
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDivergent ring-opening coupling between cyclopropanols and alkynes under cobalt catalysis†
Junfeng
Yang
,
Yixiao
Shen
,
Yang Jie
Lim
and
Naohiko
Yoshikai
 *
*
Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore. E-mail: nyoshikai@ntu.edu.sg
First published on 16th July 2018
Abstract
Cobalt–diphosphine catalysts promote ring-opening coupling reactions between cyclopropanols and unactivated internal alkynes, affording either β-alkenyl ketones or multisubstituted cyclopentenol derivatives in good yields with good to excellent regioselectivities. The chemoselectivity between these β-alkenylation and [3 + 2] annulation reactions, which likely share a cobalt homoenolate as a key catalytic intermediate, is exquisitely controlled by the reaction conditions, with the solvent being a major controlling factor. The reactions are proposed to involve ring opening of cobalt cyclopropoxide into homoenolate, migratory insertion of the alkyne into the Co–C bond, and protodemetalation or intramolecular carbonyl addition of the resulting alkenylcobalt species. The feasibility of these reaction steps was supported by DFT calculations.
Introduction
The strain-driven C–C bond cleavage of small ring compounds offers a unique entry for the synthesis of linear or larger-ring compounds.1 Among strained carbocycles, cyclopropanols and related compounds have received considerable attention as readily accessible starting materials that can undergo ring opening with the aid of various reagents such as bases, acids, and metal complexes.2 In this context, the chemistry of metal homoenolates (or β-metallocarbonyl compounds) generated from siloxycyclopropanes represents a significant milestone (Scheme 1a).3 Exposure of siloxycyclopropanes to Lewis acidic metal salts such as ZnCl2 and TiCl4 causes ring-opening to generate the corresponding homoenolates, which are amenable to trapping with typical organic electrophiles such as aldehydes, enones, and organic halides in the presence of appropriate catalysts or additives, affording β-functionalized carbonyl compounds. A major drawback of this chemistry is, however, that it often involves a preformed homoenolate and thus inevitable consumption of a stoichiometric metal salt.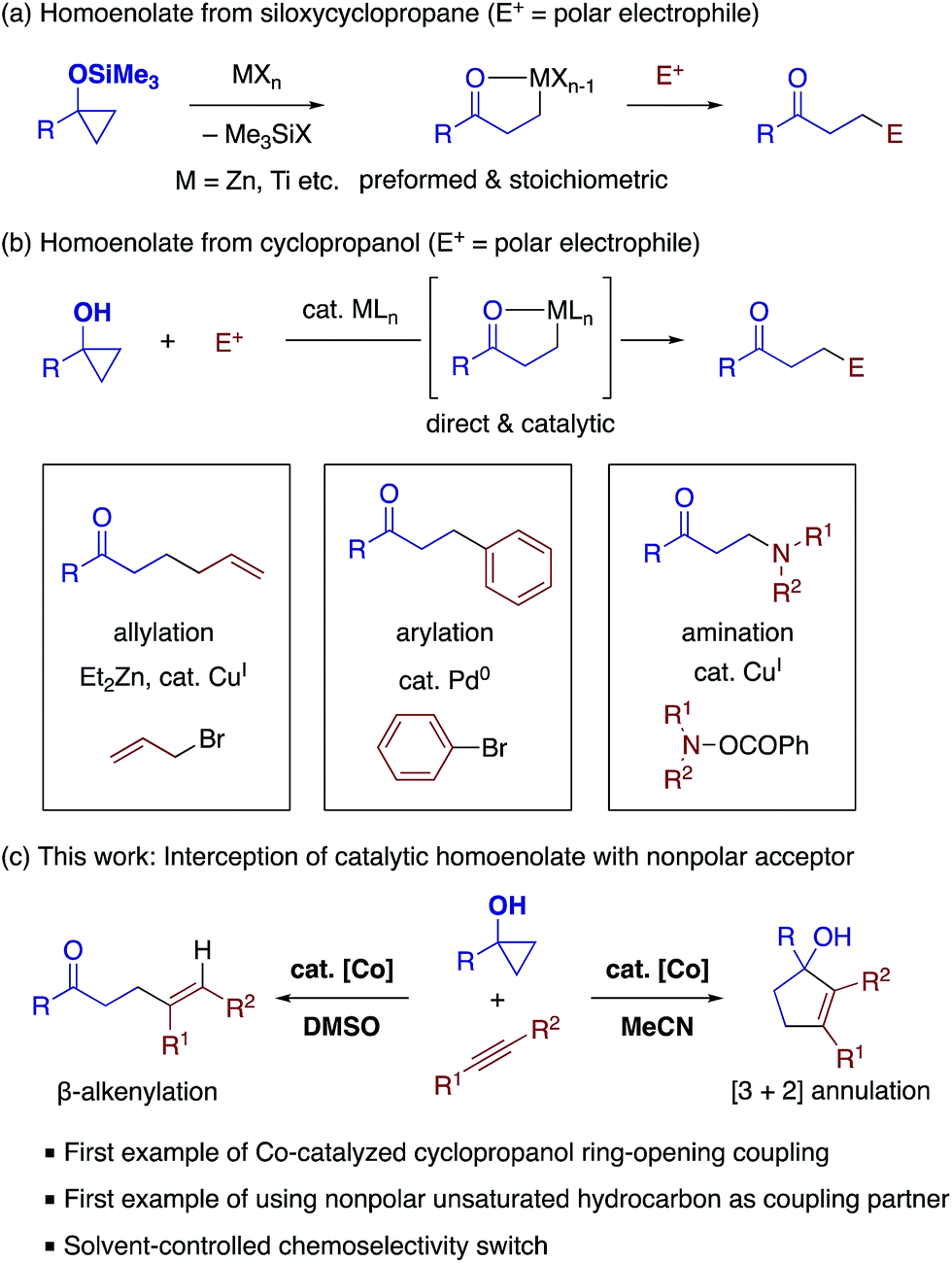 | ||
| Scheme 1 Ring-opening transformations of cyclopropanol derivatives via homoenolate. | ||
Recently, significant progress has been made in the generation of homoenolates from unmasked cyclopropanols via metal cyclopropoxides and their use as catalytic intermediates for cross-coupling with electrophiles (Scheme 1b).4 This strategy allows for direct and often fully catalytic conversion of cyclopropanols into a variety of β-functionalized ketones. Notable examples in this respect include allylation via copper catalysis of zinc homoenolates,5 arylation via palladium homoenolates,6,7 and amination via copper homoenolates,8 among others.9 Besides the homoenolate-based approach, recent years have also witnessed other mechanistically distinct types of ring-opening transformations of cyclopropanols, which employ coupling partners such as (fluoro)alkyl halides,10 alkynylbenziodoxoles,11 diazo compounds,12 and so on4 to access the corresponding β-functionalized ketones. Despite all the progress in the stoichiometric and catalytic cross-couplings via metal homoenolates, viable coupling partners have thus far been limited to polar organic electrophiles, and interception of a homoenolate with a nonpolar unsaturated hydrocarbon as a reaction partner has been elusive.
Herein, we report that a cobalt catalyst promotes ring-opening coupling between a cyclopropanol and a nonpolar internal alkyne to afford a β-alkenyl ketone or a cyclopentenol derivative, with the solvent being a major selectivity-controlling factor (Scheme 1c). These divergent β-alkenylation and [3 + 2] annulation reactions likely involve a cobalt homoenolate as a common catalytic intermediate, and mark a sharp contrast with previously reported ring-opening alkenylation and [3 + 2] annulation of siloxycyclopropanes with activated, ester-substituted alkynes via stoichiometric homoenolates.13 While intramolecular reactions between cyclopropanol and alkyne moieties of 1-alkynylcyclopropanols have been explored using Co, Au, or Ru catalysts to achieve ring-opening rearrangement into cyclopentenones or methylenecyclobutanes,14 the present reactions represent the first example of intermolecular coupling between cyclopropanol and unfunctionalized alkyne. Featuring broad scope and high stereo- and regioselectivity with respect to alkynes, the present reactions offer unique approaches to γ,δ-unsaturated ketones and cyclopentenols, which would complement existing approaches including reductive coupling/cycloaddition between enones and alkynes.15,16
Results and discussion
Pursuing our continuing interest in cobalt-catalyzed C–C bond formation and transformation of unactivated alkynes,17–19 the present study commenced with exploration of the coupling between 1-phenylcyclopropanol (1a) and 5-decyne (2a) (Table 1). Through extensive experimentation, the feasibility of the β-alkenylation was revealed first. A catalytic system comprised of CoBr2 (10 mol%), 1,2-bis(diphenylphosphino)ethane (dppe, 10 mol%), Zn dust (50 mol%), and 1,4-diazabicyclo[2.2.2]octane (DABCO, 150 mol%) promoted the reaction in dimethylsulfoxide (DMSO) at 80 °C, affording β-alkenyl ketone 3aa in 97% yield with exclusive syn-selectivity (entry 1). While 1,3-bis(diphenylphosphino)propane (dppp) was equally effective as dppe (entry 2), later, dppe proved to show better performance than other diphosphines for 1-benzylcyclopropanol (Table S1†). The reaction became sluggish under ligand-free conditions (entry 3). Zn would play an important role as a reductant,20 as its replacement by Mn or its removal resulted in a sharp drop in the yield (entries 4 and 5). The beneficial effect of DABCO was apparent from control experiments performed with a lower loading or in its absence (entries 6 and 7).|
|
|||
|---|---|---|---|
| Entry | Deviation from std conditions | Yieldb (%) | |
| 3a | 4a | ||
| a The reaction was performed using 0.15 mmol of 1a and 0.1 mmol of 2a (0.3 M). dppe = 1,2-bis(diphenylphosphino)ethane; dppp = 1,3-bis(diphenylphosphino)propane. b Determined by GC using mesitylene as an internal standard. | |||
| 1 | None | 97 | 0 |
| 2 | dppp instead of dppe | 98 | 0 |
| 3 | dppe omitted | 29 | 0 |
| 4 | Mn instead of Zn | 30 | 0 |
| 5 | Zn omitted | 19 | 0 |
| 6 | DABCO (50 mol%) | 62 | 4 |
| 7 | DABCO omitted | 11 | 0 |
| 8 | DMF instead of DMSO | 70 | 10 |
| 9 | MeCN instead of DMSO | 8 | 80 |
| 10 | CoI2 and MeCN instead of CoBr2 and DMSO | 3 | 88 |
Interestingly, the solvent was found to have a dramatic impact on the reaction outcome. While dimethylformamide (DMF) gave rise to a small amount of cyclopentenol 4a (entry 8), the reaction in MeCN afforded 4a as the major product in 80% yield along with a small amount of 3a (entry 9). The annulation/alkenylation selectivity was further enhanced to >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by using CoI2 as the precatalyst (entry 10). Note that neither the β-alkenylation nor the [3 + 2] annulation took place in the absence of the cobalt catalyst.
:1 by using CoI2 as the precatalyst (entry 10). Note that neither the β-alkenylation nor the [3 + 2] annulation took place in the absence of the cobalt catalyst.
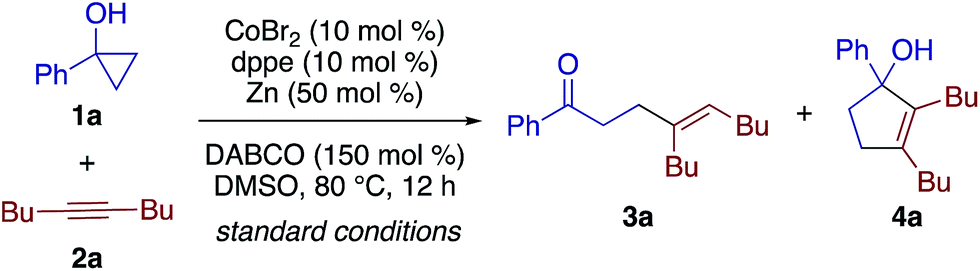
We explored the generality of both the β-alkenylation and [3 + 2] annulation reactions. Scheme 2 shows the scope of the β-alkenylation with respect to cyclopropanols. A variety of 1-aryl- and 1-alkylcyclopropanols smoothly participated in the reaction with 2a to afford the corresponding adducts 3a–3m in moderate to high yields. A partial dechlorination was observed in the reaction of 1-(4-chlorophenyl)cyclopropanol, presumably due to the oxidative addition of the C–Cl bond to the cobalt catalyst (see 3e). Tetrahydronaphthalene-fused cyclopropanol 1n underwent exclusive cleavage of the less substituted C–C bond to give the adduct 3n in 90% yield. While monocyclic 1,2-disubstituted cyclopropanol 1o reacted sluggishly under the standard conditions, the reaction under modified conditions using 1,4-bis(diphenylphosphino)butane (dppb) and MeCN afforded β-alkenyl ketone 3o in 54% yield, again via cleavage of the less substituted C–C bond. In contrast to these 1-aryl-2-alkylcyclopropanols, 1,2-diphenylcyclopropanol and 1-methyl-2-phenylcyclopropanol failed to undergo β-alkenylation reaction with 2a but decomposed into 1,3-diphenylpropan-1-one and 4-phenylbutan-2-one, respectively, as a result of cleavage of the more substituted C–C bond. This observation may be rationalized by the formation of cobalt homoenolate with the more stable benzylic C–Co bond. In addition, the reaction of a secondary cyclopropanol, 2-methyl-2-phenylcyclopropanol, with 2a failed to afford either a β-alkenylation product or [3 + 2] annulation product but resulted in decomposition into intractable products. Note that the reaction of 1a could be performed on a 3 mmol scale, affording 3a in 75% yield.
 | ||
| Scheme 2 Scope of β-alkenylation with respect to cyclopropanols. aObtained as a mixture with dechlorinated product (8%). | ||
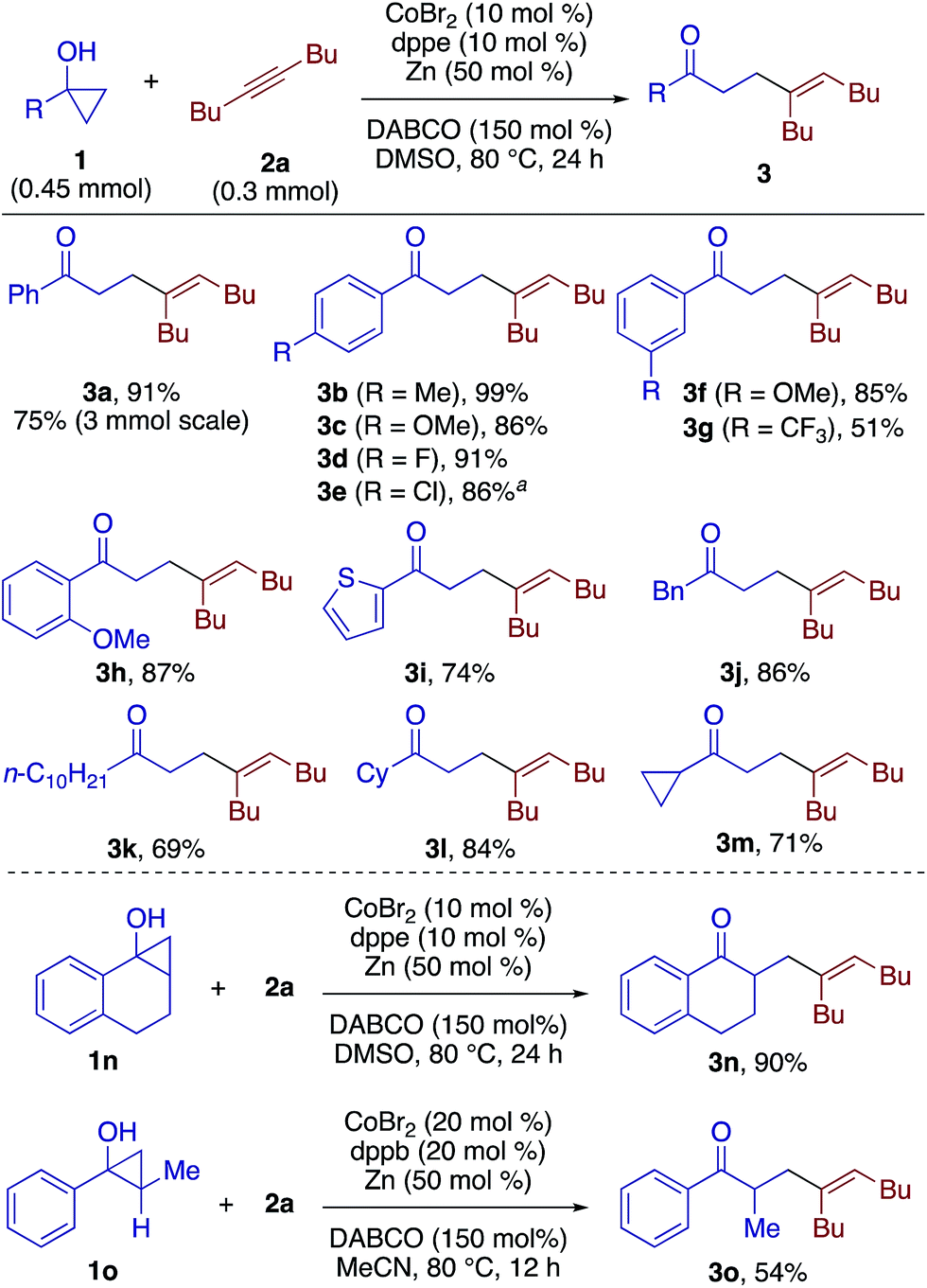
The β-alkenylation of 1a with 1-phenyl-1-butyne (2b) under the above conditions took place smoothly but with a moderate regioselectivity (4.4:1), with preferential C–C bond formation at the proximity of the ethyl group. Upon reexamination of diphosphine ligands (Table S2†), 1,6-bis(diphenylphosphino)hexane (dpphex) was found to exhibit higher regioselectivity (9:1) without compromising the yield (Scheme 3, 3p). The modified catalytic system was applicable to a variety of aryl(alkyl)alkynes, displaying moderate to high regioselectivities (3:1 to ≥20:1) and tolerance to ester (3t, 3ab), chloro (3v), trifluoromethyl (3w), bulky aryl (3z), and thienyl (3ad) groups. The reaction of an enyne substrate also took place smoothly, affording γ,δ,ε,ζ-unsaturated ketone 3ae in good yield and regioselectivity. Diphenylacetylene also took part in the reaction with 1a to give the adduct 3af with good stereoselectivity. Terminal alkynes such as phenylacetylene failed to afford any of the possible addition products.
 | ||
| Scheme 3 Scope of β-alkenylation with respect to alkynes. | ||
The scope of the [3 + 2] annulation was explored next (Scheme 4). A series of 1-arylcyclopropanols reacted with 2a to give the desired products 4a–4g in good yields except for the one bearing a para-methoxy group (4c). The low yield of 4c was the result of its facile decomposition on silica gel during purification, which could be attributed to the instability of the electron-rich tertiary alcohol moiety. 1-Alkylcyclopropanols were also amenable to the [3 + 2] annulation (see 4h and 4i). Note that the 1,2-disubstituted cyclopropanols 1n and 1o failed to undergo the annulation reaction, and also did not give any β-alkenylation products. As a substantial amount of the cyclopropanol remained unreacted, we speculate that both the homoenolate formation and the alkyne insertion steps were problematic for these cases. Upon a slight modification of the reaction conditions (Table S3†), the annulation of 1a and 1-phenyl-1-butyne using dppp (10 mol%) and DABCO (50 mol%) afforded cyclopentenol 4j in 85% yield with exclusive regioselectivity (>20:1) and good product selectivity (8:1). Likewise, a variety of aryl(alkyl)alkynes could be annulated with 1a to afford cyclopentenols 4k–4u in moderate to high yields. The enyne and diphenylacetylene also proved to be excellent substrates for the present annulation (see 4v and 4w). Note that most of the [3 + 2] annulation reactions were accompanied by the competing β-alkenylation, while the degree of competition was generally low as judged from TLC analysis of the crude mixture. For the case of 4j and 4l, the yields of the accompanying β-alkenylation products were rather low (3p, 6%; 3r, 3%).
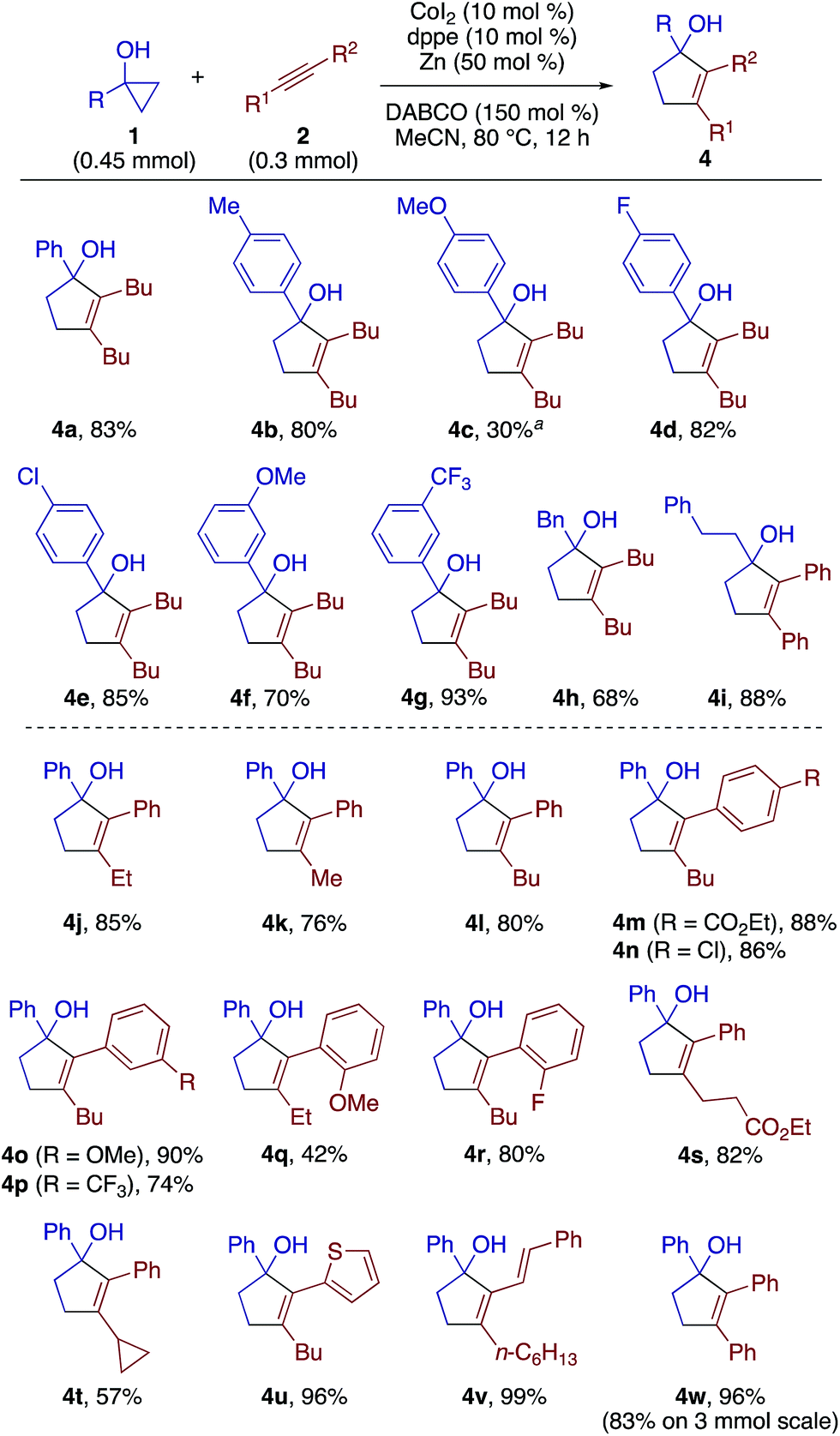 | ||
| Scheme 4 Scope of [3 + 2] annulation. The reactions for 4i–4w were performed using 10 mol% of dppp (instead of dppe) and 50 mol% of DABCO at 0.1 M. aThe yield was determined by 1H NMR. | ||
Our preliminary screening demonstrated the feasibility of an enantioselective [3 + 2] annulation using a chiral cobalt catalyst (Scheme 5; see also Table S4†). Thus, asymmetric induction in the reaction of 1a and 2a was observed using a host of chiral diphosphine ligands having different backbones, among which (R,R)-QuinoxP* displayed the highest ee of 93% albeit in a low yield. While further screening of ligands, additives, and other reaction conditions is currently underway to improve the yield and enantioselectivity, these results would serve as a testament to the reaction mechanism involving an alkenylcobalt species (vide infra).
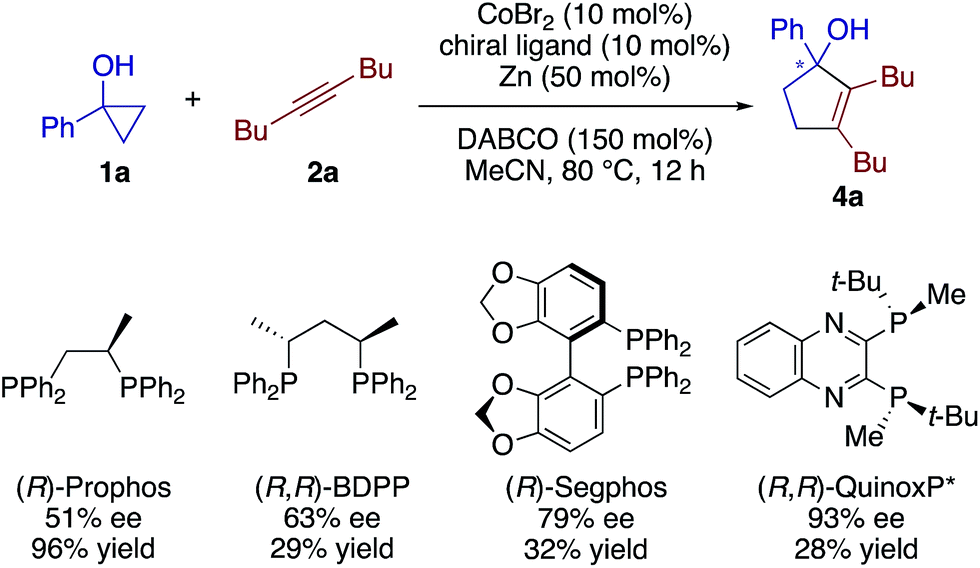 | ||
| Scheme 5 Enantioselective [3 + 2] annulation. | ||
The β-alkenyl ketone products would be amenable to a number of follow-up synthetic transformations. To demonstrate simultaneous utilization of the ketone and olefin moieties, we derivatized some of the β-alkenyl ketones into the corresponding O-pivaloyl oximes, and subjected them to the copper-catalyzed aza-Heck type reaction21 to obtain 3,4-dihydro-2H-pyrrole derivatives 6a–6c in good yields (Scheme 6a). As a demonstration of the utility of the [3 + 2] adducts, the cyclopentenol 4w was readily converted to 1,2,3-trisubstituted cyclopentadiene 7 and the corresponding cyclopentadienyl (CpX)–RhIII complex 8 (Scheme 6b). Thus, the present [3 + 2] annulation would allow for convenient access to structurally diverse CpX–M complexes, which would find many applications in catalysis.22
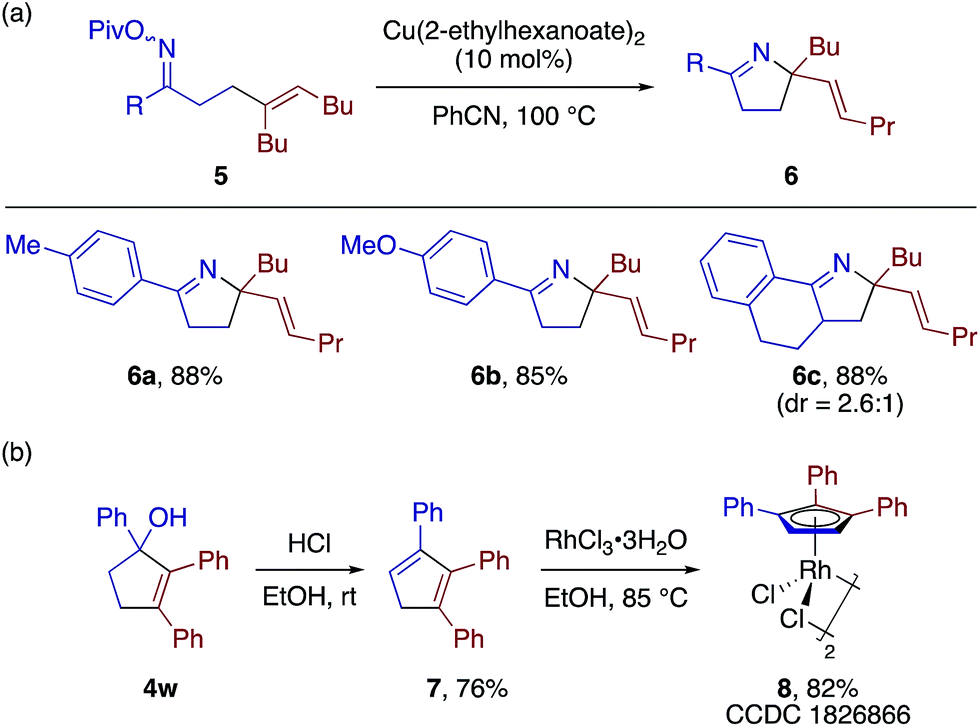 | ||
| Scheme 6 Product transformations. | ||
Several control experiments were performed to gain mechanistic insight into the β-alkenylation reaction. To probe the role of each component of the catalytic system in the ring-opening of cyclopropanol, conversion of 1-benzylcyclopropanol (1j) into 1-phenylbutan-2-one was examined under different conditions in the absence of alkyne (Scheme 7a; see also Table S5†). As a result, the ring opening was found to take place most smoothly when both CoBr2 and Zn were present, while dppe and DABCO were not indispensable for the ring opening. In addition, ZnBr2, which should be generated upon reduction of CoBr2 with Zn, only sluggishly promoted the ring opening in the presence or absence of DABCO. Thus, a cobalt(I) species generated from the cobalt(II) precatalyst and Zn20 appears to play a major role in the ring opening, and formation of a zinc homoenolate from cyclopropanol and zinc(II)9c is less likely to operate under the present conditions. The reaction of deuterium-labeled cyclopropanol 1a-d (68% D at the hydroxyl group) with 2a resulted in the adduct with partial deuterium incorporation into the vinylic and the carbonyl α-positions (Scheme 7b). A greater degree of deuterium incorporation was observed for the reaction of 1a with 2a in the presence of CD3OD (1.5 equiv.), and the degree of deuterium incorporation was further enhanced with an increased amount (10 equiv.) of CD3OD (Scheme 7c). These observations suggest that not only the alcoholic protons but also the carbonyl α-protons are responsible as the proton source for the vinylic position. We also confirmed that radical scavengers such as 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) and 1,1-diphenylethylene did not interfere with either the β-alkenylation or [3 + 2] annulation.
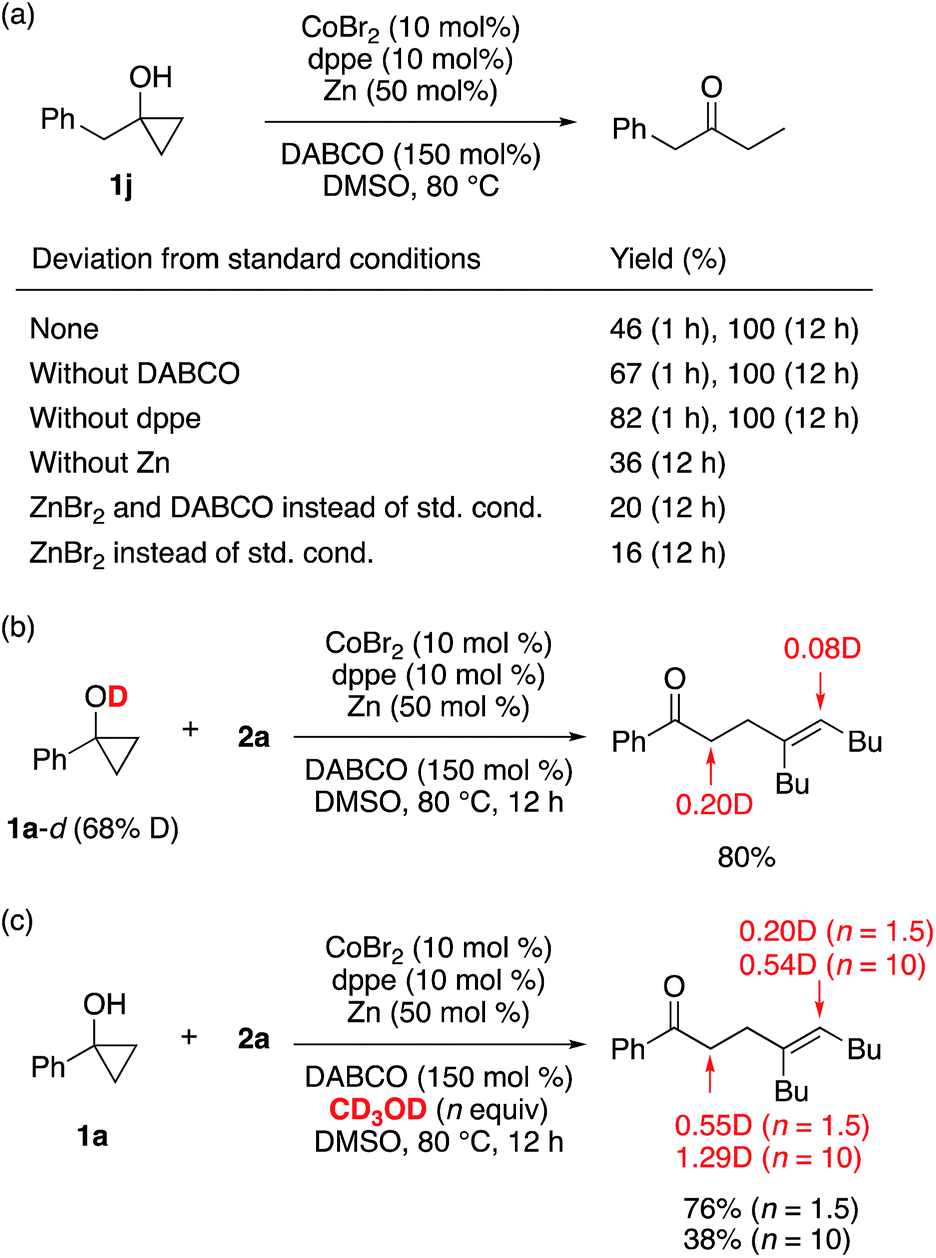 | ||
| Scheme 7 Control experiments. | ||
Scheme 8 illustrates proposed catalytic cycles for the β-alkenylation and [3 + 2] annulation reactions. We consider that these reactions share a cobalt homoenolate C, which would be formed from a cobalt(I) species A (generated from the cobalt(II) precatalyst and Zn), cyclopropanol 1, and base (DABCO) via a cobalt cyclopropoxide B, as a common intermediate. Insertion of alkyne 2 into C would give an alkenylcobalt species D,23 which would then undergo protodemetalation to furnish β-alkenyl ketone 3 or intramolecular carbonyl addition18c and protodemetalation to afford cyclopentenol 4. The proton source in the former protodemetalation step would be the alcoholic proton of cyclopropanol (or externally added alcohol) or the carbonyl α-proton of the β-alkenyl ketone 3, as suggested by the deuterium labeling experiments (Scheme 7b and c). DABCO is assumed to serve as a proton shuttle, which would facilitate the formation of B and the catalytic turnover (D to A or E to A) while being engaged in the exchange of the alcoholic protons of 1 and the carbonyl α-protons of 3. We are tempted to interpret the solvent-controlled product divergence in terms of the solvent-dependent acidity.24 Thus, the proton source (e.g., base·HX, cyclopropanol) would exhibit higher acidity in more polar solvents, and hence direct protodemetalation of the alkenylcobalt intermediate D would be favored in DMSO (ε = 46.8) rather than in MeCN (ε = 35.7). In addition, coordination of the carbonyl oxygen to the cobalt center in D might be stronger in less Lewis basic MeCN, thus assisting intramolecular carbonyl addition in MeCN. The possibility of a radical mechanism for the cyclopropanol opening and a β-keto radical intermediate may be excluded on the basis of the cleavage of the less substituted C–C bond of 1n and 1o (Scheme 2), the exclusive syn-selectivity of the alkenylation, and the lack of inhibitory effects of radical scavengers.
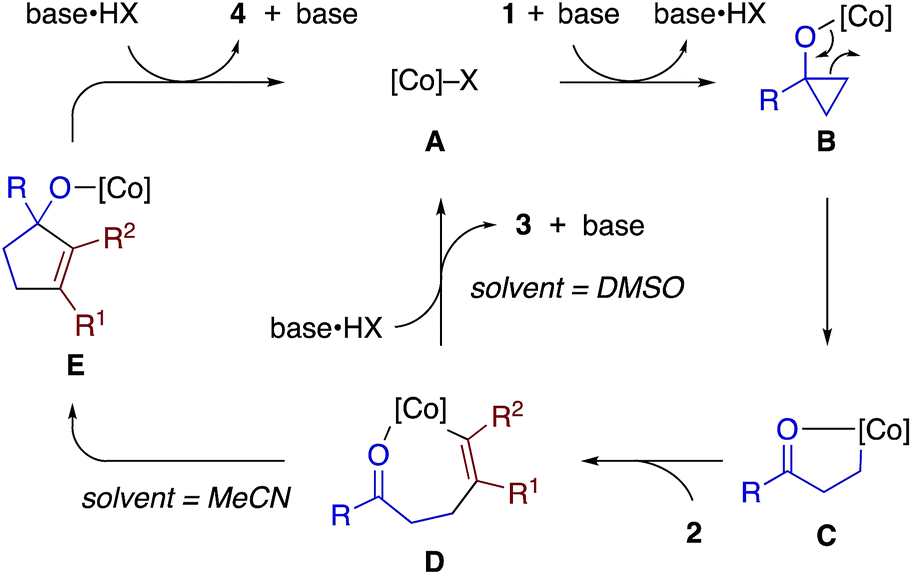 | ||
| Scheme 8 Proposed reaction pathways. | ||
To probe the feasibility of the key steps in the proposed mechanism, we performed density functional theory (DFT) calculations on a model reaction using 1-methylcyclopropanol and 2-butyne as the reactants and a (dppe)cobalt(I)-cyclopropoxide (CP1s, singlet or CP1t, triplet) as the starting complex (Scheme 9). Geometry optimization was performed in the gas phase with the M06L functional25 using the Stuttgart/Dresden effective core potential (SDD)26 for cobalt and the 6-31G(d) basis set for all other atoms. For the optimized structures, single-point energy calculations were performed with the M06L functional and the def2-TZVP basis set27 for all atoms using the SMD solvation model28 for acetonitrile to obtain free energy profiles.29 While CP1t was found to be more stable than CP1s by 11.2 kcal mol−1, viable reaction pathways for the [3 + 2] annulation could be identified on both the singlet and triplet surfaces.30 On the singlet surface, CP1s undergoes very facile and exergonic ring opening (TS1s, ΔG‡ = 0.7 kcal mol−1) to give cobalt homoenolate CP2s with a square-planar geometry. This is followed by complexation of the alkyne (CP3s) and slightly endergonic syn-migratory insertion into the Co–C bond (TS2s; ΔG‡ = 22.2 kcal mol−1) to afford non-chelated alkenylcobalt species CP4s. Cyclization of CP4s requires very low activation energy (TS3s; ΔG‡ = 0.7 kcal mol−1) to afford the cyclized product complex CP5s. The triplet complex CP1t also undergoes facile and exergonic ring opening (TS1t, ΔG‡ = 8.4 kcal mol−1) to give homoenolate CP2t with a distorted tetrahedral geometry. Complexation of the alkyne to CP2t is endergonic (CP3t), and subsequent alkyne insertion (TS2t; ΔG‡ from CP2t = 28.3 kcal mol−1) occurs with large exergonicity to give alkenylcobalt species CP4t, which is chelated by the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group through π-coordination. Intramolecular cyclization of CP4t takes place through TS3t with modest activation energy (9.9 kcal mol−1) to afford cyclized product complex CP5t.
O group through π-coordination. Intramolecular cyclization of CP4t takes place through TS3t with modest activation energy (9.9 kcal mol−1) to afford cyclized product complex CP5t.
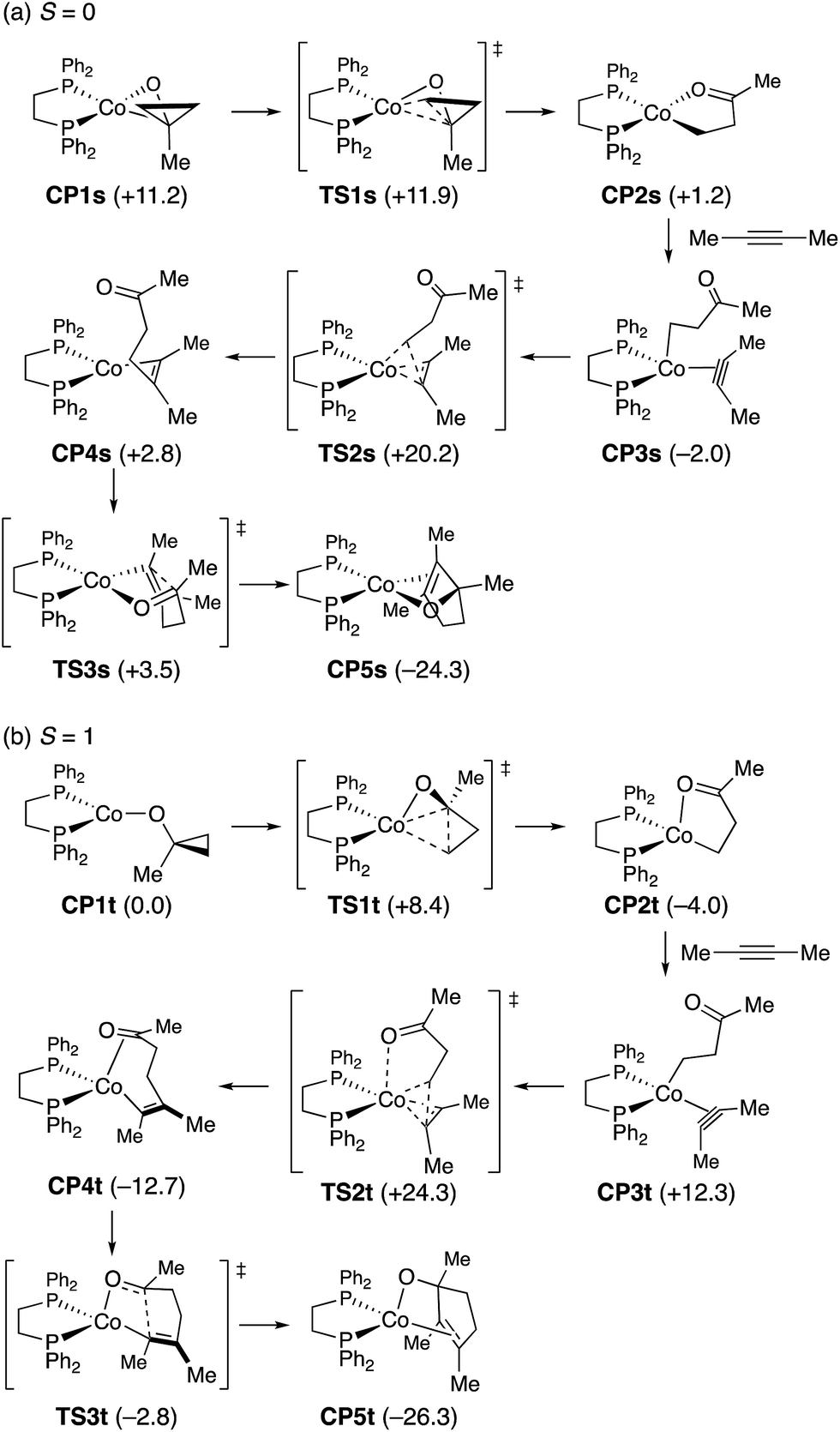 | ||
| Scheme 9 Reaction pathways for (dppe)Co(I)-mediated [3 + 2] annulation of 1-methylcyclopropanol and 2-butyne starting from cobalt cyclopropoxide species (CP1s for singlet and CP1t for triplet), obtained by DFT calculations (SMD(MeCN)-M06L/def2-TZVP//M06L/SDD(Co)-6-31G(d)). Values in the parentheses refer to free energies relative to CP1t. | ||
The energy diagrams in Fig. 1 demonstrate that there is no critically large energy gap between the singlet and triplet surfaces, and both the reaction pathways would be feasible on their own. The crossings of the singlet and the triplet surfaces suggest that spin crossover may take place multiple times throughout the reaction, e.g., upon alkyne complexation (CP2 to CP3; triplet to singlet) and alkyne insertion (TS2 to CP4; singlet to triplet). Despite this complexity, it is clear that the alkyne insertion step requires a distinctly higher activation barrier than other steps, that is, ring opening and intramolecular carbonyl addition.
 | ||
| Fig. 1 Energy diagrams for (dppe)Co(I)-mediated [3 + 2] annulation of 1-methylcyclopropanol and 2-butyne (blue, S = 0; red, S = 1). | ||
Conclusions
In summary, we have disclosed cobalt-catalyzed divergent coupling reactions between cyclopropanols and internal alkynes, which afford β-alkenyl ketones or 1,2,3-trisubstituted cyclopentenols with high syn-selectivity and good to high regioselectivity. These β-alkenylation and [3 + 2] annulation reactions likely share the same reaction steps involving ring-opening formation of a cobalt homoenolate and subsequent alkyne insertion to form an alkenylcobalt intermediate. The fate of the alkenylcobalt intermediate, i.e., the chemoselectivity of the reaction, can be exquisitely controlled by the reaction conditions, with the solvent being the major controlling factor. The feasibility of the key steps in the proposed reaction mechanism was supported by DFT calculations. Overall, by engaging nonpolar alkynes as reaction partners, the present study has expanded the scope of direct catalytic transformation of cyclopropanols under transition metal catalysis. The development of an enantioselective variant of the [3 + 2] annulation reaction and further exploration of cobalt-catalyzed cross-couplings between cyclopropanols and other nonpolar unsaturated hydrocarbons and their mechanistic underpinnings are ongoing in our laboratory and will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Ministry of Education (Singapore) and Nanyang Technological University (RG3/15, RG114/15, and MOE2016-T2-2-043). We thank Dr Rakesh Ganguly and Dr Yongxin Li (Nanyang Technological University) for their assistance with the X-ray crystallographic analysis. The High-Performance Computing Centre of NTU is acknowledged for computer resources.Notes and references
- (a) G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404 CrossRef PubMed; (b) P.-H. Chen, B. A. Billett, T. Tsukamoto and G. Dong, ACS Catal., 2017, 7, 1340 CrossRef PubMed; (c) I. Marek, A. Masarwa, P. O. Delaye and M. Leibeling, Angew. Chem., Int. Ed., 2015, 54, 414 Search PubMed; (d) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410 CrossRef PubMed; (e) M. Murakami and T. Matsuda, Chem. Commun., 2011, 47, 1100 RSC.
- O. G. Kulinkovich, Chem. Rev., 2003, 103, 2597 CrossRef PubMed.
- I. Kuwajima and E. Nakamura, Top. Curr. Chem., 1990, 133, 3 Search PubMed.
- A. Nikolaev and A. Orellana, Synthesis, 2016, 48, 1741 CrossRef.
- (a) P. P. Das, K. Belmore and J. K. Cha, Angew. Chem., Int. Ed., 2012, 51, 9517 CrossRef PubMed; (b) B. B. Parida, P. P. Das, M. Niocel and J. K. Cha, Org. Lett., 2013, 15, 1780 CrossRef PubMed; (c) R. V. N. S. Murali, N. N. Rao and J. K. Cha, Org. Lett., 2015, 17, 3854 CrossRef PubMed.
- (a) N. Nithiy, D. Rosa and A. Orellana, Synthesis, 2013, 45, 3199 CrossRef; (b) D. Rosa, A. Nikolaev, N. Nithiy and A. Orellana, Synlett, 2015, 26, 441 CrossRef.
- (a) D. Rosa and A. Orellana, Org. Lett., 2011, 13, 110 CrossRef PubMed; (b) D. Rosa and A. Orellana, Chem. Commun., 2013, 49, 5420 RSC; (c) K. Cheng and P. J. Walsh, Org. Lett., 2013, 15, 2298 CrossRef PubMed; (d) N. Nithiy and A. Orellana, Org. Lett., 2014, 16, 5854 CrossRef PubMed.
- (a) Z. Ye and M. Dai, Org. Lett., 2015, 17, 2190 CrossRef PubMed; (b) Y. Li, Z. Ye, T. M. Bellman, T. Chi and M. Dai, Org. Lett., 2015, 17, 2186 CrossRef PubMed.
- (a) D. C. Davis, K. L. Walker, C. Hu, R. N. Zare, R. M. Waymouth and M. Dai, J. Am. Chem. Soc., 2016, 138, 10693 CrossRef PubMed; (b) X. Zhou, S. Yu, L. Kong and X. Li, ACS Catal., 2016, 6, 647 CrossRef; (c) L. R. Mills, L. M. B. Arbelaez and S. A. L. Rousseaux, J. Am. Chem. Soc., 2017, 139, 11357 CrossRef PubMed; (d) P. Wu, M. Jia, W. Lin and S. Ma, Org. Lett., 2018, 20, 554 CrossRef PubMed.
- Z. Ye, K. E. Gettys, X. Shen and M. Dai, Org. Lett., 2015, 17, 6074 CrossRef PubMed.
- (a) S. Wang, L.-N. Guo, H. Wang and X.-H. Duan, Org. Lett., 2015, 17, 4798 CrossRef PubMed; (b) K. Jia, F. Zhang, H. Huang and Y. Chen, J. Am. Chem. Soc., 2016, 138, 1514 CrossRef PubMed; (c) C.-Y. Wang, R.-J. Song, Y.-X. Xie and J.-H. Li, Synthesis, 2016, 48, 223 Search PubMed.
- H. Zhang, G. Wu, H. Yi, T. Sun, B. Wang, Y. Zhang, G. Dong and J. Wang, Angew. Chem., Int. Ed., 2017, 56, 3945 CrossRef PubMed.
- (a) I. Ryu, K. Matsumoto, Y. Kameyama, M. Ando, N. Kusumoto, A. Ogawa, N. Kambe, S. Murai and N. Sonoda, J. Am. Chem. Soc., 1993, 115, 12330 CrossRef; (b) M. T. Crimmins and P. G. Nantermet, J. Org. Chem., 1990, 55, 4235 CrossRef; (c) M. T. Crimmins, P. G. Nantermet, B. W. Trotter, I. M. Vallin, P. S. Watson, L. A. Mckerlie, T. L. Reinhold, A. W. H. Cheung, K. A. Stetson, D. Dedopoulou and J. L. Gray, J. Org. Chem., 1993, 58, 1038 CrossRef.
- (a) N. Iwasawa, T. Matsuo, M. Iwamoto and T. Ikeno, J. Am. Chem. Soc., 1998, 120, 3903 CrossRef; (b) J. P. Markham, S. T. Staben and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 9708 CrossRef PubMed; (c) B. M. Trost, J. Xie and N. Maulide, J. Am. Chem. Soc., 2008, 130, 17258 CrossRef PubMed; (d) E. Gyanchander, S. Ydhyam, N. Tumma, K. Belmore and J. K. Cha, Org. Lett., 2016, 18, 6098 CrossRef PubMed.
- (a) A. Herath, B. B. Thompson and J. Montgomery, J. Am. Chem. Soc., 2007, 129, 8712 CrossRef PubMed; (b) W. Li, A. Herath and J. Montgomery, J. Am. Chem. Soc., 2009, 131, 17024 CrossRef PubMed; (c) H.-T. Chang, T. T. Jayanth, C.-C. Wang and C.-H. Cheng, J. Am. Chem. Soc., 2007, 129, 12032 CrossRef PubMed.
- (a) A. Herath and J. Montgomery, J. Am. Chem. Soc., 2006, 128, 14030 CrossRef PubMed; (b) A. D. Jenkins, A. Herath, M. Song and J. Montgomery, J. Am. Chem. Soc., 2011, 133, 14460 CrossRef PubMed; (c) C.-H. Wei, S. Mannathan and C.-H. Cheng, Angew. Chem., Int. Ed., 2012, 51, 10592 CrossRef PubMed.
- (a) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208 CrossRef PubMed; (b) N. Yoshikai, Bull. Chem. Soc. Jpn., 2014, 87, 843 CrossRef.
- (a) J. Yang and N. Yoshikai, J. Am. Chem. Soc., 2014, 136, 16748 CrossRef PubMed; (b) J. L. Wu and N. Yoshikai, Angew. Chem., Int. Ed., 2016, 55, 336 CrossRef PubMed; (c) J. Yan and N. Yoshikai, ACS Catal., 2016, 6, 3738 CrossRef; (d) J. Yang, A. Rerat, Y. J. Lim, C. Gosmini and N. Yoshikai, Angew. Chem., Int. Ed., 2017, 56, 2449 CrossRef PubMed.
- For selected reviews on cobalt-catalyzed C–C bond formation, see: (a) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498 CrossRef; (b) P. Röse and G. Hilt, Synthesis, 2016, 48, 463 Search PubMed; (c) P. Gandeepan and C.-H. Cheng, Acc. Chem. Res., 2015, 48, 1194 CrossRef PubMed; (d) G. Cahiez and A. Moyeux, Chem. Rev., 2010, 110, 1435 CrossRef PubMed; (e) W. Hess, J. Treutwein and G. Hilt, Synthesis, 2008, 3537 Search PubMed.
- L. Fiebig, J. Kuttner, G. Hilt, M. C. Schwarzer, G. Frenking, H. G. Schmalz and M. Schafer, J. Org. Chem., 2013, 78, 10485 CrossRef PubMed.
- A. Faulkner, N. J. Race, J. S. Scott and J. F. Bower, Chem. Sci., 2014, 5, 2416 RSC.
- T. Piou, F. Romanov-Michailidis, M. Romanova-Michaelides, K. E. Jackson, N. Semakul, T. D. Taggart, B. S. Newell, C. D. Rithner, R. S. Paton and T. Rovis, J. Am. Chem. Soc., 2017, 139, 1296 CrossRef PubMed.
- (a) P.-S. Lin, M. Jeganmohan and C.-H. Cheng, Chem.–Eur. J., 2008, 14, 11296 CrossRef PubMed; (b) K. Murakami, H. Yorimitsu and K. Oshima, Org. Lett., 2009, 11, 2373 CrossRef PubMed; (c) B.-H. Tan, J. Dong and N. Yoshikai, Angew. Chem., Int. Ed., 2012, 51, 9610 CrossRef PubMed.
- E. Rossini and E. W. Knapp, J. Comput. Chem., 2016, 37, 1082 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215 Search PubMed.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866 CrossRef.
- (a) A. Shafer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 CrossRef; (b) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem., 2009, 113, 6378 CrossRef PubMed.
- See the ESI† for computational details.
- We did not examine protonation of the alkenylcobalt intermediates CP4s and CP4t (i.e., β-alkenylation pathway) because of the ambiguity of the proton source, which would make quantitatively meaningful comparison of the two competing pathways difficult.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1826866. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc02074d |
| This journal is © The Royal Society of Chemistry 2018 |