
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLive-cell labeling of endogenous proteins with nanometer precision by transduced nanobodies†
A.
Klein
a,
S.
Hank
a,
A.
Raulf
b,
E. F.
Joest
a,
F.
Tissen
a,
M.
Heilemann
 b,
R.
Wieneke
*a and
R.
Tampé
*ac
b,
R.
Wieneke
*a and
R.
Tampé
*ac
aInstitute of Biochemistry, Biocenter, Goethe University Frankfurt, Max-von-Laue-Str. 9, 60438 Frankfurt/Main, Germany. E-mail: wieneke@em.uni-frankfurt.de
bInstitute of Physical and Theoretical Chemistry, Goethe University Frankfurt, Max-von-Laue-Str. 7, 60438 Frankfurt/Main, Germany
cCluster of Excellence – Macromolecular Complexes, Goethe University Frankfurt, Max-von-Laue-Str. 9, 60438 Frankfurt/Main, Germany
First published on 20th August 2018
Abstract
Accurate labeling of endogenous proteins for advanced light microscopy in living cells remains challenging. Nanobodies have been widely used for antigen labeling, visualization of subcellular protein localization and interactions. To facilitate an expanded application, we present a scalable and high-throughput strategy to simultaneously target multiple endogenous proteins in living cells with micro- to nanometer resolution. For intracellular protein labeling, we advanced nanobodies by site-specific and stoichiometric attachment of bright organic fluorophores. Their fast and fine-tuned intracellular transfer by microfluidic cell squeezing enabled high-throughput delivery with less than 10% dead cells. This strategy allowed for the dual-color imaging of distinct endogenous cellular structures, and culminated in super-resolution imaging of native protein networks in genetically non-modified living cells. The simultaneous delivery of multiple engineered nanobodies does not only offer exciting prospects for multiplexed imaging of endogenous protein, but also holds potential for visualizing native cellular structures with unprecedented accuracy.
Introduction
The localization of endogenous proteins in a native cell environment is fundamental for the quantitative spatiotemporal delineation of cellular homeostasis and dysfunction. Live-cell imaging is highly desired to visualize timely balanced protein networks, avoiding contingent artifacts by fixation or permeabilization procedures.1 In this regard, genetic fusion of fluorescent proteins (FPs) enables the visualization of proteins of interest (POIs).2 However, the fusion of FPs or other bulky enzyme-based tags, e.g. SNAPf tag, can provoke protein misassembly, mistargeting, or other artifacts.3,4 Current efforts in the visualization of endogenous proteins focus on the insertion of small epitope tags, such as split-GFP, SunTag, or His-tag, into endogenous genomic loci.5,6 Although genomic tagging via CRISPR/Cas9 dispels concerns about overexpression, it is still difficult and time-consuming for a number of applications. Antibodies or recombinant binders are prominent alternatives to trace unmodified proteins.7–10 Targeting by antibodies has the advantage of detecting endogenous proteins rather than heterologously expressed ones, and thus facilitates a correct localization or proper expression of the POI. Yet, labeling with conventional antibodies is limited to fixed cells since their disulfide bonds are reduced in the cytosol. To overcome this downside, antibody fragments have been developed for protein targeting.11,12In this regard, the variable domains of heavy-chain-only antibodies (VHHs) of camelids, named nanobodies, are prime candidates (Scheme 1).13,14 They bind with nanomolar or even picomolar affinities to their targets, are small in size (≈13 kDa), exhibit great solubility, and can functionally be produced in the reducing cytosolic milieu of mammalian cells. Nanobodies fused to fluorescent proteins, termed ‘chromobodies’, have been used to label various POIs.14–22 Nonetheless, visualization of intracellular proteins is still impeded because fused FPs triple the size of the nanobody, and FPs entail suboptimal photophysical properties (Scheme 1). An additional drawback is that the expression level of chromobodies is hard to control, evoking high background and deteriorated signal-to-background staining, which in turn makes the observation of low abundant proteins, in particular by super-resolution microscopy, very difficult or impossible. Recently, an α-GFP nanobody was delivered into cells by attaching a cyclic cell-permeable peptide sequence.23 In addition, nanobodies directed against FPs were equipped with quantum dots or organic dyes to trace kinesin by single-particle tracking.24,25 However, most approaches still have some limitations restricting their broad applicability: (i) two or more antigens in the same cell are difficult to address, (ii) the intracellular concentration of the nanobody is very difficult to control, and (iii) endogenous proteins are not readily targeted by super-resolution microscopy. The latter two obstacles are mainly caused by the overexpression of a genetically modified POI for recruiting the nanobody as well as an imprecise regulation of the nanobody delivery to match its intracellular level to that of the target antigen. Likewise, it cannot be excluded that nanobody binding impairs the POIs' dynamic, function, or localization as observed for fusion tags. Despite all efforts, the application of nanobodies for visualizing endogenous proteins by single-molecule localization microscopy in non-modified living cells is still underexplored.
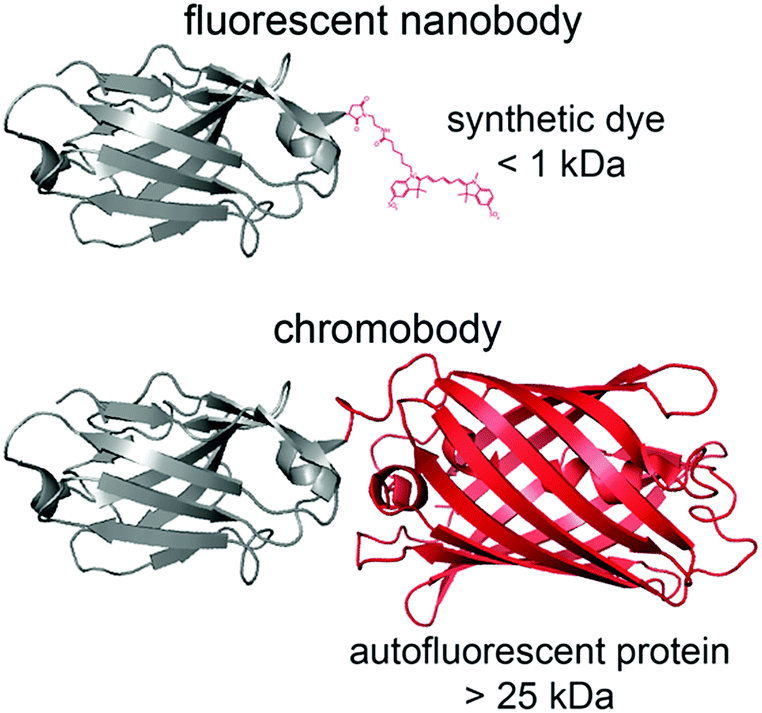 | ||
| Scheme 1 Size of a fluorescent nanobody compared to nanobodies labeled with a fluorescent protein, termed chromobody. Bulky fluorescent proteins (>25 kDa) triple the size of the nanobody (≈13 kDa). In comparison, the minimal size of fluorophores (<1 kDa) is unlikely to disturb the nanobody and offers a higher quantum yield as well as photostability. | ||
To bypass several limitations, we advanced nanobodies with bright, organic fluorophores in stoichiometric ratio. Using a vector-free microfluidic device, these fluorescent nanobodies (fNbs) were delivered at defined nanomolar concentrations via a direct, non-endocytic transfer. The approach precluded intracellular excess of fluorescently labeled nanobodies and thus resulted in the dual-color imaging of endogenous target proteins, e.g. vimentin and lamin, with high fidelity and excellent signal-to-background ratios. For multiplexing, different fNbs were delivered in parallel at defined nanomolar concentrations. Ultimately, the endogenous nuclear lamina was resolved at nanometer precision by live-cell super-resolution microscopy in native, genetically non-modified cells.
Results and discussion
Specific protein labeling
The quantitative and stoichiometric fluorophore labeling is a crucial factor for advanced fluorescence microscopy as it holds the potential to investigate absolute protein copy numbers. In order to achieve a high labeling specificity and efficiency, as well as to strategically position small synthetic fluorophores, we engineered nanobodies with single free cysteines either at the N terminus, position 9, or the C terminus. Compared to auto-fluorescent proteins, brighter synthetic fluorophores are easier to detect as they provide an enhanced signal-to-background ratio. Moreover, the devised strategy offers the possibility to easily tune the spectral properties of different fNbs. Our goal was to create fluorescent nanobodies that preserve their specific and high-affinity antigen binding combined with negligible label displacement. To this aim, our approach provides simple customization and velocity in adapting the spectral properties for advanced microscopy or other spectroscopy techniques.As a first showcase, we modified the α-GFP nanobody,26–28 and then generalized the approach to target different endogenous proteins using the cognate fNbs. Furthermore, its respective antigen (GFP) allows for immediate read-out by co-localization studies in vitro as well as in vivo, and thus to test fNb function after transfer into GFP-expressing cells. After expression and affinity purification, nanobodies with a single free cysteine were labeled via maleimide coupling with high-quantum yield fluorophores, namely sulfo-Cyanine 3 (sCy3), sCy5, or ATTO655 (Scheme 1a).29 For comparison, random labeling of lysines was also examined. Specific conjugation of the fluorophore was confirmed by SDS-PAGE in-gel fluorescence and by fluorescence-detection size-exclusion chromatography, yielding molar labeling ratios of ≈0.9 for α-GFPsCy3 and ≈0.8 for α-GFPsCy5 (Fig. S1 and S2†).
As an initial screen for fNb function, we evaluated the specific binding of the α-GFP nanobodies (fNbs) in fixed cells prior to live-cell applications. We expected to identify nanobodies that tightly bind to the target and hence will intracellularly co-localize with the target protein. The equilibrium dissociation constant for the α-GFP nanobody was reported to be ≈1 nM.27 Different intracellular assemblies were selected as targets, namely the transporter associated with antigen processing TAP (TAP1mVenus) in the ER membrane, histone 2B (H2BEGFP) in the nucleus, and lamin A (mEGFPLamin A) as a component of the nuclear envelope. The respective POIs were expressed in human HeLa Kyoto cells, labeled with the α-GFP fNbs (50–200 nM) and analyzed by confocal laser scanning microscopy (CLSM). All engineered α-GFP fNbs retained high specificity with negligible background staining, which was not affected by the positioning of the fluorophores (Fig. 1A and S3, S4†). An excellent co-localization was observed as depicted by Pearson's coefficients ranging from 0.948 to 0.986, independent of the attached synthetic dyes (Fig. 1A and S3, S4†). Specific fNb binding was further demonstrated by the intensity distribution, which highly correlated with the POI expression level (Fig. 1B and S3–S5†).
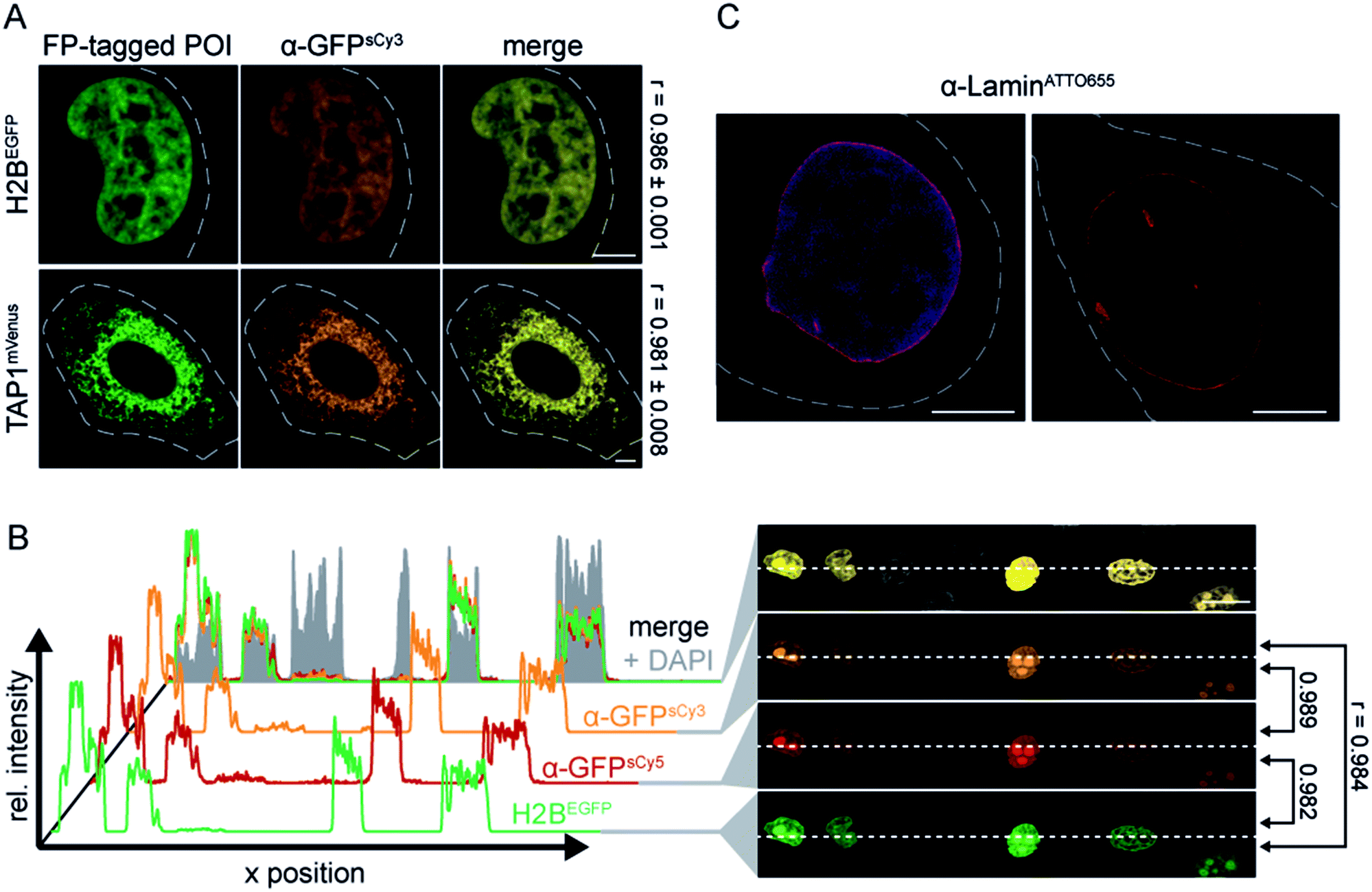 | ||
| Fig. 1 Specific binding of site-specifically labeled nanobodies. (A) Specific labeling of FP-tagged proteins in chemically arrested cells. HeLa Kyoto cells expressing H2BEGFP or TAP1mVenus (green) were labeled with 100 nM of α-GFPsCy3 fNb (orange). A co-localization (merge) between the fNbs and both FP-tagged POIs was revealed. Pearson's coefficients were determined from 9–11 individual cells (right). (B) Fixed HeLa Kyoto cells expressing H2BEGFP were stained with α-GFPsCy3 (orange), α-GFPsCy5 (red, 100 nM each), and DAPI (blue). Cross-sections of relative fluorescence intensity profiles of both fNbs (along dashed line) highly correlated with the expression level of H2BEGFP. Low background fluorescence was detected in untransfected cells (only DAPI positive), supported by the Pearson's coefficients (r) between different channels (right). (C) After fixation, the endogenous nuclear lamina of HeLa Kyoto cells was visualized by 100 nM of α-LaminATTO655 fNb (red). The fNb specifically decorated the nuclear envelope (DNA visualized via DAPI staining, blue). Images were taken by CLSM with the Airy Scan detector (C). Dashed lines indicate the cell border. Scale bars: 5 μm (A, C) and 20 μm (B). | ||
Next, we transferred the findings to engineer an α-Lamin fNb. As basic constituents of the nuclei, lamins were assembled in a filamentous meshwork, providing structural stability. Since α-Lamin traces the endogenous nuclear envelope, the engineered fNb can avoid limitations typically entailed by (over-)expression of fusion proteins. For nanobody production, we here employed an engineered E. coli strain that promotes disulfide bond formation in the cytosol and yields an optimized coupling procedure. To suppress transient exposure of inner scaffold cysteines by thermal breathing, covalent dye attachment was performed at 4 °C (Fig. S1†).29
Specific labeling of the endogenous nuclear lamina was observed in the in vitro screen of fNb function (Fig. 1C and S6†). By this means, we generated α-Lamin fNbs equipped with sCy5 or ATTO655 at labeling ratios of 0.8 to 1.0, respectively. Noteworthy, different labeling procedures led to successful dye conjugation but were accompanied by a loss in specificity (Fig. S7†). Similar results were obtained when lysine residues on the nanobody surface were stochastically modified via N-hydroxysuccinimide Alexa647 (Fig. S7a†). Unlike the very robust α-GFP fNb, which always retained its high target specificity independently of all described routes for production and labeling, the α-Lamin fNb was drastically affected by the procedures (Fig. S8†). Since the highest signal-to-background ratio was achieved after dye-conjugation to the N-terminal cysteine of the α-Lamin fNb (Fig. 1C; ESI Video 1 and Fig. S6 and S7b†), labeling at this position was employed for all further experiments. Regarding site-specific labeling, the proper site-specific functionalization as well as the coupling procedure are important as either the nanobody function or specificity can be impeded. This was particularly evident for the α-Lamin nanobody targeting endogenous protein networks.
Live-cell protein labeling at nanomolar concentrations
After validating the target specificity in chemically arrested cells, we focused on live-cell intracellular labeling. A central restriction in the in-cell application of nanobodies is their inability to penetrate the cell membrane. This has substantially limited their scope of application and often restricted their use in fixed cells. Alternative strategies are required to transfer fNbs, which allow for a controlled, non-endocytic delivery of multiple binders into living cells. To this end, we made use of microfluidic cell squeezing for the fast and efficient fNb intracellular transfer. This promising technique enables the fNb delivery into the cytosol of mammalian cells by mechanically disrupting the plasma membrane and allowing diffusion through transient pores (Fig. S9†). Cell squeezing bypasses the endosomal uptake and offers precise control of the transduced cargo concentration combined with high-throughput delivery (up to one million cells per s) and resulted in less than 10% dead cells.30,31 Based on these unique signatures for cell transduction, we decided to utilize this methodology to deliver engineered fNbs. We first aimed to validate the correct antigen specificity with the α-GFP fNbs, as its delivery provides a direct read-out through the recognition of its fluorescent antigen GFP. Again, HeLa Kyoto cells expressing the three different FP-tagged proteins were chosen as targets. After cell squeezing in the presence of nanomolar concentrations of the fNbs (100–500 nM), the intracellular fNb distribution was analyzed one to three hours after cell transfer via co-localization CLSM analyses. For the diverse arrays of target proteins tested, the fluorescence of the α-GFP fNbs showed a high degree of co-localization with FP-tagged H2B, TAP1, and lamin A, reflected by Pearson's coefficients ranging from 0.892 to 0.911 (Fig. 2A and S10a†). For example, an excellent correlation between the fluorescence intensity profiles of TAP1mVenus and the α-GFPsCy3 fNb was observed (Fig. 2B and C; Pearson's coefficient of 0.926). High-throughput and efficient nanobody delivery was apparent by the high number of transduced cells (Fig. S11†). Notably, all transduced cells displayed a very low background staining, and the intensity of the fNb reflected the variability in the POI expression level. We observed an increased fNb binding in cells with high levels of FP-tagged POIs, whereas cells expressing low amounts of the POIs displayed weaker fluorescence intensities (Fig. S11†). One hypothesis for this correlation can be found in the retrograde diffusion of free binders before membrane resealing as well as in the nanomolar concentration of fNbs used in cell squeezing, both synergistically contributing to a high signal-to-background staining (Fig. 2D).25 Untransfected cells showed a very low level of uniform intracellularly distributed fNbs (Fig. S12†), which is in line with the localization of the α-GFP-mCherry chromobody or mCherry expressed in HeLa Kyoto cells (Fig. S13a and b†). In contrast, the plasmid-driven expression of α-GFP-mCherry chromobodies together with FP-tagged POIs often resulted in an overabundance of the binder due to high and unmatched expression levels (Fig. S13c–e†). Here, a high signal-to-background labeling is impaired in cells with high expression levels of chromobodies, while very low levels entail high sensitivity to photobleaching. To accurately report on the localization of endogenous proteins, it is essential and indispensable to minimize the excess of nanobodies and hence background signal. By utilizing microfluidic fNb transfer, these drawbacks and impediments were not observed.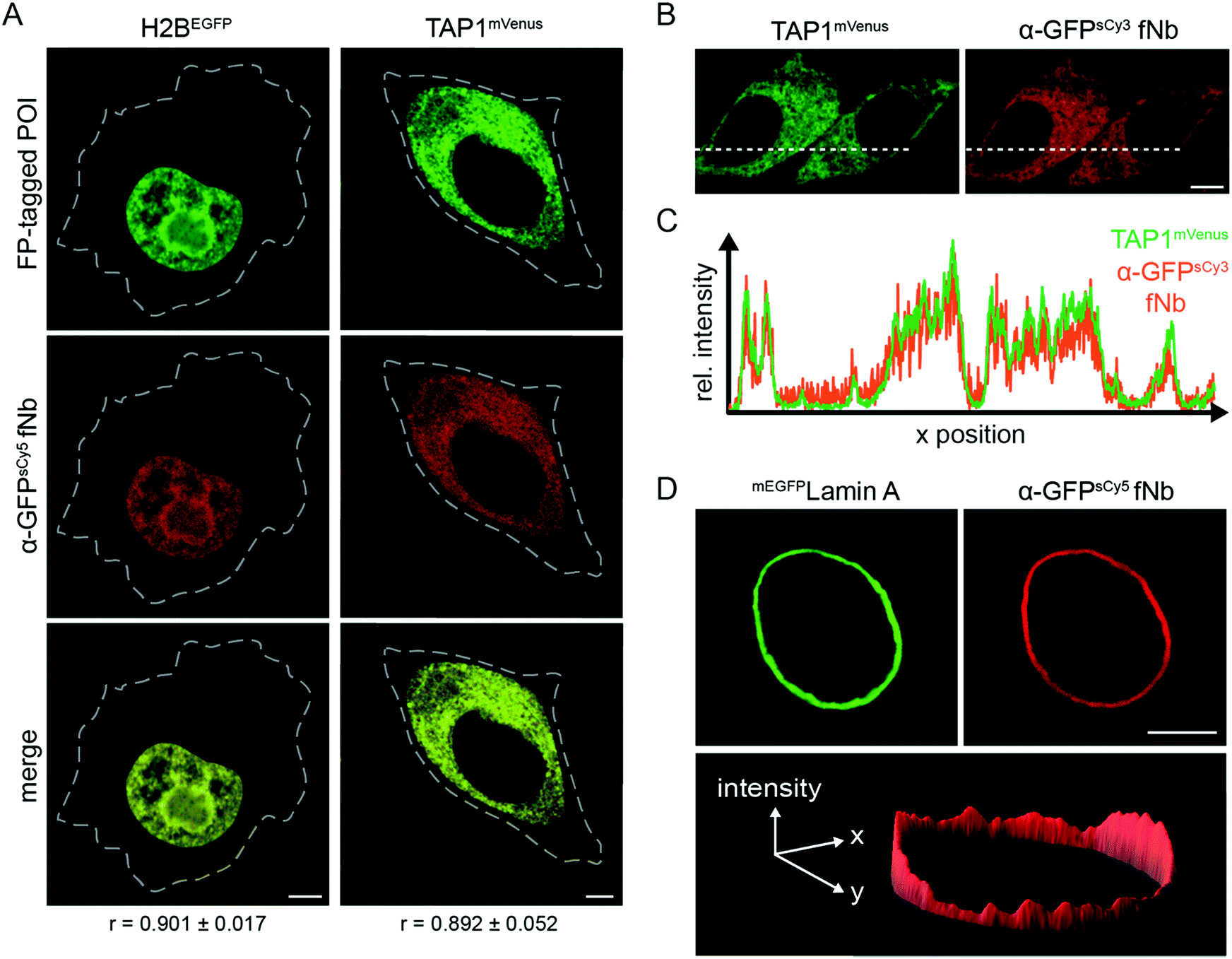 | ||
| Fig. 2 Live-cell protein labeling by fluorescent nanobodies. (A) Protein labeling in living cells at different subcellular localizations. The α-GFPsCy5 fNb (200 nM, red) was delivered via cell squeezing into HeLa Kyoto cells, expressing H2BEGFP or TAP1mVenus (green). Excellent co-localization (merge channel) of the fNbs with both FP-tagged POIs was observed 3 h post squeezing by CLSM. The Pearson's coefficients (r, bottom) ranging from 0.892 to 0.901 were determined from 8–10 individual cells. Dashed lines indicate cell border. (B) Live-cell visualization of TAP1mVenus (green) by α-GFPsCy3 fNb (orange) 20 h after squeezing. (C) Cross-sections of relative fluorescence intensity profiles of TAP1mVenus and α-GFPsCy3 (along dashed line) showed specific and low-background labeling in live cells by the α-GFP fNb. Pearson's coefficient: 0.926. (D) 3D intensity plot of a representative HeLa Kyoto cell expressing mEGFPLamin A (green) was stained by α-GFPsCy5 fNb (red) and showed a high signal-to-background ratio. All images were taken by CLSM. Scale bar: 5 μm. | ||
We also addressed the long-term stability and persistence of fNb labeling. Remarkably, even 20 h after delivery, imaging fNb transduced cells showed that the POIs were still decorated by α-GFP fNbs, highlighting their long-term specificity and in-cell robustness (Fig. 2B). After a prolonged period, however, cytosolic punctae and a higher background signal were detected, indicating incipient fNb degradation (Fig. S10b†). Overall, the adaptive, fine-tuned intracellular delivery of fNbs by cell squeezing promoted low-background labeling of genetically tagged proteins.
High signal-to-background labeling of endogenous proteins in live mammalian cells
To fully exploit the scalable and high-throughput delivery of fNbs we next focused on live-cell labeling of endogenous proteins. With regard to their extenuated abundance compared to overexpressed proteins, the utilization of high-performance fluorescent or fluorogenic probes is essential to enhance sensitivity. Therefore, we selected two intermediate filaments as targets. Specifically, we used the engineered α-Lamin fNb to visualize the native lamin meshwork of the nuclear envelope and α-VimentinATTO488 in order to label vimentin as a cytoskeletal component, crucial for organelle positioning. In living cells, the selective labeling of the native cytoskeleton of vimentin was so far only facilitated by overexpression of vimentin fusion proteins,32,33 genetic code expansion34 or endogenous tagging of vimentin with a SNAP or Halo-tag, respectively.35Based on the target specificity of both binders in fixed cells (α-LaminATTO655, Fig. 1C and S6;† α-VimentinATTO488, Fig. S14†), we aimed to label endogenous protein networks by fNb delivery. Live, non-transfected HeLa Kyoto cells were squeezed in the presence of 200 nM α-LaminATTO655 fNb to visualize the native lamin meshwork. 3 h after delivery, specific decoration of the endogenous nuclear envelope with a concomitant negligible background was observed by means of live-cell CLSM (Fig. 3A). Even in the crowded environment, the expected localization of the α-LaminATTO655 fNb was clearly resolved by CLSM and visualized as endogenous lamin. The observed morphology was similar to the one obtained in fixed cells (Fig. 1C), demonstrating that α-LaminATTO655 specifically recognizes its intracellular antigen even at subnanomolar concentrations. Besides this, the fNb delivery enabled the targeting of native protein networks with marginal background signal, which constitutes to the best of our knowledge the first labeling of an endogenous protein inside living cells by exogenously derived fNbs. By contrast, in live HeLa Kyoto cells expressing α-Lamin-EGFP chromobodies, decoration of the nuclear lamina was accompanied by high background staining and cytosolic punctae, presumably due to saturation of the protein synthesis machinery and aggregation (Fig. S15†). Worthy of note is that even 20 h after cell squeezing the labeling of the nuclear lamina was still persistent.
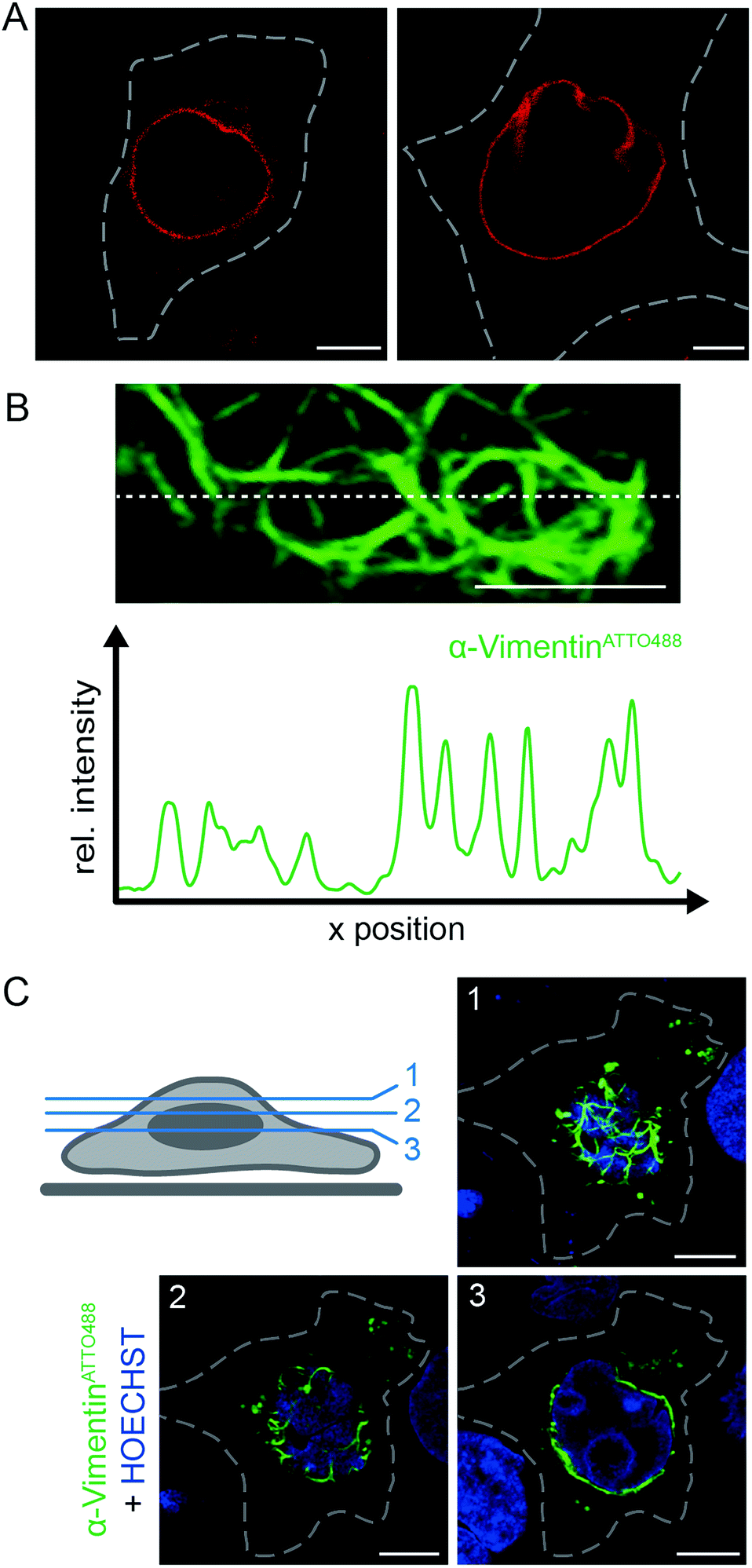 | ||
| Fig. 3 Live-cell labeling of endogenous targets. (A) The α-LaminATTO655 fNb (200 nM) was transferred into HeLa Kyoto cells via squeezing. Confocal imaging after 3 h showed specific labeling of the native nuclear lamina in living cells with high signal-to-background ratio. (B) HeLa Kyoto cells were squeezed with 50 μg ml−1 of α-VimentinATTO488. Fluorescence intensity cross-section of a live HeLa Kyoto cell transduced with α-VimentinATTO488 (along dashed line) showed specific and distinct decoration of the endogenous vimentin meshwork with high signal-to-background ratio. The respective network was localized beneath the nucleus (corresponding to z-plane 2 in C). (C) The fNb decorated vimentin network was observed 3 h post squeezing and visualized as a dense meshwork around the nucleus (HOECHST-positive, blue) in different confocal z-planes (1–3; indicated in scheme, left). Images were taken by CLSM with the Airy Scan detector. Dashed lines indicate the cell border. Scale bars: 5 μm. | ||
Nevertheless, higher background, mainly as cytosolic punctae, was observed (Fig. S16†), which is in line with the results obtained for the α-GFP fNb (Fig. S10b†). Notably, specific targeting was unaffected by the utilized fluorophore (ATTO655 or sCy5; Fig. 3A and S16†).
To further broaden the range of applications, α-VimentinATTO488 was delivered into live HeLa Kyoto cells. Following cell squeezing, live-cell CLSM was employed to image the filamentous structure of endogenous vimentin (Fig. 3B, C and S17†). 3 h as well as 20 h after cell transduction, the well-defined native vimentin cytoskeleton was observed at high signal-to-background ratio as reflected in the fluorescence intensity profile (Fig. 3B). In addition, we were able to visualize the native vimentin meshwork at various confocal z-planes (Fig. 3C). Specific and distinct decoration of vimentin at endogenous level was observed throughout the cell. In each z-plane, an explicit fNb accumulation at the intermediate filament was present. The accurate representation of the endogenous target structure was facilitated by the combination of efficient and scalable fNb delivery (Fig. S18†). A well-controlled fNbs delivery at nanomolar concentrations without overloading the cytosol allowed for a very good signal-to-noise ratio and thus enhanced sensitivity, compared to cell-permeable nanobodies.23 Taken together, our methodology provides straightforward access to employ fNbs for live-cell microscopy.
Multiplex and super-resolution analysis of native intracellular protein networks
A main limitation for multiplexing in living cells is the simultaneous delivery of two or more labeling probes at defined concentrations. To meet that challenge, we intended to demonstrate how multiple fNbs can be used for multi-color live-cell imaging. Thus, we performed dual-color imaging of fNbs transduced cells. When α-LaminATTO655 and α-VimentinATTO488 fNbs were present at nanomolar concentrations during cell squeezing, the simultaneous delivery of both fNbs into native HeLa Kyoto cells was easily facilitated. CLSM imaging of double transduced cells, indeed, demonstrated that both fNbs were successfully transferred and specifically localized with their target proteins (Fig. 4A). By dual-color labeling, both individual protein networks were clearly visible, and we were able to simultaneously image the nuclear lamina (Fig. 4A, red) and vimentin filaments (Fig. 4A, green) at endogenous level. The resolutions in representative z-planes were high enough to discernibly visualize the vimentin cytoskeleton next to the nuclear envelope. The protein networks were closely spaced, but with spatially different distributions. 3D mapping of cell topology by multiplexed imaging further confirmed the specific decoration of both endogenous antigens by fNb targeting. Due to the high target specificity and precise localization of the fNbs conjugated to bright organic dyes, we thus enabled live-cell immunofluorescence imaging. Notably, the fluorescence background in double transduced cells was negligible and substantially lower compared to plasmid-driven expression of chromobodies (Fig. S15†). | ||
| Fig. 4 Dual-color live-cell labeling of endogenous targets and live-cell super-resolution imaging by fluorescent nanobodies. (A) Dual-color labeling of endogenous lamin and vimentin in living cells. α-LaminATTO655 and α-VimentinATTO488 were simultaneously transferred into HeLa Kyoto cells. 3 h after cell squeezing, spatially distinct labeling of endogenous lamin (red) and vimentin (green) was visualized by multiplexed imaging. Magnification (box, top) of the α-VimentinATTO488 stained vimentin network showed the three-dimensional organization around the nucleus. The respective confocal z-planes are indicated in the scheme. (B) Reconstructed dSTORM image of endogenous lamin specifically labeled with α-LaminATTO655 fNb (5 h after squeezing). The sensitive and high degree of fNb labeling allowed for visualization of the native nuclear envelope with nanometer precision. (C) Comparison of dSTORM analysis with the averaged fluorescence intensity. Cross section of the intensity profiles (along the dashed line in B) displayed the enhanced resolution by dSTORM. Images were taken by CLSM with the Airy Scan detector or dSTORM. Dashed lines indicate the cell border. Scale bars: 5 μm. | ||
A low background fluorescence is also a key requirement for super-resolution microscopy. We therefore examined the efficacy of fNbs for high-resolution imaging. By combining the precisely adjusted delivery via cell squeezing with the small site-specifically labeled fNbs, we aimed to perform live-cell super-resolution microscopy. In particular, the bright-fluorescent dye delivery via fNbs provides an adequate labeling efficiency and specificity, combined with close proximity of the dye to the target structure. For demonstration, α-LaminATTO655 was delivered into genetically non-modified HeLa Kyoto cells. Subsequently, live cells were subjected to direct stochastic optical reconstruction microscopy (dSTORM).36 We used highly inclined laminar optical sheet (HILO) microscopy with laser intensities in the range of 0.5–2 kW cm−2.37 We recorded 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 images at a frame rate of 33 Hz. After reconstructing the dSTORM images, the endogenous nuclear lamina was visualized with sub-diffractional spatial resolution in live cells. An average resolution of ≈55 nm (localization precision of 23.4 ± 8.3 nm; Fig. 4B) was obtained with significantly increased contrast compared to wide-field imaging (Fig. 4C). Strikingly, dSTORM analysis of α-LaminATTO655 fNb in fixed cells resulted in a similar average localization precision, but an increased signal-to-background ratio was obtained in live cells (Fig. S19†). Nevertheless, this underscores the applicability of our approach for an efficient, scalable and controlled nanobody delivery. In summary, fNbs containing bright, high-quantum yielding dyes allowed the resolution of finer details with high fidelity and low concentration in super-resolution microscopy, thus making them suitable for high-end microscopy. To this end, the fNb labeling will help to visualize dynamic processes in living cells by avoiding problems with regard to overexpression as well as fixation.
000 images at a frame rate of 33 Hz. After reconstructing the dSTORM images, the endogenous nuclear lamina was visualized with sub-diffractional spatial resolution in live cells. An average resolution of ≈55 nm (localization precision of 23.4 ± 8.3 nm; Fig. 4B) was obtained with significantly increased contrast compared to wide-field imaging (Fig. 4C). Strikingly, dSTORM analysis of α-LaminATTO655 fNb in fixed cells resulted in a similar average localization precision, but an increased signal-to-background ratio was obtained in live cells (Fig. S19†). Nevertheless, this underscores the applicability of our approach for an efficient, scalable and controlled nanobody delivery. In summary, fNbs containing bright, high-quantum yielding dyes allowed the resolution of finer details with high fidelity and low concentration in super-resolution microscopy, thus making them suitable for high-end microscopy. To this end, the fNb labeling will help to visualize dynamic processes in living cells by avoiding problems with regard to overexpression as well as fixation.
Conclusions
In general, the lower level of endogenous proteins renders their specific and high signal-to-background labeling quite difficult. To circumvent this constraint, we established a method for live-cell imaging of endogenous proteins at nanometer resolution, together with minimal disturbance and high-throughput delivery (up to one million cells per s). This robust and generic technique is versatile in the choice of the nanobody and bright organic fluorophore, and allows for low-background labeling of high as well as low abundant native protein networks in living cells. In case of the selected target, fNb decoration was persistent for several hours after fNb transfer. Increased background after 20 h, however, suggests that long-term protein labeling can be affected. This obstacle is experimentally manageable, depending on the turnover rate of the POI as well as the rate of cell division. Alternative approaches to tag endogenous proteins, such as the generation of stable cell lines utilizing e.g. CRISPR/Cas9 for low chromobody levels, are highly promising, yet very labor-intensive and hardly feasible for multiple knock-ins. Other transduction methods including electroporation or semi-permeabilization are promising for cargo delivery. But, so far, they suffer from low in vivo efficiency, low throughput, batch-to-batch variations, or elaborated chemical synthesis.24,25,38,39 Our strategy, however, involves the transient disruption of the cell plasma membrane followed by entry of the target material before resealing.All in all, our devised strategy strikes a new path for the live-cell visualization of endogenous targets by high quantum yield fluorescent nanobodies to study their precise intracellular localization and trafficking. The simplicity and robustness of the methodology can be extended to deliver simultaneously multiple probes for multiplexed imaging and has potential for other types of microscopy (e.g. cryo-EM tomography). By cell squeezing, the fNb targeting of endogenous proteins in patient-derived primary immune cells or embryonic stem cells is feasible. Moreover, our method provides access for exogenously derived, high-affinity binding scaffolds (e.g. fibronectin-based binding proteins or designed ankyrin repeat proteins) to 3D and multi-color super-resolution imaging in native cells, and it opens avenues to label cellular ultrastructures at endogenous level for correlated light and electron microscopy (CLEM) or lattice light-sheet microscopy. These advantages of our approach outweigh potential limitations such as modalities encountered by membrane disruption. Especially the labeling of different proteins in parallel will be valuable to simultaneously study multiple proteins by imaging as well as non-imaging techniques, given the speed, flexibility and efficiency of the delivery protocol. In addition, the nanobody delivery will directly benefit from the progress in the development of functional nanobodies and opens opportunities for the comprehensive study of low-abundant proteins in their native cellular environment. Overall, the viable expansion of the methodology will pioneer the non-genetically encoded visualization of cellular networks and subcellular structures, and hence provide complementary and unique information with unprecedented precision. To this effect, our approach allows researchers to rapidly and precisely visualize the localization of endogenous proteins in various cell types, and it provides an alternative platform for antigen labeling in living cells via transduction of exogenously derived antigen binders.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The German Research Foundation (Cluster of Excellence EXC 115 to R. W., M. H., and R. T.; SPP 1623, GRK 1986 to R. T. and SFB 807 and 902 to M. H. and R. T.) and the Volkswagen Foundation (91067-9 to M. H., R. W., and R. T.) supported this work. We thank Dr Eric Geertsma (Goethe University Frankfurt, Germany) for providing the sequence coding for the α-GFP nanobody and helpful discussions. α-VimentinATTO488 was a kind gift from Dr Ulrich Rothbauer (Eberhard Karls Universität Tübingen, Germany). We thank Dr Heinrich Leonhardt (LMU Munich, Germany) for generously providing the plasmid coding for α-Lamin-EGFP.References
- U. Schnell, F. Dijk, K. A. Sjollema and B. N. Giepmans, Nat. Methods, 2012, 9, 152–158 CrossRef PubMed.
- A. B. Cubitt, R. Heim, S. R. Adams, A. E. Boyd, L. A. Gross and R. Y. Tsien, Trends Biochem. Sci., 1995, 20, 448–455 CrossRef PubMed.
- A. Gautier, A. Juillerat, C. Heinis, I. R. Correa, Jr, M. Kindermann, F. Beaufils and K. Johnsson, Chem. Biol., 2008, 15, 128–136 CrossRef PubMed.
- A. E. El-Husseini, E. Schnell, D. M. Chetkovich, R. A. Nicoll and D. S. Bredt, Science, 2000, 290, 1364–1368 Search PubMed.
- M. D. Leonetti, S. Sekine, D. Kamiyama, J. S. Weissman and B. Huang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E3501–E3508 CrossRef PubMed.
- D. Kamiyama, S. Sekine, B. Barsi-Rhyne, J. Hu, B. Chen, L. A. Gilbert, H. Ishikawa, M. D. Leonetti, W. F. Marshall, J. S. Weissman and B. Huang, Nat. Commun., 2016, 7, 11046 CrossRef PubMed.
- M. Brauchle, S. Hansen, E. Caussinus, A. Lenard, A. Ochoa-Espinosa, O. Scholz, S. G. Sprecher, A. Plückthun and M. Affolter, Biol. Open, 2014, 3, 1252–1261 CrossRef PubMed.
- E. Karatan, M. Merguerian, Z. Han, M. D. Scholle, S. Koide and B. K. Kay, Chem. Biol., 2004, 11, 835–844 CrossRef PubMed.
- P. A. Nygren, FEBS J., 2008, 275, 2668–2676 CrossRef PubMed.
- A. Skerra, J. Mol. Recognit., 2000, 13, 167–187 CrossRef PubMed.
- R. E. Bird, K. D. Hardman, J. W. Jacobson, S. Johnson, B. M. Kaufman, S. M. Lee, T. Lee, S. H. Pope, G. S. Riordan and M. Whitlow, Science, 1988, 242, 423–426 CrossRef PubMed.
- J. S. Huston, D. Levinson, M. Mudgett-Hunter, M. S. Tai, J. Novotny, M. N. Margolies, R. J. Ridge, R. E. Bruccoleri, E. Haber and R. Crea, et al. , Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 5879–5883 CrossRef.
- S. Muyldermans, J. Biotechnol., 2001, 74, 277–302 Search PubMed.
- J. Helma, M. C. Cardoso, S. Muyldermans and H. Leonhardt, J. Cell Biol., 2015, 209, 633–644 CrossRef PubMed.
- E. Caussinus, O. Kanca and M. Affolter, Nat. Struct. Mol. Biol., 2011, 19, 117–121 CrossRef PubMed.
- J. Maier, B. Traenkle and U. Rothbauer, Sci. Rep., 2015, 5, 13402 CrossRef PubMed.
- K. Zolghadr, J. Gregor, H. Leonhardt and U. Rothbauer, Methods Mol. Biol., 2012, 911, 569–575 Search PubMed.
- U. Rothbauer, K. Zolghadr, S. Tillib, D. Nowak, L. Schermelleh, A. Gahl, N. Backmann, K. Conrath, S. Muyldermans, M. C. Cardoso and H. Leonhardt, Nat. Methods, 2006, 3, 887–889 CrossRef PubMed.
- S. Schornack, R. Fuchs, E. Huitema, U. Rothbauer, V. Lipka and S. Kamoun, Plant J., 2009, 60, 744–754 CrossRef PubMed.
- J. C. Tang, E. Drokhlyansky, B. Etemad, S. Rudolph, B. Guo, S. Wang, E. G. Ellis, J. Z. Li and C. L. Cepko, eLife, 2016, 5, e15312 CrossRef PubMed.
- M. Plessner, M. Melak, P. Chinchilla, C. Baarlink and R. Grosse, J. Biol. Chem., 2015, 290, 11209–11216 CrossRef PubMed.
- M. Melak, M. Plessner and R. Grosse, J. Cell Sci., 2017, 130, 525–530 CrossRef PubMed.
- H. D. Herce, D. Schumacher, A. F. L. Schneider, A. K. Ludwig, F. A. Mann, M. Fillies, M. A. Kasper, S. Reinke, E. Krause, H. Leonhardt, M. C. Cardoso and C. P. R. Hackenberger, Nat. Chem., 2017, 9, 762–771 CrossRef PubMed.
- E. A. Katrukha, M. Mikhaylova, H. X. van Brakel, P. M. van Bergen En Henegouwen, A. Akhmanova, C. C. Hoogenraad and L. C. Kapitein, Nat. Commun., 2017, 8, 14772 CrossRef PubMed.
- K. W. Teng, Y. Ishitsuka, P. Ren, Y. Youn, X. Deng, P. Ge, S. H. Lee, A. S. Belmont and P. R. Selvin, eLife, 2016, 5, e20378 CrossRef PubMed.
- J. Ries, C. Kaplan, E. Platonova, H. Eghlidi and H. Ewers, Nat. Methods, 2012, 9, 582–584 CrossRef PubMed.
- A. Kirchhofer, J. Helma, K. Schmidthals, C. Frauer, S. Cui, A. Karcher, M. Pellis, S. Muyldermans, C. S. Casas-Delucchi, M. C. Cardoso, H. Leonhardt, K. P. Hopfner and U. Rothbauer, Nat. Struct. Mol. Biol., 2010, 17, 133–138 CrossRef PubMed.
- J. Guizetti, L. Schermelleh, J. Mantler, S. Maar, I. Poser, H. Leonhardt, T. Muller-Reichert and D. W. Gerlich, Science, 2011, 331, 1616–1620 CrossRef PubMed.
- T. Pleiner, M. Bates, S. Trakhanov, C. T. Lee, J. E. Schliep, H. Chug, M. Bohning, H. Stark, H. Urlaub and D. Görlich, eLife, 2015, 4, e11349 CrossRef PubMed.
- A. Kollmannsperger, A. Sharei, A. Raulf, M. Heilemann, R. Langer, K. F. Jensen, R. Wieneke and R. Tampé, Nat. Commun., 2016, 7, 10372 CrossRef PubMed.
- A. Sharei, J. Zoldan, A. Adamo, W. Y. Sim, N. Cho, E. Jackson, S. Mao, S. Schneider, M. J. Han, A. Lytton-Jean, P. A. Basto, S. Jhunjhunwala, J. Lee, D. A. Heller, J. W. Kang, G. C. Hartoularos, K. S. Kim, D. G. Anderson, R. Langer and K. F. Jensen, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2082–2087 CrossRef PubMed.
- A. N. Butkevich, G. Y. Mitronova, S. C. Sidenstein, J. L. Klocke, D. Kamin, D. N. Meineke, E. D'Este, P. T. Kraemer, J. G. Danzl, V. N. Belov and S. W. Hell, Angew. Chem., Int. Ed., 2016, 55, 3290–3294 CrossRef PubMed.
- A. N. Butkevich, V. N. Belov, K. Kolmakov, V. V. Sokolov, H. Shojaei, S. C. Sidenstein, D. Kamin, J. Matthias, R. Vlijm, J. Engelhardt and S. W. Hell, Chemistry, 2017, 23, 12114–12119 CrossRef PubMed.
- E. Kozma, G. Estrada Girona, G. Paci, E. A. Lemke and P. Kele, Chem. Commun., 2017, 53, 6696–6699 RSC.
- A. N. Butkevich, H. Ta, M. Ratz, S. Stoldt, S. Jakobs, V. N. Belov and S. W. Hell, ACS Chem. Biol., 2018, 13, 475–480 CrossRef PubMed.
- M. Heilemann, S. van de Linde, M. Schuttpelz, R. Kasper, B. Seefeldt, A. Mukherjee, P. Tinnefeld and M. Sauer, Angew. Chem., Int. Ed., 2008, 47, 6172–6176 CrossRef PubMed.
- M. Tokunaga, N. Imamoto and K. Sakata-Sogawa, Nat. Methods, 2008, 5, 159–161 CrossRef PubMed.
- R. Röder, J. Helma, T. Preiss, J. O. Radler, H. Leonhardt and E. Wagner, Pharm. Res., 2017, 34, 161–174 CrossRef PubMed.
- P. Zhang, D. S. He, P. M. Klein, X. W. Liu, R. Roder, M. Doblinger and E. Wagner, Adv. Funct. Mater., 2015, 25, 6627–6636 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02910e |
| This journal is © The Royal Society of Chemistry 2018 |