
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePyrophosphorylation via selective phosphoprotein derivatization†
Alan M.
Marmelstein
 ab,
Jeremy A. M.
Morgan
a,
Martin
Penkert
ac,
Daniel T.
Rogerson
d,
Jason W.
Chin
d,
Eberhard
Krause
a and
Dorothea
Fiedler
*ac
ab,
Jeremy A. M.
Morgan
a,
Martin
Penkert
ac,
Daniel T.
Rogerson
d,
Jason W.
Chin
d,
Eberhard
Krause
a and
Dorothea
Fiedler
*ac
aLeibniz-Forschungsinstitut für Molekulare Pharmakologie, Robert-Rössle Str. 10, 13125 Berlin, Germany. E-mail: fiedler@fmp-berlin.de
bDepartment of Chemistry, Princeton University, Washington Road, Princeton, New Jersey 08544, USA
cInstitut für Chemie, Humboldt Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany
dMedical Research Council Laboratory of Molecular Biology, Francis Crick Avenue, Cambridge, UK
First published on 12th June 2018
Abstract
An important step in elucidating the function of protein post-translational modifications (PTMs) is gaining access to site-specifically modified, homogeneous samples for biochemical characterization. Protein pyrophosphorylation is a poorly characterized PTM, and here a chemical approach to obtain pyrophosphoproteins is reported. Photo-labile phosphorimidazolide reagents were developed for selective pyrophosphorylation, affinity-capture, and release of pyrophosphoproteins. Kinetic analysis of the reaction revealed rate constants between 9.2 × 10−3 to 0.58 M−1 s−1, as well as a striking proclivity of the phosphorimidazolides to preferentially react with phosphate monoesters over other nucleophilic side chains. Besides enabling the characterization of pyrophosphorylation on protein function, this work highlights the utility of phosphoryl groups as handles for selective protein modification for a variety of applications, such as phosphoprotein bioconjugation and enrichment.
Introduction
Post-translational modifications (PTMs) of proteins – as exemplified by kinase-mediated phosphorylation – constitute critical conduits for signal transduction, affecting nearly every cellular process.1,2 Although phosphorylation of serine, threonine, and tyrosine residues has been characterized most thoroughly to date, to date, histidine, cysteine, and arginine phosphorylation has also been described.3–5 Elucidating the functions of these P–N and P–S bond-containing phosphorylation marks has been challenging due to their labile nature.6,7 Synthetic access to peptides bearing these modifications was critical for characterizing their chemical stability, validating the specificity of antibodies, and developing mass spectrometry-based detection methods.8 These new tools are expected to reveal the specific effects of labile phosphorylation sites on protein function and activity, in much the same way that established techniques have done so for serine, threonine and tyrosine phosphorylation.Among the most recent additions to the list of labile modes of phosphorylation is protein pyrophosphorylation, a modification putatively mediated non-enzymatically by a class of intracellular messengers termed inositol pyrophosphates (PP-InsPs). Specifically, the β-phosphoryl group from 5PP-InsP5 (5-diphosphoinositol-1,2,3,4,6-pentakisphosphate) can be transferred to protein substrates in a Mg2+-dependent fashion (Fig. 1a).9 The PP-InsPs are conserved across eukaryotic organisms and are important for sensing the metabolic state of the cell and maintaining phosphate homeostasis.10–14 In mammals, PP-InsPs are necessary for proper insulin secretion and sensitivity,15 modulating weight gain through the regulation of adipogenesis,16 and controlling cell motility.17
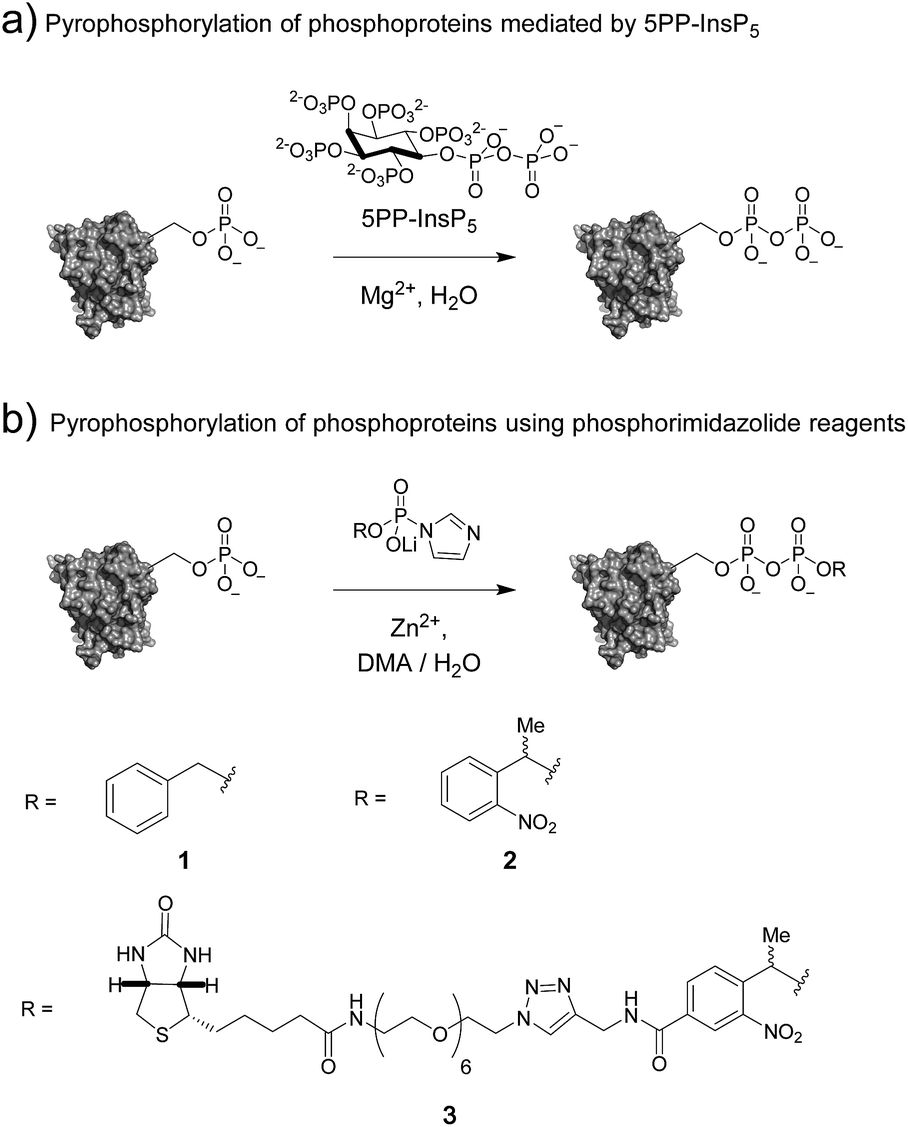 | ||
| Fig. 1 (a) Protein pyrophosphorylation by 5PP-InsP5, and (b) chemical pyrophosphorylation using phosphorimidazolide reagents. | ||
The molecular mechanisms underlying these phenotypes have not been fully elucidated, however a possible explanation is a bimodal mechanism of action: protein binding and protein pyrophosphorylation.12,18,19 Previously, our group developed synthetic methodologies to access pyrophosphorylated peptides from phosphorylated precursors using phosphorimidazolide 1 (Fig. 1b).20,21 While the peptides facilitated the development of analytical tools,22,23 a long-term goal is to demonstrate the effect of pyrophosphorylation on protein structure and function. The ability to produce site-specifically and stoichiometrically pyrophosphorylated proteins would provide the opportunity for full biochemical and biophysical characterization of this modification. Here, we show that phosphorimidazolide reagents (also termed phosphorimidazolidates or P-imidazolides) can be used for covalent modification of pre-existing phosphoryl groups on full-length protein substrates (Fig. 1b). Remarkably, the high selectivity and favorable reaction kinetics of these reagents suggest that this method will be suitable for new bioconjugation strategies.
Results and discussion
Synthesis of phosphorimidazolide reagents
In our earlier report, benzyl-protected P-imidazolide 1 (Fig. 1b, Scheme S1†) was employed to convert phosphopeptides to pyrophosphopeptides. This method, however, was incompatible with cysteine residues since the palladium catalyst required for subsequent hydrogenolysis binds to sulfhydryl groups irreversibly. Therefore, we now pursued a photo-labile P-imidazolide reagent with an o-nitrophenylethyl (o-NPE) group (2, Fig. 1b), since this protecting group had been reported to release phosphate monoesters in complex biological samples and in the presence of cysteine residues.24,25 The o-NPE protected P-imidazolide was synthesized by adapting a phosphitylation and oxidation protocol26,27 to yield phosphate triester 5, followed by deprotection of the cyanoethyl groups providing o-NPE phosphate monoester 6. Compound 6 was then converted to the corresponding P-imidazolide 2 and isolated via precipitation as a lithium salt (Scheme 1a). Reagent 2 was validated by using it in a pyrophosphorylation reaction with a cysteine-containing model peptide and then irradiating the resulting pyrophosphate diester with 355 nm light. The photo-deprotection proceeded to high conversion and was not accompanied by significant side-product formation (Fig. S1†).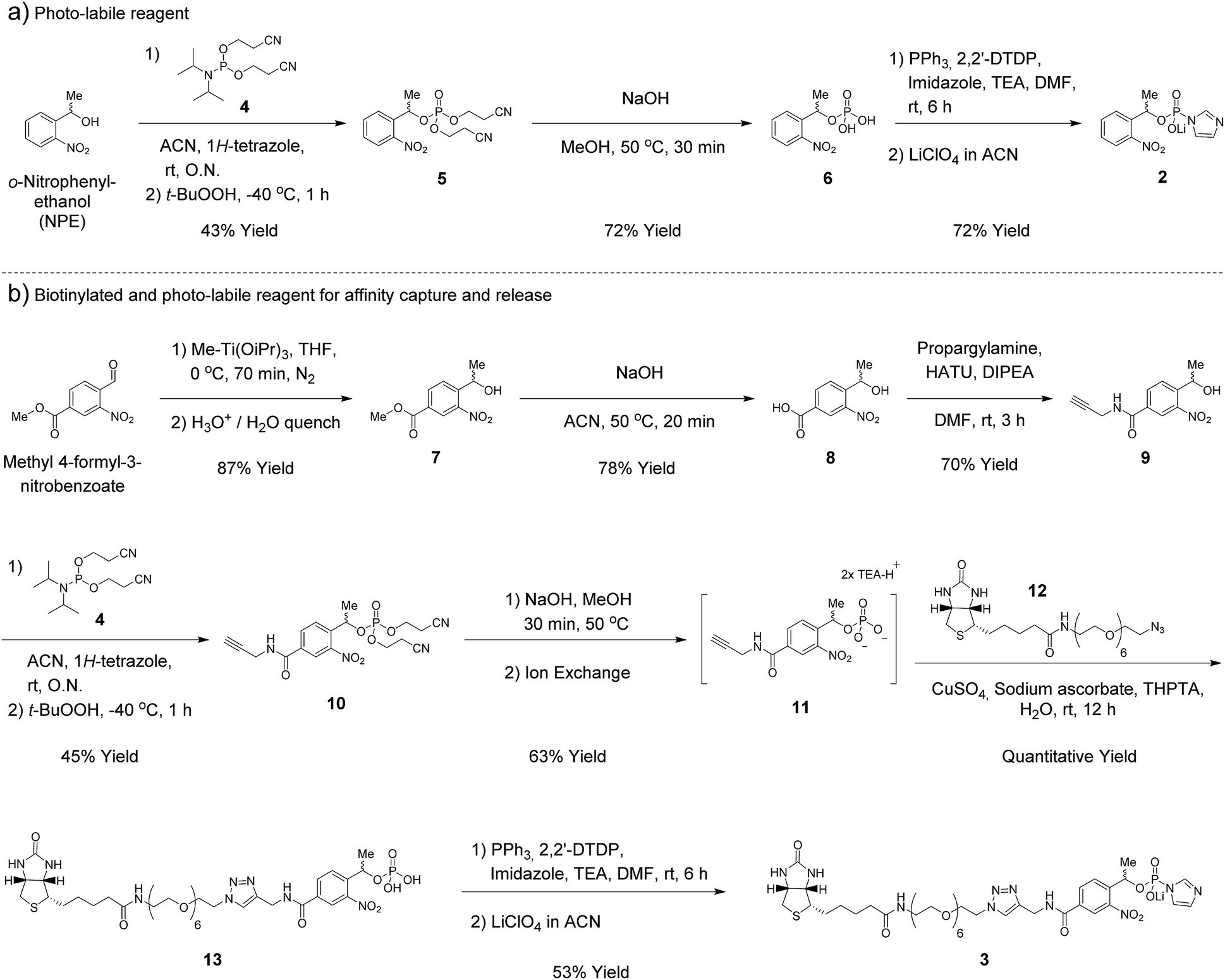 | ||
| Scheme 1 Synthesis of phosphorimidazolide reagents. | ||
For added functionality, we next synthesized an NPE-based P-imidazolide reagent with a biotin-tag connected via a PEG linker (3, Fig. 1b) to enable both affinity capture and photo-release of phosphoprotein substrates. To start, the aldehyde of methyl 4-formyl-3-nitrobenzoate was selectively reduced using a freshly-prepared organotitanium reagent to yield alcohol 7.28,29 Base hydrolysis of the methyl ester provided carboxylic acid 8, and a terminal alkyne was appended via amide bond formation (9). The phosphoryl group was installed using the synthetic sequence described above yielding phosphate ester 11. Biotin-PEG6-Et-N3 (12) was then attached to the terminal alkyne using CuAAC chemistry30 to provide triazole 13 in quantitative yield. Gratifyingly, 13 could be converted to biotin-PEG6-Tz-NPE-P-imidazolide (3) using the PPh3 and 2,2′-DTDP activation protocol and the desired product was subsequently isolated by selective precipitation as the lithium salt (Scheme 1b).
Kinetic characterization of peptide pyrophosphorylation
We next evaluated the kinetic properties of the pyrophosphorylation reaction to determine whether the bimolecular reaction is rapid enough and has sufficient functional functional group compatibility to conveniently modify low-abundance protein substrates. The reaction between a model phosphopeptide (14) and P-imidazolide reagent 1 (Fig. 2a) was monitored using an HPLC-based assay (Fig. S2†). Peptide 14 possesses a phosphoryl group as its only reactive moiety, and was consumed most rapidly in DMA at 45 °C with a rate constant of 0.58 M−1 s−1 under pseudo-first order conditions. The rate constants decreased notably as the polarity of the solvent increased (Fig. 2b and S3†).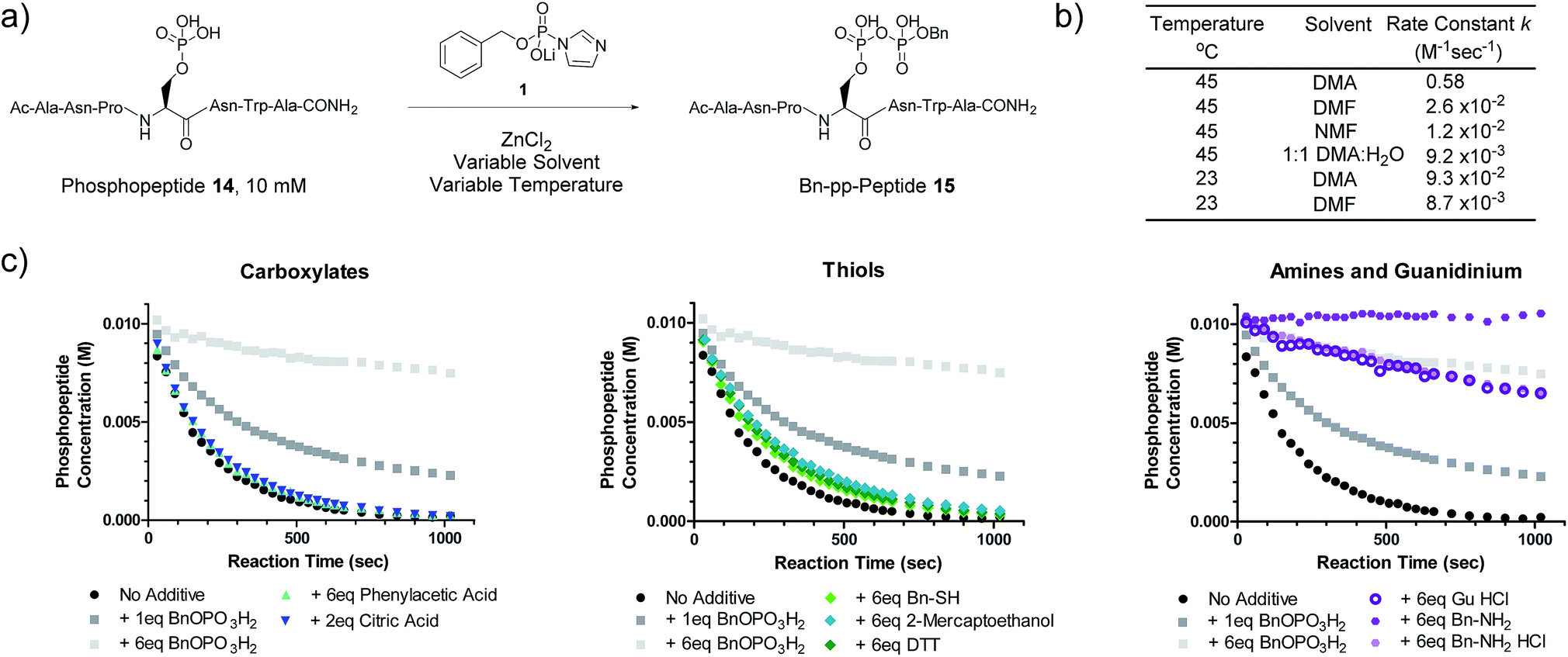 | ||
Fig. 2 Kinetic characterization of pyrophosphorylation reactions. (a) Reaction of model phosphopeptide 14 with P-imidazolide 1. (b) Tabulated pseudo-first order kinetic data. Conditions: 100 mM P-imidazolide 1, 10 mM phosphopeptide 14, 267 mM ZnCl2. (c) Reaction progress kinetic data for the reaction of phosphopeptide 14 with reagent 1 in the presence of various additives. Data points represent averages from three replicate reactions (except for runs with added 2-mercaptoethanol, DTT, and guanidine HCl). Error bars are omitted for clarity but are depicted in Fig. S7.† Processed RPKA data is shown in Fig. S8.† Conditions: 10 mM peptide 4, 30 mM P-imidazolide 1, 80 mM ZnCl2, in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 H2O:DMA at 45 °C. :9 H2O:DMA at 45 °C. | ||
To assess the extent of potential competing side reactions with other nucleophiles, a series of reaction progress kinetic analyses (RPKA) were conducted on the pyrophosphorylation of model peptide 14 by 3 eq. of reagent 1 (at 45 °C in 1:9 H2O:DMA). Zn2+-activated P-imidazolide reagent 1 exhibited a half-life of approx. 13 hours under the reaction conditions (Fig. S4†), suggesting that hydrolysis is negligible over the timespan of the reactions. Kinetic experiments without any additive went to complete conversion rapidly, while those reactions with added monobenzylphosphate slowed down in a concentration-dependent manner (Fig. 2c). Excesses of carboxylic acid additives and thiols appeared to have little effect on the rate of the reaction, indicating minimal competition of these functionalities with phosphopeptide 14 (Fig. 2c). Alcohols also exhibit minimal reactivity towards P-imidazolide reagents31,32 and the reaction can be run in methanol.20 By contrast, addition of HCl salts of a primary amine or guanidine caused the reaction to slow down dramatically, and the free base of benzylamine inhibited the reaction entirely (Fig. 2c). Extended monitoring of reactions with added amine HCl salts showed that peptide 14 is eventually transformed to 15 with high conversion, while reactions with added monobenzylphosphate remained stalled (Fig. S5, Table S1†). To determine the cause of this rate deceleration, NMR spectra of Zn2+-activated reagent 1 were recorded before and after addition of various amines in neat DMA (Fig. S8 and S9†). No signs of phosphoramidate formation were observed, indicating that amines inhibit P-imidazolide pyrophosphorylation by interacting non-covalently with the Zn2+-activated reagent. Therefore, while longer reaction times may be required for pyrophosphorylation in the presence of amines, the amines themselves do not appear to react readily with reagent 1 under these conditions.
Pyrophosphorylation of phosphoproteins
The most important test of the utility and selectivity of the P-imidazolide reagents is the modification of a pre-phosphorylated full-length protein. Achieving selectivity in this context is much more challenging than on peptide substrates: due to the larger number and greater diversity of potentially reactive residues on proteins, even low levels of off-target reactivity will result in over-functionalization of a significant proportion of the substrate population. To best quantify the conversion of the reaction, a stoichiometrically phosphorylated protein substrate would be optimal. We therefore utilized a recently reported amber codon suppression system for the incorporation of phosphoserine into recombinantly expressed proteins.33 At this point, no protein known to be pyrophosphorylated (and thus pre-phosphorylated) has been expressed using the amber codon suppression system for pSer. Consequently, ubiquitin was chosen as the first generic substrate in which serine 65 is quantitatively phosphorylated (Ub-S65pS). Ub-S65pS (29 μM) was treated with P-imidazolide 1 and ZnCl2 (in 1:9 H2O:DMA at 45 °C) and the reaction was monitored using ESI-MS (Fig. 3). After 6 hours, Ub-S65pS was consumed to yield a protein with a mass corresponding to a single pyrophosphorylation event as the primary product (Fig. S10†). Importantly, wild-type ubiquitin (Ub-wt) was recalcitrant to derivatization under the same reaction conditions, exhibiting only similar levels of off-target modification (Fig. S11†).
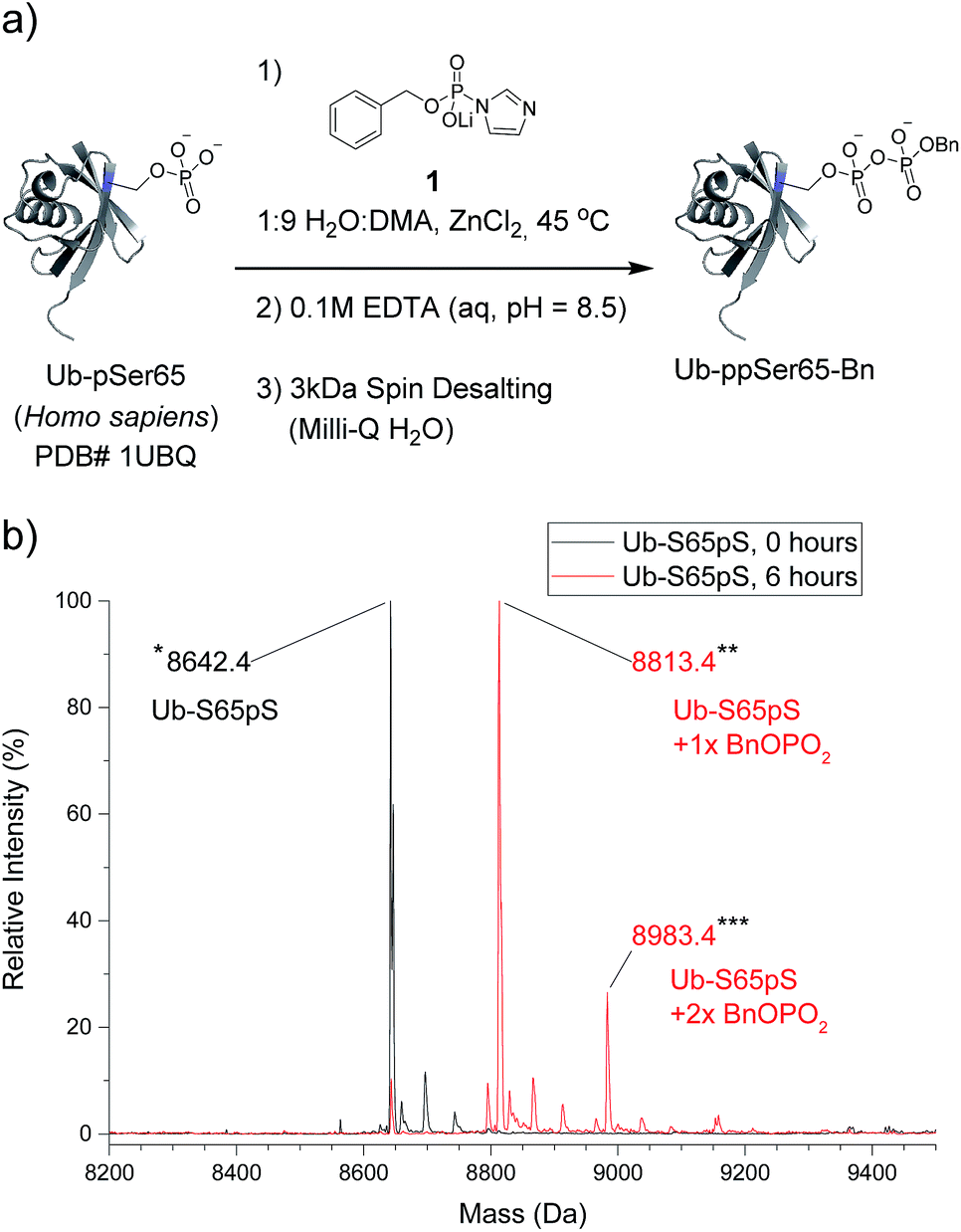 | ||
| Fig. 3 Pyrophosphorylation and characterization of ubiquitin-S65pS. (a) Pyrophosphorylation procedure for Ub-S65pS. 306 mM ZnCl2, 61.2 mM reagent 1 and 29 μM protein. (b) ESI-MS of Ub-S65pS before pyrophosphorylation (black) and after 6 hours under pyrophosphorylation reaction conditions (red). Mass error of 10 ppm; mass resolution of 0.1 Da. | ||
We next wanted to explore a larger, more challenging substrate, and chose Physeter catodon (sperm whale) myoglobin, in which Asp 127 was mutated to pSer (Myo-D127pS). Early attempts to pyrophosphorylate Myo-D127pS gave poor yields due to precipitation during the aqueous workup. Building on previously-reported refolding protocols,34–36 we developed a workup procedure from which the protein could be recovered in approximately 30% overall yield. To assess refolding of myoglobin after pyrophosphorylation with reagent 1, Circular Dichroism (CD) spectra were recorded. A comparison of molar ellipticities of pyrophosphorylated and non-pyrophosphorylated proteins (Fig. 4b) revealed that the spectra were essentially superimposable, indicating that the refolding protocol was sufficient to allow the protein to recover its secondary structure. Furthermore, the pyrophosphorylation modification itself does not appear to perturb the protein structure. Monitoring of the protein derivatization by ESI-MS indicated that the starting phosphoprotein was completely consumed after 90–100 minutes to yield primarily the desired product and a 22% relative abundance peak corresponding to the pyrophosphorylated product with one off-target modification (Fig. 4c and S12†). This was consistent with the level of off-target modification observed in the sample of non-phosphorylated wild-type myoglobin (Myo-wt) (Fig. S13†). Since wild-type myoglobin lacks cysteine residues, two cysteine-containing mutants (Myo-G154C, and Myo-D127pS-G154C) were expressed so that the impact of this residue on off-target modification could be assessed. In agreement with the kinetic experiments conducted in the presence of thiols, no significant increase in off-target modification was detected in these cysteine mutants (Fig. S14 and S15†).
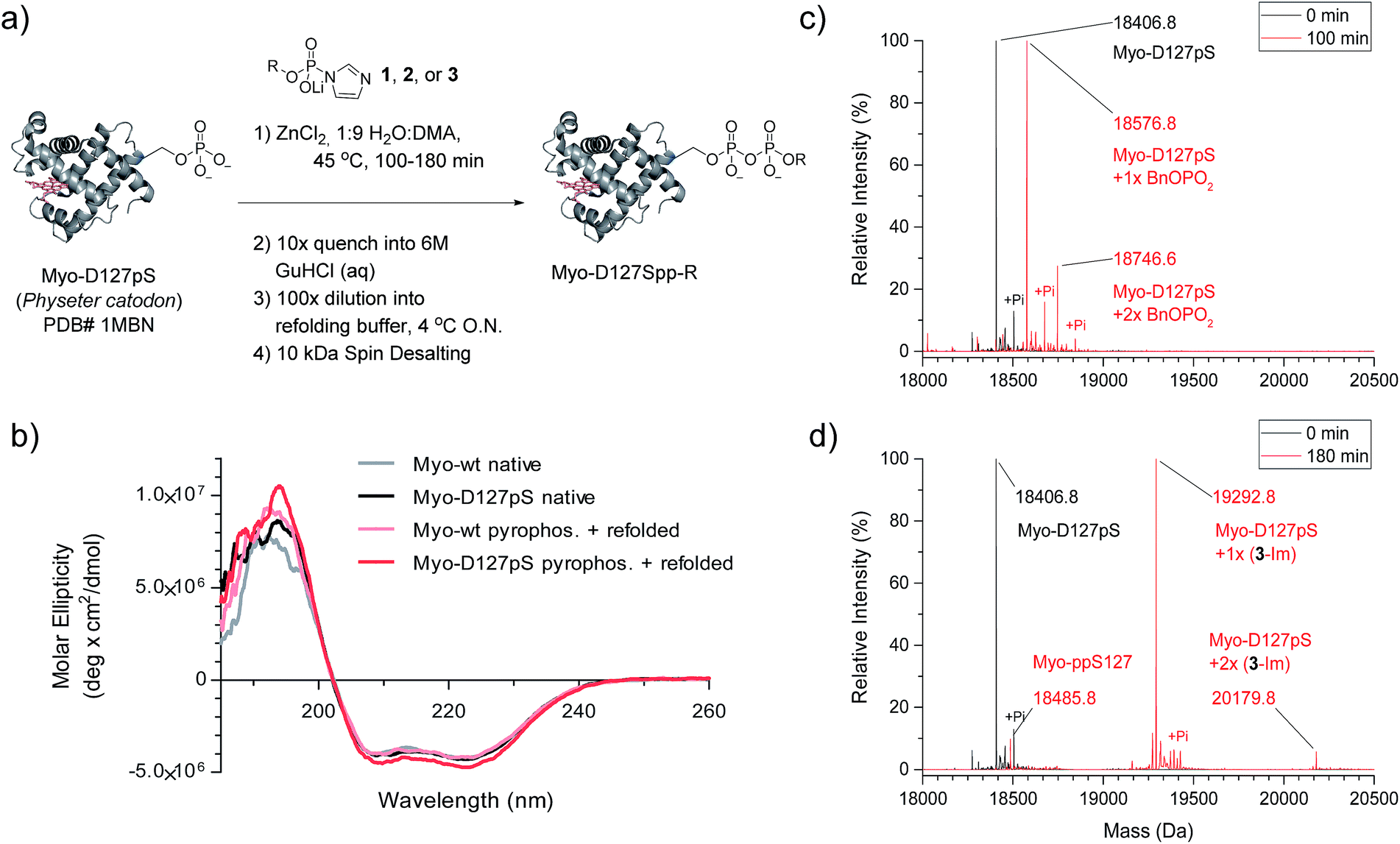 | ||
| Fig. 4 Pyrophosphorylation of myoglobin-D127pS and subsequent characterization. (a) Pyrophosphorylation and refolding protocol for Myo-D127pS. Condition: 306 mM ZnCl2, 61.2 mM reagent 1, 2, or 3, and 49 μM protein. (b) CD spectra of myoglobin before (native) and after the pyrophosphorylation reaction (pyrophos.) according to (a) with reagent 1. (c) ESI-MS of Myo-D127pS pyrophosphorylated with reagent 1 for 100 min and refolded. (d) ESI-MS of Myo-D127pS pyrophosphorylated with reagent 3 for 180 min and refolded. Mass error of 4 ppm; mass resolution of 0.2 Da. | ||
Selective affinity capture and release of phosphorylated myoglobin
Given the successful protein derivatization with reagent 1, the newly synthesized P-imidazolides 2 and 3 were explored for modification of Myo-D127pS. Interestingly, both NPE-protected P-imidazolide 2 (Fig. S16†) and biotin-PEG6-NPE P-imidazolide reagent 3 (Fig. 4d and S17†) reacted more slowly with Myo-D127pS, reaching complete conversion between 2.5 and 3 hours while providing lower levels of off-target phosphorylation (5–10% relative abundance). The o-NPE protected Myo-D127pS was subsequently irradiated with 360 nm UV light for 40 min to release the free pyrophosphorylated protein without an appreciable amount of side product formation (Fig. S18†). Finally, biotinylated and photo-labile reagent 3 was applied to selectively modify and affinity capture a pre-phosphorylated protein selectively, and subsequently release the pyrophosphorylated product via photo-cleavage (Fig. 5a). Accordingly, samples of Myo-D127pS and Myo-wt were subjected to the pyrophosphorylation reaction conditions and then either incubated over BSA-blocked streptavidin beads, or directly loaded onto an SDS-PAGE gel. A distinct gel shift corresponding to the attachment of one equivalent of reagent (+887 Da) could be observed on Myo-D127pS (Fig. 5b). Faint bands corresponding to additional reagent attachment were also visible for Myo-wt and Myo-D127pS, in line with the low degree of off-target modification observed via mass spectrometry. Gratifyingly, only the pre-phosphorylated protein treated with 3 was pulled-out of solution (Fig. 5b) with the streptavidin beads. The captured protein could then be released into solution by irradiating the re-suspended bead aliquots with 360 nm light for 3 h (Fig. 5c). Again, only the beads which were exposed to 3-treated Myo-D127pS released a significant amount of protein, underscoring the utility of 3 for the selective affinity capture and release of a pre-phosphorylated protein substrate.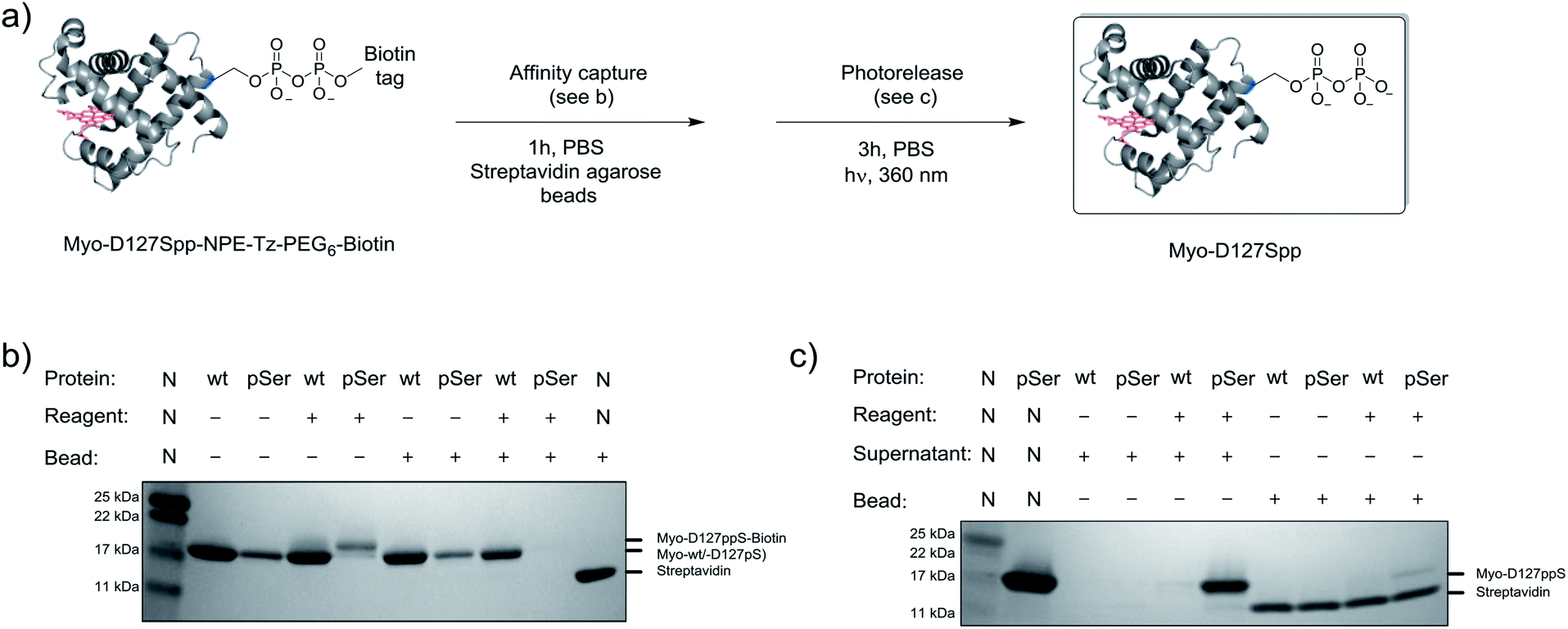 | ||
| Fig. 5 Modification of Myo-D127pS, affinity-capture, and release. (a) General workflow for affinity capture and photorelease. (b) Following treatment with reagent 3, Myo-wt or Myo-D127pS were exposed to streptavidin beads to affinity capture the conjugate. (c) Irradiation releases the free pyrophosphoproteins from the beads into the supernatant. See Fig. S21† for full gel images. PBS; phosphate buffered saline. | ||
Considering that the use of organic solvents may lead to denaturation of protein substrates, we sought to investigate the water compatibility of the selective pyrophosphorylation reaction. We subjected an aqueous solution of Myo-D127pS to either compound 1 (3 h, Fig. S19†) or 3 (a 24 h time course, Fig. S20†) at lower reagent concentrations (10 mM P-imidazolide reagents, 32 mM ZnCl2; the solubility limit for the Zn–phosphorimidazolide complex in water), and at 37 °C. We were pleased to observe that the reaction indeed occurred in straight water, albeit with lower conversion. An increase in the reaction time promoted the conversion but at the expense of increased off-target modification.
Bottom-up mass spectrometry analysis of pyrophosphorylated myoglobin
While the data illustrate the high propensity of P-imidazolide reagents to preferentially react with phosphorylated residues, the characterization of apparent off-target sites remained necessary. Therefore, chemically pyrophosphorylated and photo-deprotected myoglobin (Myo-D127ppS) was digested with trypsin and submitted to bottom-up proteomic analysis using nanoLC tandem mass spectrometry. Peptides were fragmented by electron-transfer/higher-energy dissociation (EThcD) to allow for reliable localization of different types of phosphorylation while enabling high sequence coverage.7,22,37 The analysis confirmed the pyrophosphorylation of myoglobin at S127 (Fig. 6a) and over 90% conversion of the starting monophosphorylated protein. Although multiple off-target phosphorylation sites were found on Ser, Thr, His, and Lys residues, the peptides bearing these modifications made up only 0.9% of the total phosphorylated ions detected (Fig. 6b and Table S2†). The desired pyrophosphorylated peptide was by far the most abundant species, comprising 93.5% of the total phosphorylated ions, again confirming the efficacy of the P-imidazolide reagents described here.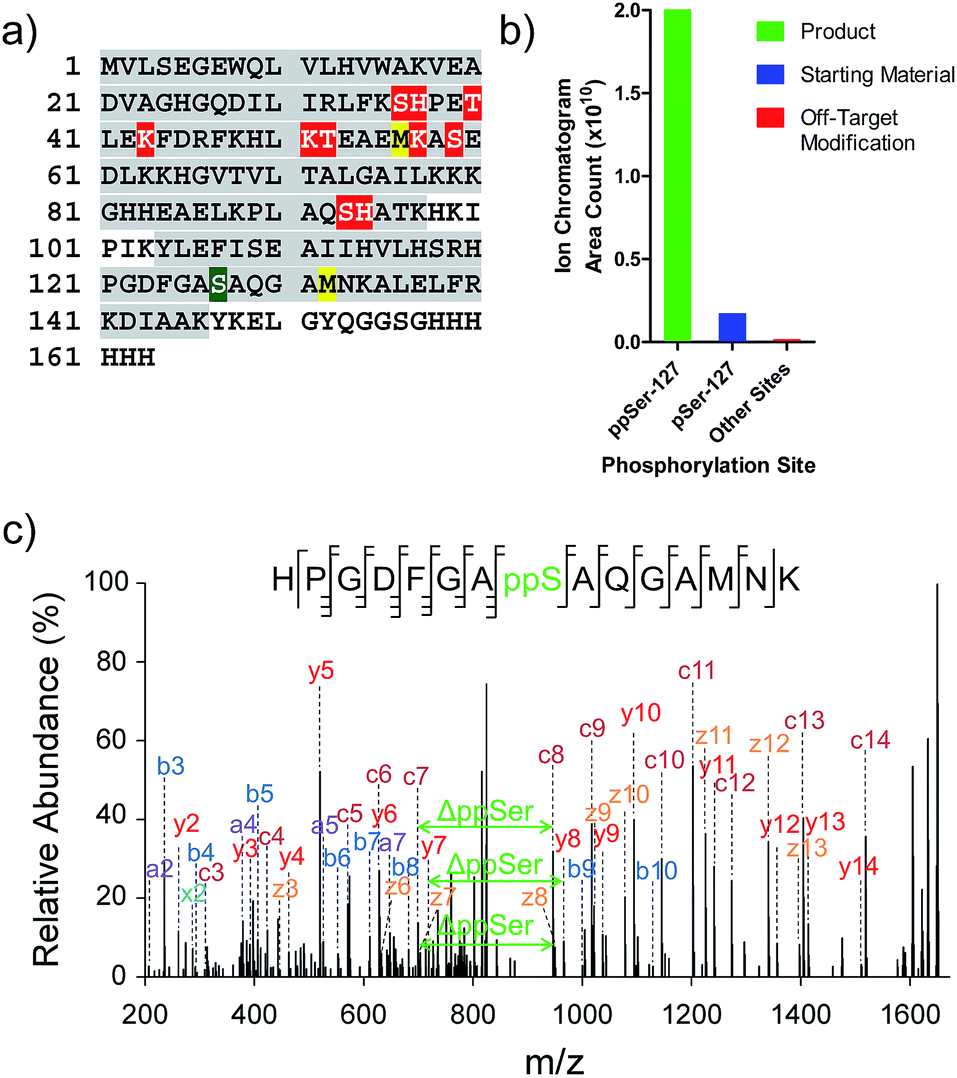 | ||
| Fig. 6 Characterization of Myo-D127ppS, and off-target phosphorylation sites. (a) Sequence of myoglobin with MS/MS coverage highlighted in grey. Red residues: off-target phosphorylation sites; green residue: pyrophosphorylation site (methionine residues highlighted in yellow were found to be partially oxidized). (b) Ion abundances of peptides bearing ppS127 product (green), pS127 starting material (blue), and off-target phosphorylation sites (red). See Table S2† for peptide sequences and ion counts, Fig. S22† for a detailed graphical representation of the off-target ion abundances, and Fig. S23† for single ion chromatograms of pS and ppS-bearing peptides. (c) EThcD MS/MS of the pyrophosphorylated peptide fragment ion. | ||
Conclusions
In sum, we present a set of functionalized P-imidazolide reagents for the site-selective derivatization of phosphoproteins. P-imidazolide reagents have a rich history in the field of nucleotide chemistry,31,38–44 and as model compounds for investigating enzymatic phosphorylation reactions.45,46 In recent years, a number of synthetic endeavours have built on the proclivity of P-imidazolides and related activated phosphoryl groups to form new phosphoanhydride bonds,31,47–49 which we exploited here for the site-specific conversion of full-length phosphoproteins to pyrophosphoproteins.Despite the evidence for protein pyrophosphorylation in vitro, demonstrating the functional consequences of this modification in a biochemical setting has remained a challenge. We have developed reagents which enable the production of stoichiometrically modified proteins in assay-scale quantities, by virtue of fast reaction kinetics and high functional group compatibility. Moreover, the capture and isolation of the modified proteins facilitated by the tagged P-imidazolide not only simplifies the purification procedure but is also a testament to the multifunctional reagent platform. With these tools, the means to investigate the biochemical and biophysical effects of pyrophosphorylation on protein substrates are now available. Myoglobin and ubiquitin were selected as model substrates to allow for the development of broadly compatible reaction conditions. We intend to apply these reagents to known pyrophosphorylation substrates in the future to investigate the function of this modification.
The current necessity of using solvent composed predominantly of DMA to achieve satisfactory pyrophosphorylation reaction rates raises the question of how broadly this methodology can be utilized for protein substrates. Organic solvents are normally avoided during the chemical modification of proteins in order to maintain secondary and tertiary structure. However, many proteins are routinely misfolded in the course of their recombinant overexpression, and can regain their activities after a refolding step.35 Our success in mainly organic media suggests that proteins which can be refolded could be candidates for modification in organic solvent. Future work will focus on increasing the rate of the reaction in water and improving selectivity. The effect of varying the protecting group on the phosphorimidazolide also merits further exploration, as the different reagents described in this study exhibited distinct rates of off-target modification.
To date, the chemical biology community has targeted many of the naturally occurring amino acids and protein N-termini as handles for bioconjugation.50–54 However, the potential of a phosphorylated side chain to act as a site for covalent attachment or derivatization has not been explored so far. Given the ubiquitous occurrence of protein phosphorylation and the intense interest it attracts, a selective chemical modification strategy for phosphorylated residues could be of great utility in a variety of applications. For example, in the field of bioconjugation, pyrophosphate diesters were recently shown to be ideal linkers for antibody–drug conjugates (ADCs).55 Phosphoproteomics workflows include a step for the enrichment of phosphorylated peptides. Although several enrichment techniques are in use, they have been shown to isolate distinct subsets of the phosphoproteome,56 suggesting that additional techniques for enriching and isolating phosphopeptides could be useful. A method using P-imidazolides for the covalent capture of phosphopeptides appears feasible, in which bound peptides would subsequently be released via the direct hydrolysis of the phosphoanhydride bond. Alternatively, bead release could be achieved by photocleavage, followed by hydrolysis of pyrophosphopeptides to phosphopeptides.
Overall, in combination with an efficient amber codon suppression system for phosphoserine incorporation, P-imidazolide reagents provide a powerful tool for further studying protein pyrophosphorylation and exploring applications of selective functionalization of protein phosphoryl groups.
Experimental
All materials and methods are reported in the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We would like to thank the Muir, MacMillan, Sorensen, Yang, Doyle and Hackenberger labs for sharing chemicals, equipment, and expertise. Dr Istvan Pelczer and Dr Peter Schmieder assisted with NMR spectroscopy. Heike Nikolenko and Heike Stephanowitz are gratefully acknowledged for providing an assistance with CD spectroscopy and MS/MS analysis respectively. Core funding from the FMP and Princeton University start-up funds are gratefully acknowledged.Notes and references
- C. T. Walsh, Posttranslational Modifications of Proteins: Expanding Nature's Inventory, Roberts and Company Publishers, Greenwood Village, Colorado, 2006 Search PubMed.
- T. Hunter, Philos. Trans. R. Soc., B, 2012, 367, 2513–2516 CrossRef PubMed.
- P. V. Attwood, M. J. Piggott, X. L. Zu and P. G. Besant, Amino Acids, 2007, 32, 145–156 CrossRef PubMed.
- P. G. Besant, P. V. Attwood and M. J. Piggott, Curr. Protein Pept. Sci., 2009, 10, 536–550 CrossRef PubMed.
- P. V. Attwood, P. G. Besant and M. J. Piggott, Amino Acids, 2011, 40, 1035–1051 CrossRef PubMed.
- J. Bertran-Vicente, R. A. Serwa, M. Schümann, P. Schmieder, E. Krause and C. P. R. Hackenberger, J. Am. Chem. Soc., 2014, 136, 13622–13628 CrossRef PubMed.
- J. Bertran-Vicente, M. Penkert, O. Nieto-Garcia, J.-M. Jeckelmann, P. Schmieder, E. Krause and C. P. R. Hackenberger, Nat. Commun., 2016, 7, 12703 CrossRef PubMed.
- R. C. Oslund, J. M. Kee, A. D. Couvillon, V. N. Bhatia, D. H. Perlman and T. W. Muir, J. Am. Chem. Soc., 2014, 136, 12899–12911 CrossRef PubMed.
- R. Bhandari, A. Saiardi, Y. Ahmadibeni, A. M. Snowman, A. C. Resnick, T. Z. Kristiansen, H. Molina, A. Pandey, J. K. Werner, K. R. Juluri, Y. Xu, G. D. Prestwich, K. Parang and S. H. Snyder, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15305–15310 CrossRef PubMed.
- M. S. C. Wilson, T. M. Livermore and A. Saiardi, Biochem. J., 2013, 452, 369–379 CrossRef PubMed.
- S. B. Shears, Adv. Biol. Regul., 2015, 57, 203–216 CrossRef PubMed.
- R. Wild, R. Gerasimaite, J.-Y. Jung, V. Truffault, I. Pavlovic, A. Schmidt, A. Saiardi, H. J. Jessen, Y. Poirier, M. Hothorn and A. Mayer, Science, 2016, 352, 986–990 CrossRef PubMed.
- C. D. Cordeiro, A. Saiardi and R. Docampo, Mol. Microbiol., 2017, 106(2), 319–333 CrossRef PubMed.
- R. Gerasimaite, I. Pavlovic, S. Capolicchio, A. Hofer, A. Schmidt, H. J. Jessen and A. Mayer, ACS Chem. Biol., 2017, 12, 648–653 CrossRef PubMed.
- R. Bhandari, K. R. Juluri, A. C. Resnick and S. H. Snyder, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2349–2353 CrossRef PubMed.
- A. Chakraborty, M. A. Koldobskiy, N. T. Bello, M. Maxwell, J. J. Potter, K. R. Juluri, D. Maag, S. Kim, A. S. Huang, M. J. Dailey, M. Saleh, A. M. Snowman, T. H. Moran, E. Mezey and S. H. Snyder, Cell, 2010, 143, 897–910 CrossRef PubMed.
- F. Rao, J. Xu, C. Fu, J. Y. Cha, M. M. Gadalla, R. Xu, J. C. Barrow and S. H. Snyder, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1773–1778 CrossRef PubMed.
- M. Wu, L. S. Chong, D. H. Perlman, A. C. Resnick and D. Fiedler, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E6757–E6765 CrossRef PubMed.
- M. Wu, L. S. Chong, S. Capolicchio, H. J. Jessen, A. C. Resnick and D. Fiedler, Angew. Chem., Int. Ed., 2014, 53, 7192–7197 CrossRef PubMed.
- A. M. Marmelstein, L. M. Yates, J. H. Conway and D. Fiedler, J. Am. Chem. Soc., 2014, 136, 108–111 CrossRef PubMed.
- L. M. Yates and D. Fiedler, ChemBioChem, 2015, 16, 415–423 CrossRef PubMed.
- M. Penkert, L. M. Yates, M. Schuemann, D. H. Perlman, D. Fiedler and E. Krause, Anal. Chem., 2017, 89, 3672–3680 CrossRef PubMed.
- J. H. Conway and D. Fiedler, Angew. Chem., Int. Ed., 2015, 54, 3941–3945 CrossRef PubMed.
- B. N. Goguen, A. Aemissegger and B. Imperiali, J. Am. Chem. Soc., 2011, 133, 11038–11041 CrossRef PubMed.
- N. Kotzur, B. Briand, M. Beyermann and V. Hagen, J. Am. Chem. Soc., 2009, 131, 16927–16931 CrossRef PubMed.
- E. J. Nurminen, J. K. Mattinen and H. Lönnberg, J. Chem. Soc., Perkin Trans. 2, 1998, 1621–1628 RSC.
- J. E. T. Corrie, G. P. Reid, D. R. Trentham, M. B. Hursthouse and M. A. Mazid, J. Chem. Soc., Perkin Trans. 1, 1992, 1015–1019 RSC.
- B. Weidmann and D. Seebach, Helv. Chim. Acta, 1980, 63, 2451–2454 CrossRef.
- R. Imwinkelried and D. Seebach, Org. Synth., 1989, 67, 180 CrossRef.
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., Int. Ed., 2009, 48, 9879–9883 CrossRef PubMed.
- M. Strenkowska, P. Wanat, M. Ziemniak, J. Jemielity and J. Kowalska, Org. Lett., 2012, 14, 4782–4785 CrossRef PubMed.
- H. Tanaka, Y. Yoshimura, M. R. Jürgensen, J. A. Cuesta-Seijo and O. Hindsgaul, Angew. Chem., Int. Ed., 2012, 51, 11531–11534 CrossRef PubMed.
- D. T. Rogerson, A. Sachdeva, K. Wang, T. Haq, A. Kazlauskaite, S. M. Hancock, N. Huguenin-Dezot, M. M. K. Muqit, A. M. Fry, R. Bayliss and J. W. Chin, Nat. Chem. Biol., 2015, 11, 496–503 CrossRef PubMed.
- Z. Yang, L. Zhang, Y. Zhang, T. Zhang, Y. Feng, X. Lu, W. Lan, J. Wang, H. Wu, C. Cao and X. Wang, PLoS One, 2011, 6, 1–8 Search PubMed.
- K. L. Maxwell, D. Bona, C. Liu, C. H. Arrowsmith and A. M. Edwards, Protein Sci., 2003, 12, 2073–2080 CrossRef PubMed.
- R. Feng and Y. Konishi, J. Am. Soc. Mass Spectrom., 1993, 4, 638–645 CrossRef PubMed.
- C. K. Frese, H. Zhou, T. Taus, A. F. M. Altelaar, K. Mechtler, A. J. R. Heck and S. Mohammed, J. Proteome Res., 2013, 12, 1520–1525 CrossRef PubMed.
- D. E. Hoard and D. G. Ott, J. Am. Chem. Soc., 1965, 87, 1785–1788 CrossRef PubMed.
- R. Lohrmann and L. E. Orgel, Nature, 1973, 244, 418–420 CrossRef PubMed.
- H. Sawai, H. Wakai and M. Shimazu, Tetrahedron Lett., 1991, 32, 6905–6906 CrossRef.
- M. Kadokura, T. Wada, C. Urashima and M. Sekine, Tetrahedron Lett., 1997, 38, 8359–8362 CrossRef.
- B. J. Weimann, R. Lohrmann, L. E. Orgel, H. Schneider-Bernloehr and J. E. Sulston, Science, 1968, 161, 387 CrossRef PubMed.
- T. Walton and J. W. Szostak, J. Am. Chem. Soc., 2016, 138, 11996–12002 CrossRef PubMed.
- L. Li, N. Prywes, C. P. Tam, D. K. O'Flaherty, V. S. Lelyveld, E. C. Izgu, A. Pal and J. W. Szostak, J. Am. Chem. Soc., 2017, 139, 1810–1813 CrossRef PubMed.
- G. J. Lloyd and B. S. Cooperman, J. Am. Chem. Soc., 1971, 93, 4883–4889 CrossRef PubMed.
- G. J. Lloyd, C.-M. Hsu and B. S. Cooperman, J. Am. Chem. Soc., 1971, 93, 4889–4892 CrossRef PubMed.
- Q. Sun, J. P. Edathil, R. Wu, E. D. Smidansky, C. E. Cameron and B. R. Peterson, Org. Lett., 2008, 10, 1703–1706 CrossRef PubMed.
- S. Mohamady, A. Desoky and S. D. Taylor, Org. Lett., 2012, 14, 402–405 CrossRef PubMed.
- Q. Sun, S. Gong, J. Sun, S. Liu, Q. Xiao and S. Pu, J. Org. Chem., 2013, 78, 8417–8426 CrossRef PubMed.
- A. C. Obermeyer, J. B. Jarman, C. Netirojjanakul, K. El Muslemany and M. B. Francis, Angew. Chem., Int. Ed., 2014, 53, 1057–1061 CrossRef PubMed.
- S. Lin, X. Yang, S. Jia, A. M. Weeks, M. Hornsby, P. S. Lee, R. V. Nichiporuk, A. T. Iavarone, J. A. Wells, F. D. Toste and C. J. Chang, Science, 2017, 355, 597–602 CrossRef PubMed.
- C. B. Rosen and M. B. Francis, Nat. Chem. Biol., 2017, 13, 697–705 CrossRef PubMed.
- N. Krall, F. P. da Cruz, O. Boutureira and G. J. L. Bernardes, Nat. Chem., 2015, 8, 1–11 Search PubMed.
- B. G. Davis, X. Shang, G. DeSantis, R. R. Bott and J. B. Jones, Bioorg. Med. Chem., 1999, 7, 2293–2301 CrossRef PubMed.
- J. C. Kern, M. Cancilla, D. Dooney, K. Kwasnjuk, R. Zhang, M. Beaumont, I. Figueroa, S. C. Hsieh, L. Liang, D. Tomazela, J. Zhang, P. E. Brandish, A. Palmieri, P. Stivers, M. Cheng, G. Feng, P. Geda, S. Shah, A. Beck, D. Bresson, J. Firdos, D. Gately, N. Knudsen, A. Manibusan, P. G. Schultz, Y. Sun and R. M. Garbaccio, J. Am. Chem. Soc., 2016, 138, 1430–1445 CrossRef PubMed.
- B. Bodenmiller, L. N. Mueller, M. Mueller, B. Domon and R. Aebersold, Nat. Methods, 2007, 4, 231–237 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01233d |
| This journal is © The Royal Society of Chemistry 2018 |