. Matsumoto and M. Inouye, J. Org. Chem., 2008, 73, 4650 CrossRef PubMed;
(c) H.-B. Wang, B. P. Mudraboyina, J. Li and J. A. Wisner, Chem. Commun., 2010, 46, 7343 RSC;
(d) H.-B. Wang, B. P. Mudraboyina and J. A. Wisner, Chem.–Eur. J., 2012, 18, 1322 CrossRef PubMed;
(e) B. P. Mudraboyina and J. A. Wisner, Chem.–Eur. J., 2012, 18, 14157 CrossRef PubMed.
-
(a) G. Struckmeier, U. Thewalt and J. H. Fuhrhop, J. Am. Chem. Soc., 1976, 98, 278 CrossRef;
(b) W. S. Sheldrick and J. Engel, J. Chem. Soc., Chem. Commun., 1980, 5 RSC;
(c) R. G. Khoury, L. Jaquinod and K. M. Smith, Tetrahedron, 1998, 54, 2339 CrossRef;
(d) M. Bröring, S. Link, C. D. Brandt and E. Cónsul Tejero, Eur. J. Inorg. Chem., 2007, 1661 CrossRef;
(e) T. Hashimoto, T. Nishimura, J. M. Lim, D. Kim and H. Maeda, Chem.–Eur. J., 2010, 16, 11653 CrossRef PubMed;
(f) H. Maeda, T. Nishimura, R. Akuta, K. Takaishi, M. Uchiyama and A. Muranaka, Chem. Sci., 2013, 4, 1204 RSC;
(g) H. Maeda, T. Nishimura, A. Tsujii, K. Takaishi, M. Uchiyama and A. Muranaka, Chem. Lett., 2014, 43, 1078 CrossRef.
- G. R. Kiel, S. C. Patel, P. W. Smith, D. S. Levine and T. Don Tilley, J. Am. Chem. Soc., 2017, 139, 18456 CrossRef PubMed.
-
(a) M. Bröring and C. D. Brandt, Chem. Commun., 2001, 499 RSC;
(b) M. Bröring and C. D. Brandt, Chem. Commun., 2003, 2156 RSC;
(c) M. Bröring, C. D. Brandt and S. Stellwag, Chem. Commun., 2003, 2344 RSC;
(d) M. Bröring, S. Prikhodovski and C. D. Brandt, Inorg. Chim. Acta, 2004, 357, 1733 CrossRef;
(e) M. Bröring, S. Prikhodovski, C. D. Brandt and E. Cónsul Tejero, Chem.–Eur. J., 2007, 13, 396 CrossRef PubMed;
(f) M. Bröring, S. Prikhodovski, E. Cónsul Tejero and S. Köhler, Eur. J. Inorg. Chem., 2007, 1010 CrossRef;
(g) R. Gautam, J. J. Loughrey, A. V. Astashkin, J. Shearer and E. Tomat, Angew. Chem., Int. Ed., 2015, 54, 14894 CrossRef PubMed.
-
(a) A. Jaumà, J.-A. Farrera and J. M. Ribó, Monatsh. Chem., 1996, 127, 935 CrossRef;
(b) S. P. Rath, M. M. Olmstead, L. Latos-Grażyński and A. L. Balch, J. Am. Chem. Soc., 2003, 125, 12678 CrossRef PubMed;
(c) H. Furuta, H. Maeda and A. Osuka, Inorg. Chem. Commun., 2003, 6, 162 CrossRef;
(d) S. D. Roth, T. Shkindel and D. A. Lightner, Tetrahedron, 2007, 63, 11030 CrossRef PubMed;
(e) S. K. Dey, S. Datta and D. A. Lightner, Monatsh. Chem., 2009, 140, 1171 CrossRef;
(f) S. Bahnmuller, J. Plotzitzka, D. Baabe, B. Cordes, D. Menzel, K. Schartz, P. Schweyen, R. Wicht and M. Bröring, Eur. J. Inorg. Chem., 2016, 4761 CrossRef.
- M. Bröring, S. Prikhodovski and E. Cónsul Tejero, Chem. Commun., 2007, 876 RSC.
- M. Gałęzowski, J. Jaźwiński, J. P. Lewtak and D. T. Gryko, J. Org. Chem., 2009, 74, 5610 CrossRef PubMed.
-
(a) T. Rohand, M. Baruah, W. Qin, N. Boens and W. Dehaen, Chem. Commun., 2006, 266 RSC;
(b) A. R. Sekhar, M. A. Kaloo and J. Sankar, Chem.–Asian J., 2014, 9, 2422 CrossRef PubMed;
(c) X. Zhou, Q. Wu, Y. Feng, Y. Yu, C. Yu, E. Hao, Y. Wei, X. Mu and L. Jiao, Chem.–Asian J., 2015, 10, 1979 CrossRef PubMed;
(d) T. Satoh, M. Minoura, H. Nakano, K. Furukawa and Y. Matano, Angew. Chem., Int. Ed., 2016, 55, 2235 CrossRef PubMed.
- For the characterization of α-anilino-α′-bromotripyrrin S1, see ESI.† We assume that two equivalents of aniline are necessary for one substitution because an equimolar amount of hydrogen bromide is generated.
- The solid-state structures of anilinotripyrrin 5 for both the monomer and dimer were revealed by X-ray crystallographic analysis (Fig. S5-4 and S5-5; ESI†).
- The helical pitch was defined as the distance between the two nitrogen atoms of the aniline segments.
- Despite the different conjugation modes, the NH tautomerism does not cause a significant change in their electronic state in view
of the MO aptitude (see Fig. S7 and S6; ESI†).
- The ratio of 7:8 :[7 + 8] was roughly estimated to be 1:1.4:7.7 by considering the 1H NMR peak intensities and symmetry.
- In some solvents, the absorption intensity does not follow the Beer–Lambert law, implying a decent concentration effect even in 10−5 M order.
-
(a)
Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, vol. 28 Search PubMed;
(b) L.-L. Li and E. W.-G. Diau, Chem. Soc. Rev., 2013, 42, 291 RSC;
(c) M. Taniguchi and J. S. Lindsey, Chem. Rev., 2017, 117, 344 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Atsuhiro
Osuka
* and
Atsuhiro
Osuka