
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlass wool: a novel support for heterogeneous catalysis†
Ayda
Elhage
 ,
Bowen
Wang
,
Nancy
Marina
,
M. Luisa
Marin‡
,
Menandro
Cruz
,
Anabel E.
Lanterna
* and
Juan C.
Scaiano
*
,
Bowen
Wang
,
Nancy
Marina
,
M. Luisa
Marin‡
,
Menandro
Cruz
,
Anabel E.
Lanterna
* and
Juan C.
Scaiano
*
Department of Chemistry and Biomolecular Science, Centre for Advanced Materials Research (CAMaR), University of Ottawa, 10 Marie Curie, Ottawa, ON K1N 6N5, Canada. E-mail: titoscaiano@mac.com; anabel.lanterna@icloud.com
First published on 12th July 2018
Abstract
Heterogeneous catalysis presents significant advantages over homogeneous catalysis such as ease of separation and reuse of the catalyst. Here we show that a very inexpensive, manageable and widely available material – glass wool – can act as a catalyst support for a number of different reactions. Different metal and metal oxide nanoparticles, based on Pd, Co, Cu, Au and Ru, were deposited on glass wool and used as heterogeneous catalysts for a variety of thermal and photochemical organic reactions including reductive de-halogenation of aryl halides, reduction of nitrobenzene, Csp3–Csp3 couplings, N–C heterocycloadditions (click chemistry) and Csp–Csp2 couplings (Sonogashira couplings). The use of glass wool as a catalyst support for important organic reactions, particularly C–C couplings, opens the opportunity to develop economical heterogeneous catalysts with excellent potential for flow photo-chemistry application.
Introduction
Heterogeneous catalytic processes have several advantages over the equivalent homogeneous ones, specifically, ease of catalyst separation, minimal product contamination1 and the distinct possibility of reuse.2 Quite frequently these catalysts are in the form of nanometric or micrometric powders decorated with active nanostructures such as metal or metal oxides.3,4 Although they are easy to separate after batch reactions, they may not be ideal for flow chemistry, a strategy that enables easy scale up of reactions.5 In catalysis, the term support is used with a wide range of meanings, from the passive support that simply restricts the mobility of the active catalyst, to cases where the support is an integral part of the catalyst and its performance; frequently, this is the case with semiconductors such as TiO2.4,6,7 Further, in the case of photocatalysis the absorption and scattering properties of the support are also important. With this in mind, we explored the possibility of using fibrous materials as catalyst supports, as they would be easy to separate after batch reactions and have the potential for flow chemistry applications where a static catalyst would act on flowing solutions, also facilitating photocatalytic processes. Glass wool (GW) is inexpensive, readily available with a variety of surface properties and easy to modify to provide physical or chemical affinity towards many catalytic materials. Glass wool is widely employed for thermal and noise insulation in homes, appliances and instrumentation. In chemistry, GW is commonly used as a filter, packing material in GC columns, purge trap and adsorbent bed,8 where it is normally regarded as a fairly robust inert material towards many chemicals, showing advantages including good pH tolerance. There are some examples where GW is used as an attachment surface for applications in biology, including bactericidal studies,9,10 but its applications in organic chemistry are virtually unexplored. The use of glass fibers and cloths for catalysis was reviewed in 2002;11 their applications at the time dealt with gas and liquid processes, mostly oxidations, but interestingly it was recognized that these materials could avoid the technological problems and limitations associated with handling and separation of powders. While this contribution was in preparation, Barelko et al.12 published a review with the significant technological advances achieved during the last 15 years. Yet, with the exception of a nitro-compound reduction, no applications to organic chemistry were reported, in particular, not a single example on C–C bond formation, a key reaction in organic synthesis and drug-development applications. Some concerns about the inert nature of the glass wool were noted many years ago by Hayes and Macdonald, but no further studies were found in the literature.13 Apart from these limited examples in catalysis, where high temperature14 or redox harsh conditions are usually employed,12 it is hard to find GW uses other than the ones mentioned above,8 largely excluding organic catalysis. There are few studies focused on depositing metals on glass, mostly dedicated to redox reactions,15–17 however to the best of our knowledge, no complex organic reactions – such as C–C couplings – have been explored utilizing glass as a catalyst support.In this contribution, we report a number of metal and metal oxide nanostructures supported on commercially available – sometimes modified – GW and how they perform on a variety of catalytic processes. We explored individual reactions emphasizing the use of classic reactions with diverse novel materials. Our work on glass wool was initiated with the assumption that this support would be of the passive type. Notice that the use of these materials extremely facilitates the catalyst separation from the reaction vessel, and thus a regular pair of tweezers can be used to remove the GW as shown in Fig. 1. Further purification by filtration is straightforward.
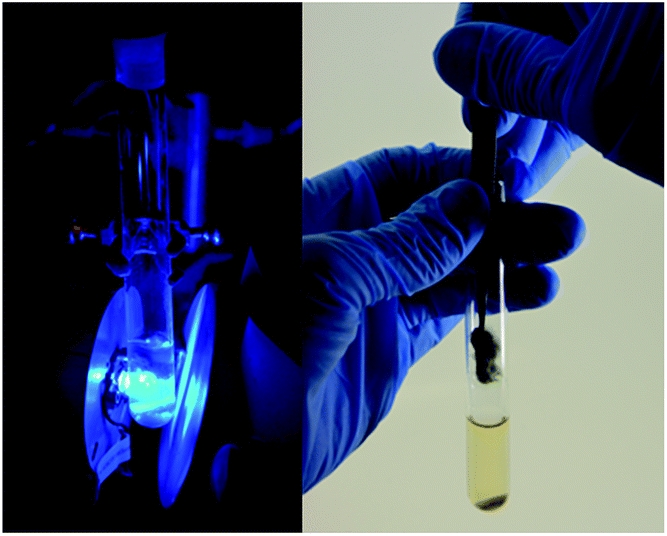 | ||
| Fig. 1 Picture showing the reaction setup used under LED irradiation and continuous stirring (left), and the easy removal of the GW material from the reaction vessel utilizing a pair of tweezers (right). | ||
Results and discussion
Materials characterization
Two different types of commercial glass wool were used in this work: non-silanized (NGW) and silanized (SGW) glass wool. They were decorated with different metal and metal oxide nanoparticles, namely Au, Pd, Ru, Co and Cu, utilizing photoinduced and chemical methods as described in the ESI.† The optimal synthetic method was chosen based on the observed catalytic activity and stability of the new materials prepared. Thus, GWs were subjected to different pre-treatments before incorporating the metal/metal oxide nanoparticles in order to efficiently attach the metal to the GW surface. The experimental section summarizes several different trials and the rationale behind the selection of the preferred pre-treatment method. In particular, APTES treatment was selected to add a source of amino groups on the glass surface. These groups not only help to attach metal nanoparticles to the glass surface but can also be used as reducing agents for in situ formation of metal nanoparticles under dark conditions (see ESI†).18 In general, non-silanized and silanized GW show no difference in their catalytic activity. In particular, NGW is preferred in cases were modification with APTES is required. When only acid pre-treatment is used, SGW shows better reactivity (vide infra). Fig. 2 shows the materials before and after different surface modifications. | ||
| Fig. 2 Pictures of different materials used in this work: (a) pristine SGW, (b) pristine NGW, (c) HCl-treated SGW, d) APTES-functionalized NGW (NGW*), (e) Pd@SGW, (f) Cu@NGW, (g) Co@SGW, (h) Ru@NGW* and (i) Au@NGW*. | ||
The microscopic analysis of the GW samples was also performed. As shown in Fig. 3a, the commercial GW is constituted by glass fibers of about 10 μm diameter. The crystalline structures present together with the fibers correspond to NaCl as determined by a careful EDS analysis of different areas of the image (Fig. 3b and c). Upon derivatization, the presence of metal or metal oxides nanoparticles can be easily distinguished by SEM. Fig. 3d and e show the surface of a glass fiber covered by CoxOy NPs and the corresponding EDS spectra. The same characterization was performed for each of the different metal-derivatized GWs as shown in Fig. S5–S8.†
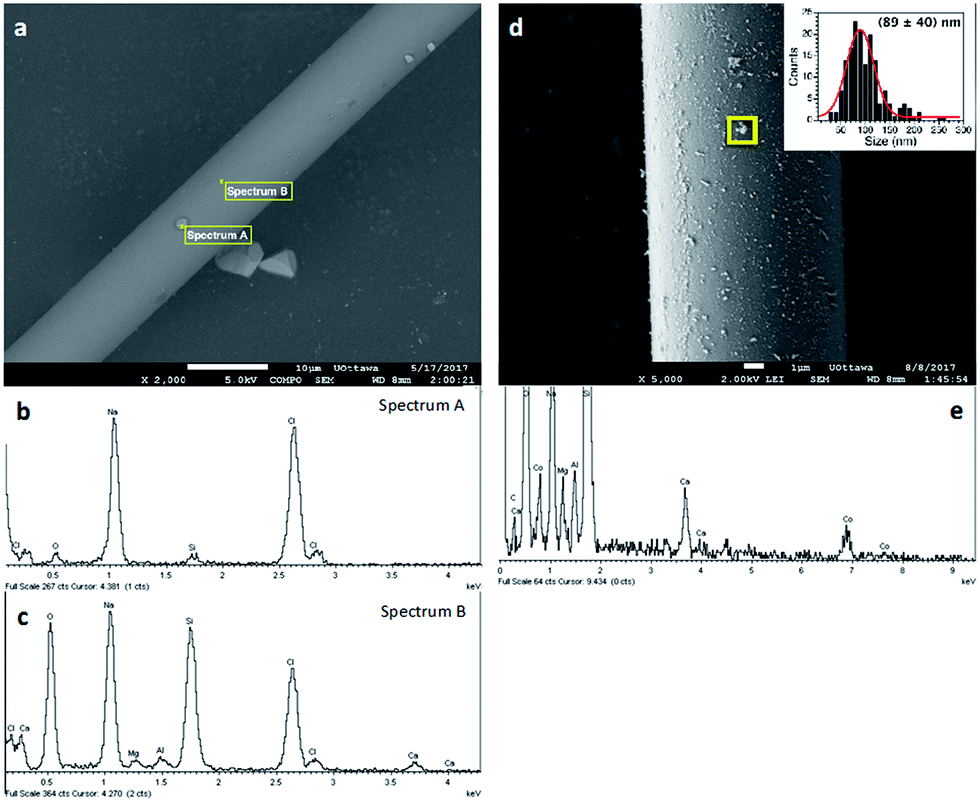 | ||
| Fig. 3 SEM images (top) and EDS spectra (bottom) in the marked areas of untreated SGW (a–c) and of Co@SGW (d and e). Notice that the particulates on the SGW fiber are mostly composed of NaCl. Agglomerated particles were not considered to determine particle size distribution. | ||
The materials were further characterized by ICP-OES, diffuse reflectance (DR) and X-ray photoelectron spectroscopy (XPS). The amount of metal loaded on each material as well as particle size distribution and calculated surface area are reported in Table 1. Fig. 4 (left) shows the absorption profile of SGW before and after Co-derivatization, where slight changes on the absorption of Co@SGW can account for the contribution of CoxOy species in the material. The different absorption profiles of other metal-derivatized GWs are presented in Fig. S9–S12,† where Pd, Au and Ru-derivatized GW show higher absorption in the visible region compared to the non-derivatized GW. XPS deconvolution analyses were performed for each material in order to determine the oxidation state of each metal. Fig. 4 (right) shows the deconvoluted Co 2p HR-XPS spectrum obtained for Co@SGW, whereas the rest of the materials are described in Fig. S13–S16.† Characteristic peaks for Co 2p were fitted using the spin–orbit split constituted by Co 2p3/2 (781.2 eV) and Co 2p1/2 (797.7 eV) separated by ∼16.5 eV. The corresponding Co(II) satellites (787.4 and 803.5 eV) further prove the presence of Co oxides (CoxOy).19 XPS analysis of the Pd@NGW* exposes a Pd 3d core-level spectrum deconvoluted by using two spin–orbit split Pd 3d5/2 and Pd 3d3/2 components centered at 337 eV and 342.4 eV and separated by ∼5.4 eV, which more likely correspond to PdO.4 In contrast, more reduced Pd species are found in the Pd@SGW material with spin–orbit components at 335 eV and 340.4 eV. The XPS fitting for the Ru 3p in Ru@NGW reveals the presence of two main peaks at 463.1 and 485.5 eV, which according to the literature correspond to RuO2,20 although other reports suggest a mixture between metallic Ru(0), RuO2 and RuO2·xH2O.21,22 In the case of Cu@NGW, the two spin–orbit split Cu 2p3/2 (934.0 eV) and Cu 2p1/2 (953.7 eV) components (Δ ∼ 19.7 eV) and the noticeable Cu(II) satellites indicate that CuO23 is present in this material. Finally, for Au@NGW* the peaks corresponding to Au 4f7/2 (84.5 eV) and Au 4f5/2 (88.0 eV) suggest the presence of Au(0). The additional features at lower BE (∼82 eV) can account for low-coordinated atoms on the Au surface.24
| Materiala | Metal amountb (wt%) | Particle sizec (nm) | Surface aread (cm2 g−1) |
|---|---|---|---|
| a APTES treated GW are denoted with *. b By ICP-OES analysis. None of the metals mentioned were detected on NGW*, pristine NGW or pristine SGW. c Particle size distributions were calculated without considering agglomerated particles. d Metal surface per gram of glass wool; considering that the mean diameter of the GW fibers used is 10 μm, the GW (d = 2.2 g cm−3) surface area is approximated to 1820 cm2 g−1. | |||
| Pd@SGW | 0.16 ± 0.03 | 19 ± 8 | 425 |
| Pd@NGW* | 0.54 ± 0.05 | 22 ± 9 | 1184 |
| Co@SGW | 0.070 ± 0.005 | 89 ± 40 | 53 |
| Ru@NGW* | 0.030 ± 0.001 | 63 ± 30 | 23 |
| Cu@NGW* | 1.1 ± 0.2 | 100 ± 60 | 737 |
| Au@NGW* | 0.9 ± 0.2 | 23 ± 10 | 1215 |
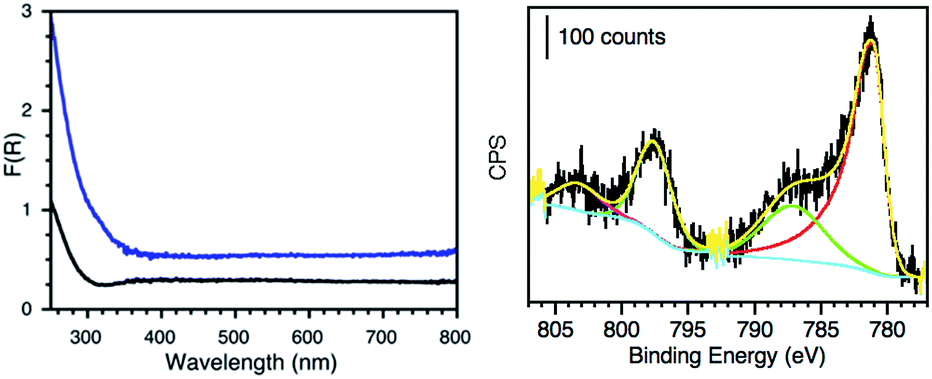 | ||
| Fig. 4 Left: diffuse reflectance spectrum of HCl-treated SGW (black) and Co@SGW (blue). Right: deconvoluted Co 2p HR-XPS spectrum of the Co@SGW catalyst. | ||
Catalytic activity
In order to determine if these new materials would perform as catalysts we selected different reactions based on some of our previous reports.4,6,7,25Scheme 1 summarizes the reactions tested that were successfully catalyzed utilizing the various new materials. | ||
| Scheme 1 Scope of reactions tested with the modified glass wool composites. Only the reactions that were successfully catalyzed are listed here. | ||
Conditions explored for each reaction vary from thermal control to photo-induced catalysis. Table 2 shows a summary of the reactivity of different GW-based materials toward different organic transformation, where the last column shows the table number and detailed entry for relevant examples. To discuss their activity we divided the following sections according to the organic transformation under study.
| Entry | Materiala | Reaction | % Yield | Table (entry) | |
|---|---|---|---|---|---|
| a The star (*) indicates APTES-treated materials. | |||||
| i | Co@SGW | (A) Reductive de-halogenation | >99 | 3 (i, v) | |
| ii | Pd@SGW | >99 | 3 (ii, vi) | ||
| iii | Ru@SGW* (NGW*) | (B) Reduction of nitrobenzene to aniline | 71 | 4 (ii, iv) | |
| iv | Au@NGW* | (B′) Reduction of nitrobenzene to azobenzene | 72 | 5 (ii) | |
| v | Au@NGW* | (C) C–C coupling (sp3–sp3) | 80 | 6 (ii) | |
| vi | Cu@NGW (NGW*) | (D) N–C heterocycloaddition | 92 | 7 (i) | |
| vii | Pd@NGW* (SGW*) | (E) C–C coupling (sp–sp2) (Sonogashira) | 90 | 8 (i) | |
Reductive de-halogenation of aryl halides
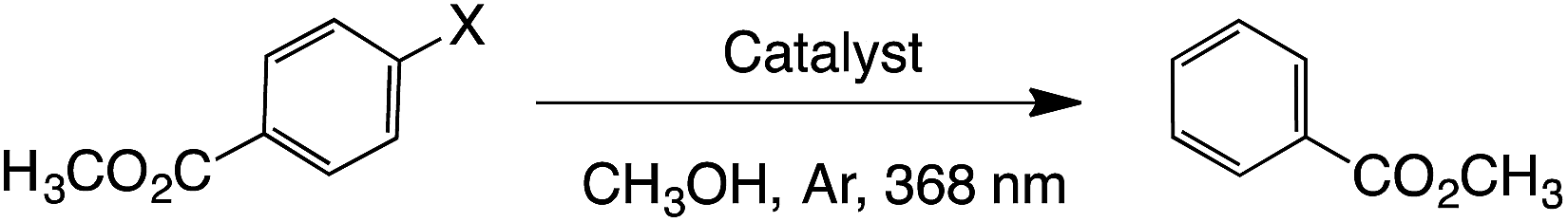
De-halogenation processes are usually dictated by the C–halogen bond strength; thus, C–Cl bonds are harder to break than C–Br bonds and C–I bonds, which is usually reflected under harsher reaction conditions to achieve de-halogenation.26,27 Recent reports on dehalogenation processes involved the use of either high pressure conditions (H2, 30 bar),28 or toxic and expensive iridium complexes.29 Here we performed photodehalogenation of methyl 4-halogenobenzoates catalyzed by GW-based materials, particularly, Pd@SGW, Co@SGW and SGW. Both Pd- and Co-based NPs are known to work as photocatalysts under UV-visible light.30–33The results summarized in Table 3 show that the reaction in the presence of Co@SGW proceeds under irradiation and can be accelerated under an inert atmosphere. Pd@SGW and SGW show the same reactivity as Co@SGW when methyl 4-iodobenzoate is used. As expected, the reaction slows down moving from iodide to bromide and to chloride reagents. This tendency is more evident with catalysts such as Pd@SGW and SGW, whereas Co@SGW has shown exceptional yields, being able to reduce methyl 4-chlorobenzoate within 3 h (entry v, see ESI† for kinetic analysis). Additionally, Co@SGW showed great reusability; thus, after 3 catalytic cycles >99% yield of reductive compound was achieved within 4 h of irradiation of methyl 4-chlorobenzoate (Table S1†).
|
|
||||
|---|---|---|---|---|
| Xb (1) | Catalyst | Time (h) | % Yieldc | |
| a Reaction conditions: 0.2 mmol of substrate 1, 0.36 mmol of K2CO3, 5 mL methanol, and 60 mg catalyst under Ar. b When X = I, yield of 99% is reached in the presence of Co@SGW (or SGW) in 15 min of irradiation under Ar or 1 h under air. Only 15% yield was detected after 24 h of reaction in the dark at 47 °C. c Yields were determined by GC-FID using t-stilbene as the external standard. | ||||
| i | Br– | Co@SGW | 2 | >99 |
| ii | Br– | Pd@SGW | 2 | >99 |
| iii | Br– | SGW | 5 | >99 |
| ix | Br– | None | 5 | 98 |
| v | Cl– | Co@SGW | 3 | >99 |
| vi | Cl– | Pd@SGW | 20 | >99 |
| vii | Cl– | SGW | 24 | 37 |
| viii | Cl– | None | 24 | 36 |
Reduction of nitro compounds
The photoreduction of nitrocompounds has been studied as a method to synthesize aniline derivatives under mild conditions.34,35 Based on our previous work18 we decided to test the ability of Ru- and Au-derivatized GW as catalysts for the reduction of nitrobenzene to produce aniline. We used the optimized conditions previously reported for this type of organic transformation18,36 (see the Experimental section). Table 4 summarizes the results of the nitrobenzene reduction by Ru-derivatized GWs. We noticed that using NGW* or SGW* as supports shows no difference in the catalytic performance of the Ru catalyst. It is worth mentioning that Au-derivatized GW showed no catalytic activity under the same experimental conditions. The reaction kinetics are depicted in Fig. S3.†|
|
|||
|---|---|---|---|
| Catalyst | Time (h) | % Yieldb | |
| a Reaction conditions: 25 mg of catalyst, 1.5 mmol nitrobenzene, 6 eq. hydrazine, and 5 mL ethanol. b Yields were determined by GC-FID using 1,3,5-trimethoxybenzene as an internal standard. c After 4 days 85% of the desired product was detected. d After 4 days 79% of the desired product was detected. | |||
| i | Ru@SGW* | 24 | 28 |
| ii | Ru@SGW* | 64 | 71c |
| iii | Ru@NGW* | 24 | 28 |
| iv | Ru@NGW* | 64 | 66d |
| v | NGW* | 24 | ND |
| vi | None | 24 | ND |

The formation of azo compounds as products of this reaction is also possible. However, their formation under illumination is rarely found as the reductive environment forces the reaction toward the corresponding amine.37 There are just a few examples where the azo-compounds form under illumination of AuNP.36,38,39 Here we show that excitation of AuNPs deposited on GW can also catalyze this reaction under green light irradiation for 24 h with moderate yields (Table 5). Briefly, the catalyst was mixed together with the nitrocompound in i-propanol under an inert atmosphere (Ar) in the presence of KOH and irradiated with 532 nm LEDs. The formation of the azocompound was monitored by UV-Vis spectroscopy (Fig. S4†) following the azobenzene absorption band at ∼350 nm. As expected when visible light is utilized only trans azobenzene is detected as the reaction product.40
|
|
||||
|---|---|---|---|---|
| Catalyst | Time (h) | % Convb | % Yieldb | |
| a Reaction conditions: 0.3 mmol of substrate, 0.03 mmol of KOH, 5 mL i-propanol, and 60 mg catalyst under Ar. b Conversion and yields were determined by UV-Vis spectroscopy. c In the dark at 85 °C. d Under air. | ||||
| i | Au@NGW* | 24 | 68 | 65 |
| ii | Au@NGW* | 48 | 73 | 72 |
| iii | Au@NGW*c | 24 | 40 | 35 |
| iv | Au@NGW*d | 24 | ND | ND |
| v | NGW* | 24 | ND | ND |
| vi | None | 24 | ND | ND |
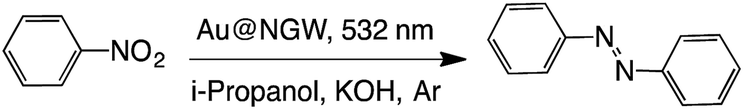
Combining the data presented in Tables 3 and 4 it becomes clear that GW-based catalysts offer an excellent opportunity of product and selectivity control by tuning the metal, its loading and the type of activation used (i.e. thermal or photochemical activation).
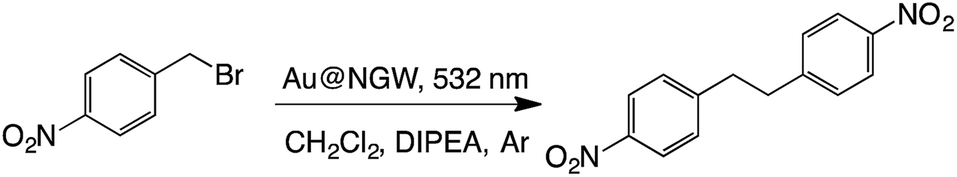
C–C coupling (sp3–sp3)
The reductive dimerization of benzyl bromides can be photoinduced using supported AuNPs and green light excitation.6 This reaction is known as a radical reaction in which light intensity can lead the selectivity towards the dimer product, and thus, higher intensity yields more dimer. Much to our surprise the same reaction proceeds with excellent yields if AuNPs are deposited on GW (Table 6). It is worth highlighting that a noticeable product yield is obtained in the absence of catalyst under these irradiation conditions. These have been previously reported as a contribution from the UV contamination of the green light source.6 The material also shows reasonable reusability (see Table S1†).|
|
||||
|---|---|---|---|---|
| Catalyst | Time (h) | % Convb | % Yieldc | |
| a Reaction conditions: 0.1 mmol of substrate, 2 eq. of DIPEA, and 30 mg of catalyst in 4 mL of CH2Cl2. b By-product: 4-nitrotoluene. c Yields and conversions were calculated by 1H-NMR using caffeine as the external standard. d Dark reaction at 39 °C. e From ref. 6. | ||||
| i | Au@NGW* | 5 | 77 | 64 |
| ii | Au@NGW* | 7 | 100 | 80 |
| iii | Au@NGW*d | 48 | ND | ND |
| iv | NGW* | 24 | 36 | 26 |
| v | Nonee | 5 | — | 25 |
| vi | Nonee | 5 | — | ND |
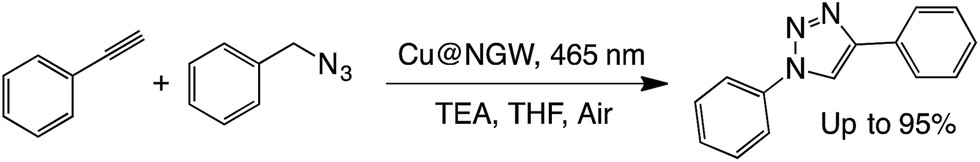
N–C heterocycloaddition (click reaction).
We have recently reported the photocatalyzed click reaction using supported CuxO nanoparticles on TiO2 and Nb2O5.7 The reaction can proceed within 6 h under both UV and visible light. The unexpected reactivity of the material under visible light suggests that the CuxO particles play an important light harvesting role. With this in mind, we decided to test the CuxO activity when deposited on GW. The efficiency of the catalyst was compared to the reactivity of a commercial Cu@charcoal catalyst, which is one of the preferred catalysts to perform click chemistry under thermal conditions. As seen in Table 7, the new material showed great reactivity and high efficiency under both visible light irradiation or dark conditions at 55 °C (the same temperature as reached upon illumination). Unfortunately, the catalyst showed poor reusability, probably due to the visible leaching of copper species into the solution.|
|
||||
|---|---|---|---|---|
| Catalyst | Condition | Time (h) | % Yieldb,c | |
a Reaction conditions: 15 mg of catalyst, azide/alkyne/TEA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1), 6 h under air.
b Yields were calculated by 1H-NMR analysis in CDCl3 using caffeine as the external standard.
c Values between brackets are yields obtained after 6 h of reaction using supported Cu@NGW*.
d Mixture of two isomers. :1:1), 6 h under air.
b Yields were calculated by 1H-NMR analysis in CDCl3 using caffeine as the external standard.
c Values between brackets are yields obtained after 6 h of reaction using supported Cu@NGW*.
d Mixture of two isomers.
|
||||
| i | Cu@NGW | 465 nm, 55 °C | 6 | 92 (95) |
| ii | Cu@NGW | Dark, 55 °C | 6 | 93 (94) |
| iii | Cu@NGW | Dark, RT | 6 | 8 (25) |
| iv | Cu@NGW | Dark, RT | 24 | 75 |
| iv | Cu@charcoal | Dark, 55 °C | 6 | 21 |
| iv | Cu@charcoal | Dark, 55 °C | 24 | 73 |
| v | NGW | Dark, 55 °C | 6 | ND |
| vi | NGW | Dark, 55 °C | 24 | 2d |
C–C coupling (sp–sp2) (Sonogashira)
One of our recent reports demonstrated that PdNP-decorated materials such as TiO2, nanodiamonds and Nb2O5 can selectively catalyze the cross-coupling of iodobenzene and phenylacetylene under both reflux and visible light irradiation.32 Here we show that the same reaction can be catalyzed by the new Pd@SGW* and Pd@NGW* catalysts and selectively lead to the cross-coupling products showing similar activity. Briefly, the selective cross coupling reaction of iodobenzene and phenylacetylene has been studied with methanol as solvent, K2CO3 as base and supported PdNPs on the activated and/or functionalized surface of GW (Table 8). Control reactions in the presence of GW and in the absence of catalyst did not show any product after 24 h. The photocatalyst can be reused at least two times with excellent performance (see Table S1†), showing the same reactivity as the previously reported materials.32|
|
||||
|---|---|---|---|---|
| Catalyst | Time (h) | % Conv | % Yieldb | |
| a Reactions conditions: 1 eq. of iodobenzene, 1.3 eq. of phenylacetylene, 2 eq. of K2CO3, 4 mL methanol, and 60 mg Pd@NGW*. b Yields were determined by GC-FID using t-butylbenzene as an external standard. c Under dark conditions, the reaction was run at 42 °C. Values between brackets are yields obtained after 5 h of reaction using supported Pd@SGW. | ||||
| i | Pd@NGW* | 5 | >99(>99) | 90 (88) |
| iic | Pd@NGW* | 5 | 0 | ND |
| ii | NGW* | 24 | 0 | ND |
| iv | None | 24 | 0 | ND |
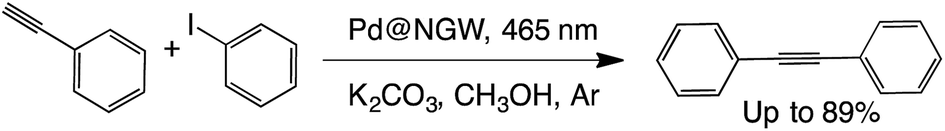
Conclusions
We show the versatility of the use of glass wool as a very inexpensive, widely available and easily handled material for heterogeneous catalysis. We demonstrate that despite the different nature of the glass wool utilized, the efficacies of at least two commercial materials are similar. We have explored different methods to activate the glass surface in order to better anchor the catalytic species to the material. Also, low surface loadings (ranging from 1 down to 0.07 wt%) with different types of metal or metal oxide nanoparticles were tested, to illustrate how easy and reliable these materials are to use. The materials excel in both thermal and light-induced catalysis in a variety of different organic transformations ranging from reductions to C–C couplings and cyclizations, showing adaptability to different reaction media and conditions. Also important, the material can be easily separated from the reaction mixture eliminating tedious workup. Notice that optimization of the metal loadings could indeed open the door for more exploratory studies of these materials. This is a first effort on the use of GW as a catalytic support for fine organic reactions and more complex and interesting systems can be developed from here. We envision this as a first step towards an easy way to recover heterogeneous catalysts, and to develop catalysts with great flow chemistry potential.Experimental
General
Unless otherwise specified, all chemicals were purchased from Sigma-Aldrich or Fisher Scientific and used without further purification. Particularly, the following glass wools were used: non-treated GW (Aldrich catalog #: 20384) and silanized GW (Aldrich catalog #: 20411). Irgacure-2959 (I-2959) was purchased from Ciba.Scanning electron microscopy (SEM) images were obtained using a JEOL JSM-1600 SE microscope working at an accelerating voltage of 2 kV. X-ray photoelectron spectroscopy (XPS) was performed on a Kratos analytical model Axis Ultra DLD, using monochromatic aluminum Kα X-rays at 140 W. XPS data were analyzed using CasaXPS software, Version 2.3.15. All spectra were calibrated at the C 1s signal at 284.8 eV and fittings obtained using a Gaussian 30% Laurentian and a Shirley baseline. The amount of metal loaded onto the materials was determined by Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES), using an Agilent Vista Pro ICP Emission Spectrometer. Approximately 10 mg portions were accurately weighed in triplicate and digested with aqua regia. Solutions were further diluted and measured by ICP-OES. The following emission lines were used for quantification when applicable: Pd 229.65 nm, Au 267.59 nm, Cu 327.00 nm, Co 228.00 nm, and Ru 245.55 nm. Diffuse reflectance measurements were carried out using an Agilent Cary 7000 UV-Vis-NIR Universal Measurement Spectrophotometer coupled with an Agilent praying Mantis accessory. UV-Vis spectroscopy was carried out using an Agilent Cary 60 UV-Vis Spectrophotometer.
UV irradiation used for catalyst synthesis was performed in a Luzchem photoreactor equipped with UVA bulbs (typically operated with 14 bulbs, corresponding to ∼0.029 W cm−2 (with ∼4% spectral contamination). Light-emitting diodes (LEDs) of 10 W from LedEngin were used for photocatalytic reactions in the visible region (centered at 465 and 532 nm, respectively) and in the UV region (centered at 368 nm), Fig. S1.†
Quantification was carried out using a Perkin Elmer, Claurus Gas Chromatograph coupled to a Flame Ionization Detector (FID) and using a DB-5 column (30 m length, 0.320 mm diameter, 0.25 μm film), helium as a carrier gas and t-butyl benzene as the external standard. GC-MS analyses were performed on an Agilent 6890-N Gas Chromatograph with an Agilent 5973 mass selective detector calibrated with acetophenone. All 1H NMR spectra were recorded on a Bruker AVANCE 400 spectrometer expressing the chemical shifts in ppm relative to the H-signal of tetramethylsilane (TMS).
Catalyst preparation
Although some metals are in the form of metal oxides upon deposition on the GW surface, we will refer to all materials as M@GW. In many cases metal oxide nanoparticles are formed spontaneously following ambient exposure of metal nanoparticles.
Catalytic reactions
Aniline formation.
Using Ru@NGW* or Ru@SGW*. 25 mg of catalyst was placed in a centrifuge tube together with 1.5 mL of nitrobenzene (1 M), 1.7 mL of EtOH, 0.3 mL of internal standard 1,3,5-trimethoxy benzene (0.5 M) and 1.5 mL of NH2NH2 solution (6 M). The tube was tightly closed and immersed in an oil bath at 78 °C under constant stirring. Aliquots of 15 μL were taken from the reaction mixture, and diluted with 1.5 mL of ethylacetate prior to GC-FID quantification.
Using Au@NGW*. 60 mg of Au@NGW* was dispersed in 5 mL of EtOH in a clean dry round bottom flask then 1 mmol of nitrobenzene and 5 mmol of hydrazine were added. The resulting mixture was heated up to 78 °C under continuous stirring. The progress of the reaction was monitored by UV-visible spectroscopy. No reaction was detected under these conditions.
Thermal-induced isomerization. 50 mg of Ru@NGW* (or Ru@SGW*) was dispersed in 8 mL of i-propanol by sonication ca. for 5 min in a clean round bottom flask, and then 25 μL (0.16 mmol) of reactant was added. The reaction mixture was heated up to 85 °C (solvent boiling point) under air with continuous stirring for 24 hours. The progress of the reaction was monitored by GC-FID. The solid catalyst was separated by filtration and the crude product was obtained after solvent evaporation. Quantification was done by GC-FID.
Light-induced isomerization or hydrogenation. 60 mg of Pd@NGW*(or Pd@SGW) was dispersed in 4 mL of methanol in a clean glass tube, and then 0.14 mmol of estragol was added. The reaction mixture was purged with argon for 15 min and then irradiated with a 465 nm LED (a 368 nm LED for hydrogenation) for 24 hours at room temperature. The progress of the reaction was monitored by GC-FID. The solid catalyst was separated by filtration, and the crude product was obtained after solvent evaporation. Quantification was done either by 1H-NMR or by GC-FID.
Unfortunately, no reaction was detected under neither of these conditions.
Thermal-induced Sonogashira C–C coupling. 60 mg of Pd@NGW* (Pd@SGW or Ru@NGW) was dispersed in 4 mL of methanol by sonication for about 5 min in a clean round bottom flask. Then 0.12 mmol of iodobenzene, 0.16 mmol of phenylacetylene and 0.24 mmol of K2CO3 were added. The reaction mixture was purged with Ar and heated up to 42 °C (temperature of the reaction mixture under blue LED irradiation) with continuous stirring for 5 hours. The solid catalyst was separated by filtration and the crude product was obtained after solvent evaporation. Quantification was done by GC-FID.
Light-induced Sonogashira C–C coupling. 60 mg of Pd@NGW* (Pd@SGW or Ru@NGW) was dispersed in 4 mL of methanol in a 10 mL clean glass tube, and then 0.12 mmol of iodobenzene, 0.16 mmol of phenylacetylene and 0.24 mmol of K2CO3 were added. The reaction mixture was purged with Ar for 15 min and then irradiated with a 465 nm LED light source setup at 1 W cm−2 for 5 hours. The solid catalyst was separated by filtration and the crude product was obtained after solvent evaporation. Quantification was done by GC-FID. Control experiments were carried out as mentioned above. Notice that no reaction was detected under these conditions when using Ru@NGW as the catalyst; only Pd@NGW* was effective.
N–C heterocycloaddition. We followed a previously reported protocol.7 Briefly, 15 mg of Cu@NGW (or Cu@NGW*) was dispersed in 1.5 mL of THF in a quartz test tube and mixed with azide (0.5 mmol), alkyne (0.5 mmol), and amine (0.5 mmol). The reaction mixture was stirred and irradiated with a 465 nm LED at room temperature and under air for 6 h. The solid catalyst was separated by filtration and the pure product was obtained after vacuum evaporation. Yields were calculated by 1H NMR in CDCl3.
Control experiments were performed under dark conditions at room temperature or at 55 °C (maximum temperature reached upon illumination) under air.
C–C coupling (sp3–sp3): dimerization of benzyl bromides. We followed a previously reported protocol.7 Briefly, 30 mg of Au@NGW* was dispersed in 4 mL of dichloromethane (DCM) in a glass test tube and then 0.1 mmol of 4-nitrobenzylbromide and 0.2 mmol of diisopropyl ethylamine (DIPEA) were added. The resulting mixture was purged with argon for 15 minutes prior to 532 nm LED irradiation with a 4xLED system each working at 0.56 W cm−2. The solid catalyst was separated by filtration and the product was obtained after vacuum evaporation. Yields and conversions were calculated by 1H NMR in CDCl3 using caffeine as the external standard. Control experiments were carried out as mentioned above.
Catalyst recyclability
Co@SGW, Pd@NGW* and Au@NGW* catalysts were tested for potential reusability in the direct, selective and efficient reductive dehalogenation, Sonogashira coupling and 4-nitrobenzyl bromide dimerization respectively, using the conditions described above within at least two reusability cycles. The catalyst was removed from the reaction mixture by filtration, washed twice with methanol and 3 times with water, and then dried in an oven at 100 °C overnight prior to reuse (Table S1†).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada, the Canada Foundation for Innovation, the Canada Research Chairs Program and funding from Canada's International Development Research Centre (IDRC). Thanks are due to the RISE program for the scholarship awarded to M. C. and the Generalitat Valenciana (BEST/2017/049) for the financial support granted to M. L. M. The authors would like to thank Dr Yun Liu for helping on the acquisition of the SEM images.References
- I. W. Davies, L. Matty, D. L. Hughes and P. J. Reider, J. Am. Chem. Soc., 2001, 123, 10139–10140 CrossRef PubMed.
- R. Schlçgl, Angew. Chem., Int. Ed., 2015, 54, 3465–3520 CrossRef PubMed.
- W. C. Conner and J. L. Falconer, Chem. Rev., 1995, 95, 759–788 CrossRef.
- A. Elhage, A. E. Lanterna and J. C. Scaiano, ACS Catal., 2017, 7, 250–255 CrossRef.
- D. Cambie, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noel, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed.
- A. E. Lanterna, A. Elhage and J. C. Scaiano, Catal. Sci. Technol., 2015, 5, 4336–4340 RSC.
- B. Wang, J. Durantini, J. Nie, A. E. Lanterna and J. C. Scaiano, J. Am. Chem. Soc., 2016, 138, 13127–13130 CrossRef PubMed.
- Sigma-Aldrich, Glass Wool, http://www.sigmaaldrich.com/catalog/product/supelco/20411?lang=en%26region=CA, accessed September, 2017, 2017.
- B. Steyn, M. C. Oosthuizen, R. MacDonald, J. Theron and V. S. Brozel, Proteomics, 2001, 1, 871–879 CrossRef PubMed.
- M. Nisnevitch, M. Kolog-Gulco, D. Trombka, B. S. Green and M. A. Firer, J. Chromatogr. B: Biomed. Sci. Appl., 2000, 738, 217–223 CrossRef.
- Y. Matatov-Meytal and M. Sheintuch, Appl. Catal., A, 2002, 231, 1–16 CrossRef.
- V. V. Barelko, M. V. Kuznetsov, V. G. Dorokhov and I. Parkin, Russ. J. Phys. Chem. B, 2017, 11, 606–617 CrossRef.
- R. W. Macdonald and K. E. Hayes, J. Chem. Soc., Chem. Commun., 1972, 1030 RSC.
- G. K. Ramaswamy, A. Somasundaram, B. K. Kuppuswamy and M. Velayudham, J. Chin. Chem. Soc., 2013, 60, 97–102 CrossRef.
- H. T. Yang, Z. P. Fang, X. Y. Fu and L. F. Tong, Chin. J. Catal., 2007, 28, 947–952 CrossRef.
- B. S. Bal'zhinimaev, A. P. Suknev, Y. K. Gulyaeva and E. V. Kovalyov, Catalysis in Industry, 2015, 7, 267–274 CrossRef.
- L. G. Simonova, V. V. Barelko, A. V. Toktarev, A. F. Chernyshov, V. A. Chumachenko and B. S. Bal'zhinimaev, Kinet. Catal., 2002, 43, 61–66 CrossRef.
- A. I. Carrillo, K. G. Stamplecoskie, M. L. Marin and J. C. Scaiano, Catal. Sci. Technol., 2014, 4, 1989–1996 RSC.
- B. H. R. Suryanto, X. Y. Lu, H. M. Chan and C. Zhao, RSC Adv., 2013, 3, 20936–20942 RSC.
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef.
- K. C. Park, I. Y. Jang, W. Wongwiriyapan, S. Morimoto, Y. J. Kim, Y. C. Jung, T. Toya and M. Endo, J. Mater. Chem., 2010, 20, 5345 RSC.
- C. Bock, C. Paquet, M. Couillard, G. A. Botton and B. R. MacDougall, J. Am. Chem. Soc., 2004, 126, 8028–8037 CrossRef PubMed.
- J. P. Espinos, J. Morales, A. Barranco, A. Caballero, J. P. Holgado and A. R. Gonzalez-Elipe, J. Phys. Chem. B, 2002, 106, 6921–6929 CrossRef.
- A. Y. Klyushin, T. C. R. Rocha, M. Havecker, A. Knop-Gericke and R. Schlogl, Phys. Chem. Chem. Phys., 2014, 16, 7881–7886 RSC.
- C. S. Higman, A. E. Lanterna, M. L. Marin, J. C. Scaiano and D. E. Fogg, ChemCatChem, 2016, 8, 2446–2449 CrossRef.
- R. B. Bedford, C. S. J. Cazin and D. Holder, Coord. Chem. Rev., 2004, 248, 2283–2321 CrossRef.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, Taylor and Francis Group, Boca Raton, FL, 88th edn, 2007, p. 2640 Search PubMed.
- B. Sahoo, A. E. Surkus, M. M. Pohl, J. Radnik, M. Schneider, S. Bachmann, M. Scalone, K. Junge and M. Belle, Angew. Chem., 2017, 129, 11394–11399 CrossRef.
- J. J. Devery, J. D. Nguyen, C. Dai and C. R. J. Stephenson, ACS Catal., 2016, 6, 5962–5967 CrossRef.
- L. B. Liao, Q. H. Zhang, Z. H. Su, Z. Z. Zhao, Y. N. Wang, Y. Li, X. X. Lu, D. G. Wei, G. Y. Feng, Q. K. Yu, X. J. Cai, J. M. Zhao, Z. F. Ren, H. Fang, F. Robles-Hernandez, S. Baldelli and J. M. Bao, Nat. Nanotechnol., 2014, 9, 69–73 CrossRef PubMed.
- A. S. Hainer, J. S. Hodgins, V. Sandre, M. Vallieres, A. E. Lanterna and J. C. Scaiano, ACS Energy Lett., 2018, 3, 542–545 CrossRef.
- A. Elhage, A. E. Lanterna and J. C. Scaiano, ACS Sustainable Chem. Eng., 2018, 6, 1717–1722 CrossRef.
- A. Elhage, A. E. Lanterna and J. C. Scaiano, ACS Catal., 2017, 7, 250–255 CrossRef.
- P. Roy, A. P. Periasamy, C. T. Liang and H. T. Chang, Environ. Sci. Technol., 2013, 47, 6688–6695 CrossRef PubMed.
- B. W. Zhou, J. L. Song, H. C. Zhou, L. Q. Wu, T. B. Wu, Z. M. Liu and B. X. Han, RSC Adv., 2015, 5, 36347–36352 RSC.
- H. Y. Zhu, X. B. Ke, X. Z. Yang, S. Sarina and H. W. Liu, Angew. Chem., Int. Ed., 2010, 49, 9657–9661 CrossRef PubMed.
- K. Selvam, H. Sakamoto, Y. Shiraishi and T. Hirai, New J. Chem., 2015, 39, 2856–2860 RSC.
- S. Sarina, E. R. Waclawik and H. Y. Zhu, Green Chem., 2013, 15, 1814–1833 RSC.
- L. B. Wang, X. Q. Pan, Y. Zhao, Y. Chen, W. Zhang, Y. F. Tu, Z. B. Zhang, J. Zhu, N. C. Zhou and X. L. Zhu, Macromolecules, 2015, 48, 1289–1295 CrossRef.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- K. L. McGilvray, M. R. Decan, D. Wang and J. C. Scaiano, J. Am. Chem. Soc., 2006, 128, 15980–15981 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02115e |
| ‡ On leave from Instituto Universitario Mixto de Tecnología Química (UPV-CSIC), Universitat Politècnica de València. Avenida de los Naranjos s/n, 46022 Valencia. Spain. |
| This journal is © The Royal Society of Chemistry 2018 |