
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective C–H halogenation over hydroxylation by non-heme iron(IV)-oxo†
Sujoy
Rana
a,
Jyoti Prasad
Biswas
a,
Asmita
Sen
a,
Martin
Clémancey
b,
Geneviève
Blondin
b,
Jean-Marc
Latour
 *b,
Gopalan
Rajaraman
*a and
Debabrata
Maiti
*a
*b,
Gopalan
Rajaraman
*a and
Debabrata
Maiti
*a
aDepartment of Chemistry, IIT Bombay, Powai, Mumbai-400076, India
bUniversity of Grenoble Alpes, LCBM/PMB and CEA, IRTSV/CBM/PMB and CNRS, LCBM UMR 5249, PMB, 38000 Grenoble, France
First published on 15th August 2018
Abstract
Non-heme iron based halogenase enzymes promote selective halogenation of the sp3-C–H bond through iron(IV)-oxo-halide active species. During halogenation, competitive hydroxylation can be prevented completely in enzymatic systems. However, synthetic iron(IV)-oxo-halide intermediates often result in a mixture of halogenation and hydroxylation products. In this report, we have developed a new synthetic strategy by employing non-heme iron based complexes for selective sp3-C–H halogenation by overriding hydroxylation. A room temperature stable, iron(IV)-oxo complex, [Fe(2PyN2Q)(O)]2+ was directed for hydrogen atom abstraction (HAA) from aliphatic substrates and the iron(II)-halide [FeII(2PyN2Q)(X)]+ (X, halogen) was exploited in conjunction to deliver the halogen atom to the ensuing carbon centered radical. Despite iron(IV)-oxo being an effective promoter of hydroxylation of aliphatic substrates, the perfect interplay of HAA and halogen atom transfer in this work leads to the halogenation product selectively by diverting the hydroxylation pathway. Experimental studies outline the mechanistic details of the iron(IV)-oxo mediated halogenation reactions. A kinetic isotope study between PhCH3 and C6D5CD3 showed a value of 13.5 that supports the initial HAA step as the RDS during halogenation. Successful implementation of this new strategy led to the establishment of a functional mimic of non-heme halogenase enzymes with an excellent selectivity for halogenation over hydroxylation. Detailed theoretical studies based on density functional methods reveal how the small difference in the ligand design leads to a large difference in the electronic structure of the [Fe(2PyN2Q)(O)]2+ species. Both experimental and computational studies suggest that the halide rebound process of the cage escaped radical with iron(III)-halide is energetically favorable compared to iron(III)-hydroxide and it brings in selective formation of halogenation products over hydroxylation.
Introduction
High-valent iron-oxo species serve as the key intermediates for performing different natural transformations like halogenation, hydroxylation and olefin epoxidation.1 Several oxygenases including Cyt P450, Rieske oxygenases, α-keto gluterate-dependent oxygenases and various halogenases exhibit their activity via formation of an iron-oxo intermediate.2 Among these mononuclear iron based enzymes, α-KG-dependent halogenases carry out biosynthesis of several halogen based natural products by implementing selective halogenation of sp3 C–H bonds by overriding hydroxylation.3 These enzymes contain α-KG as the key structural motif which is coordinated to an iron(II) cofactor in a facial triad fashion along with two histidine moieties. Other halogenases such as CytC3 and SyrB2 differ structurally compared to α-KG dependent halogenases, in a sense wherein halide is coordinated to the iron centre instead of carboxylate from α-KG. Although these enzymes differ structurally, their mode of action towards C–H halogenation is very much similar. First, the iron(II) cofactor performs O2 activation resulting in a high-spin (S = 2) cis-iron(IV)-oxo-halide species. Subsequently, it performs the hydrogen atom abstraction (HAA) from the C–H bond. The corresponding nascent radical and iron(III)-halide are placed in such a way that the radical selectively undergoes rebound with halide to generate halogenated products.A biomimetic non-heme iron(III)-halide complex, [FeIII(TPA)Cl2]+, was first synthesized and judiciously employed by Que and co-workers for sp3 C–H halogenation.4 The iron(V)-oxo-halide, [FeV(TPA)(O)(Cl)]2+, was proposed as the key intermediate for the halogenation reaction. Recent studies showed that substrate positioning between halide and hydroxide is a key factor to ensure selectivity for halogenation.5 Later on Comba reported C–H halogenations with [FeII(bispidine)(Cl2)] where iron(IV)-oxo-halide, [FeIV(bispidine)(O)(Cl))]2+, was proposed as the key intermediate.6 Subsequently Costas synthesized cis-iron(IV)-oxo-halide, [FeIV(O)(X)(Pytacn)]+ (X, Cl and Br), for pursuing sp3 C–H halogenation.7 Later Paine and co-workers reported halogenation of aliphatic and benzylic C–H bonds where similar iron(IV)-oxo-halide, [TPPhFeIV(O)(Cl)]2+, was proposed as the key intermediate.8
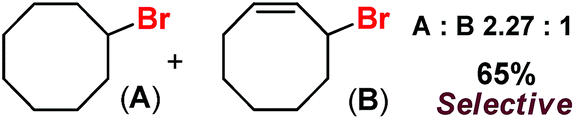
Very recently Que and co-workers employed high-spin (S = 2) iron(IV)-oxo-halide complexes, [FeIV(TQA)(O)(X)]2+ (X = Cl and Br) for sp3 C–H halogenations.9 These elegant explorations demonstrated that despite having the potential for promoting selective halogenation chemistry, the formation of the hydroxylation product often remained as the bottleneck for the synthesis of selectively halogenated compounds (Fig. 1). A clear opportunity for synthetic chemists, therefore, resides on discovering a selective halogenation protocol by utilizing the potential of the non-heme iron(IV)-oxo complex.
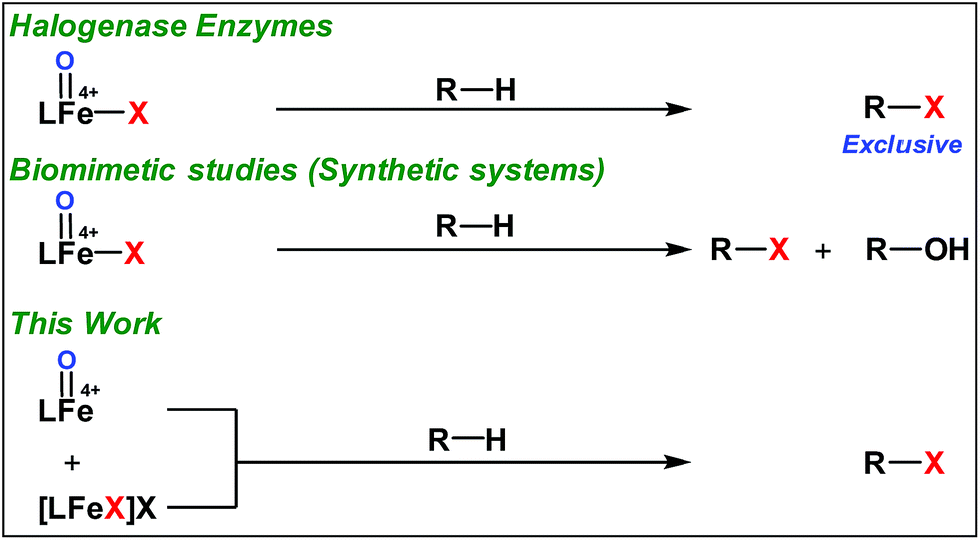 | ||
| Fig. 1 sp3-C–H halogenation by non-heme complexes. | ||
Results and discussion
We have explored a new strategy for the halogenation reaction involving a non-heme iron complex with the expectation to discover a selective sp3-C–H halogenation protocol by overriding hydroxylation chemistry, along with what is observed in α-KG-dependent halogenases. We planned to employ a room temperature stable pentacoordinated iron(IV)-oxo species [FeIV(2PyN2Q)(O)]2+ (2) supported by the 2PyN2Q ligand, (1,1-di(pyridin-2-yl)-N,N-bis(quinolin-2-ylmethyl)methanamine, Fig. 2).10 The corresponding iron(II)-complex was synthesized by reacting Fe(OTf)2(CH3CN)2 with the ligand, 2PyN2Q in acetonitrile. The synthesized complex [FeII(2PyN2Q)(OTf)2], 1, showed UV-vis bands at 368 nm (ε ∼ 1954 M−1 L−1) and 470 nm (ε ∼ 887 M−1 L−1). Complex 1 was also characterized by ESI-MS ([FeII(2PyN2Q)(OTf)]+, m/z = experimental 672.09; calculated = 672.09). Further, 1 showed a paramagnetic shift in 1H NMR spectroscopy (shift from −60 to 140 ppm). Complex 1 was also characterized by X-ray crystallography (Fig. 2). Synthesis of the reactive iron(IV)-oxo ([FeIV(2PyN2Q)(O)]2+) complex 2 was performed by adding MesI(OAc)2/mCPBA (1.5 equiv.) in acetonitrile and it showed a characteristic UV-vis band at 770 nm (ε ∼ 250 M−1 L−1). Complex 2 was further characterized by FT-IR spectroscopy where FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching appeared at 832.5 cm−1.10c,11
O stretching appeared at 832.5 cm−1.10c,11
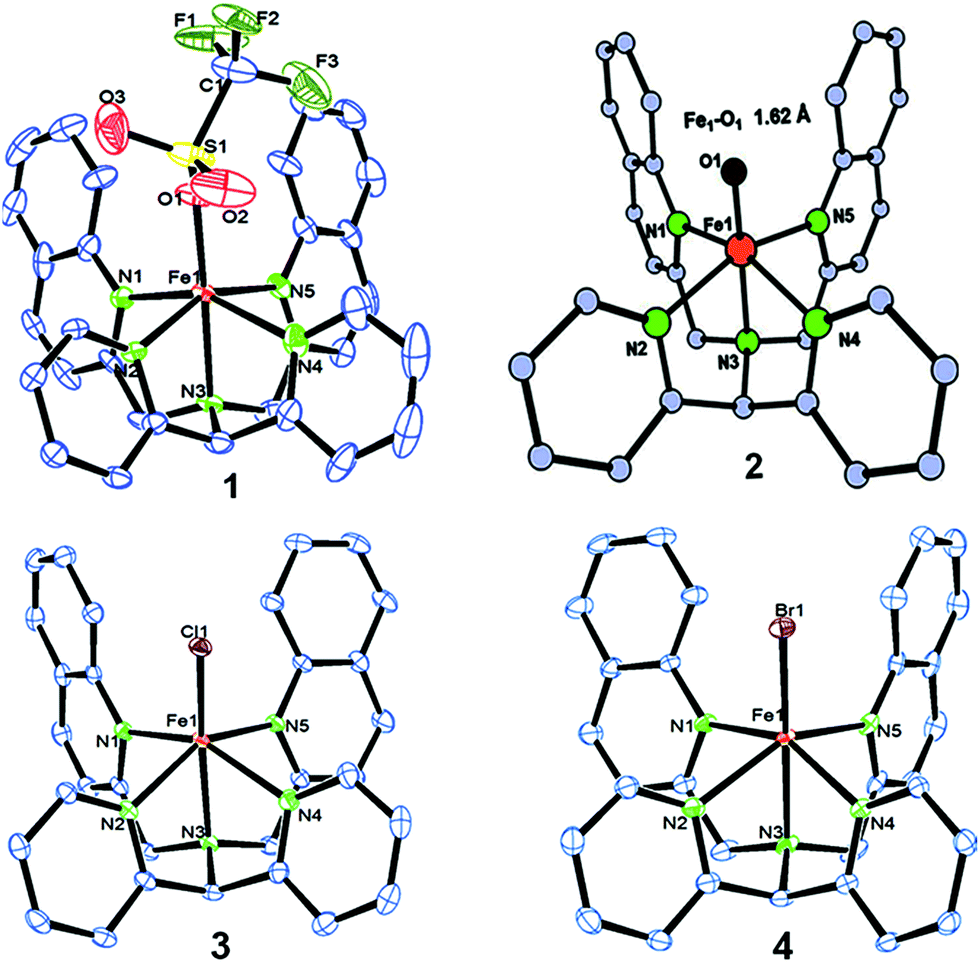 | ||
| Fig. 2 ORTEP diagram of the triflate anion coordinated iron(II)-complex 1 (CCDC 1505984), the DFT optimized structure of iron(IV)-oxo 2, [FeIV(2PyN2Q)(O)]2+, and the ORTEP diagram of iron(II)-halide complexes 3 (1505989) and 4 (1505986). | ||
Further, complex 2 was characterized by Mössbauer spectroscopy. Fig. 3a presents the spectrum recorded at 80 K with a small field (60 mT) applied parallel to the γ rays. The center of the spectrum is dominated by two overlapping quadrupole doublets and a broad component can be discerned on both sides and positive velocities. Application of a high parallel magnetic field (7 T, Fig. 3b bottom) turns the latter component into a sextet extending from −7.4 to 8 mms−1. By contrast, the two central features are slightly broadened. These two spectra can be simulated simultaneously by considering the presence of three components (Table S1†).11 Component A (red line in the simulations), which amounts to 35% of the total iron, is a spin S = 1 species with an isomer shift δ = 0.043 mms−1, a quadrupole splitting ΔEQ = 0.57 mms−1 and a D value ca. 26 cm−1. These parameters are consistent with an FeIVO species. Component B (blue line in the simulations, 12% of the total iron) is a μ-oxodiferric S = 0 species with δ = 0.5 mm s−1 and a quadrupole splitting ΔEQ = 1.5 mms−1. The additional magnetic component (C, green line) can be accounted for by considering that it is a high spin FeIII species (53%, S = 5/2, δ = 0.56 mms−1, ΔEQ = 0.72 mms−1). These Mössbauer experiments thus show the formation of an iron(IV)oxo, FeIVO, species which is reactive at room temperature forming a μ-oxo diferric species together with another ferric species.
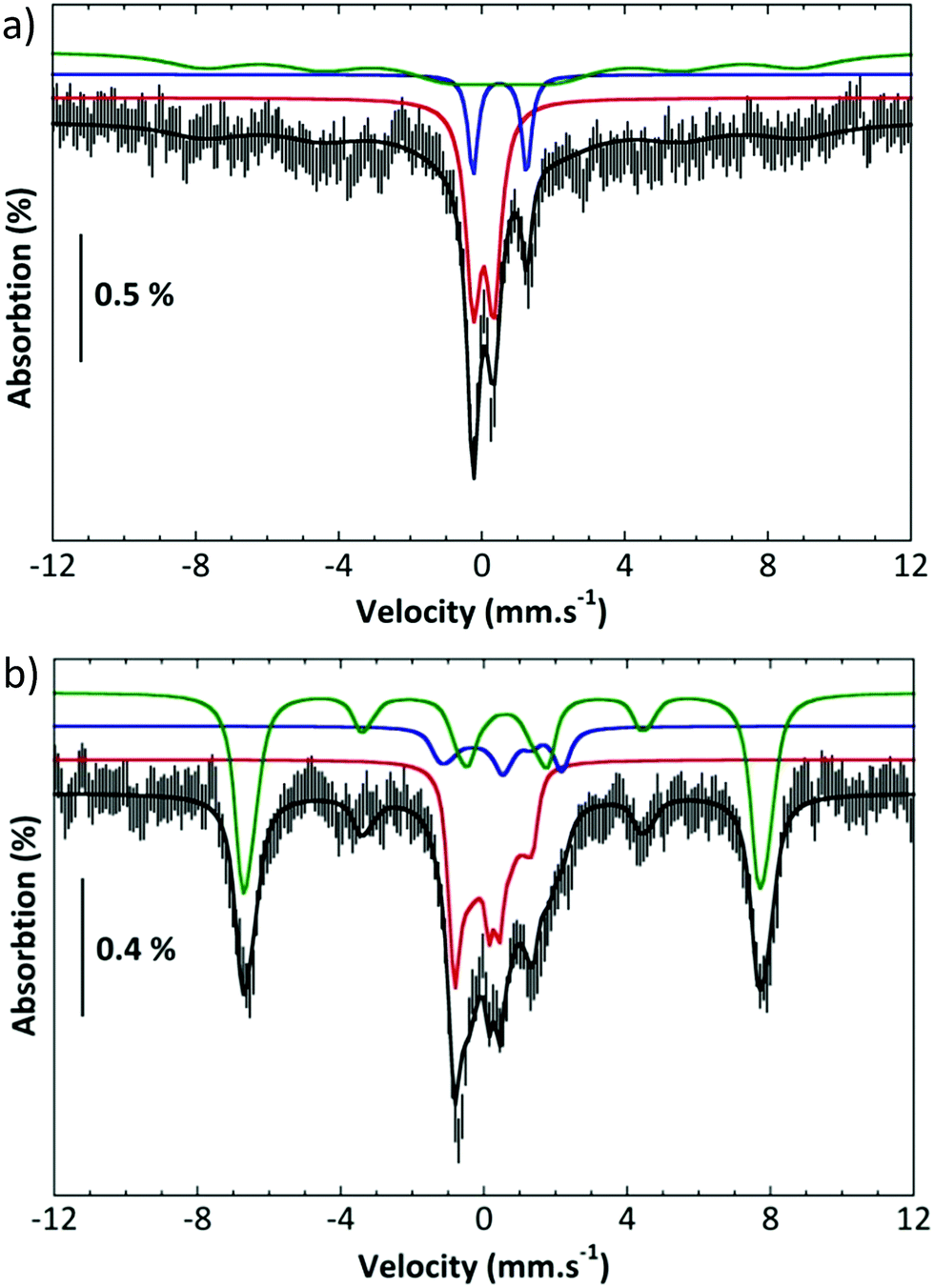 | ||
| Fig. 3 (a) Mössbauer spectra recorded at 80 K and 60 mT top and (b) at 5.5 K and 7 T (bottom). Experimental spectra: hatched bars; solid black line: simulation; colored lines: contributions of species A (red), B (blue) and C (green). | ||
Initially we opted for demonstrating the feasibility of the HAA step by the iron(IV)-oxo species, [FeIV(2PyN2Q)(O)]2+ (2) during C–H oxidation. Substrates like ethylbenzene, toluene and cyclohexane yielded the corresponding oxidation products when reacted with the room temperature stable iron(IV)-oxo species 2.
Kinetic studies of C–H oxidation reactions were performed (UV-vis, Fig. 4a and b) under pseudo first order conditions. The second order rate constant (k2) values were found to be 20 times higher when compared to the previously reported rates of C–H oxidation by penta-coordinated nitrogen containing iron(IV)-oxo species,12 which could be due to the steric bulk originating from the quinoline moiety.13
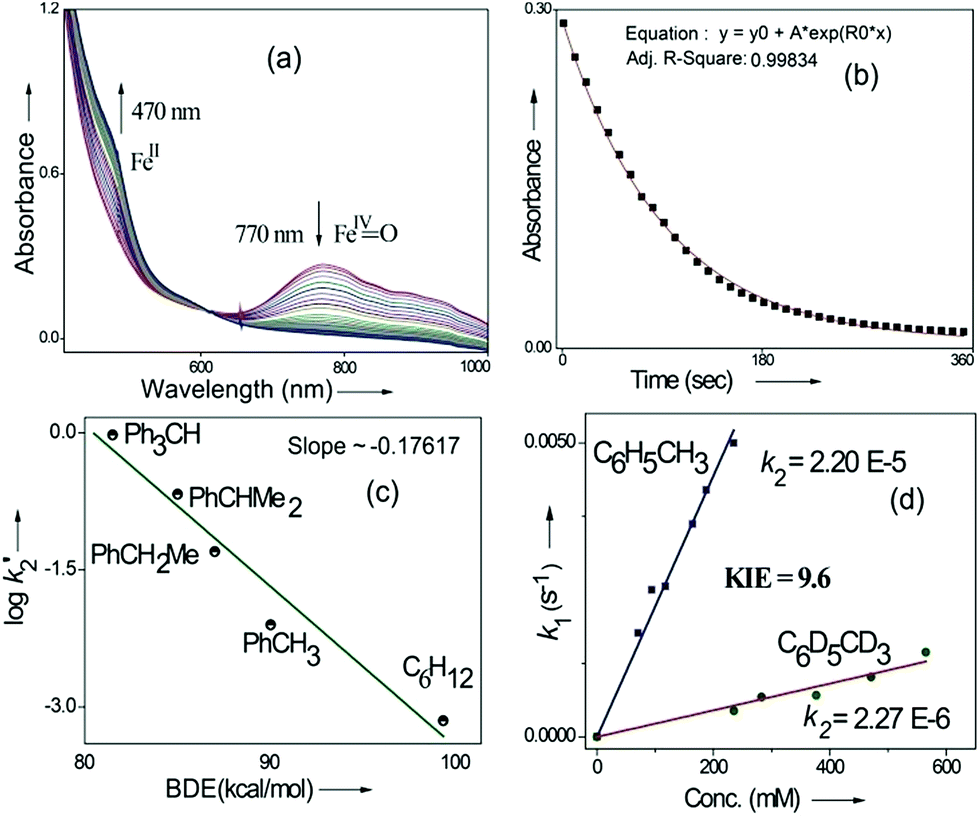 | ||
| Fig. 4 (a) UV-vis change during C–H oxidation of ethylbenzene, (b) time trace of 2 at 770 nm, (c) bond dissociation energy (BDE) correlation plot: Bell–Evans–Polyani plot during C–H oxidation by 2 and (d) kinetic isotope effect study (kinetic studies were carried out under a N2 atmosphere at 25 °C). | ||
The Bell–Evans–Polayni (BEP) plot (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k′2vs. BDE) showed a linear correlation (Fig. 4c). Further we studied the kinetic isotope effect (KIE, kH/kD) between C6H5CH3/C6D5CD3 and the value was found to be 9.6 (Fig. 4d). The linearity in the BEP plot and the large KIE value suggested that the initial HAA step is the rate-determining step (RDS) during C–H oxidation.14 The slope (−0.176 (kcal mol−1)−1) of the BEP plot is related to the Bronsted parameter (α) by α = [slope(RT)] and it gives α ∼ −0.10 i.e. the value is closer to 0. The lower value of the Bronsted parameter suggests that a reactant like transition state is involved during C–H oxidation reactions via the HAA step which is further supported by a DFT study.15
k′2vs. BDE) showed a linear correlation (Fig. 4c). Further we studied the kinetic isotope effect (KIE, kH/kD) between C6H5CH3/C6D5CD3 and the value was found to be 9.6 (Fig. 4d). The linearity in the BEP plot and the large KIE value suggested that the initial HAA step is the rate-determining step (RDS) during C–H oxidation.14 The slope (−0.176 (kcal mol−1)−1) of the BEP plot is related to the Bronsted parameter (α) by α = [slope(RT)] and it gives α ∼ −0.10 i.e. the value is closer to 0. The lower value of the Bronsted parameter suggests that a reactant like transition state is involved during C–H oxidation reactions via the HAA step which is further supported by a DFT study.15
We moved towards product analysis during C–H oxidation to gain insights into the mechanistic pathway. When ethyl benzene was reacted with complex 2, it provided 1-phenylethanol (34%), 1-phenylethylacetate (26%) and acetophenone (5%) under a N2 atmosphere. Control reactions were carried out between 1-phenylethanol and MesI(OAc)2 as well as between 1-phenylethanol and in situ generated complex 2. In none of these cases, 1-phenyl ethyl acetate was detected. Therefore, the formation of 1-phenylethylacetate during the reaction with ethyl benzene could occur via a reaction between the cage-escaped radical and the acetoxy radical (˙OAc) generated from MesI(OAc)2 during the reaction (Fig. 5). This observation suggests that the radical could dissociate from the solvent cage after the HAA step. Toluene and cyclohexane (N2 atmosphere) produced 48% of benzyl alcohol and 3% of benzaldehyde (Fig. 5), whereas cyclohexane provided cyclohexanol (54%) and cyclohexanone (3%). Further, experiments with O-18 labelled 2 and ethylbenzene provided 1-phenylethanol with 86% of O-18 labelling which supported 2 as the sole oxygen source during C–H oxidation.
 | ||
| Fig. 5 C–H oxidation by [FeIV(2PyN2Q)(O)]2+, 2 (reactions were carried out under a N2 atmosphere inside a glove box at 25 °C). | ||
The C–H oxidation reactions with ethylbenzene, toluene and cyclohexane under air were found out to produce alcohol/ketone (A/K) products with a ratio of 1 or <1. Additionally, radical trap experiments with CCl3Br were carried out. Expectedly, cyclohexane and toluene provided exclusively radical trapped products bromocyclohexane and benzyl bromide, respectively. Similarly, ethylbenzene provided radical trapped products (1-chloroethyl)benzene (20%), (1-bromoethyl)benzene (10%) and 1-phenylethylacetate (25%). Such observations implied dissociation of the carbon radical from the solvent cage,16 following which it undergoes a rebound with iron(III)-hydroxide to provide the C–H oxidation product. Further the cage escape behaviour of the radical was confirmed by carrying out a reaction of a pure stereoisomer of cis-1,2-dimethylcyclohexane with 2. It provided a mixture of cis/trans-1,2-dimethylcyclohexanol (46% yield, 3.7:1 ratio). The high-degree of epimerization in the products arises from a long-lived radical, which could be generated due to the dissociation of the radical from the solvent cage after HAA (Fig. 5).17
Although complex 2 was found to be suitable for HAA from the sp3-C–H bond of cyclohexane, our attempts to promote halogenation of cyclohexane by a combination of complex 2 and 1 equiv. of halide sources Bu4N+Cl− or Bu4N+Br− failed. Such an observation could be attributed to the inability of 2 to form a reactive iron(IV)-oxo-halide intermediate with the pentacoordinated 2PyN2Q ligand. Additionally, complex 2 reacted with the halide anion faster compared to abstracting the hydrogen atom from the sp3-C–H bond of cyclohexane.18 We have carried out the kinetic study for the oxidation of chloride (Cl−) and bromide (Br−) by 2. We found that the second order rate constant, k2, for Cl− and Br− oxidation is 221 and 938 M−1 s−1 respectively. The complex 2 oxidizes Br− faster than Cl−, which in turn is much faster compared to the hydrogen atom abstraction process (for the C–H oxidation of toluene and cyclohexane the second order rate constant, k2, is 0.0242 and 0.011 M−1 s−1 respectively).11
With these unsuccessful attempts, our attention returned to the existing report for the formation of a mixture of halogenation and hydroxylation products. Notably, halide rebound of the cage escaped radical with metal-halide was well documented with a Mn-catalyzed fluorination reaction by Groves and co-workers.19 We envisioned that if we can outcompete iron(III)-hydroxide (formed upon HAA of the sp3-C–H bond by 2) with iron(III)-halide, then the cage-escaping carbon centered radical will undergo rebound with iron(III)-halide. The iron(III) halide can potentially act as a suitable halide donor and can be generated in situ via one-electron oxidation (by oxidant mCPBA) of the corresponding iron(II)-halide complexes [FeII(2PyN2Q)(X)](X) [X = Cl (3) and Br (4)] (Fig. 2). Reaction of 4 with 2 and cyclohexane led to the formation of the bromocyclohexane (90%) product selectively (Table 1, entry 1, 4 as the limiting reagent under a N2 atmosphere). Use of an excess amount of 4 was detrimental to the formation of bromocyclohexane. Furthermore, we focused on the discovery and implementation of the new strategy for promoting the selective halogenation reaction by overcoming the hydroxylation chemistry described above. The chlorination reactions with cyclohexane in the presence of 2 and 3 gave chlorocyclohexane in 52% yield with 5:1 selectivity for halogenation over hydroxylation (Table 1, entry 2).
|
|
||
|---|---|---|
| Entry | Substrate (s) | Product (s) |
| a Minor hydroxylation (∼5% w.r.t.3 or 4). Reactions were carried out under a N2 atmosphere inside a glove box at 25 °C. | ||
| 1 |
|

|
| 2 |
|
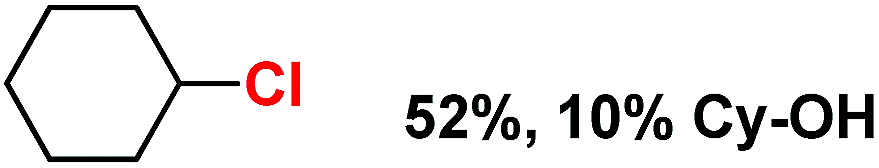
|
| 3 |

|

|
| 4 |
|

|
| 5 |
|
|
| 6 |
|

|
| 7 |
|

|
| 8 |
|
|
| 9 |
|

|
| 10 |
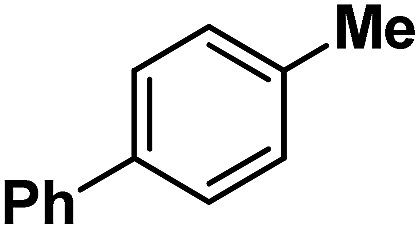
|
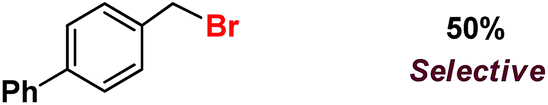
|
| 11 |
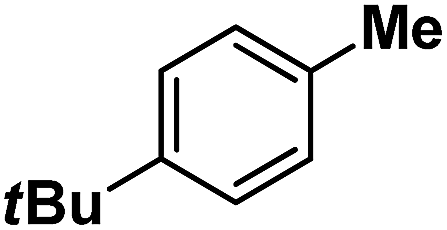
|
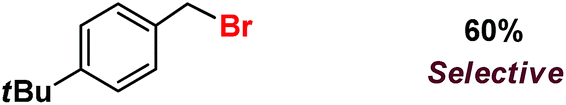
|
| 12 |
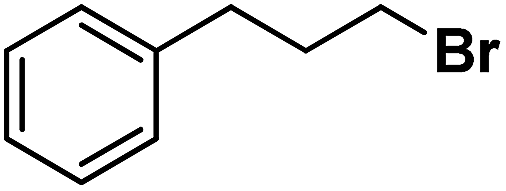
|
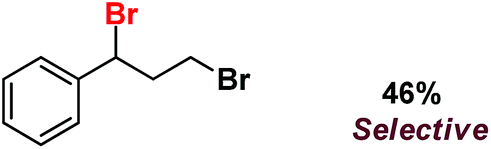
|
| 13 |
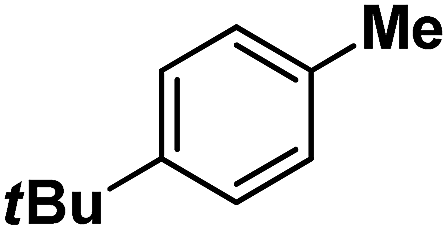
|

|
| 14 |
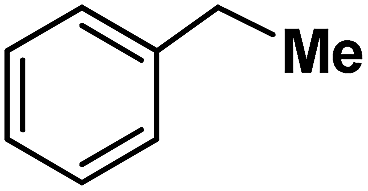
|
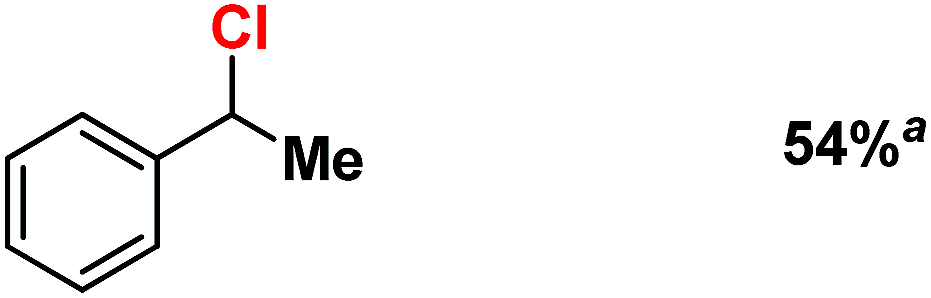
|
The appreciable levels of selectivity for the synthetic non-heme iron-oxo mediated halogenation chemistry clearly suggest that the carbon centered radical, generated upon HAA, escapes the solvent cage and combines with the free halide radical generated upon halide oxidation by 2. Similar to non-heme halogenase enzymes, this carbon radical engages the halide radical from the iron(III)-halide intermediate to yield the halogenation product preferably over hydroxylation (Fig. 6).
 | ||
| Fig. 6 Plausible mechanism of C–H halogenation by iron(IV)-oxo, 2 in the presence of iron(II)-halide complexes 3 or 4. | ||
Further in order to get mechanistic insight, we synthesized iron(II)-halide complexes with perchlorate as the counter anion, [FeII(2PyN2Q)(X)](ClO4), X = Cl, 5 (1516453), Br, 6 (1516421), and characterized these complexes by X-ray crystallography. Interestingly, when 6 was used for the bromination of cyclohexane, we selectively obtained bromocyclohexane (7:1). Similarly, employing complex 5 we selectively obtained a benzylic chlorinated product with 4-tertbutyltoluene. Moreover the solution of 4 (also 6) with 0.5 (or 1 equiv.) mCPBA provided a brominated product efficiently and therefore suggested the feasibility of one electron oxidation of these halide complexes to provide iron(III)-halide required for the halide rebound step (Fig. 6).20 Additionally, we have tested the possibility of one electron oxidation of the halide complexes by recording the EPR spectrum of 6 with 0.5 equiv. or 1 equiv. mCPBA (pre-oxidized solution) in acetonitrile at 4 k. The EPR spectra showed a rhombic signal (g1 = 1.99, g2 = 4.38, g3 = 6.24) which is characteristic of a high spin (S = 5/2) iron(III)-species (Fig. 7).21 These observations suggested that the halide complexes 5 and 6 (also 3 and 4) possibly undergo one electron oxidation in the presence of 2 equiv. mCPBA used in halogenation to provide iron(III)-halide which would undergo halide rebound with the cage escaped radical (Fig. 6). Moreover, we have measured the one electron oxidation potential of halide complexes (3–6) by a cyclic voltammetry study. The halide complexes, 3–6, showed FeII/FeIII oxidation potentials around ∼0.9–1.0 V in acetonitrile vs. SCE (saturated calomel electrode). Thus it can be oxidized by mCPBA as the oxidation potentials of peracids are around ∼1.8–2.0 V.22 However, we have not observed any peak around 770 nm in UV-vis upon portion wise addition of mCPBA to the [(L)Fe(III)X]X complexes, which indicates that for the system under study Fe(III)-X might not undergo oxidation by mCPBA (Fig. S38†).11 Notably as the iron(II)-halide complexes (3–6) undergo rapid one electron oxidation under the halogenation reaction conditions and generate iron(III)-halide species, they do not participate in the known comproportionation reaction with iron(IV)-oxo 2. However, the possibility of the comproportionation reaction cannot be completely ruled out.
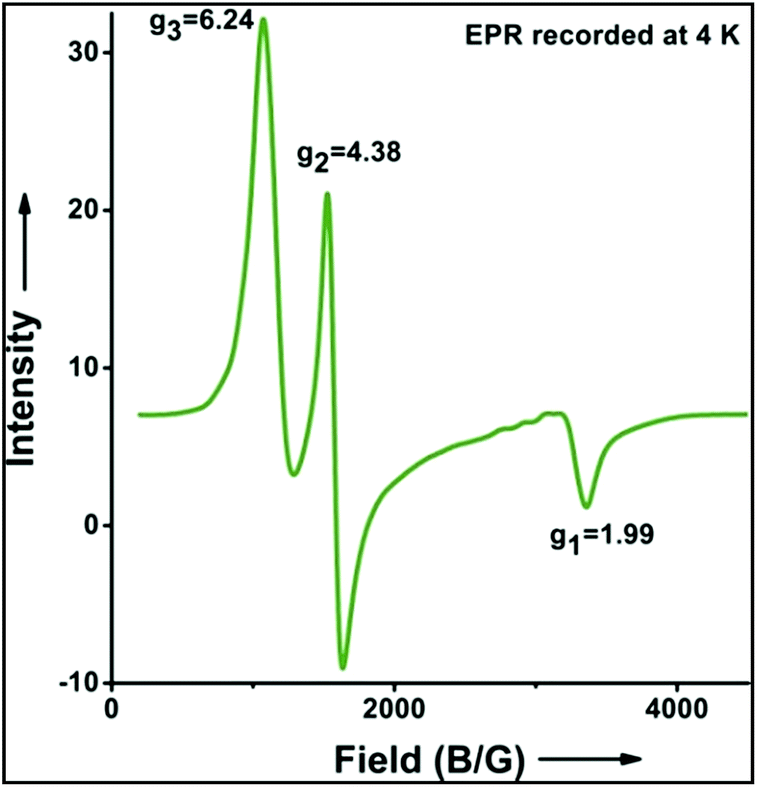 | ||
| Fig. 7 EPR spectrum showing one electron oxidation of 6 using 0.5 of mCPBA (temperature 4 K, X-band frequency 9.376 GHz, modulation amplitude 4G, modulation frequency 100 kHz, and attenuation 22 dB). | ||
The higher selectivity of bromination over chlorination can be rationalized by the ease of halide rebound of the cage escaped radical with iron(III)-bromide compared to iron(III)-chloride (Fig. 6). Additionally the ease of formation of Br˙ from Br−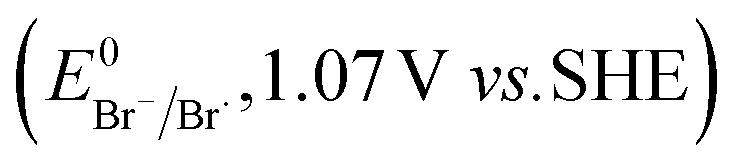 compared to Cl˙ from Cl−
compared to Cl˙ from Cl−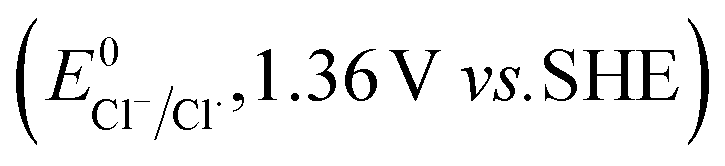 from complexes 3 and 4 during the reaction (which can combine with the cage escaped radical and can lead to the partial formation of halogenated products) can also be accounted for higher selectivity.6 The formation of the iron(III)-halide complex could also occur via substitution by the halide anion from the iron(II)-halide complex (Fig. 6). Furthermore, we observed that the use of mCPBA as an oxidant, instead of MesI(OAc)2, during the bromination reaction with cyclohexane increases the bromocyclohexane product yield from 18% to 90%. The ease of hydroxide substitution by the halide ion in the presence of H+ from mCPBA can be accounted for the higher yield. Similar substitution of hydroxide bound to the iron centre in the presence of the proton source was elegantly described by de Visser, Hillier and Paine groups (Fig. 6).11,26
from complexes 3 and 4 during the reaction (which can combine with the cage escaped radical and can lead to the partial formation of halogenated products) can also be accounted for higher selectivity.6 The formation of the iron(III)-halide complex could also occur via substitution by the halide anion from the iron(II)-halide complex (Fig. 6). Furthermore, we observed that the use of mCPBA as an oxidant, instead of MesI(OAc)2, during the bromination reaction with cyclohexane increases the bromocyclohexane product yield from 18% to 90%. The ease of hydroxide substitution by the halide ion in the presence of H+ from mCPBA can be accounted for the higher yield. Similar substitution of hydroxide bound to the iron centre in the presence of the proton source was elegantly described by de Visser, Hillier and Paine groups (Fig. 6).11,26
No halogenation products were obtained in the presence of air; rather we obtained exclusively C–H oxidation products. This phenomenon suggested that the cage escaped radical was intercepted by O2 (air).9 Further studies were carried out in order to get insights about these selective halogenation reactions. Addition of [FeII(2PyN2Q)Br](Br) (3) to a solution of [FeIV(2PyN2Q)O]2+ (2) showed the iron(IV)-oxo band at 770 nm (Fig. 8a). This experiment suggests that iron(IV)-oxo exists in solution in the presence of iron(II) halides. Iron(IV)-oxo is the key species for the initiation of the halogenation reactions. The kinetic isotope effect study between C6H5CH3 and C6D5CD3 under standard halogenation reaction conditions gave a value of 13.5 (Fig. 8c), which supported the initial HAA step as the rate determining step.
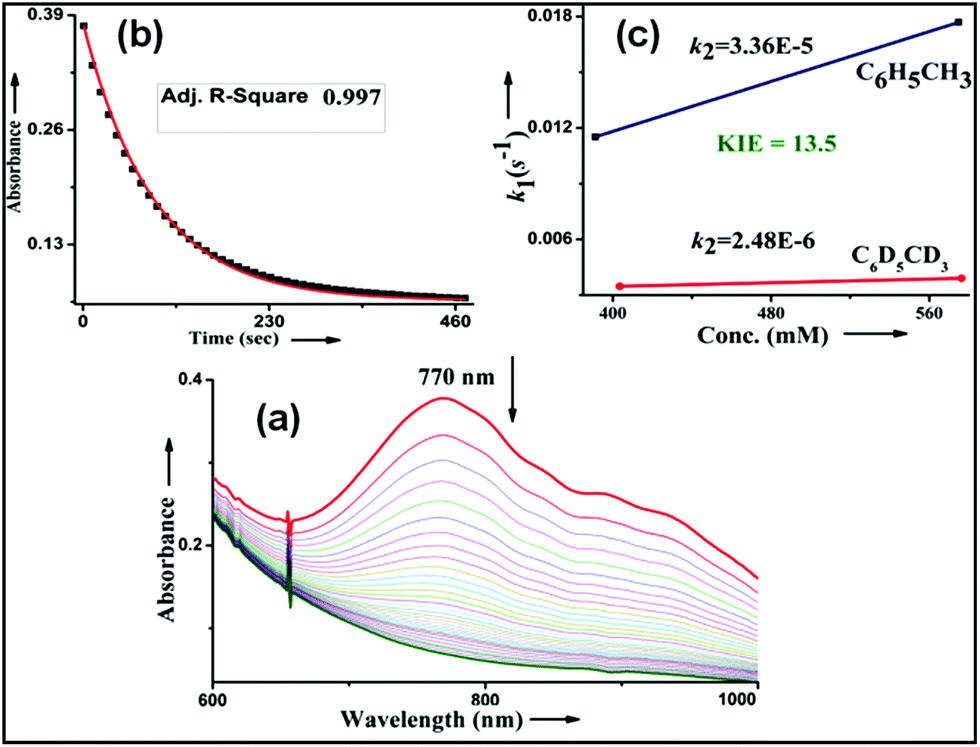 | ||
| Fig. 8 (a) UV-vis change during the bromination of toluene, (b) inset 770 nm decay plot of 2 during the bromination of toluene, (c) kinetic isotope effect study during the bromination reaction (kinetic studies were carried out under a N2 atmosphere at 25 °C). | ||
Following the above protocol, cyclopentane, cycloheptane, cyclooctane and norbornane were subjected to the same reaction conditions. Expectedly, selective formation of halogenation products was encountered in these cases (Table 1, entries 3–6). After successful halogenation of the sp3-C–H bond of aliphatic substrates, we decided to explore benzylic halogenation. Under the standard reaction protocol, toluene provided benzyl bromide in an excellent yield (97%, Table 1, entry 7). Differentially substituted benzylic substrates provided selective brominated products as well (Table 1, entries 8–12). Interestingly, 4-tertbutyltoluene showed excellent selectivity towards benzylic chlorination (Table 1, entry 13). By overriding the hydroxylation reaction, ethyl benzene was chlorinated at the benzylic position selectively (Table 1, entry 14).
Computational study
To fully comprehend the mechanistic proposal based on the experimental evidence, we have turned to computational tools where the B3LYP/LACVP//B3LYP/TZVP setup has been used to model the reaction pathway for the halogenation reaction (see computational details for elaborate discussion on the methodology employed). To begin with, calculations are performed on the putative [FeIV(2PyN2Q)O]2+(2) species. Calculations reveal S = 1 as the ground state for this species with the S = 2 and S = 0 states lying 12.7 kJ mol−1 and 121.2 kJ mol−1 higher in energy, respectively. The ground state predicted by the calculations is consistent with the Mössbauer observations and in accord with the literature reports for such pentadentate aminopyridine ligands.23 While the ground state is the same as that of the structurally similar [FeIV(N4Py)O]2+ species, the S = 2 excited state is found to lie closer in species 2 and this is attributed to the steric and electronic differences in the ligand moiety. The smaller triplet-quintet gap is known to enhance the reactivity in iron(IV)-oxo species as reported earlier.24The optimized structure of the S = 1 state is given in Fig. 9a. The Fe–O bond length is found to be 1.626 Å which is consistent with the experimental data reported for other Fe(IV)O species.23b Since an asymmetric ligand environment is maintained at the equatorial plane with two strong pyridine donors and moderate quinoline donors, two sets of Fe–N distances are visible with Fe–NQu distances slightly longer than the Fe–Npy distances (Fig. 9a). Due to the steric hindrance of the quinoline group, the ferryl oxygen atom is found to incline towards the pyridine ring and this is clearly reflected in the N(1)–Fe–O angle which has been found to be 170.1° (for most of the Fe(IV)O species reported this angle is found to be close to linearity).23b,e Other notable differences in species 2, include two strong C–H⋯O interactions between the C(8)–H-atom of the quinoline and ferryl-oxygen atom and the distances are noted as 2.016 Å. This interaction is likely to weaken the Fe–O bond in species 2 compared to the [FeIV(N4Py)O]2+ species where such interactions are absent. Besides, the steric crowding at the active site offered by the quinoline ligand could hinder or facilitate the reactions further. The spin density distribution reveals a strong oxyl-radical character at both the S = 1 and S = 2 states with the oxygen atom possessing a significant spin density. Interestingly, the H-atoms involved in the C–H⋯O interactions with the ferryl-oxygen atoms are strongly polarized with sizable negative spin density (Fig. 9b) and this is similar to the behavior observed earlier with strong H-bonding ligands.25 The Eigen-value plot of the S = 1 state computed is shown in Fig. 9c. Due to two weaker Fe–N bonds in the equatorial plane, the Fe–N anti-bonding interaction diminishes for the 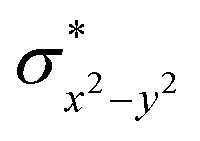 orbital leading to a smaller gap between the
orbital leading to a smaller gap between the 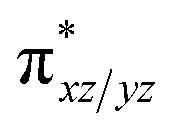 and the
and the 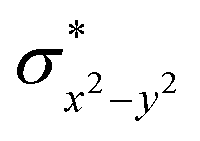 orbital compared to the [FeIV(N4Py)O]2+ species and this leads to a smaller triplet-quintet gap. Besides, the degeneracy between the
orbital compared to the [FeIV(N4Py)O]2+ species and this leads to a smaller triplet-quintet gap. Besides, the degeneracy between the 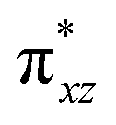 and
and 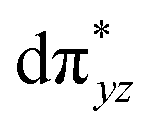 orbitals is lifted due to weak C–H⋯O interactions. With these insights into the electronic structure of the active species, we turn to study the mechanistic aspect. The mechanism adapted for DFT calculations is shown in Fig. 10. In the first step, C–H bond activation by the Fe(IV)O species is expected.
orbitals is lifted due to weak C–H⋯O interactions. With these insights into the electronic structure of the active species, we turn to study the mechanistic aspect. The mechanism adapted for DFT calculations is shown in Fig. 10. In the first step, C–H bond activation by the Fe(IV)O species is expected.
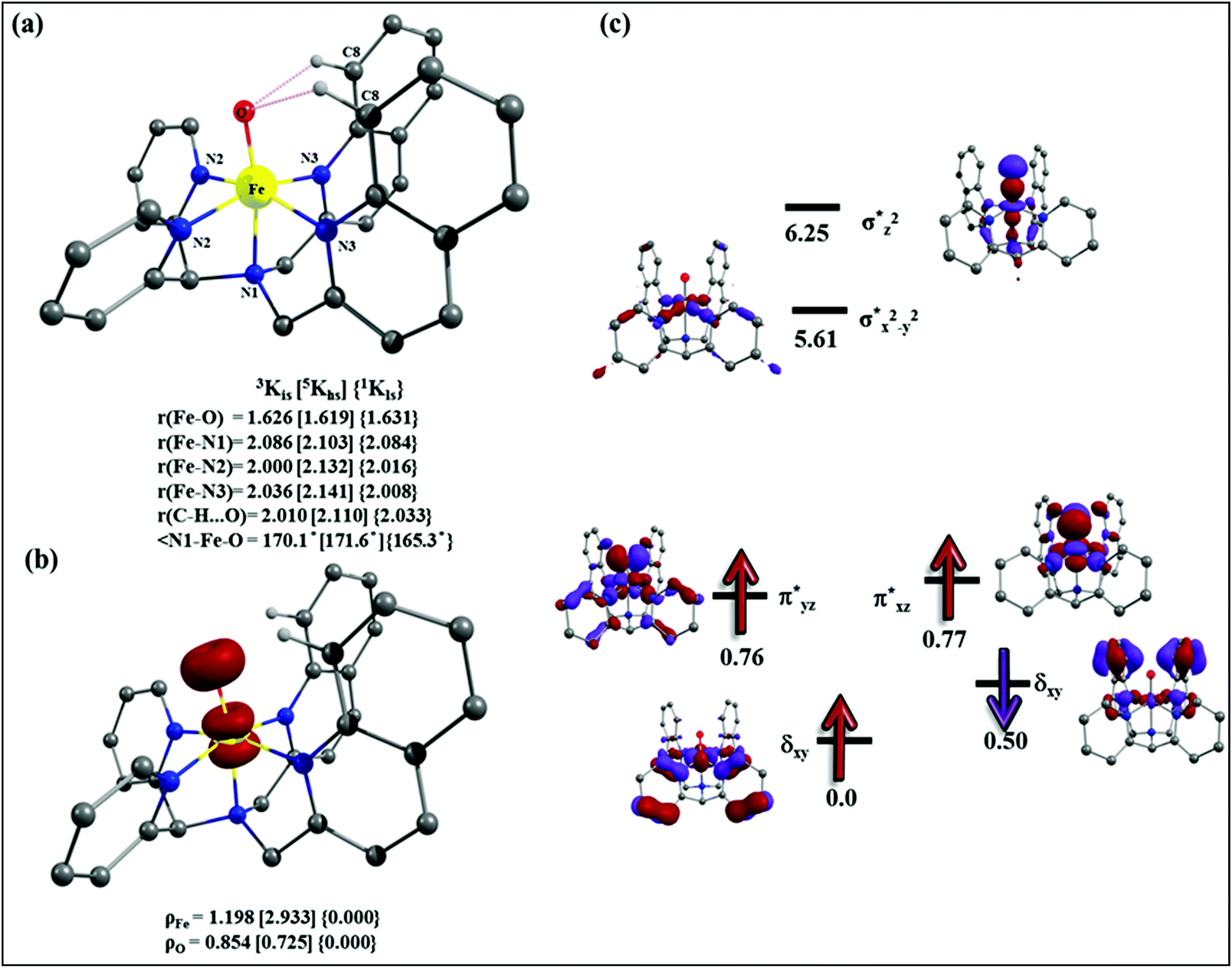 | ||
| Fig. 9 (a) Important structural parameters and (b) spin densities of B3LYP-D2 optimized structures of [FeIV(2PyN2Q)(O)]2+, and (c) key orbitals of 3K. | ||
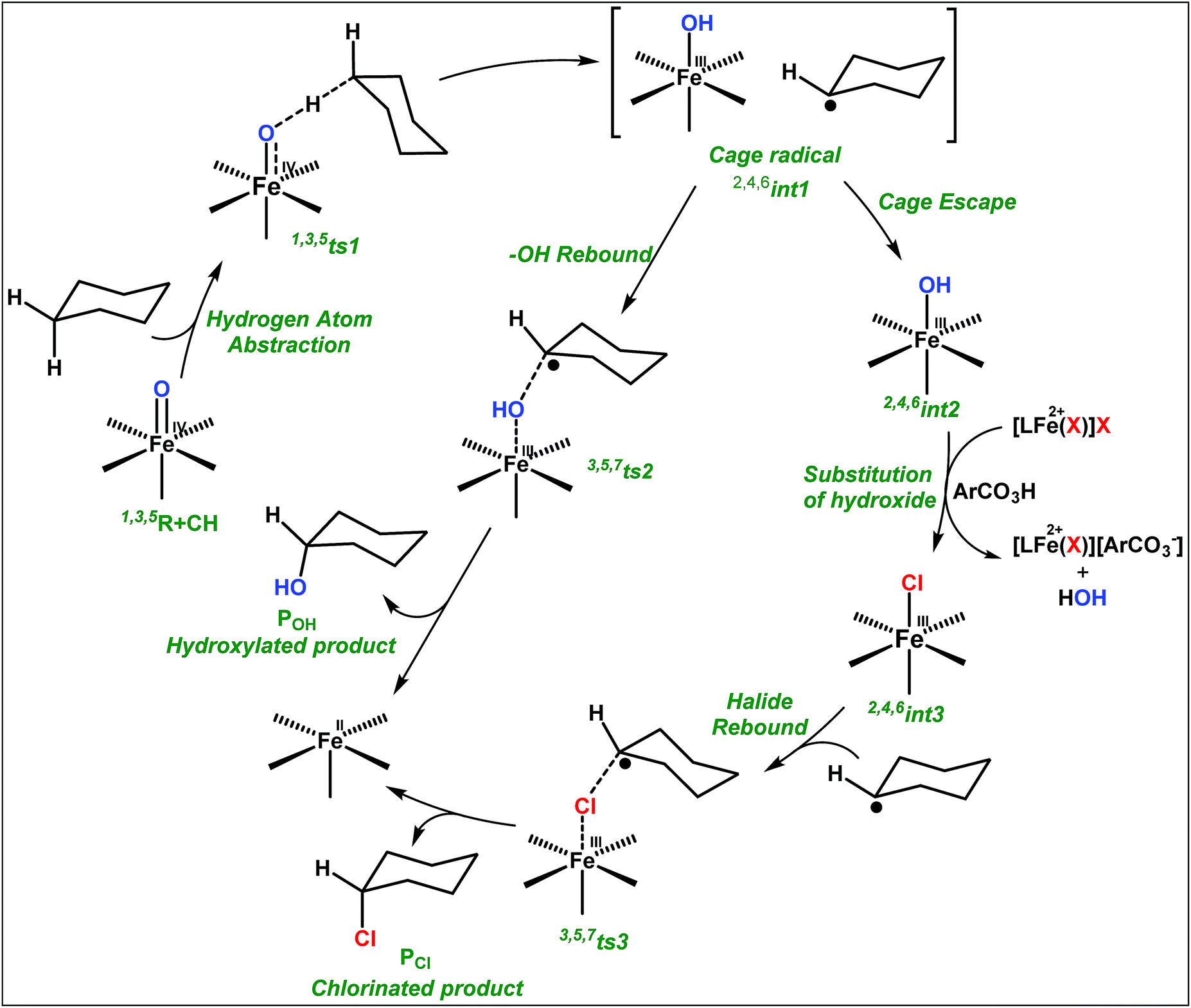 | ||
| Fig. 10 Adopted mechanism for the DFT calculation for the halogenation/hydroxylation of cyclohexane by the putative Fe(IV)–O species. | ||
Initially, the Fe(IV)O moiety abstracts the H-atom from the cyclohexane via ts1 leading to the formation of Fe(III)-hydroxide and a radical intermediate (int1) where the carbon centered radical at the cyclohexane is expected. This radical species is expected to be in the vicinity of the Fe(III)–OH units stabilized by weak non-covalent interactions (cage-radical species). For the C–H bond activation, the barrier heights are computed to be 69.4 kJ mol−1 and 96.0 kJ mol−1 in the quintet and triplet surfaces, respectively. For the S = 0 state on the other hand, the computed barrier height is prohibitively high (175.2 kJ mol−1) and rules out the possibility of S = 0 participating in the reaction mechanism. Although the triplet state is the ground state for the Fe(IV)O species, the quintet state is found to have the lowest energy barrier for the C–H bond activation and this suggests a two-state reactivity scenario as witnessed in several other cases.16a,26
The optimized structure of the transition state corresponding to S = 2 is shown in Fig. 11a. In the 5ts1, the Fe–O bond is elongated from 1.626 Å to 1.693 Å whereas the C–H bond is stretched from 1.101 Å to 1.194 Å. The distance for the newly developed O⋯H interaction is 1.432 Å. All these parameters indicate that the Fe–O double bond and C–H bond are not broken completely in the transition state suggesting a reactant like transition state. The spin density plot of 5ts1 (Fig. 11b) clearly suggests transfer of an α-electron from the substrate to the Fe-center and this is also reflected in the enhanced value of spin densities computed at the Fe centre for the transition state revealing a σ-type pathway (Fig. 12a and b) as shown in the orbital evolution diagram. In the 5ts1 transition state, the Fe–O⋯H angle is estimated to be 167.3° degrees and this indicates a σ-type pathway for the H-atom abstraction reaction. A similar scenario is witnessed also for the high-lying 3ts1 species, except for the fact that the Fe–O⋯H angle is estimated to be 127.4° degrees indicating a π-type pathway at the S = 1 surface.
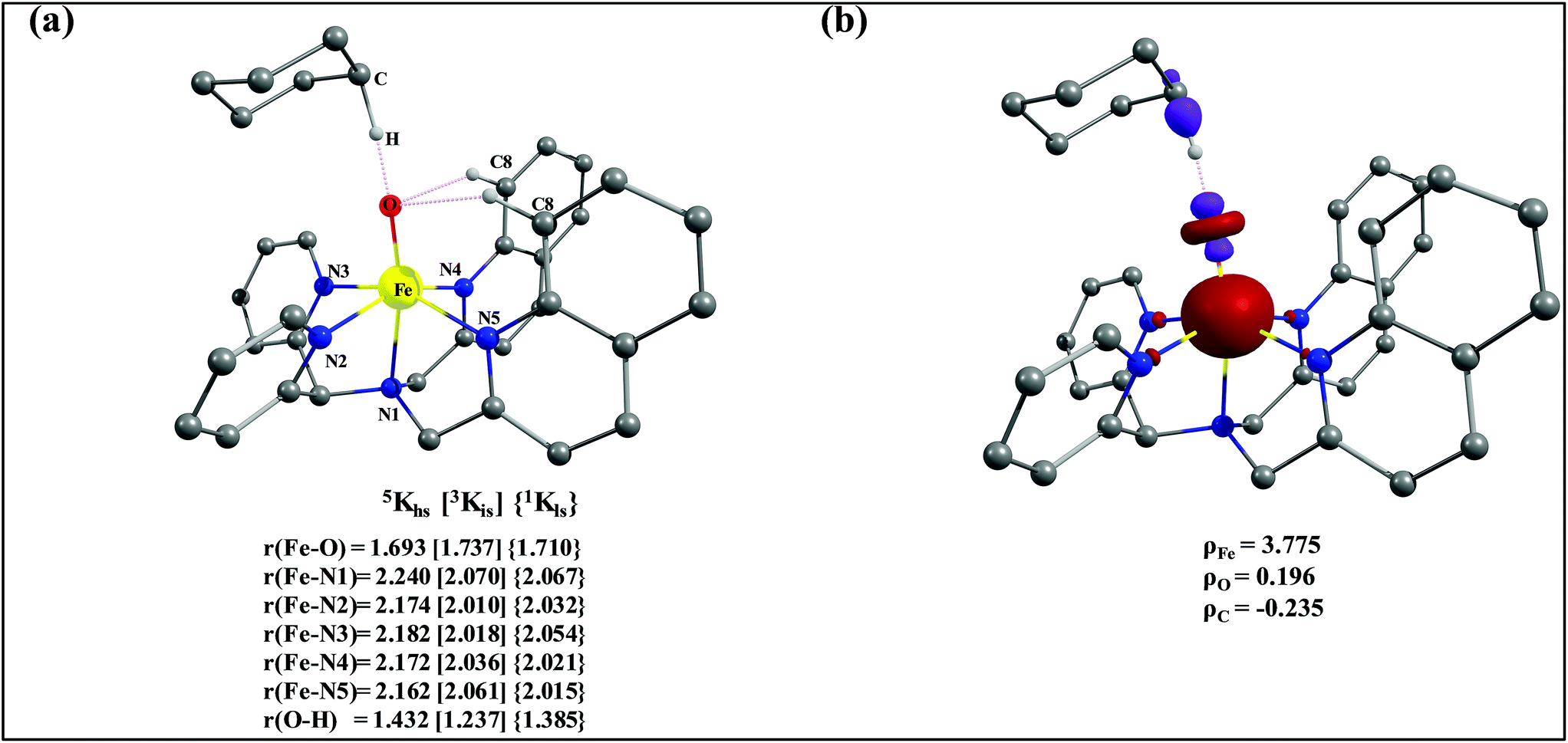 | ||
| Fig. 11 (a) Important structural parameters of the B3LYP-D2 optimized structures of the ts1 and (b) spin densities of B3LYP-D2 computed 5ts1. | ||
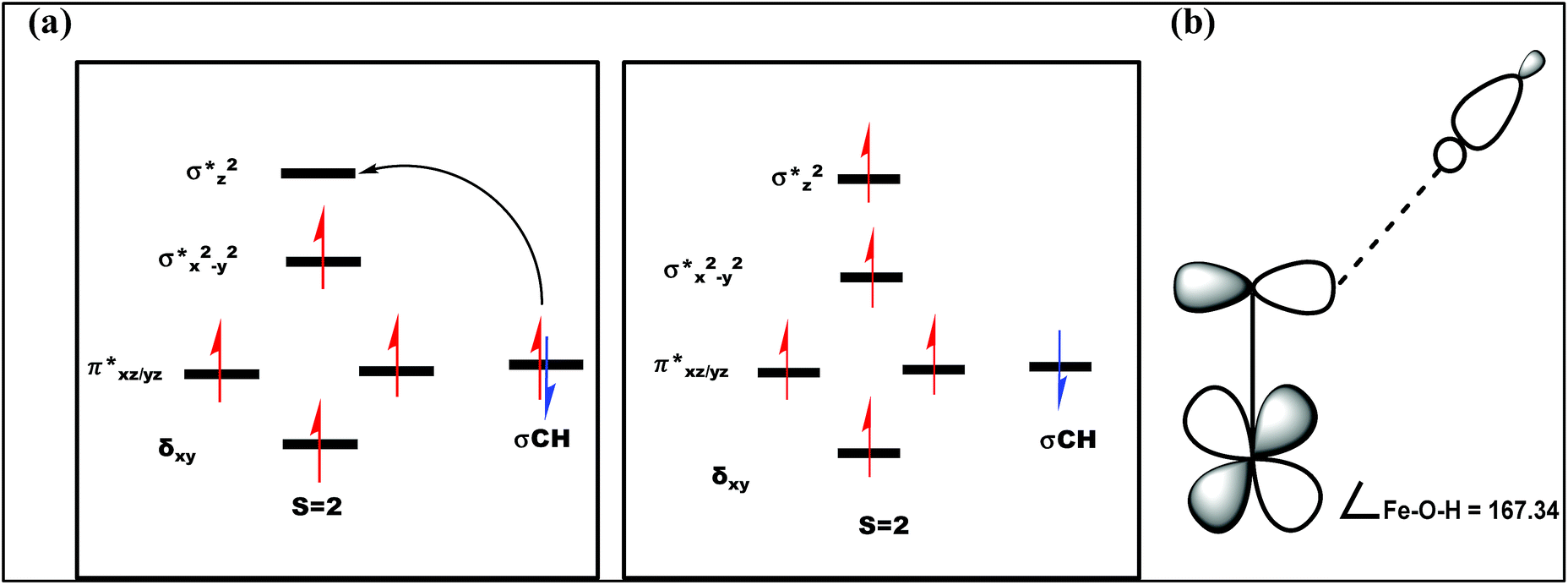 | ||
| Fig. 12 (a) Orbital occupancy diagrams for the H-abstraction process and (b) corresponding orbital selection rules for predicting transition-state structures. | ||
After the H-abstraction transition state, int1 formation is expected and this step is computed to be endothermic for all the spin states. For the int1 species, a triplet state arising from the S = 1 spin surface is found to be the lowest lying in energy (10.5 kJ mol−1) followed by the quintet state at 20.7 kJ mol−1 (5int1hs). Besides these two states, 3int1is, 7int1hs, 3int1ls and 1int1ls states are found to lie 22.4, 27.3, 61.8, and 66.5 kJ mol−1 higher in energy, respectively. The next obvious step from the int1 is the –OH rebound transition state (ts2) where the hydroxide is expected to rebound to the radical intermediate leading to the hydroxylated product. We have computed the rebound transition state at the triplet, quintet and septet states. The energetic cost associated with the hydroxylation step is estimated to be 173.0 kJ mol−1 at the quintet surface (5ts2) and this is the lowest lying transition state with other two spin surfaces found to lie even higher in energy (35.6 and 117.2 kJ mol−1 for 3ts2 and 7ts2 respectively, compared to the 5ts2 species, see Fig. 13). The barrier heights computed for the hydroxylation step are steeply higher compared to –OH rebound barriers reported for the related [FeIV(N4Py)O]2+ species. The optimized structure of the 5ts2 species along with selected structural parameters computed for other spin states is given in Fig. 14. At the transition state, the Fe–O(H) bond is elongated to 1.920 Å and the newly developed O⋯C interaction is still longer (3.268 Å). The cyclohexane attempts to approach between the pyridine and quinoline ring and this is still a sterically hindered position as evidenced from the distortions seen in the quinoline moiety which lies closer to the cyclohexane substrate. This structural deformation adds an energy penalty leading to a very large barrier height for hydroxylation to occur (see below for further discussion on this aspect). Due to the stronger C–H⋯O interactions, positioning of the hydrogen atom of the –OH group is restricted and this leads to the substrate to approach from a sterically hindered position leading to a large barrier. The formation of the hydroxylated product is exothermic with the 5POH lying at −132.5 kJ mol−1 followed by 1POH and 3POH at −82.4 and −80.5 kJ mol−1, respectively. The computed –OH rebound barrier height for the [FeIV(N4Py)O]2+ species with the cyclohexane substrate is reported to be 55.2 kJ mol−1 at the quintet surface with a slightly different computational set up.23a The hydroxylation product formation energy is reported to be −153.5 kJ mol−1 for the [FeIV(N4Py)O]2+ species and this is very similar to the one computed for 2. This clearly reveals that the additional phenyl ring attached to the ligand in 2 substantially increases the kinetic barrier height to prevent a facile hydroxylation reaction, albeit the thermodynamics of reactions remain similar.
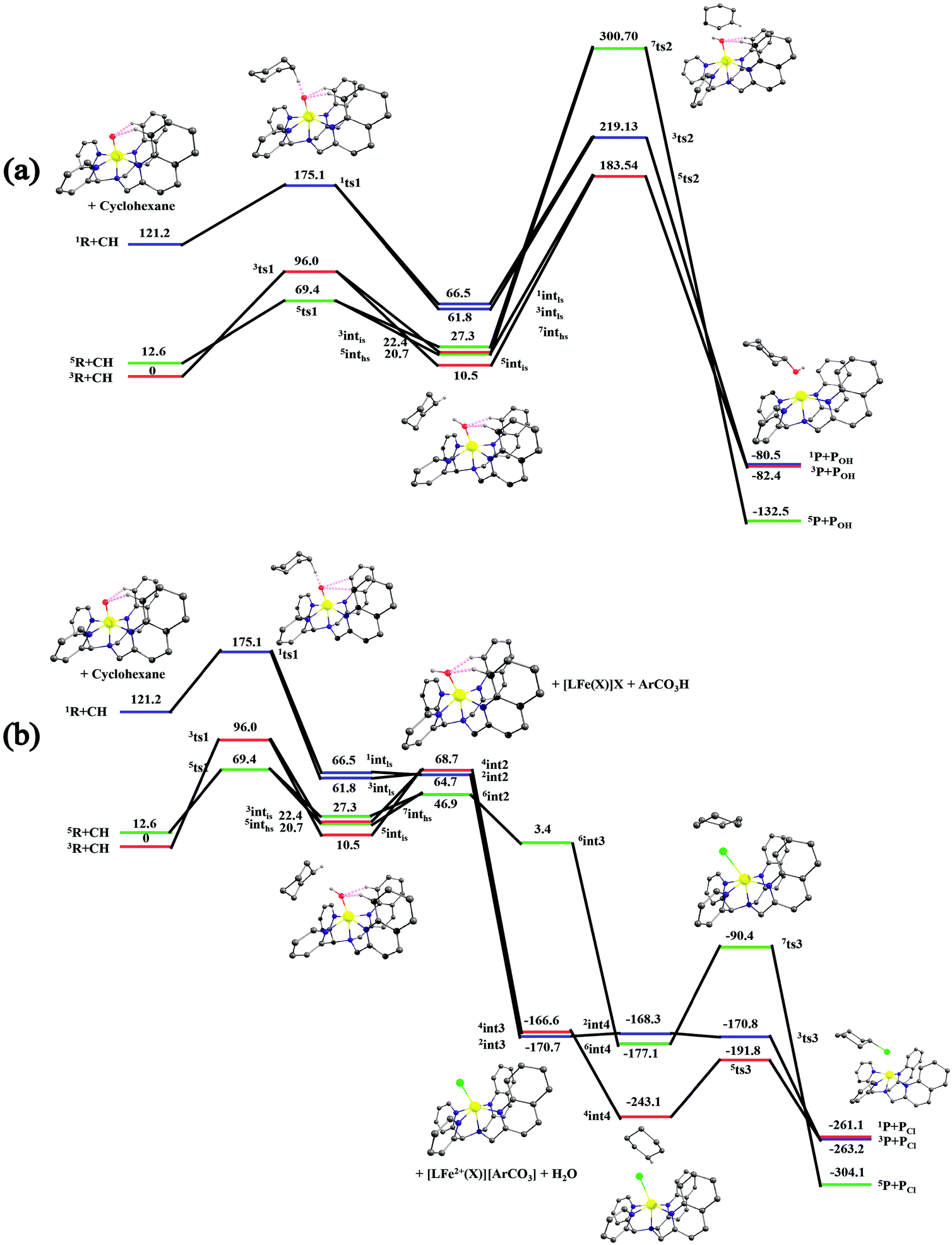 | ||
| Fig. 13 Depiction of the B3LYP-D2-computed potential energy surface (ΔG in kJ mol−1) for the C–H activation followed by (a) the O–H rebound process and (b) the cage escape and subsequent halogenation pathway. Horizontal lines represent the spin state: high spin by green color, low spin state by blue and the intermediate state by red. The C–H bond activation pathway is shown in black, with the –OH rebound leading to hydroxylation in pink and –Cl rebound leading to the halogenation pathway in orange. | ||
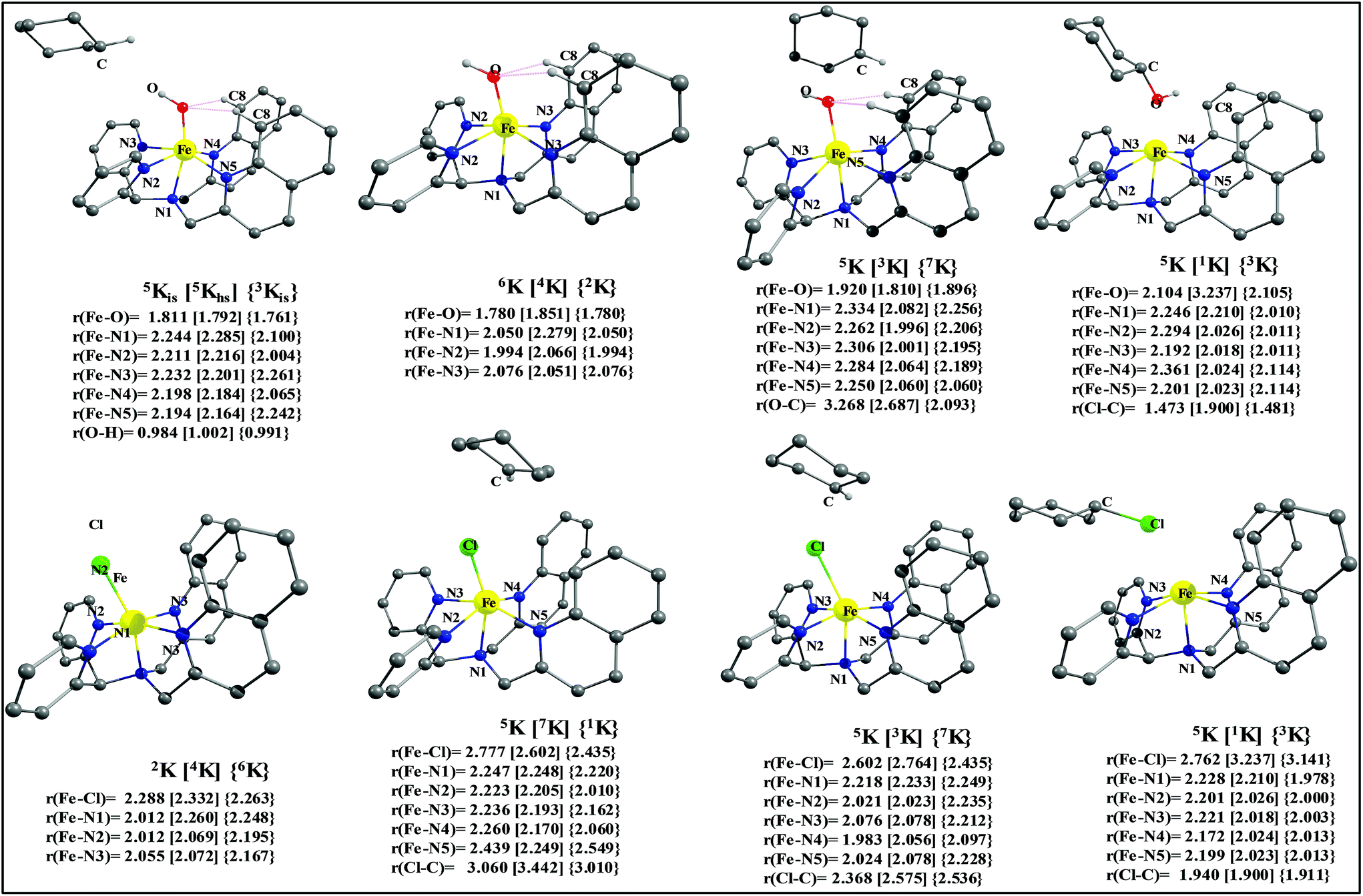 | ||
| Fig. 14 B3LYP-D2-optimized structures with selected structural parameters: (a) 5,5,3int1, (b) 2,4,6int2, (c) 3,5,7ts2, (d) 1,3,5P + POH, (e) 2,4,6int3, (f) 3,5,7int4, (g) 3,5,7ts3, and (h) 1,3,5P + PCl. | ||
Since the hydroxylation step is computed to be kinetically hindered, in the next step, the cyclohexyl radical species is assumed to leave the proximity of the Fe(III)–OH species leading to the formation of a cage-escaped radical and the Fe(III)–OH intermediate (int2). This step is computed to be endothermic in energy with the 6int2 species lying 46.9 kJ mol−1 and 4int2 and 2int2 at 64.7 and 68.7 kJ mol−1 higher in energy, respectively. The optimized structure of the 6int2 is shown in Fig. 14; here the Fe–O(H) bond distance is estimated to be 1.812 Å revealing an Fe–O single bond character and the N(1)–Fe–O angle is 164.6° revealing further inclination towards the pyridine rings as the Fe–O bond strength weakens.
In the next step, addition of Fe(II)–Cl into the reaction mixture is expected to generate Fe(III)–Cl (also can be generated by one electron oxidation of Fe(II)–Cl by the mCPBA) (int3) species by ligand exchange (Fig. 10). We have computed the thermodynamics of this reaction and the formation of int3 is substantially exothermic with the 2int3 species being the lowest in energy (−170.7 kJ mol−1) followed by the 4int3 and 6int3 species at −166.6 kJ mol−1 and +3.4 kJ mol−1 respectively. For the 2int3 species the Fe–Cl bond distance is estimated to be 2.262 Å and the –Cl atom is also found to bend towards the pyridine ring with the N(1)–Fe–Cl bond angle estimated to be 159.4° and this is smaller than that found for the corresponding Fe(III)–OH species. The Fe atom possesses a spin density of 1.048 at the ground state with some spin density delocalized to the –Cl atom (see Table S6 in the ESI†). Subsequently, the cage-escaped cyclohexyl radical species penetrate into the coordination sphere of the int3 species leading to the formation of the {Fe(III) –Cl⋯cage radical} species (int4) and this formation is also found to be excessively exothermic with respect to the Fe(III)–Cl formation by −72.4 kJ mol−1 for the quintet surface (5int4). The 7int4 and 1int4 species are found to be 66.0 and 74.8 kJ mol−1 respectively, higher compared to the 5int4 species. At the ground state, the radical species is in the vicinity of the complex with the Fe–C(radical) distance estimated to be 3.06 Å and is stabilized purely by the dispersion.
Further, the rebound of the –Cl from the {Fe(III)–Cl⋯cage radical} (int4) via ts3 is expected. Transition states corresponding to the –Cl rebound have been calculated for three different spin surfaces with the 5ts3 found to be the lowest energy transition state with an estimated barrier height of 51.3 kJ mol−1 for –Cl rebound. This is followed by two other transition states, 3ts3 and 7ts3, lying 21 kJ mol−1 and 101.4 kJ mol−1 higher in energy compared to the 5ts3 transition state. The geometry of the 5ts3 transition state is shown in Fig. 14, along with the structural parameters computed for other two transition states. At the 5ts3 transition state, the newly forming Cl⋯C(cyclohexane) distance is estimated to be 2.368 Å and this is slightly lower than that found for the corresponding intermediate. Similarly the Fe–Cl bond is elongated from 2.439 Å to 2.602 at the transition state. Although this transition state has a lower-bound barrier of 51.3 kJ mol−1, a significant gain in energy upon replacing the –OH by –Cl leads to overall negative energy for the transition state 5ts3 from the Fe(IV)O reactant, suggesting that the reaction is expected to take place very rapidly without any significant kinetic barrier. In the next step, the formation of the chlorinated product takes place and this is computed to be exothermic in nature with {5P + PCl} being the ground state with the formation energy of −61.0 kJ mol−1. The {1P + PCl} and {3P + PCl} spin states of the products are higher in energy by 40.9 kJ mol−1 and 43.0 kJ mol−1 from the ground state {5P + PCl} species.
It is worth comparing the computed mechanistic aspects to the experimental findings. To begin with, the triplet-quintet gap for species 2 is drastically lower compared to the [FeIV(N4Py)O]2+species due to the difference in the geometry as discussed earlier. The C–H bond activation barrier computed at the S = 2 surface for species 2 is the rate determining step of the halogenation reaction. This barrier is estimated to be 69.4 kJ mol−1 for species 2, while the same is computed to be 79.9 kJ mol−1 for the [FeIV(N4Py)O]2+species.23a As the Fe–N bonds present in the equatorial planes are unsymmetrical and longer for species 2 compared to [FeIV(N4Py)O]2+, the estimated barrier for C–H bond activation is lower. This is also reflected in the control experiments performed with cyclohexane where species 2 is found to react 20 times faster compared to the [FeIV(N4Py)O]2+ species. Secondly, upon H-atom abstraction, a cage-radical formation takes place, which is slightly endothermic in nature. However there is a substantial barrier for the –OH rebound from the Fe(III)–OH species. This is correlated with the difference in the ligand structure (both steric and electronic effects). Although a significant barrier for –OH rebound has also been computed for [FeIV(N4Py)O]2+,23a in our case the barrier is much larger suggesting a possibility of cage-escape. This is also supported by the control experiments performed with species 2 and cyclohexane, toluene and ethyl benzene as substrates.
The radical escaping from the Fe(III)–OH species is computed to be slightly endothermic (by 46.9 kJ mol−1) and suggests that such an event is certainly possible with species 2. This is essentially due to the fact that the additional quinoline present in the ligand moiety exerts steric repulsion to the radical species and the phenyl rings of the quinoline are also not in favorable orientation for a C–H⋯π interaction with the radial species. Thus strong anchoring of the radical species with the Fe(III)–OH species is absent leading to the cage-escape being the favored pathway. In such a scenario, the radical is expected to have a longer life time leading to stereochemical scrambling and this is also witnessed in the control experiments performed with species 2. Addition of the Fe(II)–Cl complex into the solution, however, changes drastically the scenario as the exchange between –OH and –Cl is found to an energetically favourable exothermic process. Besides the radical cage formation with the Fe(III)–Cl is found to be further exothermic suggesting the clear possibility of the cage-escaped radical to preferably penetrate into the coordination sphere of the Fe(III)–Cl species. It is highly informative to compare the cage-radical species {cyclohexylradical⋯Fe(III)–Cl} and {cyclohexylradical⋯Fe(III)–OH} from the corresponding Fe(III)–Cl and Fe(III)–OH. The formation of {cyclohexylradical⋯Fe(III)–OH} from the Fe(III)–OH is exothermic by 36.4 kJ mol−1 while the formation of {cyclohexylradical⋯Fe(III)–Cl} from Fe(III)–Cl is exothermic by 72.4 kJ mol−1. Thus the energetic gain associated with the formation of {cyclohexylradical⋯Fe(III)–Cl} is more than compensated by the energetic loss due to the cage-escape process from the {cyclohexylradical⋯Fe(III)–OH}. The stabilization of {cyclohexylradical⋯Fe(III)–Cl} compared to the corresponding Fe(III)–OH is also correlated with the variation in the electronic structure. The cyclohexylradical in the {cyclohexylradical⋯Fe(III)–Cl} caged species lies in closer proximity to the complex and the carbon radical is found to possess a smaller spin density suggesting a long range electron-transfer from the radical to the {Fe(III)–Cl} complex leading to a variation in the electronic structure and hence the energetic gain. This electron transfer is supported by the high electron affinity difference between the two species {EA[Fe(III)–Cl] = −122.95 kCal mol−1 and EACy˙ = −47.43 kCal mol−1). This is also in agreement with the ionization potential of the two.27 Again the highest occupied molecular orbital of the Fe(III)–Cl complex lies lower in energy than that of the radical system, indicative of the feasibility of electron transfer.
Now we turn to analyze the transition state corresponding to the hydroxylation and halogenation reaction to ascertain why halogenation is favoured with species 2. The lower-bound barrier for –OH rebound from the Fe(III)–OH species is 173 kJ mol−1 while for –Cl rebound from the Fe(III)–Cl is estimated to be 51.3 kJ mol−1. Both the lower-bound barriers are for the quintet surface and hence the difference due to the nature of spin-states can be ruled out. Particularly, the –OH rebound barrier is substantially higher and this is correlated with the approach of the cyclohexyl radical towards the hydroxyl group. Strong deformation at both the complexes and the substrate is noted for this transition state with the total deformation energy computed to be 111.1 kJ mol−1 for the quintet transition state. As the barrier computed is 173 kJ mol−1, the difference of 61.6 kJ mol−1 is due to electronic contribution to the transition state. The deformation energy clearly reveals that the steric factor dominates the barrier height and is the primary reason for such a large barrier height. The deformation energy computed for the –Cl rebound transition state is much lower (40.0 kJ mol−1), although here as well the steric factor seems to dictate the terms of the kinetic outcome. A careful look at the geometries of both the transition states reveals that the cyclohexyl radical needs to approach closer to the Fe centre in the Fe(III)–OH rebound transition state as the Fe(III)–OH distance is estimated to be 1.920 Å at the transition state. For the –Cl rebound transition state, however, the Fe(III)–Cl distances are much longer (2.602 Å). Therefore, a very close approach (causing steric repulsion with the bulky quinoline ligand) is avoided. This leads to a lower barrier height. Additionally, the energetic gain upon exchanging the –OH with –Cl eases out the energetic cost of the chlorination, since the –Cl rebound is a barrier-less process. A facile chlorination is likely to happen once the C–H bond abstraction completes. It is important to note that the cyclohexyl radial species formed upon hydrogen atom abstraction needs to escape from the Fe(III)–OH cage and penetrate back in to the Fe(III)–Cl species to complete the chlorination.
The cage-escaped radical could choose to react via other possible routes. This is consistent with the experimental observations that in the presence of air, due to the auto-oxidation process, other products such as ketone and alcohol are witnessed.
As steric congestion of the quinoline at the rebound barrier determines the rate of the reaction, one can rationalize the faster bromination reactions compared to chlorination that are witnessed in the experiments. As the Fe(III)–Br distances are expected to be longer than the Fe(III)–Cl bond, the rebound of –Br at the transition state is expected to have less steric repulsion than the corresponding –Cl rebound transition state. This is expected to reduce the barrier even further leading to faster reactivity for bromination compared to chlorination.
Conclusions
In summary, we have developed a new strategy for selective halogenation by using a room-temperature stable non-heme iron(IV)-oxo complex in the presence of an iron(II)-halide complex. The interplay of non-heme iron(IV)-oxo and iron(II)-halide complexes provided selective halogenation. The present strategy and detailed theoretical investigations are expected to have immense importance for further development of selective halogenation methods.Experimental section
Materials and methods
All 1H NMR spectra were reported in units of parts per million (ppm) and measured relative to the signals for residual chloroform (7.26 ppm) in CDCl3/and for residual CH3CN in CD3CN at 1.96 ppm, unless otherwise stated. All 13C NMR spectra were reported in ppm relative to CDCl3 (77.23 ppm), unless otherwise stated and were obtained with 1H decoupling. Acetonitrile, CD3CN, FeIICl2, FeIIBr2, and trimethylsilyltriflate (TMSOTf) were purchased from Sigma Aldrich. Diethyl ether was procured from Spectrochem chemicals. Ethylbenzene was bought from TCI. H218O was purchased from Sigma Aldrich and ICON-isotope. 2-(Chloromethyl)quinoline hydrochloride was purchased from TCI and di(2-pyridyl)ketone was purchased from Alfa Aesar. All the products were analyzed by GC-MS analysis. GC-MS was performed on a Thermo Scientific ISQ QD Mass Spectrometer attached with a Thermo Scientific TRACE 1300 gas chromatograph using an HP-5ms capillary column (30 m × 0.25 mm × 0.25 μm, J&W Scientific) with helium as the carrier gas. The product yields were calculated from GC traces by comparing with the area percentage of standard products.Kinetic studies
All the kinetic studies were carried out at 25 °C under a N2 atmosphere. First order rate constants (k1) were calculated based on non-linear exponential fit in OriginPro8 software. All the kinetic studies were carried out under pseudo-first-order conditions. The rate constant (k1) was calculated based on the decay pattern of the iron(IV)-oxo complex at 770 nm by nonlinear curve fitting, [y = y0 + A*exp(R0*x)], (where x is the time, t and R0 is the rate constant, k1, and y is the absorbance) and showed a good fit with the rate constant value within 10% error. The resulting k1 values varied linearly with substrate concentration to give the second-order rate constant k2. All the sp3 C–H oxidation and halogenation reactions were carried out at 25 °C under a N2 atmosphere inside a glove box.Instrumentation
NMR spectra were recorded on a Bruker 400/500/750 MHz. ESI-MS spectra were recorded on a Bruker QTOF ESI-MS instrument. UV-vis kinetic studies were performed using an Agilent 8453 diode array based UV-vis spectrophotometer. Single crystals were diffracted using a Rigaku X-ray single crystal diffractometer. GC-MS was performed on a Thermo Scientific ISQ QD Mass Spectrometer attached with a Thermo Scientific TRACE 1300 gas chromatograph using an HP-5ms capillary column (30 m × 0.25 mm × 0.25 μm, J&W Scientific) with helium as the carrier gas and an Agilent 7890A GC system connected with a 5975C inert XL EI/CI MSD (with a triple axis detector). EPR was recorded with a Bruker EMX plus series instrument using an X-band at 4 K. Mössbauer spectra were recorded at 5.5 K or 80 K on a lowfield Mössbauer spectrometer equipped with a Janis CCR cryostat and at 5.5 K on a strong-field Mössbauer spectrometer equipped with an Oxford Instruments Spectromag 4000 cryostat containing an 8 T split-pair superconducting magnet. Both spectrometers were operated in a constant acceleration mode in transmission geometry. The isomer shifts were referenced against that of a metallic iron foil at room temperature. Analysis of the data was performed with the program WMOSS (WMOSS4 Mössbauer Spectral Analysis Software, www.wmosss.org, 2009–2015) and a home-made program.27Computational details
All calculations were performed using the Gaussian 09 suite of program. The geometries were optimized using the B3LYP-D2 functional which incorporates the dispersion proposed by Grimme et al.28 The geometry optimization was performed using the LACVP basis set, comprising the LanL2DZ double ζ-quality basis set with the Los Alamos effective core potential for Fe and the 6-31G* basis set for the other atoms. Single point energy calculations were performed on the optimized geometries using a TZVP basis set on all atoms. The PCM model was used to incorporate solvent effects on the geometries by including the role of solvent on the geometries and energies of the optimized structures. Here we have used acetonitrile as a solvent for our computation. We have computed frequency for all the geometries and confirmed the minima/transition state nature. All the transition states were characterized by one imaginary frequency corresponding to the expected reaction coordinate and verified by animating the vibration mode using Chemcraft. All the other species possess only positive frequencies. All the energies reported are free energies including the zero-point, enthalpy and entropic contributions unless specified otherwise.Synthesis and characterization of the 2PyN2Q ligand
Hydroxylamine hydrochloride (750.5 mg, 10.8 mmol) and sodium acetate (NaOAc) (886 mg, 10.8 mmol) were heated in 10 mL of distilled water at 60 °C for one hour. After heating the solution for one hour, a solution of di(2-pyridyl)ketone (1 g, 5.43 mmol) in 5 mL MeOH was added. The resulting mixture was stirred at 80 °C overnight. Consequently, the pink colored solidified oxime was obtained. The product oxime was washed with methanol (MeOH) and the solvent was dried over a rotary evaporator. The crude oxime, a pink solid, was used in the next step without further purification. The above prepared oxime (1 g, 5 mmol), ammonium acetate (NH4OAc, 655 mg, 8.5 mmol), ammonia (NH3, 25% aqueous solution, 15 mL), and ethanol (20 mL) were mixed along with 10 mL of water and heated at 80 °C. Activated Zn dust (1.47 g, 22.5 mmol) was then added to the reaction mixture in small amounts for a duration of 30 minutes. The resulting mixture was refluxed for 4 hours and then stirred at room temperature overnight. The mixture was filtered and the residue was washed with methanol (MeOH) and water. The filtrate was concentrated and the resulting aqueous solution was made strongly alkaline with 10 (M) sodium hydroxide (NaOH) solution. The amine was then extracted with dichloromethane and the organic phase was then washed with brine, dried over Na2SO4 and concentrated in a rotary evaporator and vacuum to afford a brown oil. 1H NMR (400 MHz, CDCl3, δ): 8.48 (m, 2H, Py), 7.55 (m, 2H, Py), 7.31 (d, 2H), 7.04 (m, 2H), 5.25 (s, 1H, CH), 2.43 (s, 2H, NH2). 2-(Chloromethyl)-quinoline hydrochloride (9.9 mmol, 2.2 g) was added to an aqueous solution of sodium hydroxide (NaOH) (5 mL, 5 M) at 0 °C. After stirring for 10 minutes, the solution was added to bis(2-pyrimidyl) methylamine (0.97 g; 5.23 mmol) and another portion of an aqueous solution of NaOH (5 M, 5 mL).29 The solution was allowed to stir for 36 hours at room temperature and then concentrated perchloric acid (HClO4) was added dropwise to get a sticky brownish precipitate, which was recrystallized from hot water. Treatment of this perchlorate salt with 2.5 (M) NaOH solutions and extraction with dichloromethane yielded a yellowish solid 2PyN2Q in 40% yield. The synthesized ligand was thoroughly characterized by 1H, 13C NMR and ESI-MS analysis. The obtained NMR data for the ligand, 2PyN2Q: 1H NMR (500 MHz, CDCl3, δ): 4.21 (s, 4H), 5.452 (s, 1H), 7.11 (2H), 7.45 (2H), 7.60–7.68 (6H, m), 7.73 (2H), 7.80 (2H), 8.01 (4H), and 8.58 (2H). 13C NMR (125 MHz, CDCl3): δ 58.40, 72.69, 121.51, 122.40, 124.51, 126.20, 127.36, 127.55, 129.06, 129.44, 136.33, 136.57, 147.60, 149.44, 160.54, and 160.02. ESI-MS ([M + Na]: observed, 490.197; calculated for C31H25N5Na: 490.20).Synthesis and characterization of iron(II)-complex 1, [FeII(2PyN2Q)](OTf)2
The ligand 2PyN2Q (1.2 mmol) and iron(II)-precursor complex Fe(OTf)2·2CH3CN30,31 (1.0 mmol) were reacted overnight in an excess amount of acetonitrile (40 mL) inside a glove box. The reaction mixture was concentrated by applying vacuum. Then excess dry diethyl ether was added to the reaction mixture and the Schlenk was shaken vigorously to get the precipitate of the complex. The mixture was kept undisturbed to settle down the precipitate of the complex at the bottom of the Schlenk flask. The clear solution part above the precipitate was decanted off. Then the precipitate was dried properly by applying vacuum and kept under a N2 atmosphere in a glove box. The resultant complex was crystallized from a dichloromethane and diethyl ether solvent mixture. The corresponding triflate anion coordinated single crystal was grown from the acetonitrile–toluene or acetonitrile–diethylether solvent combination. The obtained complex was characterized spectroscopically. UV-vis (MeCN): 368 nm (ε ∼ 1954 L mol−1 cm−1) and 470 nm (ε ∼ 1954 L mol−1 cm−1). ESI-MS (observed, 688.090; calculated, 688.093), elemental anal. (calculated for C33H25F6FeN5O6S2) C, 48.25; H, 3.07; N, 8.52; S, 7.80; found: C, 48.145; H, 3.30; N, 8.256; S, 7.65.Synthesis and characterization of iron(IV)-oxo-complex 2, [FeIV(2PyN2Q)(O)]2+
The complex 1 (1.21 mM solution in acetonitrile) was reacted with 1.5–2 equiv. of MesI(OAc)2 or mCPBA in acetonitrile. The resulting solution forms iron(IV)-oxo complex 2, within 1 minute of addition of the oxidant. The corresponding iron(IV)-oxo complex showed a UV-vis band at 770 nm (d–d transition) with a molar extinction coefficient of ε ∼ 250 L mol−1 cm−1. The formation of the iron(IV)-oxo complex 2 was monitored by UV-vis. The complex 2 showed the half-life around ∼30 minutes. The iron(IV)-oxo species 2 was characterized by ESI-MS (Calculated for [FeIV(2PyN2Q)(O)(OTf)]+, m/z 688.093, obtained 688.090).Synthesis and characterization of iron(II)-complex 3, [FeII(2PyN2Q)(Cl)]Cl32
The ligand 2PyN2Q (1 mmol) and iron(II)-chloride, FeCl2 (1.2 mmol), were reacted for 12 h in tetrahydrofuran (THF) and acetonitrile inside a glove box at 25 °C. The reaction mixture was concentrated by applying vacuum. Excess dry hexane was added to the reaction mixture and shaken to get the precipitate of the complex. The mixture was allowed to settle down. The clear solution above the precipitate was decanted off. Then the precipitate was dried properly by applying vacuum and kept under a N2 atmosphere in a glove box. The single crystal of the corresponding chloro complex was obtained from acetonitrile. The crystallized complex was characterized spectroscopically. UV-vis (MeCN): λmax = 358 nm, 454 nm, ESI-MS (observed, 558.10; calculated, 558.11), elemental anal. (calculated for -C31H25Cl2FeN5) C, 62.65; H, 4.24; N, 11.78; found: C, 62.50; H, 4.20; N, 11.71.Synthesis and characterization of iron(II)-complex 4, [FeII(2PyN2Q)(Br)]Br
The ligand 2PyN2Q (1.2 mmol) and iron(II)-bromide, FeBr2 (1.0 mmol), were reacted for 12 h in an excess amount of tetrahydrofuran (40 mL) and acetonitrile inside a glove box. The reaction mixture was concentrated by applying vacuum. Then excess dry hexane was added to the reaction mixture and shaken to get the precipitate of the Fe complex. The mixture was left undisturbed to settle down the precipitate of the complex at the bottom of the Schlenk flask. The clear solution part above the precipitate was decanted off. Then the precipitate was dried properly by applying vacuum and kept under a N2 atmosphere in a glove box. UV-vis (MeCN): λmax = 352 nm, 460 nm, ESI-MS (observed, m/z, 558.10; calculated, 558.11), elemental anal. (calculated for C31H25Br2FeN5) C, 54.50; H, 3.69; N, 10.25; found: C, 54.41; H, 3.60; N, 10.10.Synthesis and characterization of iron(II)-complex [FeII(2PyN2Q)(X)](ClO4) (5 and 6)33
The above-synthesized complexes 3 and 4 were reacted with 2.5 equiv. of NaClO4 in MeOH–MeCN (20 mL). The resulting solution was stirred for 4 hours. This process was repeated for a second time. The complex solution was dried to get an orange powder complex. Then the residue was dissolved in dichloromethane (DCM) to get the complex in the solution. The remaining white residue remained at the bottom of the Schlenk flask. The solution was kept idle for 1 hour to settle down the white precipitate. Then the clear red solution was decanted off and filtered through Whatman filter paper fitted in the sintered funnel. The filtrate was dried to get an orange powder [FeII(2PyN2Q)(X)](ClO4). The complexes were crystallized from chloroform/hexane and used for halogenation reactions. The anion exchanges were carried out for both the complexes 3 and 4. The perchlorate anion containing complexes 5 and 6 showed similar UV-vis data to complexes 3 and 4. Further the complexes were characterized by X-ray crystallography. For complex 5, UV-vis (MeCN): λmax = 355 nm, 466 nm, ESI-MS (observed, m/z 558.103; calculated, 558.115), elemental anal. (calculated for C31H25Cl2FeN5O4) C, 56.56; H, 3.83; N, 10.64; found: C, 56.46, H, 3.70, N, 10.50. For complex 6, UV-vis (MeCN): λmax = 355 nm, 445 nm, ESI-MS (observed, m/z 602.056; calculated, 602.064), elemental anal. (calculated for C31H25BrClFeN5O4) C, 52.98; H, 3.59; N, 9.97; found: C, 52.75; H, 3.47; N, 9.73.General procedure for sp3 C–H oxidation reactions
From the stock solution of complex 1, (1.21 mM acetonitrile solution), 1.5 mL was taken in a 20 mL vial along with a stir bar. 1.5 equiv. of MesI(OAc)2 were added under stirring conditions. After 1 minute, an excess amount (>100 equiv.) of substrate was added to the reaction mixture and the reaction was stirred for 30 min. Finally, the reactions mixtures were analyzed by GC/GC-MS studies. The yields of the hydroxylation products were calculated based on the area of the standard products.General procedure for the 18-O labelling study for sp3 C–H oxidation reactions
To a solution of complex 2 (1.21 mM), MesI(OAc)2 (2 equiv.) was added to generate the iron(IV)-oxo complex 2 inside a glove box. Subsequently 50 μL of H218O was added to this solution. The resulting solution was kept in a −35 °C freezer inside a glove box for 40 minutes. The solution was taken out from the fridge, ethylbenzene was added and the reaction was continued for 30 minutes. Reaction mixtures were analyzed by GC-MS to detect the percentage of 18-O labelling in the products.General procedure for sp3 C–H halogenation reactions
From a stock solution of complex 1, (4 mM in acetonitrile), 1 mL of solution was taken in a 20 mL vial with a stir bar. Subsequently 2 equiv. of mCPBA were added under stirring conditions. Immediately a solution of the halide complex 3, [Fe(2PyN2Q)(Cl)](Cl), or 4, [Fe(2PyN2Q)(Br)](Br), was added to it (40 mol% w.r.t. complex 2 concentration). The substrate was added to the reaction mixture and the reaction was stirred for 30 minutes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This activity is supported by SERB, India (EMR/2015/000164). Financial support has been received from CSIR-India (fellowship to S.R., J.P.B. and A.S.).Notes and references
- (a) F. H. Vaillancourt, E. Yeh, D. A. Vosburg, S. Garneau-Tsodikova and C. T. Walsh, Chem. Rev., 2006, 106, 3364–3378 CrossRef PubMed; (b) D. G. Fujimori and C. T. Walsh, Curr. Opin. Chem. Biol., 2007, 11, 553–560 CrossRef PubMed; (c) J. Rittle and M. T. Green, Science, 2010, 330, 933 CrossRef PubMed; (d) C. Krebs, D. Galonić Fujimori, C. T. Walsh and J. M. Bollinger, Acc. Chem. Res., 2007, 40, 484–492 CrossRef PubMed; (e) R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239–2314 CrossRef PubMed; (f) S. Sahu and D. P. Goldberg, J. Am. Chem. Soc., 2016, 138, 11410–11428 CrossRef PubMed; (g) M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841–2888 CrossRef PubMed; (h) S. A. Cook, E. A. Hill and A. S. Borovik, Biochemistry, 2015, 54, 4167–4180 CrossRef PubMed; (i) S. Kal and L. Que Jr, J. Biol. Inorg Chem., 2017, 22(2–3), 339–365 CrossRef PubMed; (j) X. Huang and J. T. Groves, J. Biol. Inorg Chem., 2016, 22(2–3), 185–207 Search PubMed; (k) S. Rana, A. Modak, S. Maity, T. Patra and D. Maiti, in Progress in Inorganic Chemistry, John Wiley & Sons, Inc., 2014, vol. 59, pp. 1–188 Search PubMed.
- (a) L. C. Blasiak, F. H. Vaillancourt, C. T. Walsh and C. L. Drennan, Nature, 2006, 440, 368–371 CrossRef PubMed; (b) F. H. Vaillancourt, J. Yin and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10111–10116 CrossRef PubMed; (c) E. I. Solomon, S. D. Wong, L. V. Liu, A. Decker and M. S. Chow, Curr. Opin. Chem. Biol., 2009, 13, 99–113 CrossRef PubMed; (d) S. M. Barry and G. L. Challis, ACS Catal., 2013, 3, 2362–2370 CrossRef PubMed; (e) E. G. Kovaleva and J. D. Lipscomb, Nat. Chem. Biol., 2008, 4, 186–193 CrossRef PubMed; (f) V. Siitonen, B. Selvaraj, L. Niiranen, Y. Lindqvist, G. Schneider and M. Metsä-Ketelä, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5251–5256 CrossRef PubMed.
- (a) S. H. Knauer, O. Hartl-Spiegelhauer, S. Schwarzinger, P. Hänzelmann and H. Dobbek, FEBS J., 2012, 279, 816–831 CrossRef PubMed; (b) R. P. Hausinger, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 21–68 CrossRef PubMed; (c) S. Martinez and R. P. Hausinger, J. Biol. Chem., 2015, 290(34), 20702–20711 CrossRef PubMed.
- T. Kojima, R. A. Leising, S. Yan and L. Que Jr, J. Am. Chem. Soc., 1993, 115, 11328–11335 CrossRef.
- (a) S. D. Wong, M. Srnec, M. L. Matthews, L. V. Liu, Y. Kwak, K. Park, C. B. Bell III, E. E. Alp, J. Zhao, Y. Yoda, S. Kitao, M. Seto, C. Krebs, J. M. Bollinger and E. I. Solomon, Nature, 2013, 499, 320–323 CrossRef PubMed; (b) M. L. Matthews, C. S. Neumann, L. A. Miles, T. L. Grove, S. J. Booker, C. Krebs, C. T. Walsh and J. M. Bollinger, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17723–17728 CrossRef PubMed.
- P. Comba and S. Wunderlich, Chem.–Eur. J., 2010, 16, 7293–7299 CrossRef PubMed.
- O. Planas, M. Clemancey, J.-M. Latour, A. Company and M. Costas, Chem. Commun., 2014, 50, 10887–10890 RSC.
- S. Chatterjee and T. K. Paine, Angew. Chem., Int. Ed., 2016, 55, 7717–7722 CrossRef PubMed.
- M. Puri, A. N. Biswas, R. Fan, Y. Guo and L. Que Jr, J. Am. Chem. Soc., 2016, 138, 2484–2487 CrossRef PubMed.
- (a) W. K. C. Lo, C. J. McAdam, A. G. Blackman, J. D. Crowley and D. A. McMorran, Inorg. Chim. Acta, 2015, 426, 183–194 CrossRef; (b) A. A. Massie, M. C. Denler, L. T. Cardoso, A. N. Walker, M. K. Hossain, V. W. Day, E. Nordlander and T. A. Jackson, Angew. Chem., Int. Ed., 2017, 56, 4178–4182 CrossRef PubMed; (c) W. Rasheed, A. Draksharapu, S. Banerjee, G. Young Victor, R. Fan, Y. Guo, M. Ozerov, J. Nehrkorn, J. Krzystek, J. Telser and L. Que Jr, Angew. Chem., Int. Ed., 2018, 57, 9387–9391 CrossRef PubMed.
- See ESI†.
- (a) J. Kaizer, E. J. Klinker, N. Y. Oh, J.-U. Rohde, W. J. Song, A. Stubna, J. Kim, E. Münck, W. Nam and L. Que Jr, J. Am. Chem. Soc., 2004, 126, 472–473 CrossRef PubMed; (b) K. B. Cho, X. Wu, Y. M. Lee, Y. H. Kwon, S. Shaik and W. Nam, J. Am. Chem. Soc., 2012, 134, 20222–20225 CrossRef PubMed; (c) S. Rana, A. Dey and D. Maiti, Chem. Commun., 2015, 51, 14469–14472 RSC; (d) J. Serrano-Plana, W. N. Oloo, L. Acosta-Rueda, K. K. Meier, B. Verdejo, E. García-España, M. G. Basallote, E. Münck, L. Que Jr, A. Company and M. Costas, J. Am. Chem. Soc., 2015, 137, 15833–15842 CrossRef PubMed; (e) S. Paria, L. Que Jr and T. K. Paine, Angew. Chem., Int. Ed., 2011, 50, 11129–11132 CrossRef PubMed; (f) M. Mitra, H. Nimir, S. Demeshko, S. S. Bhat, S. O. Malinkin, M. Haukka, J. Lloret-Fillol, G. C. Lisensky, F. Meyer, A. A. Shteinman, W. R. Browne, D. A. Hrovat, M. G. Richmond, M. Costas and E. Nordlander, Inorg. Chem., 2015, 54, 7152–7164 CrossRef PubMed.
- (a) M. Puri and L. Que Jr, Acc. Chem. Res., 2015, 48, 2443–2452 CrossRef PubMed; (b) X. Engelmann, I. Monte-Pérez and K. Ray, Angew. Chem., Int. Ed., 2016, 55, 7632–7649 CrossRef PubMed; (c) K. Ray, F. F. Pfaff, B. Wang and W. Nam, J. Am. Chem. Soc., 2014, 136, 13942–13958 CrossRef PubMed; (d) S. Sahu, L. R. Widger, M. G. Quesne, S. P. de Visser, H. Matsumura, P. Moënne-Loccoz, M. A. Siegler and D. P. Goldberg, J. Am. Chem. Soc., 2013, 135, 10590–10593 CrossRef PubMed; (e) S. Sahu, M. G. Quesne, C. G. Davies, M. Dürr, I. Ivanović-Burmazović, M. A. Siegler, G. N. L. Jameson, S. P. de Visser and D. P. Goldberg, J. Am. Chem. Soc., 2014, 136, 13542–13545 CrossRef PubMed.
- (a) J. R. Bryant and J. M. Mayer, J. Am. Chem. Soc., 2003, 125, 10351–10361 CrossRef PubMed; (b) J. M. Mayer, Acc. Chem. Res., 1998, 31, 441–450 CrossRef; (c) M. Ghosh, K. K. Singh, C. Panda, A. Weitz, M. P. Hendrich, T. J. Collins, B. B. Dhar and S. Sen Gupta, J. Am. Chem. Soc., 2014, 136, 9524–9527 CrossRef PubMed; (d) M. Ghosh, Y. L. Nikhil, B. B. Dhar and S. Sen Gupta, Inorg. Chem., 2015, 54, 11792–11798 CrossRef PubMed.
- (a) J. M. Mayer, Acc. Chem. Res., 2011, 44, 36–46 CrossRef PubMed; (b) H. Mayr and R. Ofial Armin, Angew. Chem., Int. Ed., 2006, 45, 1844–1854 CrossRef PubMed; (c) A. A. Massie, A. Sinha, J. D. Parham, E. Nordlander and T. A. Jackson, Inorg. Chem., 2018, 57, 8253–8263 CrossRef PubMed.
- (a) K.-B. Cho, H. Hirao, S. Shaik and W. Nam, Chem. Soc. Rev., 2016, 45, 1197–1210 RSC; (b) S. N. Dhuri, K.-B. Cho, Y.-M. Lee, S. Y. Shin, J. H. Kim, D. Mandal, S. Shaik and W. Nam, J. Am. Chem. Soc., 2015, 137, 8623–8632 CrossRef PubMed.
- (a) A. C. Lindhorst, S. Haslinger and F. E. Kuhn, Chem. Commun., 2015, 51, 17193–17212 RSC; (b) S. H. Bae, M. S. Seo, Y.-M. Lee, K.-B. Cho, W.-S. Kim and W. Nam, Angew. Chem., Int. Ed., 2016, 55, 8027–8031 CrossRef PubMed.
- (a) A. K. Vardhaman, C. V. Sastri, D. Kumar and S. P. de Visser, Chem. Commun., 2011, 47, 11044–11046 RSC; (b) A. K. Vardhaman, P. Barman, S. Kumar, C. V. Sastri, D. Kumar and S. P. de Visser, Chem. Commun., 2013, 49, 10926–10928 RSC; (c) A. K. Vardhaman, P. Barman, S. Kumar, C. V. Sastri, D. Kumar and S. P. de Visser, Angew. Chem., Int. Ed., 2013, 52, 12288–12292 CrossRef PubMed.
- (a) W. Liu and J. T. Groves, Angew. Chem., Int. Ed., 2013, 52, 6024–6027 CrossRef PubMed; (b) W. Liu, X. Huang, M.-J. Cheng, R. J. Nielsen, W. A. Goddard and J. T. Groves, Science, 2012, 337, 1322 CrossRef PubMed.
- K. Ray, S. M. Lee and L. Que Jr, Inorg. Chim. Acta, 2008, 361, 1066–1069 CrossRef PubMed.
- (a) Y. Zang, J. Kim, Y. Dong, E. C. Wilkinson, E. H. Appelman and L. J. Que, J. Am. Chem. Soc., 1997, 119, 4197–4205 CrossRef; (b) T. J. Hubin, J. M. McCormick, N. W. Alcock and D. H. Busch, Inorg. Chem., 2001, 40, 435–444 CrossRef PubMed; (c) S. Rana, S. Bag, T. Patra and D. Maiti, Adv. Synth. Catal., 2014, 356, 2453–2458 CrossRef.
- T. Luukkonen and S. O. Pehkonen, Crit. Rev. Environ. Sci. Technol., 2017, 47, 1–39 CrossRef.
- (a) K.-B. Cho, X. Wu, Y.-M. Lee, Y. H. Kwon, S. Shaik and W. Nam, J. Am. Chem. Soc., 2012, 134, 20222–20225 CrossRef PubMed; (b) D. Kumar, H. Hirao, L. Que Jr and S. Shaik, J. Am. Chem. Soc., 2005, 127, 8026–8027 CrossRef PubMed; (c) A. Ansari, A. Kaushik and G. Rajaraman, J. Am. Chem. Soc., 2013, 135, 4235–4249 CrossRef PubMed; (d) P. Comba, G. Rajaraman and H. Rohwer, Inorg. Chem., 2007, 46, 3826–3838 CrossRef PubMed; (e) A. Ansari and G. Rajaraman, Phys. Chem. Chem. Phys., 2014, 16, 14601–14613 RSC.
- (a) P. Comba, M. Maurer and P. Vadivelu, Inorg. Chem., 2009, 48, 10389–10396 CrossRef PubMed; (b) M. R. Bukowski, P. Comba, A. Lienke, C. Limberg, C. Lopez de Laorden, R. Mas-Ballesté, M. Merz and L. Que Jr, Angew. Chem., Int. Ed., 2006, 45, 3446–3449 CrossRef PubMed.
- E. A. Hill, A. C. Weitz, E. Onderko, A. Romero-Rivera, Y. Guo, M. Swart, E. L. Bominaar, M. T. Green, M. P. Hendrich, D. C. Lacy and A. S. Borovik, J. Am. Chem. Soc., 2016, 138, 13143–13146 CrossRef PubMed.
- (a) H. Hirao, D. Kumar, L. Que Jr and S. Shaik, J. Am. Chem. Soc., 2006, 128, 8590–8606 CrossRef PubMed; (b) B. Pandey, M. Jaccob and G. Rajaraman, Chem. Commun., 2017, 53, 3193–3196 RSC; (c) M. Ansari, N. Vyas, A. Ansari and G. Rajaraman, Dalton Trans., 2015, 44, 15232–15243 RSC; (d) S. Shaik, H. Hirao and D. Kumar, Nat. Prod. Rep., 2007, 24, 533–552 RSC.
- C. Charavay, S. Segard, F. Edon, M. Clémancey and G. Blondin, SimuMoss software, CEA/DRF/BIG, CNRS, Université Grenoble-Alpes Search PubMed.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef PubMed.
- M. Lubben, A. Meetsma, E. C. Wilkinson, B. Feringa and L. Que Jr, Angew. Chem., Int. Ed., 1995, 34, 1512–1514 CrossRef.
- J.-U. Rohde, S. Torelli, X. Shan, M. H. Lim, E. J. Klinker, J. Kaizer, K. Chen, W. Nam and L. Que Jr, J. Am. Chem. Soc., 2004, 126, 16750–16761 CrossRef PubMed.
- J. R. Hagadorn, L. Que Jr and W. B. Tolman, Inorg. Chem., 2000, 39, 6086–6090 CrossRef PubMed.
- (a) S. Rana, S. Bag, T. Patra and D. Maiti, Adv. Synth. Catal., 2014, 356, 2453–2458 CrossRef; (b) D. Mandon, A. Machkour, S. Goetz and R. Welter, Inorg. Chem., 2002, 41, 5364–5372 CrossRef PubMed.
- G. Roelfes, M. Lubben, K. Chen, R. Y. N. Ho, A. Meetsma, S. Genseberger, R. M. Hermant, R. Hage, S. K. Mandal, V. G. Young, Y. Zang, H. Kooijman, A. L. Spek, L. Que Jr and B. L. Feringa, Inorg. Chem., 1999, 38, 1929–1936 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1516421, 1516453, 1505984, 1505986, and 1505989. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc02053a |
| This journal is © The Royal Society of Chemistry 2018 |