
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRatiometric Raman imaging reveals the new anti-cancer potential of lipid targeting drugs†
Lauren E. Jamieson ,
Corinna Wetherill,
Karen Faulds and
Duncan Graham*
,
Corinna Wetherill,
Karen Faulds and
Duncan Graham*
Centre for Molecular Nanometrology, WestCHEM, Department of Pure and Applied Chemistry, Technology and Innovation Centre, University of Strathclyde, 99 George Street, Glasgow, G1 1RD, UK. E-mail: duncan.graham@strath.ac.uk
First published on 31st July 2018
Abstract
De novo lipid synthesis is upregulated in cancer cells and inhibiting these pathways has displayed anti-tumour activity. Here we use Raman spectroscopy, focusing solely on high wavenumber spectra, to detect changes in lipid composition in single cells in response to drugs targeting de novo lipid synthesis. Unexpectedly, the beta-blocker propranolol showed selectively towards cancerous PC3 compared to non-cancerous PNT2 prostate cells, demonstrating the potential of this approach to identify new anti-cancer drug leads. A unique and simple ratiometric approach for intracellular lipid investigation is reported using statistical analysis to create phenotypic ‘barcodes’, a globally applicable strategy for Raman drug-cell studies. High wavenumber spectral analysis is compatible with low cost glass substrates, easily translatable into the cytological work stream. The analytical strength of this technique could have a significant impact on cancer treatment through vastly improved understanding of cancer cell metabolism, and thus guide drug design and enhance personalised medicine strategies.
Cancer is one of the most prevalent and researched diseases in the world today.1 In the fight against cancer, scientists are turning to a variety of diverse resources to identify new targets for therapy that could provide vital information in combating the disease. In recent years attention has turned to metabolic transformation in cancerous cells and the potential to base new therapeutic approaches on these metabolic changes.2 It is widely accepted that there are characteristic changes in cellular metabolism in cancerous compared to non-cancerous cells. In addition to providing a new therapeutic target, this could hold the key to selectivity in cancer therapy. One of the major challenges for current therapeutic approaches is lack of selectivity towards cancerous over non-cancerous cells, causing major negative side effects since drugs designed to destroy cancerous cells will also destroy healthy cells.3
Otto Warburg pioneered cancer metabolomics by observing that cancer cells exhibited a shift in energy production, with vastly increased aerobic glycolysis and production of lactate, as opposed to mitochondrial respiration.4,5 This metabolic shift, commonly associated with anaerobic conditions, was observed in cancer cells, even in the presence of normal oxygen levels, and was coined the Warburg effect. More recently, Warburg's initial observations have been challenged with the proposal of a reverse Warburg effect, where cancer cells and cancer associated fibroblasts become metabolically coupled.4,6,7 In addition, the characteristic ‘Hallmarks of Cancer’ as defined by Hanahan and Weinberg,8 give way to various metabolic characteristics of cancer cells, for example, increased nucleic acid synthesis, amino acid production, and lipid biosynthesis, in order to fulfill the cancer cells ability to proliferate rapidly. By understanding these metabolic transformations at the biochemical level, drugs could be targeted specifically to exploit these characteristics and selectively destroy cancerous cells.
One key metabolic characteristic of cancer cells is the upregulation of de novo lipid synthesis.9–11 In healthy cells, lipids are generally acquired exogenously and de novo synthesis is regulated and kept minimal.9 It has been observed that cancerous cells display a marked upregulation in de novo lipid synthesis, and, therefore, by targeting enzymes in this pathway, cancer cells could be selectively targeted.2,12 It has already been reported that various drugs, in particular targeting fatty acid synthase (FASN), have displayed anti-tumour properties.13
In order to exploit metabolic transformation in cancer cells as a drug target, the biochemical basis of the metabolic shifts must be understood. This requires analytical techniques to elucidate the underlying biochemistry. Traditionally molecular biology approaches analyse lysed and isolated cell components using, for example, the polymerase chain reaction, electrophoresis and western blotting to distinguish DNA, RNA and protein composition.14 High resolution molecular level imaging techniques that allow analysis of biological systems in their native environment and which are minimally invasive are highly desirable. In vitro cell imaging has evolved largely around the use of fluorescent dye molecules to specifically target biomolecules including DNA, proteins and lipids.15 However, cellular lipid analysis and imaging is limited, largely as a result of the lack of innate fluorescence from cellular lipids, and fluorescent dyes for lipid staining are large molecules which can seriously alter innate distribution and dynamics.16 Additionally, many lipid dyes require chemical fixation prior to staining resulting in lengthy and complex sample preparation.17
In order to allow drugs to be optimally designed to target de novo lipid synthesis, an analytical technique providing a detailed understanding of the biochemistry and the effect particular drugs have on the underlying upregulated lipid synthesis is required. This requires a detection strategy that enables an accurate, easily interpretable and meaningful phenotypic readout for drug action and efficacy that offers potential as a platform for translation to high throughput drug screening. Advanced lipid imaging techniques have focused largely on the use of mass spectrometry, however, the major downfall of this technique is its destructive nature.18 Coherent anti-Stokes Raman spectroscopy (CARS) has been reported for cellular lipid imaging, showing promising results, however the information gained is limited by tuning into specific bond vibrations, as opposed to acquiring a full spectrum.17,19 Here we propose that intracellular imaging using high wavenumber Raman spectroscopy can give a detailed, high resolution insight into lipid distribution and metabolism in cancer cells at subcellular resolution in addition to a phenotypic drug response. High wavenumber Raman spectroscopy has been studied previously with application to biological samples, however there are currently very few studies using this approach and none with specific application to lipids in cells.20 The information gained in this study gives a detailed insight into lipid distribution and dynamics in in vitro cell models and leads the way to new capability to enhance the matching of drug to cancer treatment. More globally, this study develops a statistically meaningful strategy for analysis of biological Raman data that could be adopted throughout the field of vibrational spectroscopy, in particular for characterising phenotypic cell response to potential drug candidates for drug screening as this preliminary study demonstrates. Combined with the use of cheap glass substrates and the non-destructive and label free nature of the technique with potential for easy translation to analysis of live cells, the developed strategy could be a vital step forward for translation of Raman spectroscopy into cytological work streams.
Experimental methods
Cell culture
PC3 human prostate cancer cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) and PNT2 human normal immortalized prostate epithelium cells were cultured in Rosewell Park Memorial Institute Medium (RPMI) in both cases supplemented with 1% penicillin/streptomycin (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units per ml), 1% fungizone, and 10% heat-inactivated foetal bovine serum (FBS). Cells were incubated at 37 °C and 5% CO2 in a humidified incubator.
000 units per ml), 1% fungizone, and 10% heat-inactivated foetal bovine serum (FBS). Cells were incubated at 37 °C and 5% CO2 in a humidified incubator.
Cells were seeded (ca. 5 × 105 cells) in 35 mm glass bottomed imaging dishes (Ibidi) (or ca. 1.1 × 105 cells onto a 13 mm coverslip in a 24 well plate for replicate measurements as reported in ESI†). After overnight incubation at 37 °C and 5% CO2 in a humidified incubator, media was removed and replaced with a sufficient volume of stock solutions of 10 mM propranolol in DMSO, 10 mM cyclosporin in DMSO, 40 mM orlistat in DMSO, 200 μM CAY10566 in DMSO, 2 mM TOFA in DMSO or an equal volume of DMSO in medium, resulting in final concentrations of 30 μM propranolol, 30 μM cyclosporin, 100 μM orlistat, 500 nM CAY10566 and 5 μM TOFA. Final concentration of DMSO was 0.3% v/v for cyclosporin and propranolol and 0.25% v/v for control, orlistat, CAY10566 and TOFA. The drug concentrations were selected based on consideration of those used in previously reported studies and IC50 values for each drug, and in the case of propranolol and cyclosporin based on the concentration suggested in the HCS LipidTOX™ Phospholipidosis and Steatosis Detection Kit (Thermo Fisher). After 48 h incubation at 37 °C and 5% CO2 in a humidified incubator, cells were washed (×4) with phosphate buffered saline (PBS) before adding 4% paraformaldehyde for ca. 30 min. 4% Paraformaldehyde was removed and cells were washed with PBS followed by distilled water and left to air dry.
Raman measurements
Raman spectra were acquired on a Renishaw inVia Raman microscope equipped with a 532 nm Nd:YAG laser giving a maximum power of 500 mW, 1800 l mm−1 grating, and a Leica 50×/NA 0.75 N PLAN EPI objective. Fixed cells were mapped using a step size of 1 μm in x and y, with 0.5 s acquisition time, 15 mW laser power and a spectral center of 3000 cm−1. Three maps were acquired per condition.Data processing, analysis and statistics
Renishaw Wire 4.1 was used to perform basic pre-processing steps using the inbuilt functions in the software. Cosmic rays were removed followed by noise filtering and baseline subtraction.Custom MATLAB® scripts were then used to perform further analysis. Cell regions were selected based on the total spectral intensity for the map and all associated spectra were extracted for comparison between conditions. False colour images for the cell regions were created based on the following peak intensity ratios:
• 2851 cm−1/(2851 cm−1 + 2933 cm−1) = ratio 1
• 2881 cm−1/(2881 cm−1 + 2933 cm−1) = ratio 2
• 2974 cm−1/(2974 cm−1 + 2933 cm−1) = ratio 3
• 2851 cm−1/(2851 cm−1 + 2974 cm−1) = ratio 4
• 3013 cm−1/(3013 cm−1 + 2851 cm−1) = ratio 5
• 3064 cm−1/(3064 cm−1 + 2933 cm−1) = ratio 6.
All spectra extracted from the cells were cut to 2800 cm−1 to 3100 cm−1. A quality control step was performed removing all spectra outside one standard deviation of the mean for each cell map. Average spectra generated from all three maps per condition were compared after scaling spectra to the peak at 2933 cm−1. Intensity ratios 1–6 were then calculated for all the remaining spectra for each condition and used to create bar charts depicting the mean and standard deviation values in GraphPad Prism 7. GraphPad Prism 7 was used to perform one-way ANOVA tests comparing each of the drug treated conditions to the vector control (DMSO) for the respective cell line. Effect size for each drug treated condition in comparison to the DMSO control for the respective cell line was calculated using eqn (1) and (2), where NE = number of data points in the experimental (drug) group, NC = number of data points in the control (DMSO) group, SDE = standard deviation of the experimental (drug) group, and SDC = standard deviation of the control (DMSO) group.
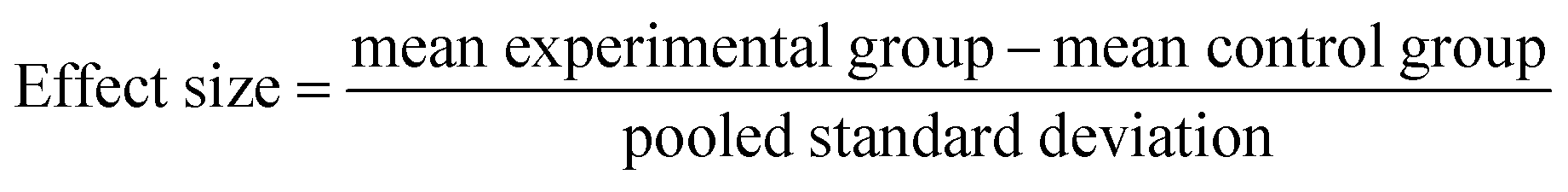 | (1) |
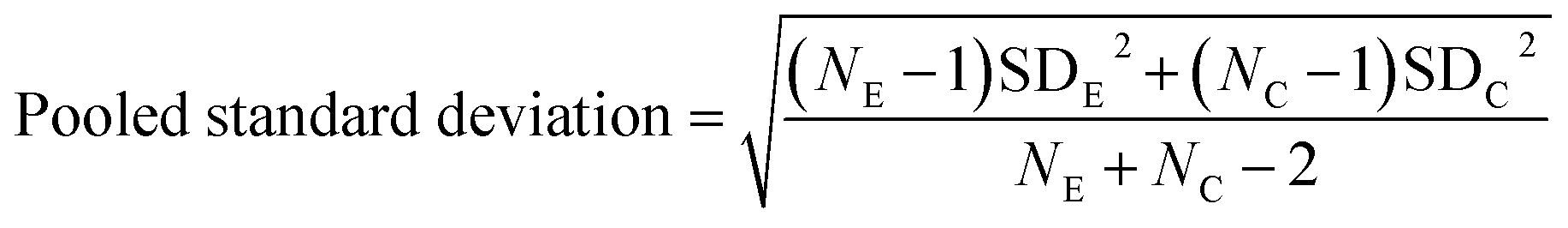 | (2) |
‘Barcodes’ for each drug treatment on each cell line were created by assigning a whole number value between −3 and 3 for each ratio in each case such that effect size:
• ≤−1 = −3
• >−1 and ≤−0.5 = −2
• >−0.5 and ≤−0.1 = −1
• >−0.1 and <0.1 = 0
• ≥0.1 and <0.5 = 1
• ≥0.5 and <1 = 2
• ≥1 = 3.
This formed a barcode which was represented pictorially using a bar chart format where the size and colour of the bars reflected the numerical value assigned, and also using a numerical tag where the values corresponded to the following:
ratio 1, ratio 2, ratio 3, ratio 4, ratio 5, ratio 6, sum of magnitude of all values.
Results
Raman spectroscopy was used to analyse cancerous and non-cancerous cells in a label-free, non-destructive, chemically specific and highly spatially resolved manner. The primary aim was to demonstrate the capability of Raman spectroscopy to revolutionise understanding of drug induced changes to cellular lipid metabolism and therefore the focus was on mapping the high wavenumber (HWN) spectral region between ca. 2800 cm−1 and 3100 cm−1. In addition to the large volume of lipid specific information contained in the HWN spectrum, the signal intensity in this region is higher than in the low wavenumber (LWN) fingerprint region for biomolecules. In contrast to the LWN spectra, there is also minimal interference from glass background signal. Raman spectroscopic analysis of cell samples is therefore made cost-effective and amenable to integration into cytological work streams.21 Raman mapping of fixed cells was carried out using a 1 μm step size in the x and y directions, resulting in a highly spatially resolved map of chemically specific signals relating to biochemical bond vibrations. This HWN Raman measurement process was used for assessment of subcellular lipid distribution, and localised and global response to treatment with drugs that are known to inhibit key enzymes in de novo lipid synthesis. Raman mapping was carried out using a laser power of 15 mW at the sample, which showed no evidence of burning at the sample, and was therefore non-destructive to the sample. As the cells were fixed prior to analysis, the interaction of the laser with the sample was unlikely to cause any disruption to the preserved cell samples. To further evidence the non-destructiveness of this method, an independent experiment on live cells showed no change to cell viability in PC3 and PNT2 cells when viability was measured using fluorescence spectroscopy in samples not subject to Raman analysis, and in samples stained after Raman analysis (ESI Fig. S1†).Fig. 1 shows the average spectra obtained from PC3 and PNT2 cells treated with different drugs. The broad peak present in the HWN cellular spectra originates from the multitude of overlapping C–H bond vibrations from a number of cellular biomolecules. Due to the chemical nature of lipid molecules which contain a number of C–H bond vibrations from their hydrocarbon backbone, this region contains extensive information on the cellular lipid composition. One of the key band assignments is the relative intensity of the signal at ca. 2850 cm−1, as this is thought to reflect the overall lipid abundance.22,23 Prostate cell lines, PC3 (cancerous) and PNT2 (non-cancerous), were used in this particular study due to the well-researched role of lipids in prostate cancer and correlation between lipids and a malignant phenotype.24–26 Research has shown that FASN overexpression is a common and early characteristic in prostate cancer27 and cholesterol levels have also been correlated with prostate cancer incidence.28
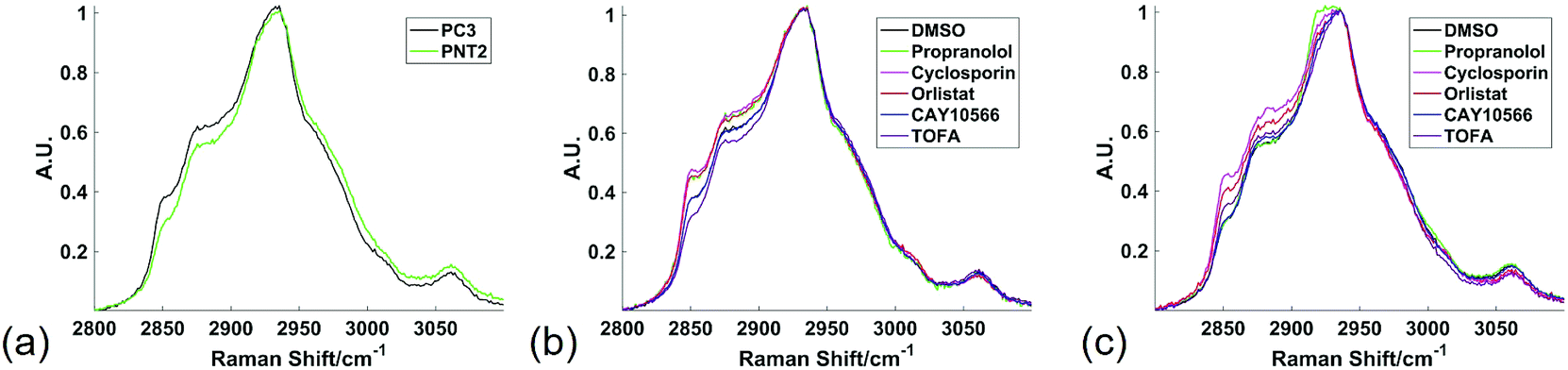 | ||
| Fig. 1 High wavenumber spectral response to lipid altering drugs. Average Raman spectra from the high wavenumber spectral region from all spectra extracted from cell regions of three different Raman maps were compared between PC3 and PNT2 control (DMSO) cells (a); PC3 cells treated with DMSO (control), or a lipid altering drug (b); and PNT2 cells treated with DMSO (control), or a lipid altering drug (c). Cells were mapped using 532 nm, 0.5 s acquisition, 15 mW laser power, 1 μm step size in x and y and a spectral center of 3000 cm−1. | ||
Cellular response to treatment with three drugs known to interfere with different stages of de novo lipid biosynthesis was monitored. Orlistat (Xenival®) was selected as an inhibitor of fatty acid synthase (FASN), one of the key enzymes in lipid biosynthesis, which forms palmitic acid from acetyl-CoA and malonyl-CoA.29,30 Orlistat is a commercially available ‘fat-busting’ drug, marketed as a weight loss drug. However, recent studies have revealed it to have anti-tumour activity, making it a strong candidate for repurposing for cancer therapy.29,30 CAY10566 is an inhibitor of stearoyl-CoA desaturase (SCD), an enzyme that creates mono-unsaturated fatty acids from saturated fatty acids.31 5-(Tetradecyloxy)-2-furoic acid (TOFA) inhibits acetyl CoA-carboxylase, which is responsible for the conversion of acetyl CoA to malonyl CoA, one of the first steps in de novo lipid synthesis.32,33 These compounds could therefore prove to be effective anti-cancer agents through their targeting action to a characteristic metabolic transformation in cancer cells. Fig. 2 summarises the action of the selected drugs on de novo lipid synthesis.
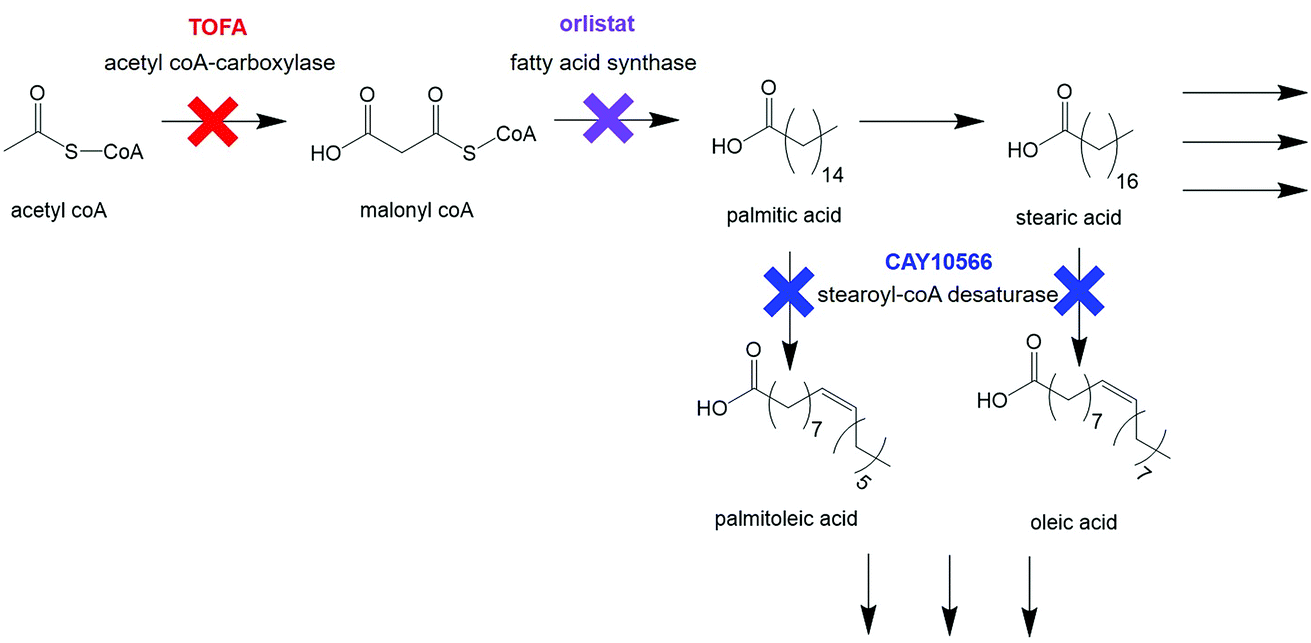 | ||
| Fig. 2 Key steps in de novo lipid synthesis. Simplified steps in de novo lipid synthesis with enzyme targets for drugs used in this study indicated. | ||
In addition to these three drugs, two control drugs, cyclosporin and propranolol, known to induce the formation of neutral lipid droplets and accumulation of phospholipid respectively, were included to allow the Raman spectral response characteristic of the cellular phenotypes associated with these drugs to be characterised.34,35Fig. 1 compares the average spectra extracted from cancerous PC3, and non-cancerous PNT2, prostate epithelial cells with each cell line treated with each of the drug compounds. Some key differences were observed upon visual inspection of the spectra, particularly in the region from ca. 2850 cm−1 to 2900 cm−1, where many of the drug treatments resulted in a relative increase of intensity in this region.
Due to the broad nature of the spectra in the HWN region, extracting species specific information is incredibly difficult. Multivariate techniques such as principal component analysis (PCA) can be used to assess whether there exists separation between groups of spectra and the major spectral contributors causing such separations, however for such broad overlapping spectra many of the data points can confound such analysis and mask key spectral changes. These techniques are also more complicated and often no consideration is given to careful analysis of the spectra themselves. Here, analysis was therefore simplified to comparison of the intensity ratio between signals at key wavenumbers in the Raman spectra, identified by visual and informed inspection of the data, thus creating a bivariate descriptor, which reported on global rather than species specific lipid changes in a simple manner. The intensity ratio between the peaks at 2851 cm−1 and 2933 cm−1 was selected as this ratio has been reported to correlate with lipid abundance, a higher ratio being reflective of a higher lipid content.22,23 The signal at 2851 cm−1 is primarily attributed to C–H stretches in CH2 groups, which are particularly abundant in the long hydrocarbon chains of lipids, and the peak at 2933 cm−1 is assigned to C–H stretches in CH3 groups, which are particularly abundant in protein polypeptide chains.36,37 While neither of these signals can be exclusively assigned to these species, the fact that they have large contributions from lipids and proteins respectively meant that this ratio semi-quantitatively reflects the lipid/protein ratio of the intracellular environment. In addition, it is clear from Fig. 1 that there are spectral variations in this ratio between control and drug treated samples, therefore even without a biological assignment this bivariate measurement could provide an important analytical descriptor for assessing cellular drug response. Fig. 3 displays false color images of one representative example of a cell under each condition created using this ratio (a total of three cells were analysed per condition and additional images can be found in ESI Fig. S2†). The particular ratio used was that of 2851 cm−1 divided by the sum of intensities at 2933 cm−1 and 2851 cm−1 (lipid/(protein + lipid)), in order to normalise the measurements.
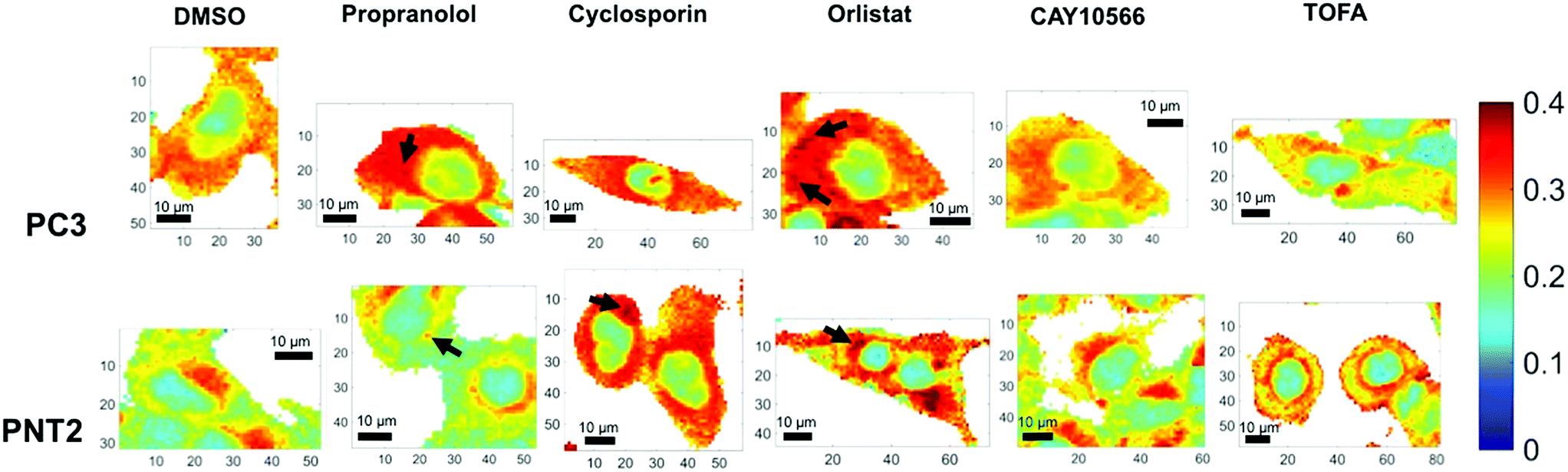 | ||
| Fig. 3 Ratiometric Raman imaging of intracellular lipid distribution. Representative examples of fixed cell regions mapped using a Raman microscope for two prostate cancer cell lines, PC3 and PNT2, treated with DMSO (control), or a lipid altering drug. After processing in Wire 4.1 and MATLAB®, false color images of the ratio of peak intensity at 2851 cm−1 and the sum of the peak intensities at 2933 cm−1 and 2851 cm−1 were created as a reflection of lipid/(protein + lipid) ratio. White areas represent background regions of the map that were omitted after selecting cellular regions. Cells were mapped using 532 nm, 0.5 s acquisition, 15 mW laser power, 1 μm step size in x and y and a spectral center of 3000 cm−1. Example regions of high intensity, potentially corresponding to lipid droplets, are indicted using arrows. Spatial coordinates on images are in μm. | ||
In addition to considering the global spectral response to drug application, it was significant to consider the detail gained in a single map in relation to subcellular lipid distribution. Considering the lipid/(protein + lipid) ratio, the variation within a single cell was vast and reflected the function of various subcellular organelles. Most significantly, the nuclear region was distinguished easily as a consequence of a markedly lower lipid/protein abundance, reflective of the known characteristics of nuclei for storage of genetic material.38 Additionally, in some cases, high intensity spots were observed. These could be attributed to lipid droplets and are reflective of storage of lipids in these regions,39,40 or regions of mitochondria, where lipids are metabolised.41 Thus, an ability to obtain high resolution information on intracellular lipid distribution was demonstrated. It is speculated that in the future higher resolution maps could be generated with even higher subcellular resolution.
Qualitative visual examination of Fig. 3 allows further trends associated with drug treatment and comparison between cancerous and non-cancerous cells to be made. Comparing vector control PC3 and PNT2 cells revealed a difference in intracellular lipid distribution, with a uniform distribution of lipid throughout the cytoplasm in PC3 cells compared to a lower level of lipids throughout the cytoplasm for PNT2 cells with some distinct regions of higher intensity, particularly in the perinuclear region. These higher intensity regions could be indicative of the Golgi body, endoplasmic reticulum and/or areas of endosomes and lysosomes. This agrees with reports that endosomal biogenesis is altered in cancerous prostate cells resulting in a more uniform cytoplasmic distribution of endosomes, which has previously been observed in prostate cancer cells.42 This could reflect an increased uptake of nutrients in cancerous cells to support their growth.
Notably, upon treatment with propranolol, cyclosporin and orlistat, the lipid abundance increased in PC3 cells, particularly in the cytoplasmic region. Spots of higher intensity could represent lipids accumulating in lipid droplets. In comparison, PNT2 cells showed a similar response after cyclosporin and orlistat treatment but no notable change in response to propranolol. Propranolol and cyclosporin were included in this study as positive controls for the phenotypes associated with neutral lipid and phospholipid accumulation. Therefore, this acts as an indicator that the response is associated with neutral lipid and/or phospholipid accumulation in PC3 cells since both compounds resulted in a marked increase in the lipid intensity in the cytoplasm. It is also significant that propranolol showed no such effect in PNT2 cells, contrary to the expected increase in phospholipids, indicating a selectivity for cancerous over non-cancerous cells. These observations were consistent over all three cells analysed per condition. It is worth noting that in addition to the changes in lipids observed with subcellular resolution, the cell images in Fig. 3 and ESI Fig. S2† allow changes in cell morphology with drug treatment to be monitored. It is expected that cell morphology would change in response to treatment with lipid altering drugs as lipids are the primary constituents of cell membranes, an important structural feature of cells. Therefore, changes in cell morphology and membrane shapes of both the cell membrane and membrane of intracellular components provide further evidence of drug action on cells. These changes are also evident in parallel fluorescent measurements performed (ESI Fig. S4†).
The increase in lipid abundance observed in the cytoplasm of both PC3 and PNT2 cells on addition of orlistat is counterintuitive to its therapeutic activity, therefore this response was attributed to a stress response from the cells. Upon inhibiting a key enzyme in lipid synthesis, it is hypothesised that the cells accumulated the lipids that they had and could acquire from the surrounding medium. Additionally, it is possible that, as the signal at 2851 cm−1 was attributed primarily to lipid C–H stretches in CH2 groups, this change could reflect an increase in long chain lipids, for example palmitate already synthesised could continue to create longer chain fatty acids through elongation steps, but no new palmitate could be synthesised, resulting in a depletion in shorter chain fatty acids.
In addition, an increase in lipids has been associated with the cellular processes ultimately leading to apoptosis, therefore it is a possibility that the increased lipid response was phenotypically characteristic of initial stages of cell death.43 In order to investigate this possibility further, cells for each condition were stained for viability (ESI Fig. S2†). This revealed that under all conditions, cells analysed i.e. those still attached to the glass substrate, maintained viability, however there were notable changes in cell confluence, particularly upon orlistat treatment, suggesting some cells were dying ultimately causing them to detach from the glass substrate. This confirmed that those cells investigated using Raman were viable and it is likely that these observations could be attributed to metabolic response to drug application. Fluorescence staining for phospholipidosis and steatosis was also in line with the observed Raman response (ESI Fig. S4†). This was an important comparison when considering the reliability of the Raman based approach, and also helped highlight the added benefits Raman provided in comparison to a commercially available fluorescence assay. The particular assay used stained for phospholipidosis (phospholipid accumulation) and steatosis (neutral lipid accumulation) and showed strong agreement with Raman measurements. Minimal staining for both phospholipidosis and steatosis was observed in the control PC3 and PNT2 cells, while significant increase in staining for phospholipidosis and/or steatosis was observed for cyclosporin and orlistat treated PC3 and PNT2 cells. Significantly, measurements for propranolol indicated the same observation made using Raman that there was a marked increase in lipid (phospholipidosis in particular) for PC3 cells but no apparent change for PNT2 cells. As with Raman, there were less pronounced changes associated with CAY10566 and TOFA. The strong agreement between these fluorescence measurements and Raman measurements served to emphasize the reliability of the Raman based approach. In addition, Raman allowed this information to be extracted without the need for external labelling regents and staining steps, including resolving cellular components at subcellular resolution e.g. nuclei were resolved without the need to fluorescently stain. Each point in the Raman map also produced a spectrum with more than 1000 data points associated with it, providing a wealth of chemically specific information not accessible via fluorescence. This particular commercially available kit also stained for particular lipid classes, while each Raman spectrum contains a wealth of information on all lipid species present in the cellular sample.
In order to compare the Raman results in a more quantitative manner, the lipid/(protein + lipid) ratios for all points in the three replicate cell maps per condition were extracted and compared (Fig. 4 and ESI Table S1†). This simple bivariate measurement revealed important biological responses to these drug molecules that could act as a simple readout for drug efficacy and selectivity. Efficacy was assessed by comparing vector to drug treated cells. Selectivity for cancerous vs. non-cancerous cells was assessed by comparing response in cancerous to non-cancerous cells. One-way ANOVA tests were performed to assess the statistical significance between control and drug treated cells for each compound tested. From Fig. 4 it was observed that in most cases there was a high level of statistical significance between control and treated cells, with most p values ≤ 0.0001. For PC3 cells treated with CAY10566 and PNT2 cells treated with propranolol, there was no significant difference from respective control cells and for PNT2 cells treated with CAY10566 the p value was ≤0.001. These results were in line with previous observations from the cell images, and confirmed the selectivity of propranolol for cancerous cells with statistical significance.
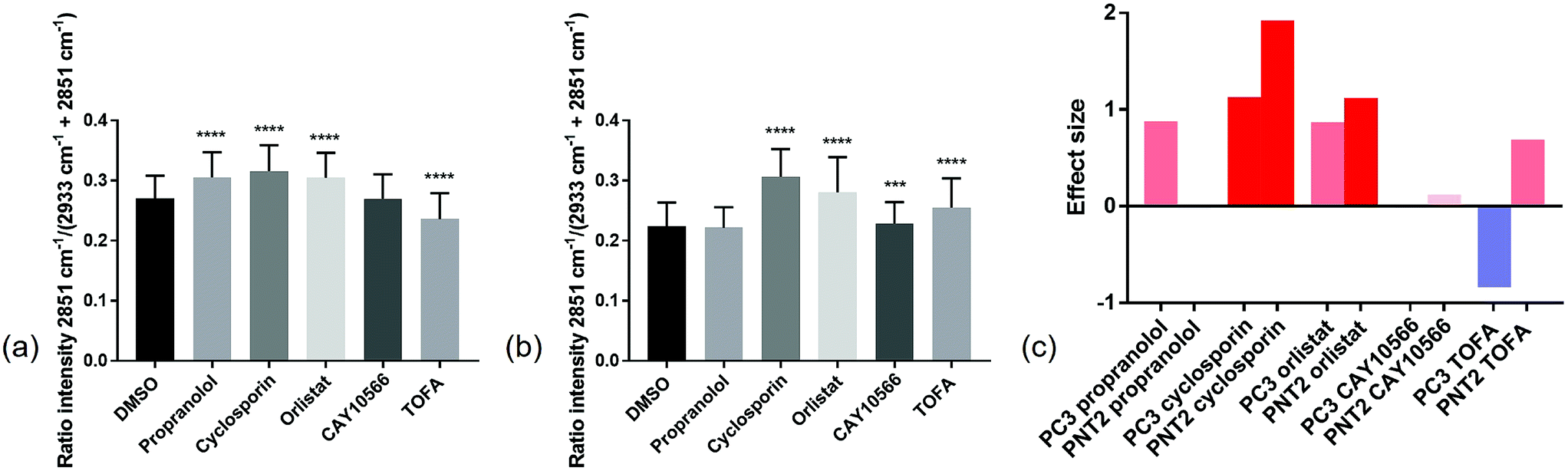 | ||
| Fig. 4 Quantitative Raman assessment of global lipid response. The intensity ratios of the Raman peak intensity at 2851 cm−1 divided by the sum of the peak intensities at 2933 cm−1 and 2851 cm−1 for spectra extracted from cell regions in a total of three different cells per condition, were compared. This ratio reflected the lipid/protein ratio in the cells, and the mean and standard deviation for control PC3 cells (DMSO) and drug treated PC3 cells (a), and control PNT2 cells (DMSO) and drug treated PNT2 cells (b) are shown. For PC3 cells and PNT2 cells there was a statistical significance between treatments determined using one-way ANOVA (F(5, 16233) = 1581, p ≤ 0.0001 and F(5, 20853) = 2037, p ≤ 0.0001 respectively). A Dunnett's multiple comparisons test revealed a statistically significant difference between control (DMSO, n = 2800) and propranolol treated (n = 3134, p ≤ 0.001), cyclosporin treated (n = 2070, p ≤ 0.001), orlistat treated (n = 2739, p ≤ 0.001) and TOFA treated (n = 3480, p ≤ 0.001) PC3 cells and no significant difference between control and CAY10566 (n = 2016, p = 0.9700) PC3 cells. A Dunnett's multiple comparisons test revealed a statistically significant difference between control (DMSO, n = 3563) and cyclosporin treated (n = 3941, p ≤ 0.001), orlistat treated (n = 3844, p ≤ 0.001), CAY10566 treated (n = 2991, p = 0.0003) and TOFA treated (n = 3998, p ≤ 0.001) PNT2 cells and no significant difference between control and propranolol (n = 2522, p = 0.2388) PNT2 cells. ***p ≤ 0.001; **** ≤ 0.0001. Effect size for each drug treatment relative to control (DMSO) was determined for both PC3 and PNT2 cells where the bar colours correspond to the direction and size of effect size (c). | ||
While one-way ANOVA tests can be used to determine statistical significance, when this test is applied to very large data sets, such as those acquired in Raman mapping experiments, it is highly likely that even the smallest differences are found to be highly statistically significant. Therefore, the results were interpreted by measuring the effect size,44 an indicator of the magnitude of this difference (Fig. 4(c)) (a useful table for interpretation of effect size is given in ESI Table S2†). This allowed quantification of phenotypic response to drug treatment, with similar effect sizes suggesting a similar phenotypic response with respect to the lipid/(protein + lipid) composition.
To strengthen this ratiometric approach, a further five different ratios were measured in the same way, ultimately giving a multivariate descriptor for each combination of cell line and drug tested (ESI Fig. S5–S10†). Again these ratios could reflect global biochemical information with regards to lipid chain length (as a function of CH2/CH3 groups) and lipid saturation (as a function of H–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) /CH2 or CH3 groups) where 2881 cm−1 and 2974 cm−1 are assigned predominantly to C–H stretches in CH3 groups and 3013 cm−1 and 3064 cm−1 associated with C–H stretch in H–C groups.37,45 Although these are tentative peak assignments, and cannot be made exclusively due to the overlapping nature of the spectra, the 6 different peak intensities selected to create the ratiometric measurements were informed by a direct inspection of spectra. This allowed key regions of maximal change between control and drug treated samples to be selected, also based on knowledge regarding the chemical origin of the spectral signatures. Thus, instead of blindly analysing the full spectral data sets using multivariate techniques such as PCA, key peaks were selected by visual inspection of the spectra themselves. A large volume of data was generated from these ratios, therefore a method to combine these results into a simple descriptor, or ‘barcode’, was devised. For each ratio the effect size was used to assign a whole number value between −3 and 3 reflective of the magnitude of the change. These numbers were then used to form a ‘barcode’ for each drug treatment on each cell line (Fig. 5). This barcode could be used as a phenotypic stamp for each drug treatment, allowing barcodes for drugs eliciting known changes to be used to build up a bank of paired barcodes for phenotype. The total sum of the absolute values of all numbers in the barcode gave the final number in each barcode and acted as an overall indicator of the extent to which any particular treatment was altering the cells. The barcodes are simple representations of selected spectral features aiding in the rapid analysis of the effects of the drugs on these cells in areas of interest e.g. lipids rather than looking at full spectral changes. Further analysis of the barcoded data can be carried out using PCA (ESI Fig. S29–S31†) to allow comparisons of this relatively small data set but serves as an example of what is possible in further studies involving much larger datasets and barcodes. The most significant overall observation from this small example study was the marked selectivity of the beta-blocker propranolol for cancerous PC3 over non-cancerous PNT2 cells. Therefore, this drug is recommended for further studies and repurposing for investigation as an anti-cancer drug, supporting some recent studies on its anti-cancer activity with new biological insight into its action on lipid metabolism and selectivity for cancerous cells.46
/CH2 or CH3 groups) where 2881 cm−1 and 2974 cm−1 are assigned predominantly to C–H stretches in CH3 groups and 3013 cm−1 and 3064 cm−1 associated with C–H stretch in H–C groups.37,45 Although these are tentative peak assignments, and cannot be made exclusively due to the overlapping nature of the spectra, the 6 different peak intensities selected to create the ratiometric measurements were informed by a direct inspection of spectra. This allowed key regions of maximal change between control and drug treated samples to be selected, also based on knowledge regarding the chemical origin of the spectral signatures. Thus, instead of blindly analysing the full spectral data sets using multivariate techniques such as PCA, key peaks were selected by visual inspection of the spectra themselves. A large volume of data was generated from these ratios, therefore a method to combine these results into a simple descriptor, or ‘barcode’, was devised. For each ratio the effect size was used to assign a whole number value between −3 and 3 reflective of the magnitude of the change. These numbers were then used to form a ‘barcode’ for each drug treatment on each cell line (Fig. 5). This barcode could be used as a phenotypic stamp for each drug treatment, allowing barcodes for drugs eliciting known changes to be used to build up a bank of paired barcodes for phenotype. The total sum of the absolute values of all numbers in the barcode gave the final number in each barcode and acted as an overall indicator of the extent to which any particular treatment was altering the cells. The barcodes are simple representations of selected spectral features aiding in the rapid analysis of the effects of the drugs on these cells in areas of interest e.g. lipids rather than looking at full spectral changes. Further analysis of the barcoded data can be carried out using PCA (ESI Fig. S29–S31†) to allow comparisons of this relatively small data set but serves as an example of what is possible in further studies involving much larger datasets and barcodes. The most significant overall observation from this small example study was the marked selectivity of the beta-blocker propranolol for cancerous PC3 over non-cancerous PNT2 cells. Therefore, this drug is recommended for further studies and repurposing for investigation as an anti-cancer drug, supporting some recent studies on its anti-cancer activity with new biological insight into its action on lipid metabolism and selectivity for cancerous cells.46
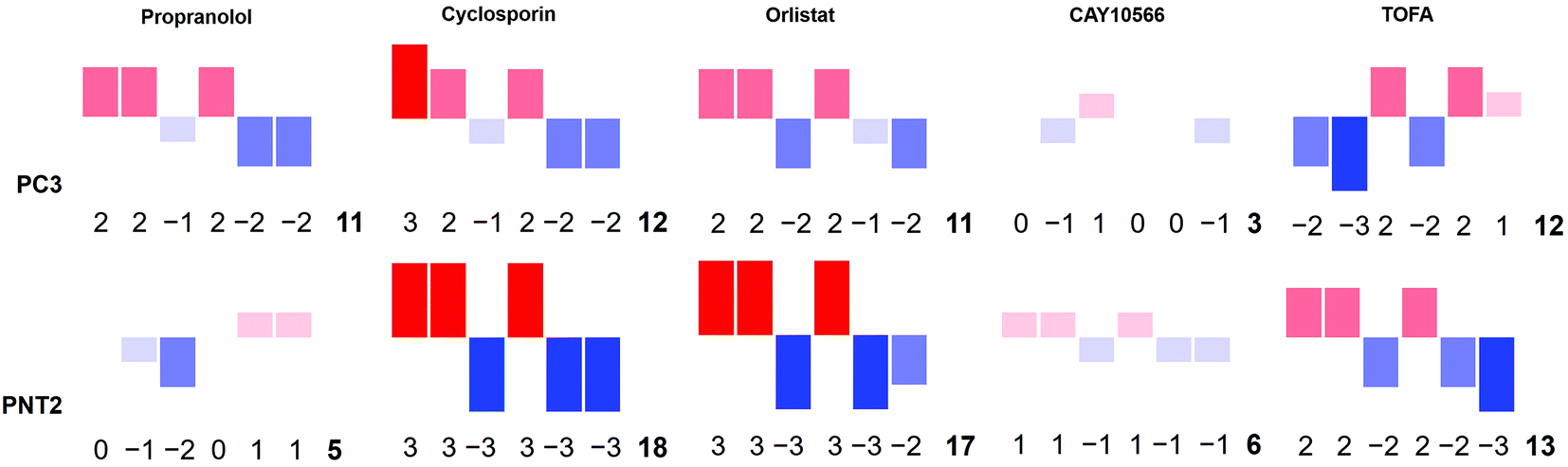 | ||
| Fig. 5 A Raman based phenotypic ‘barcode’. Barcodes were created for PC3 and PNT2 cells treated with a number of lipid altering drugs in comparison to a control cell population. Three Raman cell maps per condition per cell line were measured and for each point corresponding to a cell region, a total of six different intensity ratios were calculated. For each ratio, an effect size was calculated and a whole number value between −3 and 3 was assigned reflective of the size of this. The assigned numbers in the order corresponding to ratio 1, ratio 2, ratio 3, ratio 4, ratio 5, ratio 6 created the first six digits of the barcode. The final digit was the sum of the absolute values of all previous numbers. This was also represented pictorially where the size, direction and colour of bars corresponded to the assigned number for each ratio. Ratio 1 = 2851 cm−1/(2851 cm−1 + 2933 cm−1); ratio 2 = 2881 cm−1/(2881 cm−1 + 2933 cm−1); ratio 3 = 2974 cm−1/(2974 cm−1 + 2933 cm−1); ratio 4 = 2851 cm−1/(2851 cm−1 + 2974 cm−1); ratio 5 = 3013 cm−1/(3013 cm−1 + 2851 cm−1); ratio 6 = 3064 cm−1/(3064 cm−1 + 2933 cm−1). | ||
Conclusions
This study has demonstrated the capability of Raman spectroscopy using the high wavenumber spectral region, to study lipid distribution, metabolism and phenotypic drug response in a label free and chemically specific manner. The power of a simple ratiometric based approach was demonstrated, allowing high resolution, subcellular information regarding lipid abundance and distribution to be gained. A single bivariate descriptor extracted from Raman spectra was able to identify intracellular compartments, including nuclei, and ascertain intracellular changes to lipid abundance without the need for extrinsic markers, in contrast to fluorescence imaging which requires multiple dyes to achieve the same level of information. The reliability of the approach in addition to its added benefits was confirmed by comparison with results from a commercially available fluorescence staining kit.Comparison of ratiometric values between conditions gave a quantitative and robust measure of changes in lipid/(protein + lipid) content in cells. More universally, a statistically robust and meaningful strategy for assessing drug response was developed. Effect size as a meaningful measure of the extent of the difference between ratiometric Raman values for different conditions was highlighted. Moving from a single ratiometric value, a selection of important ratios ascertained by direct visual inspection of the spectra was used to create a phenotypic Raman ‘barcode’ for each drug response. By including control drugs with known phenotypic responses in the experiment, barcodes could be paired with phenotype for comparison to new drug candidates, and PCA could be used to determine reproducibility over multiple replicate experiments as well as what compounds elicited similar phenotypic response on individual cell lines and which compounds showed selective response between cell lines.
The developed strategy was applied to study the effect of drugs known to inhibit enzymes involved in de novo lipid synthesis as new candidates for cancer therapy. Orlistat elicited a phenotypic response characteristic of lipid accumulation in both cancerous and non-cancerous prostate cells, while CAY10566 showed very little response in both, and TOFA induced a decrease in intracellular lipids in PC3 cells in particular. Most notable was the selectivity of propranolol towards cancerous cells making this beta-blocker a strong candidate for repurposing for its selective anti-tumour action and demonstrates the power of using Raman spectroscopy to provide lead systems for further, more extensive studies.
More globally, this non-destructive label-free ratiometric and statistically meaningful Raman imaging strategy, using cost-effective glass substrates, could revolutionise understanding and analysis of drug–cell response and make this highly informative analytical technique amenable with cytological work streams. Future incorporation of this particular analytical approach into more high-throughput Raman work streams would enable a full realisation of its potential, allowing a comprehensive analysis of multiple different cell lines treated with any particular drug compound to fully elucidate efficacy and selectivity of any potential drug candidate.
Author contributions
LJ performed experiments, analysed data and prepared the manuscript. CW was responsible for cell maintenance. KF and DG contributed to the manuscript and DG was responsible for funding.Conflicts of interest
There are no conflicts of interest to declare.Data availability
Data from this work is available at DOI: 10.15129/eca751cb-8c6d-46b7-a8b0-13133e8e2526.References
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2016, 66, 7–30 CrossRef PubMed.
- D. A. Tennant, R. V. Duran and E. Gottlieb, Nat. Rev. Cancer, 2010, 10, 267–277 CrossRef PubMed.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef PubMed.
- W. H. Koppenol, P. L. Bounds and C. V. Dang, Nat. Rev. Cancer, 2011, 11, 325–337 CrossRef PubMed.
- O. Warburg, Science, 1956, 123, 309–314 CrossRef PubMed.
- S. Pavlides, D. Whitaker-Menezes, R. Castello-Cros, N. Flomenberg, A. K. Witkiewicz, P. G. Frank, M. C. Casimiro, C. Wang, P. Fortina, S. Addya, R. G. Pestell, U. E. Martinez-Outschoorn, F. Sotgia and M. P. Lisanti, Cell Cycle, 2009, 8, 3984–4001 CrossRef PubMed.
- Y. Fu, S. Liu, S. Yin, W. Niu, W. Xiong, M. Tan, G. Li and M. Zhou, Oncotarget, 2017, 8, 57813–57825 Search PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef PubMed.
- T. Mashima, H. Seimiya and T. Tsuruo, Br. J. Cancer, 2009, 100, 1369–1372 CrossRef PubMed.
- F. Baenke, B. Peck, H. Miess and A. Schulze, Dis. Models & Mech., 2013, 6, 1353–1363 Search PubMed.
- S. Beloribi-Djefaflia, S. Vasseur and F. Guillaumond, Oncogenesis, 2016, 5, e189 CrossRef PubMed.
- M. Omabe, M. Ezeani and K. N. Omabe, J. Appl. Biomed., 2015, 13, 47–59 CrossRef.
- E. S. Pizer, F. J. Chrest, J. A. DiGiuseppe and W. F. Han, Cancer Res., 1998, 58, 4611 Search PubMed.
- M. Lee, Basic Skills in Interpreting Laboratory Data, ASHP, 2009 Search PubMed.
- N. Long and W. T. Wong, The Chemistry of Molecular Imaging, Wiley, 2014 Search PubMed.
- C. Schultz, A. B. Neef, T. W. Gadella and J. Goedhart, Cold Spring Harb Protoc., 2010, 5 DOI:10.1101/pdb.top83.
- X. Nan, J. X. Cheng and X. S. Xie, J. Lipid Res., 2003, 44, 2202–2208 CrossRef PubMed.
- M. K. Passarelli and N. Winograd, Biochim. Biophys. Acta, 2011, 1811, 976–990 CrossRef PubMed.
- T. T. Le, T. B. Huff and J.-X. Cheng, BMC Cancer, 2009, 9, 42 CrossRef PubMed.
- H. J. Butler, L. Ashton, B. Bird, G. Cinque, K. Curtis, J. Dorney, K. Esmonde-White, N. J. Fullwood, B. Gardner, P. L. Martin-Hirsch, M. J. Walsh, M. R. McAinsh, N. Stone and F. L. Martin, Nat. Protoc., 2016, 11, 664–687 CrossRef PubMed.
- H. J. Byrne, M. Baranska, G. J. Puppels, N. Stone, B. Wood, K. M. Gough, P. Lasch, P. Heraud, J. Sule-Suso and G. D. Sockalingum, Analyst, 2015, 140, 2066–2073 RSC.
- A. F. García-Flores, L. Raniero, R. A. Canevari, K. J. Jalkanen, R. A. Bitar, H. S. Martinho and A. A. Martin, Theor. Chem. Acc., 2011, 130, 1231–1238 Search PubMed.
- M. Janssens, J. van Smeden, G. J. Puppels, A. P. M. Lavrijsen, P. J. Caspers and J. A. Bouwstra, Br. J. Dermatol., 2014, 170, 1248–1255 CrossRef PubMed.
- X. Wu, G. Daniels, P. Lee and M. E. Monaco, Am. J. Clin. Exp. Urol., 2014, 2, 111–120 Search PubMed.
- N. W. Clarke and M. D. Brown, Eur. Urol., 2007, 52, 3–4 CrossRef PubMed.
- J. Suburu and Y. Q. Chen, Prostaglandins Other Lipid Mediators, 2012, 98, 1–10 CrossRef PubMed.
- J. V. Swinnen, T. Roskams, S. Joniau, H. V. Poppel, R. Oyen, L. Baert, W. Heyns and G. Verhoeven, Int. J. Cancer, 2002, 98, 19–22 CrossRef PubMed.
- K. Shafique, P. McLoone, K. Qureshi, H. Leung, C. Hart and D. S. Morrison, BMC Cancer, 2012, 12, 25 CrossRef PubMed.
- S. J. Kridel, F. Axelrod, N. Rozenkrantz and J. W. Smith, Cancer Res., 2004, 64, 2070 CrossRef PubMed.
- J. A. Menendez, L. Vellon and R. Lupu, Ann. Oncol., 2005, 16, 1253–1267 CrossRef PubMed.
- Y. Uto, Chem. Phys. Lipids, 2016, 197, 3–12 CrossRef PubMed.
- C. Wang, C. Xu, M. Sun, D. Luo, D.-f. Liao and D. Cao, Biochem. Biophys. Res. Commun., 2009, 385, 302–306 CrossRef PubMed.
- E. Currie, A. Schulze, R. Zechner, T. C. Walther and R. V. Farese, Cell Metab., 2013, 18, 153–161 CrossRef PubMed.
- P. Nioi, B. K. Perry, E.-J. Wang, Y.-Z. Gu and R. D. Snyder, Toxicol. Sci., 2007, 99, 162–173 CrossRef PubMed.
- M. Grandl and G. Schmitz, Cytometry, Part A, 2010, 77, 231–242 Search PubMed.
- D. A. Orringer, B. Pandian, Y. S. Niknafs, T. C. Hollon, J. Boyle, S. Lewis, M. Garrard, S. L. Hervey-Jumper, H. J. L. Garton, C. O. Maher, J. A. Heth, O. Sagher, D. A. Wilkinson, M. Snuderl, S. Venneti, S. H. Ramkissoon, K. A. McFadden, A. Fisher-Hubbard, A. P. Lieberman, T. D. Johnson, X. S. Xie, J. K. Trautman, C. W. Freudiger and S. Camelo-Piragua, Nat. Biomed. Eng., 2017, 1, 0027 CrossRef PubMed.
- A. C. S. Talari, Z. Movasaghi, S. Rehman and I. u. Rehman, Appl. Spectrosc. Rev., 2015, 50, 46–111 CrossRef.
- B. Alberts, Molecular Biology of the Cell, Garland Pub., 1989 Search PubMed.
- T. C. Walther and R. V. Farese, Annu. Rev. Biochem., 2012, 81, 687–714 CrossRef PubMed.
- P. T. Bozza and J. P. B. Viola, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2010, 82, 243–250 CrossRef PubMed.
- W. A. Alaynick, Mitochondrion, 2008, 8, 329–337 CrossRef PubMed.
- I. R. D. Johnson, E. J. Parkinson-Lawrence, T. Shandala, R. Weigert, L. M. Butler and D. A. Brooks, Mol. Cancer Res., 2014, 12, 1851–1862 CrossRef PubMed.
- M. Jimenez-Hernandez, M. D. Brown, C. Hughes, N. W. Clarke and P. Gardner, Analyst, 2015, 140, 4453–4464 RSC.
- S. Nakagawa and I. C. Cuthill, Biol. Rev., 2007, 82, 591–605 CrossRef PubMed.
- D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli, in The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, Academic Press, San Diego, 1991, pp. 9–28, DOI:10.1016/B978-0-08-057116-4.50008-0.
- P. Pantziarka, G. Bouche, V. Sukhatme, L. Meheus, I. Rooman and V. P. Sukhatme, Ecancermedicalscience, 2016, 10, 680 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02312c |
| This journal is © The Royal Society of Chemistry 2018 |