
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrigin of metastable oligomers and their effects on amyloid fibril self-assembly†
Filip
Hasecke‡
a,
Tatiana
Miti‡
b,
Carlos
Perez
b,
Jeremy
Barton
b,
Daniel
Schölzel
ac,
Lothar
Gremer
 ac,
Clara S. R.
Grüning
a,
Garrett
Matthews
b,
Georg
Meisl
d,
Tuomas P. J.
Knowles
d,
Dieter
Willbold
ac,
Philipp
Neudecker
ac,
Henrike
Heise
ac,
Ghanim
Ullah
b,
Wolfgang
Hoyer
*ac and
Martin
Muschol
*b
ac,
Clara S. R.
Grüning
a,
Garrett
Matthews
b,
Georg
Meisl
d,
Tuomas P. J.
Knowles
d,
Dieter
Willbold
ac,
Philipp
Neudecker
ac,
Henrike
Heise
ac,
Ghanim
Ullah
b,
Wolfgang
Hoyer
*ac and
Martin
Muschol
*b
aInstitut für Physikalische Biologie, Heinrich-Heine-Universität, 40204 Düsseldorf, Germany. E-mail: wolfgang.hoyer@hhu.de
bDepartment of Physics, University of South Florida, Tampa, FL 33620, USA. E-mail: mmuschol@usf.edu
cInstitute of Complex Systems (ICS-6), Structural Biochemistry, Research Centre Jülich, Germany
dDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK
First published on 13th June 2018
Abstract
Assembly of rigid amyloid fibrils with their characteristic cross-β sheet structure is a molecular signature of numerous neurodegenerative and non-neuropathic disorders. Frequently large populations of small globular amyloid oligomers (gOs) and curvilinear fibrils (CFs) precede the formation of late-stage rigid fibrils (RFs), and have been implicated in amyloid toxicity. Yet our understanding of the origin of these metastable oligomers, their role as on-pathway precursors or off-pathway competitors, and their effects on the self-assembly of amyloid fibrils remains incomplete. Using two unrelated amyloid proteins, amyloid-β and lysozyme, we find that gO/CF formation, analogous to micelle formation by surfactants, is delineated by a “critical oligomer concentration” (COC). Below this COC, fibril assembly replicates the sigmoidal kinetics of nucleated polymerization. Upon crossing the COC, assembly kinetics becomes biphasic with gO/CF formation responsible for the lag-free initial phase, followed by a second upswing dominated by RF nucleation and growth. RF lag periods below the COC, as expected, decrease as a power law in monomer concentration. Surprisingly, the build-up of gO/CFs above the COC causes a progressive increase in RF lag periods. Our results suggest that metastable gO/CFs are off-pathway from RF formation, confined by a condition-dependent COC that is distinct from RF solubility, underlie a transition from sigmoidal to biphasic assembly kinetics and, most importantly, not only compete with RFs for the shared monomeric growth substrate but actively inhibit their nucleation and growth.
Introduction
Deposits of protein aggregates forming non-branching rigid fibrils (RFs) with a characteristic cross-β sheet architecture are closely associated with a wide range of human disorders including Alzheimer's and Parkinson's disease, as well as non-neuropathic amyloidoses such as type-II diabetes and hereditary lysozyme amyloidosis.1–9 More recently, amyloid formation has also been associated with functional biological responses.10 Beyond late-stage RFs, amyloid formation frequently involves morphologically distinct, long-lived and highly populated metastable intermediates. Globular amyloid oligomers (gOs) and their associated highly curvilinear fibrils (CFs), often referred to as protofibrils, have been observed with large numbers of amyloid proteins and over a wide range of growth conditions.11–21 Substantial evidence suggests that early-stage gOs are potent sources of cytotoxicity in amyloid diseases.19,22–30 Metastable oligomers also affect the aggregation of pharmaceuticals,31 and might hold answers to the question what distinguishes functional from pathological amyloid species.3,10 Formation of metastable precursors relates to a variety of physiochemical and biomedical problems.32 This includes metastable liquid phases as precursor of protein crystallization33 or sickle-cell hemoglobin fibrillation,34 as well as the significance of membrane-less organelles in promoting ALS fibril formation.35 Some amyloid oligomers themselves have been suggested to share characteristics of disordered liquid-like states.36,37Characterizing the mechanisms and developing solutions to kinetic schemes that replicate RF nucleation and growth kinetics in the absence of long-lived, metastable intermediates has made significant progress.38,39 These models helped identify secondary nucleation mechanisms as critical contributors to the process of fibril nucleation. Analysis of the scaling behavior of reaction half-times vs. monomer concentrations now permits quantification of the relative contributions of distinct growth mechanisms to fibril formation.39–41 In contrast, the conditions required for the formation of significant concentrations of long-lived, metastable amyloid oligomers, which are distinct from the inherently minor populations of small, on-pathway fibril seeds, remain uncertain. Similarly, the mechanisms by which these metastable oligomers are replaced by late-stage RFs continue to be elusive. The role metastable states play in the nucleation and growth of late-stage RFs has important implications for our understanding of amyloid pathogenesis and informs efforts at intervening with their formation. Yet, it remains unresolved whether metastable globular oligomers are obligatory or optional precursors of fibril growth, and whether they serve as on-pathway precursors or represent off-pathway competitors of late-stage RFs.13 Two prevalent models for the role of globular oligomers are nucleated conformational conversion (NCC) vs. nucleated polymerization with competing off-pathway oligomers (cNP) (Fig. 1A). In NCC, oligomers are on-pathway but structurally distinct precursors of RFs; restructuring of oligomers into RF seeds represents the rate-limiting nucleation step.42,43 In cNP, RFs nucleate via “classical nucleation” from the monomer pool while metastable oligomers and curvilinear fibrils are off-pathway competitors to RF nucleation and growth.44,45 It is difficult to distinguish these scenarios since both predict qualitatively identical temporal sequences of aggregate populations and growth kinetics.
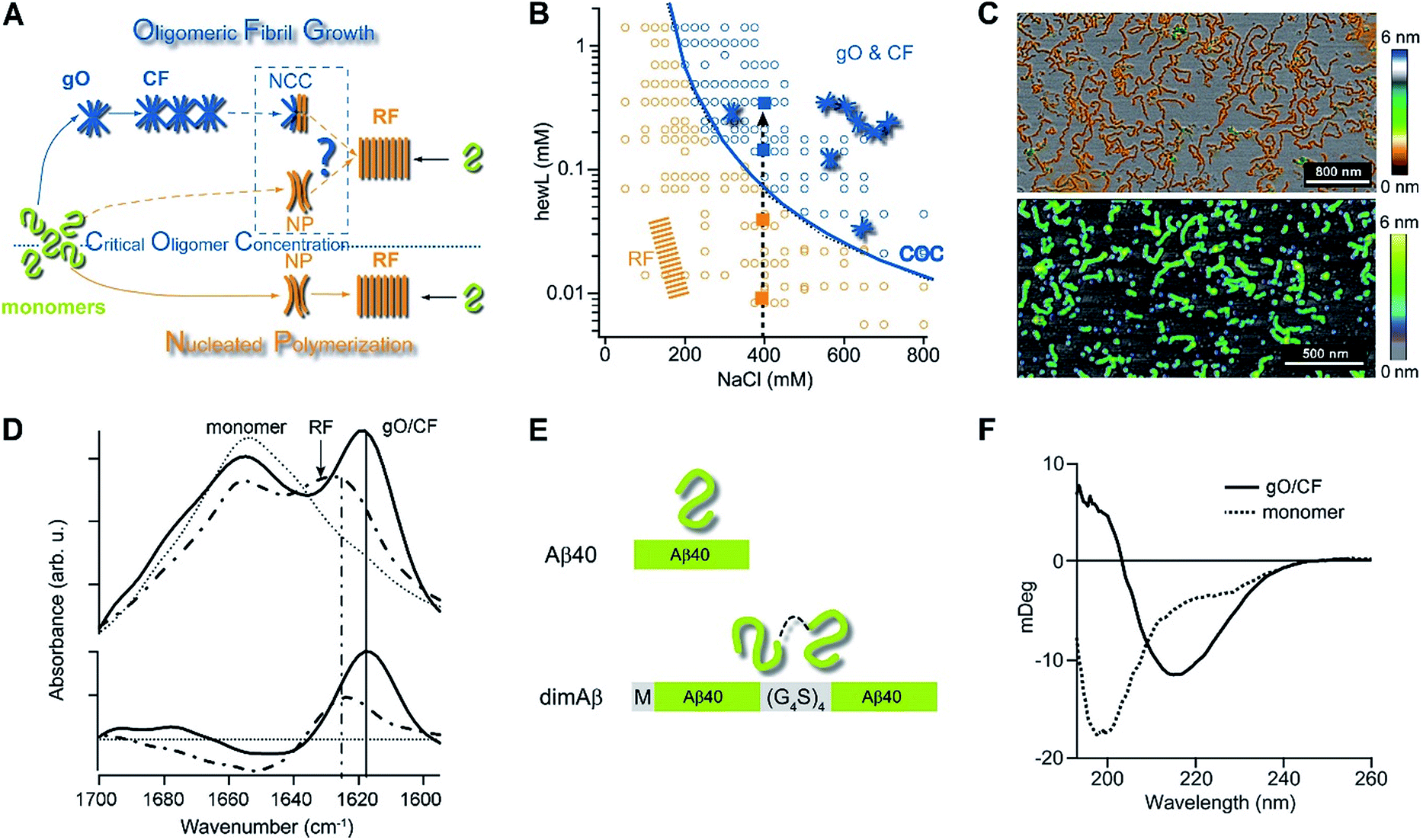 | ||
| Fig. 1 Amyloid oligomer model systems. (A) Schematic of potential roles for globular oligomers (gOs) and their curvilinear fibrils (CFs) in late-stage rigid fibril (RF) formation. (Top) Globular oligomers (gOs) and their curvilinear fibrils (CFs) are presumed to assemble prior to late-stage formation of RFs. The dashed lines represent the potential nucleation pathways for RFs either via nucleated conformational conversion (NCC) from gO/CFs or via nucleated polymerization from monomers, with RFs competing with off-pathway gO/CF formation (cNP). (Bottom) Nucleated polymerization (NP) of RFs from monomers only, i.e. in the absence of metastable gO/CFs. The transition for fibril formation in the absence or presence of gO/CF occurs upon crossing some monomer threshold called the “critical oligomer concentration” (COC, blue dotted line) (B) phase diagram for lysozyme at pH 2, 52 °C (adapted from ref. 46). Open orange circles indicate protein/salt concentrations resulting in RFs without gO/CF formation, as assessed by thioflavin T (ThT) and light scattering kinetics as well as time-resolved atomic force microscopy (AFM). Blue circles specify conditions for lag-free onset of gO/CF growth. The blue curve represents the fit from a colloidal model to the protein- and salt-dependent COC. The dashed vertical line indicates the transition from RF growth without build-up of metastable oligomeric species (below COC) to oligomeric RF growth (above COC) upon increasing monomer concentration, as applied in this study. (C) Morphologies of lysozyme (top) and dimAβ (bottom) gOs and CFs formed above their respective COCs, imaged using AFM. Color scale: height in nm. (D) Top: Amide I band infrared spectra of hewL monomers vs. gO/CFs or RFs, both after separation from monomers. Bottom: gO/CF and RF spectra after subtraction of monomer reference (adapted from ref. 54) (E) scheme of the dimeric Aβ40 construct dimAβ. Two Aβ40 units are linked in a single chain in a head-to-tail fashion. An N-terminal methionine affords recombinant expression. A flexible (G4S)4 linker was chosen to provide the Aβ40 units with conformational freedom. (F) Far-UV CD spectra of dimAβ before and after gO/CF formation, recorded at 4 °C or 20 °C, respectively, at a protein concentration of 20 μM. | ||
We have previously reported the separation of the parameter space for amyloid assembly of lysozyme in dependence of protein and salt concentration (Fig. 1B).46,50 One regime is typified by nucleated polymerization of rigid amyloid fibrils without discernible populations of metastable intermediates. The other regime results in lag-free formation of readily detectable globular oligomer (gOs) which tend to assemble into highly curvilinear fibrils (CFs), i.e. fibrils with distinctly shorter persistence lengths than their rigid fibril (RF) counterparts. A colloidal model accounting for the free-energy cost of charge repulsion among monomers upon confinement to an oligomer replicated the prominent protein- and salt-dependence of the sudden onset of gO formation in hen egg-white lysozyme (hewL) (Fig. 1B).46 Due to these strong similarities with the onset of micelle formation in charged surfactant system we labelled this transition the “critical oligomer concentration” or COC.47 Our observations also resembled a transition of β2-microglobulin amyloid assembly upon changes in solution pH.48,49 The gOs formed by hewL above the COC displayed the basic characteristics of small oligomers observed with multiple amyloid proteins, including those of β2-microglobulin, transthyretin, Aβ40/42, and α-synuclein.14,20,21,51–53 These characteristics include the globular morphology, the size of a few nanometers (Fig. 2E-I), the propensity to assemble into curvilinear fibrils (Fig. 1C, top, and Fig. 2E-II), and the muted ThT responses they elicit.54 Their IR spectra showed prominent peaks in the characteristic “amyloid band”, which were slightly but consistently shifted with respect to those of their RF counterparts (Fig. 1D).55 An additional weak peak near 1690 cm−1 hints at a potential antiparallel β-barrel architecture, now reported for multiple amyloid oligomers.56 The tinctorial and spectroscopic features of hewL gOs therefore replicate the limited number of high-resolution structures of early-stage amyloid gOs in other systems.57,58 We also confirmed that gO/CFs were metastable as RFs seeded above the COC readily grew while gO/CFs seeded below the COC slowly decayed. In short, metastable gO/CFs of hewL are confined by their COC to a subset of conditions permissive of fibril growth and are therefore nonobligatory intermediates of fibril growth (Fig. 1B).
These results raised a series of questions we set out to answer. First, is formation of metastable gO/CFs typically confined above a threshold protein concentration, i.e. a COC? Does it depend whether a folded or an intrinsically disordered protein undergoes amyloid assembly? Are these metastable gO/CFs on-pathway precursors or off-pathway byproducts of amyloid assembly? Most importantly, does the emergence of metastable gO/CFs above the COC alter RF nucleation and growth, and if so, in what ways? We chose to address these questions by comparing a single-chain Aβ dimer (dimAβ) against the behaviour of hen egg-white lysozyme (hewL). While Aβ is a disordered monomer and the key component of protein deposits associated with Alzheimer's disease, hewL is a folded protein closely related to hereditary lysozyme amyloidosis. The dimeric Aβ construct increases the local concentration of Aβ monomers and, thereby, lowers the threshold for oligomer formation in vitro. As detailed below, it also promotes the separation of timescales for gO/CF vs. RF formation and, thereby, permits separate analysis of their intrinsic kinetics. We show that, for both dimAβ and hewL, RF assembly kinetics changes from purely sigmoidal to biphasic upon crossing a protein- and condition-specific COC. The initial phase in biphasic kinetics represents the lag-free formation of gO/CFs while the second phase indicates RF nucleation and growth. Analysing the RF component, we find that the increasing levels of gO/CFs above the COC progressively slow RF formation, as evident in increasing RF lag periods. As we argue below, the formation of metastable gO/CFs therefore alters RF nucleation and growth in ways that neither of the currently dominant models of NCC and cNP fully captures.
Results
Single-chain Aβ dimer as amyloid oligomer model system
Formation of metastable gO/CFs of Aβ has been linked to neurotoxicity in Alzheimer's disease.11,12,22,24,26,28–30,59 To facilitate determination of the effects of gO/CFs on Aβ fibril formation, we introduce here a single-chain dimeric variant of Aβ40 termed dimAβ. In dimAβ, two Aβ40 units are connected through a flexible glycine–serine-rich linker in a head-to-tail fashion (Fig. 1E). A relatively long (20 amino acids), flexible linker was chosen to guarantee minimal disturbance of the conformational properties of Aβ in monomeric as well as aggregated states. This is especially important in the light of previous work on Aβ dimer constructs that suggested that short linkages via disulfide bridges restrict the accessibility of the RF state.30,60–62 In the case of dimAβ, solution NMR of the monomeric construct retrieves the resonances of monomeric Aβ40, indicating that the two Aβ subunits in dimAβ do not affect each other's largely disordered conformation (Fig. S1†). According to AFM (Fig. S2A†) and solid-state NMR spectroscopy (Fig. S2B and C†) the end product of dimAβ assembly, as for Aβ40, are RFs. A 2D INEPT MAS spectrum, which only displays mobile regions of the fibrils, almost exclusively picks up signals from glycine and serine residues. This indicates that the linker in dimAβ RFs remains flexible, while both Aβ subunits are incorporated into the RF β-sheet core (Fig. S2B†). Aβ40 and Aβ42 were previously shown to exhibit biphasic assembly kinetics at elevated protein concentrations.63–66 The linkage of two Aβ units was chosen to increase the local Aβ concentration, thereby promoting the highly concentration-dependent gO/CF formation. Compared to Aβ40 and Aβ42, this provides the separation of the two kinetics phases, i.e. the time regimes with dominant gO/CF or RF formation, at much reduced total incubation times and protein concentrations, important for the subsequent analysis of gO/CF and RF kinetics (see below). As seen by AFM and CD spectroscopy, dimAβ does readily form gO/CFs with morphologies (Fig. 1C) and β-structure (Fig. 1F) similar to those observed with hewL.Formation of gO/CFs induces switch from sigmoidal to biphasic assembly kinetics
The time courses of amyloid fibril assembly for both hewL and dimAβ undergo a discontinuous transition from purely sigmoidal to biphasic kinetics, as monitored by the amyloid indicator dye ThT (Fig. 2A and C). Below the transition (orange traces), ThT shows no discernible increase during an extended lag period lasting many hours to days. The flatness of the initial plateaus is highlighted in Fig. 2A and D by using a logarithmic axis for the ThT signal. This initial plateau is followed by a dramatic upswing in ThT emission which eventually saturates. Upon crossing a monomer concentration of about 1.5 μM dimAβ or 40 μM hewL, at their respective solution conditions, the kinetics changes discontinuously (blue traces, Fig. 2A and C). Slightly above the transition, ThT responses display a small but steady upward drift from the outset, followed by a prominent upswing at a time point consistent with RF nucleation and growth. By further raising monomer concentration the biphasic character of the kinetics becomes increasingly prominent, with the initial phase reaching a saturation plateau well before the second upswing in ThT response. The hewL traces shown in Fig. 2C emphasize another features of biphasic kinetics. The second upswing above the COC, while present, becomes less pronounced and the ThT amplitude at 96 hours decreases even though monomer concentration increases. As shown below, the progressive decrease in ThT response at this time point correlates with the increasing concentrations of residual gO/CFs. Both of these features support the model of RF assembly in the presence of gO/CFs put forth below. The ThT trace for hewL in Fig. 2E emphasize the biphasic character of hewL kinetics. The biphasic kinetics reported here extend our prior observation that the onset of a lag-free increase in ThT and light scattering kinetics in hewL coincided with the onset of gO/CF formation.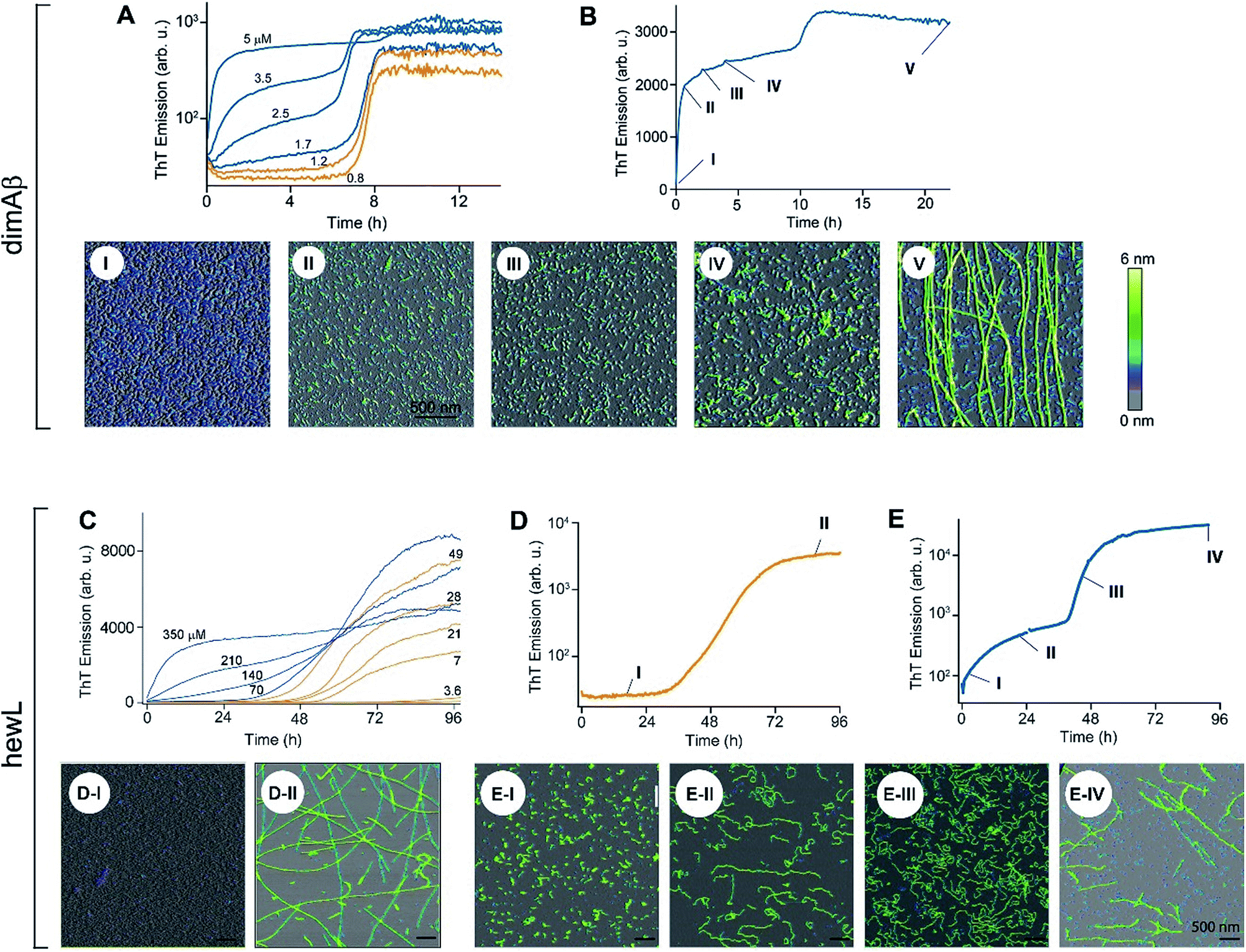 | ||
| Fig. 2 Transition from sigmoidal to biphasic growth kinetics upon oligomer formation. Transition from sigmoidal (orange) to bimodal (blue) amyloid growth kinetics of dimAβ (A, B) and hewL (C–E), as monitored by ThT fluorescence. Concentration dependent time traces of (A, B) dimAβ assembly in 50 mM Na-phosphate, 50 mM NaCl, pH 7.4, 37 °C, and (C–E) hewL assembly in 25 mM K-phosphate, pH 2.0, 52 °C, with 450 (C, E) and 500 (D) mM NaCl, respectively. Typical sigmoidal (D) and bimodal (B, E) growth kinetics correlated to AFM images of aggregate morphologies at the indicated time points (I–V) and concentrations of 20 μM dimAβ (B), 21 μM hewL (D), and 280 μM hewL (E), respectively. | ||
Using AFM imaging, we confirmed that the sigmoidal kinetics indicated RF growth without detectable intermediates while biphasic kinetics represented the sequential growth of gO/CFs during the initial phase and RF nucleation and growth during the secondary phase. AFM images of aliquots sampled for hewL growth below the COC indeed only detect monomers within the lag phase and accumulating numbers of fibrils of increasing length during the rise and subsequent plateau in ThT (Fig. 2D). The extended lag periods with no discernible fibril nucleation/growth matches well with the predictions from nucleated polymerization dominated by autocatalytic secondary nucleation mechanisms (see fits below). In contrast, AFM images of aggregate populations sampled during the initial phase of biphasic growth only show significant buildup of small gOs and CFs. This matches with the high reaction-order of gO/CF formation discussed below. Following the second upswing, in turn, AFM detects RFs in the solution (Fig. 2B and E). While AFM images of samples taken near the end of the kinetics traces clearly show RFs, significant populations of residual gO/CFs persist and their concentrations increase with monomer concentration above the COC. For hewL, for example, an additional two weeks of incubation were required before RFs had completely replaced gO/CFs. Hence, the apparent decline in the ThT amplitude above the COC (Fig. 2C) arises from the increasing populations of (weakly ThT positive) residual gO/CFs and their glacial pace of depolymerisation into (strongly ThT positive) RFs.
Inherent metastability of gO/CFs
The slow depletion of gO/CFs by RFs highlights perhaps the most distinctive feature of gO/CFs: their intrinsic metastability against RF formation.13,46 We previously corroborated the metastability of hewL gO/CFs by seeding solutions above the COC with RFs, which readily grew and replaced gO/CFs. Similarly, isolated gO/CFs, when seeded into monomeric solutions below the COC, dissolved and did so at progressively faster rates the further monomer concentrations were below the COC.46Fig. 3A exemplifies this behavior by showing the kinetics of an unseeded, RF seeded and gO/CF seeded monomer solution, with the total monomer concentration after seeding remaining below the COC. While seeding with isolated RFs induced lag-free RF elongation, seeding with isolated gO/CFs at identical concentrations caused their slow decay and increased the RF lag period compared to the unseeded sample. Moreover, we previously established that the binding protein ZAβ3, which sequesters monomeric Aβ, achieves dissolution of Aβ42 gO/CFs but not of Aβ42 RFs.65,67 Here, we used ZAβ3 as a tool to monitor the slow depletion of gO/CFs and the concomitant RF growth (Fig. 3B). When an excess of ZAβ3 is added to dimAβ assembly reactions before the second ThT upswing, fluorescence vanishes almost completely on the time scale of hours, indicating dissolution of gO/CFs at the expense of formation of the dimAβ:ZAβ3 complex. In contrast, when ZAβ3 is added after the second ThT upswing, the amplitude of the fluorescence drop progressively decreases the later ZAβ3 is added, reflecting the increasing formation of stable RFs and concomitant decrease of gO/CFs susceptible to disaggregation into the dimAβ:ZAβ3 complex (Fig. 3B). Incubation of preformed dimAβ:ZAβ3 complexes in the presence of sonicated dimAβ RF seeds resulted in the dissociation of the dimAβ:ZAβ3 complex at the expense of RF growth (Fig. S3†). Thus the dimAβ states can be ordered according to their thermodynamic stability: RF-incorporated > ZAβ3-bound > gO/CF-incorporated (Fig. 3C). This indicates substantially higher thermodynamic stability of RFs compared to gO/CFs.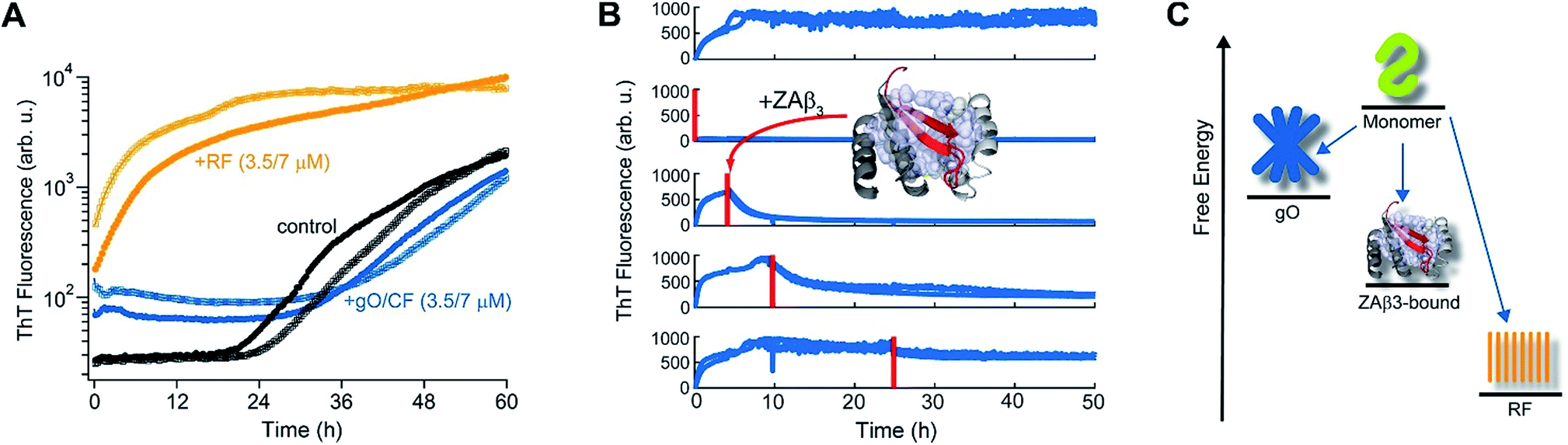 | ||
| Fig. 3 Metastability and RF seeding incompetence of gO/CFs. (A) HewL RF kinetics below the COC (38 μM hewL, 400 mM NaCl) without seeding (black), or after seeding with isolated gO/CFs (blue) or RFs (orange) at either 3.5 or 7 μM each, and incubated at pH 2, T = 52 °C. (B) ThT-detected dissociation of pre-assembled dimAβ upon addition of the binding protein ZAβ3 (grey), which sequesters monomeric dimAβ in a β-hairpin conformation (red, see ref. 65 and 67). The complex is shown in ribbon representation, hydrophobic side chains of ZAβ3 in direct contact with the Aβ40 β-hairpin are shown as spheres (Protein Data Bank entry 2OTK). The assembly of 6 μM dimAβ was monitored by ThT fluorescence, with addition of 14 μM ZAβ3 at different time points, indicated by red lines. Addition of ZAβ3 during the gO/CF formation-dominated time regime results in nearly complete loss of ThT fluorescence, while ZAβ3 addition during the RF-dominated time regime leads to a progressive decrease in the amplitude of the fluorescence drop. (C) Energy diagram illustrating the order of thermodynamic stability of different states of Aβ, above the COC and under the present experimental conditions. | ||
High reaction order of gO formation
As noted above, using the dimeric Aβ construct helped to separate the time regimes of dominant gO/CF vs. RF formation sufficiently to allow separate analysis of the gO/CF assembly kinetics (Fig. 4A and B). A global fit to the concentration-dependent time course of the initial fluorescence increase with a primary nucleation-growth model,68 assuming a common nucleus size as well as common nucleation and elongation rate constants, shows clear systematic deviations (Fig. 4A). However, it could be fit to an nth-order oligomerization reaction with a global rate constant for all concentrations (Fig. 4B). The reaction order obtained from global fits to three independent data sets was 3.3 ± 0.2, reflecting the high concentration dependence of gO/CF formation. This high reaction order explains the observability of a well-defined COC. Considering that one dimAβ molecule contains two Aβ units, the reaction order of ∼3.3 suggests an oligomer size of six to seven Aβ units, which is compatible with previous studies indicating a prominent role of hexamers in Aβ assembly.69–72 For hewL, gO/CF and RF kinetics overlapped, requiring a simultaneous fit to both gO/CF and RF growth (Fig. 4C), with the gO/CF portion better represented by first-order kinetics. The near first-order kinetics of hewL gO/CFs probably reflects the propensity of hewL gOs to assemble further into CFs (see Fig. 2E), with a simultaneous decrease in net reaction order. However, the exponential rate constant fitting the gO/CF data increased as a non-linear 3rd-order function in monomer concentration (Fig. 4C, inset), again suggesting a high reaction-order for the initial gO formation step. In both cases, the concentration dependence of gO/CF formation is much higher than the one for RF formation, for which negative scaling exponents down to only −1.7 were reported.73 These observations indicate once more that gO/CF formation occurs through a fundamentally different reaction mechanism than RF formation and rapidly becomes the initially dominant growth process above the COC.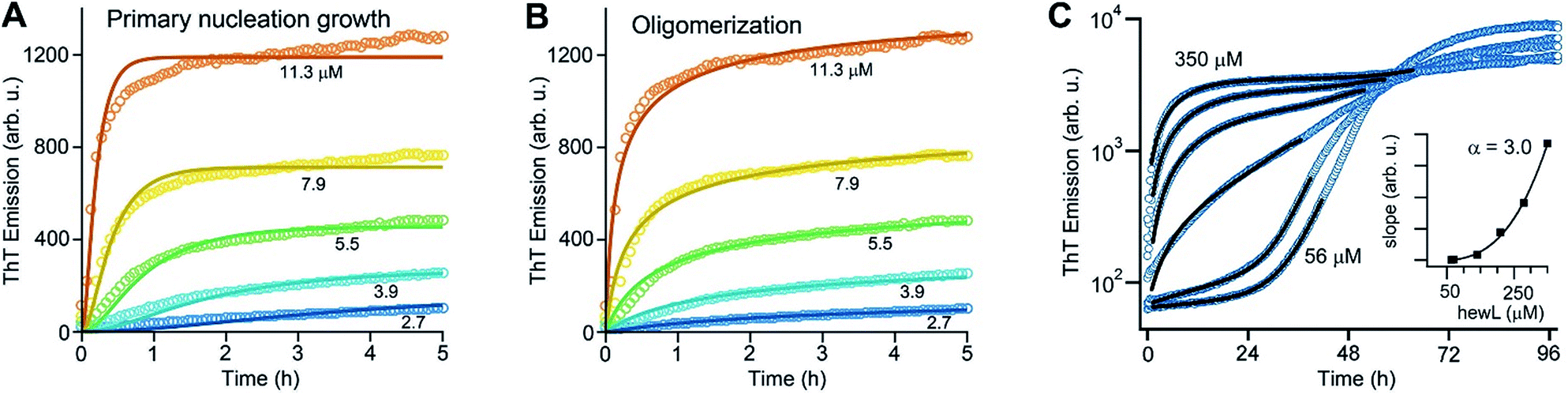 | ||
| Fig. 4 gO formation kinetics. (A, B) Concentration-dependent kinetics of gO/CF formation of dimAβ monitored by ThT fluorescence. Global fits to the data were performed using (A) a primary nucleation-growth model (eqn (2); see Materials and methods in the ESI†) or (B) a one-step oligomerization model (eqn (3)†). The reaction order obtained from the global fit to the data set in (B) was 3.4 ± 0.1. (C) Fits to the early stages of hewL ThT kinetics combining a single-exponential with the model for nucleated polymerization (eqn (4) & (1)†). The inset shows the power-law increase in the exponential growth rate required to fit the data. | ||
GO/CFs are retarding RF nucleation and growth
Crossing the COC results in a sharp transition from RF nucleation and growth in the absence of metastable gO/CFs to their concurrent growth. This provides the unique opportunity to evaluate how gO/CFs alter the mechanisms of RF nucleation and growth, while maintaining fixed solution conditions and monomer conformations. Theoretical considerations indicate that RF lag periods follow specific scaling laws as function of monomer concentration, with the magnitude of the scaling coefficients providing information about the underlying molecular mechanisms of fibril nucleation and growth.39,74 Here we investigated whether and how the presence of increasing concentrations of gO/CFs above the COC altered these scaling laws for RF nucleation and growth. It is worth mentioning that this comparison is valid irrespective of whether RFs nucleate via homogeneous or heterogeneous nucleation mechanism(s). In the absence of metastable gOs (i.e. below the COC) we fit RF kinetics with the analytical expression for nucleated polymerization with secondary mechanisms that ignores late-stage monomer depletion (early time approximation of eqn (1), see Materials and methods in the ESI† and ref. 38). Besides the quality of the individual fits (Fig. 5A), the net rate for primary and secondary nucleation extracted varied within a narrow range (Fig. 5B). The dominance of secondary over primary nucleation (Fig. 5B) reflects the highly cooperative character of RF nucleation, as already apparent from the initially completely flat ThT responses. The critical importance of autocatalytic nucleation mechanisms replicates prior observations with a multitude of amyloid proteins.38,74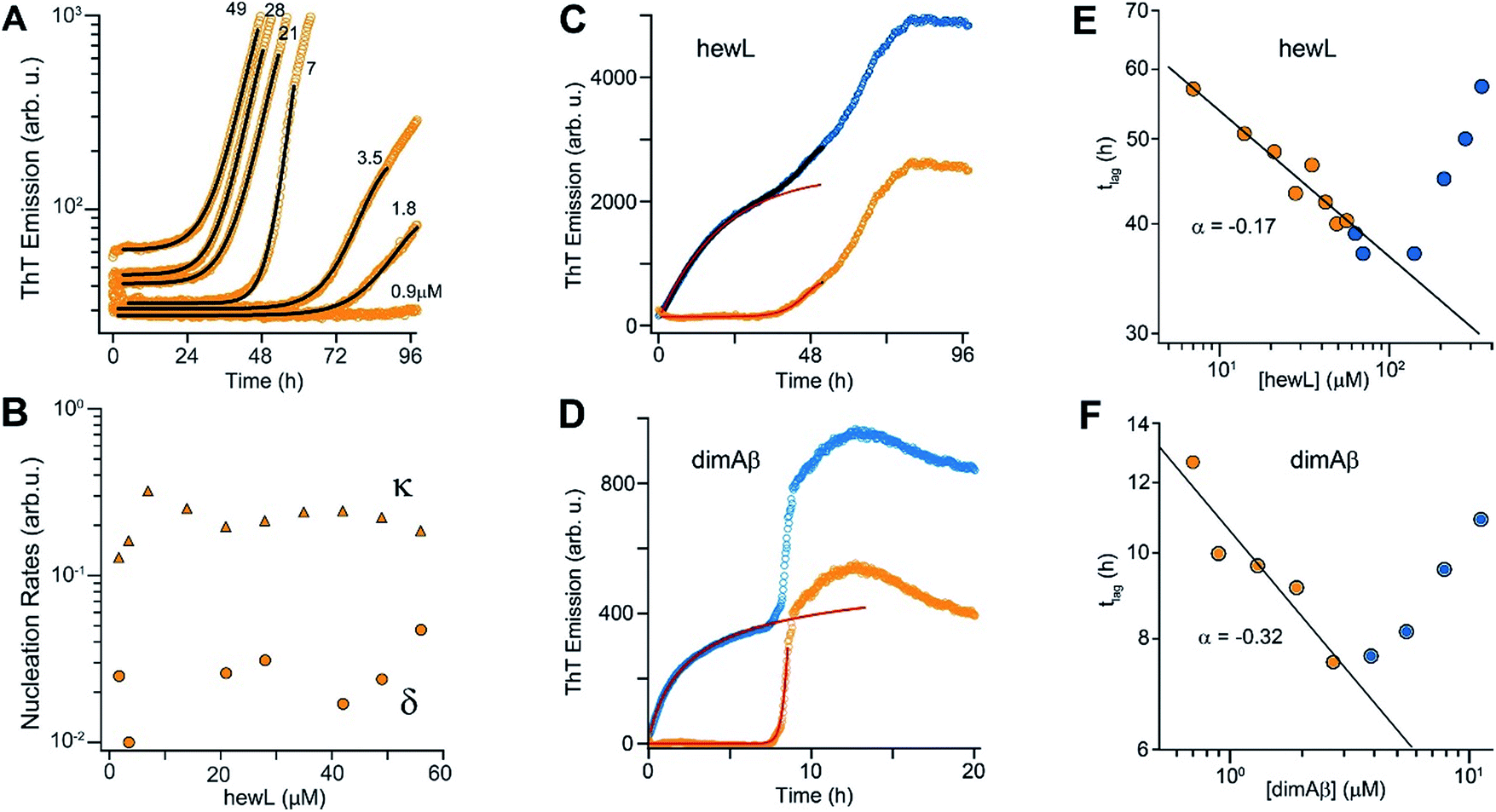 | ||
| Fig. 5 gO/CFs are retarding RF nucleation and growth. (A) Fit to ThT kinetics of hewL RFs below the COC with the analytical solution for nucleated polymerization (eqn (1)†). (B) Primary (δ) and secondary (κ) nucleation parameters obtained by the fits. (C, D) Typical ThT kinetics above the COC (blue circles), and their underlying RF kinetics (orange circles) obtained after subtracting the oligomeric portion of the fit (red line) from the data for hewL (C) and dimAβ (D). (E, F) RF lag times of hewL (E) and dimAβ (F) extracted from fits to experimental kinetics below (orange) and above (blue) the COC. The solid lines are power-law fits through these specific data sets below the COC (exponent α = −(0.17 ± 0.02) (hewL) and −(0.32 ± 0.06) (dimAβ)). Three or four, respectively, independent repeats of these measurements yielded power law values of α = −(0.24 ± 0.07) (hewL) and α = −(0.32 ± 0.05) (dimAβ). | ||
To fit RF kinetics of dimAβ above the COC, we first subtracted the global fits of the gO/CF formation time regime to an oligomerization reaction from the entire time traces (Fig. 5D). The resulting time courses represent the RF portion of the ThT signal. This treatment disregards the depletion of gO/CFs after the second ThT upswing. However, this introduces only a negligible error into the determination of lag-times, as gO/CF depletion is slow and becomes significant only long after the lag phase (see AFM images for Fig. 2B and E). Lag-times were obtained from the RF portions of the data by applying the same fits as for RF kinetics below the COC. In the case of hewL the time regimes of dominant gO/CF formation and dominant RF formation above the COC were not as clearly separated. We therefore fitted the data simultaneously to a combination of the above analytical approximation for RF growth (eqn (1)†) with a saturating exponential growth for gO/CFs (eqn (4)†). Fig. 4C indicates that the resulting fits were good when limited to the early stages of RF nucleation. Subtraction of the gO/CF component of the fits from the raw data again yielded the sigmoidal time traces expected for the RF portion of the ThT data (Fig. 5C).
Several striking features emerge from the log–log plots for RF lag time vs. protein concentration (Fig. 5E and F). The lag periods below the COC (orange) do decrease as a power law in monomer concentration, with scaling exponents yielding values of α = −(0.24 ± 0.07) and α = −(0.32 ± 0.05) for hewL and dimAβ, respectively. As recently shown, scaling exponents below a magnitude of 0.5 indicate that dock and lock steps upon monomer addition at the growing fibril end become rate-limiting of fibril elongation.39 Neglecting gO/CF formation, one would expect lag periods above the COC to continue to shorten as indicated by the extrapolation of the power law obtained below the COC (Fig. 5E and F, solid black lines). While this holds true for some data points just above the COC, rapidly increasing formation of gO/CFs above the COC not only arrests the decrease but progressively increases RF lag periods with increasing protein concentration, as shown by the blue data points. A leveling off of the lag periods would be consistent with gO/CFs as off-pathway competitors of RFs. As monomer concentrations exceed the COC, the increasingly rapid pace of gO/CF formation eventually depletes monomers down to their value at the COC. As a result RF nucleation rates from monomers would be reduced to their value at the COC. The experimentally observed rapid increase in RF lag periods, though, implies that gO/CFs actively inhibit RF nucleation beyond the capacity for off-pathway aggregates to buffer monomer concentrations.
GO/CFs act as off-pathway competitors of RFs, not as on-pathway precursors
The above changes in scaling behaviour of lag periods upon onset of oligomer formation speak to the broader question whether gO/CFs are on-pathway precursors or off-pathway competitors of RF growth. To be effective precursors of RF seed formation, gO/CFs should decrease the lag periods upon crossing the COC. Even if gO/CFs are only slowly converting to RFs, lag periods above the COC should, at best, level off. Instead, lag periods increase. Hence, nucleated conformational conversion (NCC) of the gO/CFs observed here into RF seeds is not a feasible mechanism. Our qualitative observation that AFM imaging of samples with particularly high concentrations of gO/CF failed to generate RFs for weeks further corroborates the off-pathway character of gO/CFs.As additional confirmation that hewL gO/CFs are incapable of conversion into RFs on the time scale of spontaneous RF nucleation from monomers we seeded isolated gO/CFs or RFs into solutions below the COC. As shown in Fig. 3A, controls without addition of seeds (black traces) underwent traditional nucleated polymerization. Seeding with preformed and isolated RFs (orange traces) eliminated the lag period and resulted in immediate fibril elongation. In contrast, adding identical concentrations of gO/CF seeds (blue traces) increased RF lag periods just as observed above the COC. This further supports the conclusion that gO/CFs, instead of being on-pathway for RF nucleation, retard the process of RF nucleation from monomers. The data in Fig. 3A also address the unlikely scenario that we might have missed small populations of gO/CFs already present below the COC, which in turn dominate nucleation rates under those conditions. The significant increase in the ThT baseline upon adding gO/CFs indicates that their concentrations are well above any vanishingly small levels of gO/CF present below the COC. Yet, even at those high concentrations, they do not promote RF formation. Notably, the progressively prominent inhibitory effect of gO/CF formation on RF nucleation and growth with increasing protein concentration falls outside the current versions for off-pathway oligomer formation as well.
Numerical simulations of biphasic off-pathway oligomer growth
The above data suggest that gO/CFs are off-pathway products that emerge only over the limited range of amyloid fibril growth conditions above the COC. They are non-obligatory since RFs can readily form above and below the COC. To gain a better understanding of the transition from sigmoidal to biphasic growth kinetics not just during the initial stage of RF nucleation but over the entire range of reaction time scales, we adopted the off-pathway kinetics model originally proposed by Powers and Powers,75 but modified it in two significant ways. First, off-pathway oligomer formation was limited to concentrations above the COC. In addition, we needed to include secondary nucleation mechanisms for on-pathway fibril growth in order to replicate the experimentally observed RF kinetics. Details of the numerical scheme are provided in the ESI.† An example of fitting experimental kinetics below and above the COC to simulated growth kinetics is illustrated in Fig. 6 (for further examples see Fig. S4†). This off-pathway kinetic model readily replicates the transition from sigmoidal to biphasic behavior seen in our experimental data. Equally important, it provides a natural decomposition of the ThT signal into its gO/CF and RF components. Since the current model does not include mechanisms to account for active inhibition of RFs by gO/CF formation, the parameters for RF formation rates had to be adjusted to match the individual kinetic curves.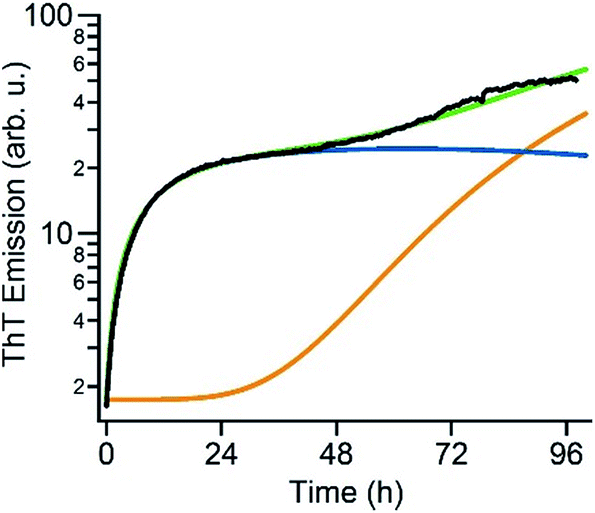 | ||
| Fig. 6 Numerical simulation of biphasic ThT kinetics. Fit of the experimental ThT kinetics for 280 μM hewL (black) to the dual-pathway assembly model (green) and corresponding decomposition into its RF (orange) and gO/CF (blue) components. | ||
Discussion
The combined kinetics and imaging data reported here show that the sharp transition in amyloid assembly from sigmoidal to biphasic kinetics coincides with the onset of gO/CF formation above a protein and solution-condition dependent COC (Fig. 2A and C). Prior reports of biphasic ThT kinetics for Aβ43,63,66 and Sup35 yeast protein42 suggest that biphasic kinetics represent a generic mode for amyloid assembly. Similarly, there are previous reports associating a micelle-like transition in Aβ40,64,76,77 amylin,78 β-microglobulin,21 and lysozyme,46 with the formation of gO/CFs. Our data indicate that biphasic kinetics represents a generic mode of amyloid assembly that is directly related to the formation of metastable gO/CF formation, and is distinct from the sigmoidal kinetics associated with nucleated polymerization of RFs from monomers. The lack of any detectable gO/CFs below the COC (Fig. 2D) and their inability to accelerate RF nucleation and growth under those conditions (Fig. 3A) indicates that metastable gO/CFs are not precursors of RFs. This distinguishes gO/CFs from any on-pathway oligomeric fibril seeds transiently formed below the COC.We presume that the commonly observed formation of gO/CFs and RFs by various amyloid proteins, and over a wide range of solution conditions, arises from two basic yet distinct features of polypeptide chains: their amphiphilic nature and their propensity to form intermolecular hydrogen bonds across their backbone. The latter is well established as the fundamental driving force underlying RF formation.79 Similarly, the amphiphilic character of polypeptide chains has been repeatedly suggested to contribute to the existence of a COC and gO formation.77,78 We have previously provided a quantitative colloidal model replicating both the salt and protein concentration dependence of the COC of hewL.46 The available structural data suggest that gOs are short anti-parallel β-barrels,80 clearly distinct from the steric zipper structure of RFs. GO/CFs arising from amphiphilic phase separation vs. RFs via intermolecular hydrogen bonding also rationalizes the existence of two distinct aggregate species formed along separate assembly pathways. Furthermore, it explains why they have distinct morphologies and mechanical rigidities (sterically constrained gOs polymerizing into weakly-linked CFs vs. mechanically rigid hydrogen-linked RFs), their relative thermodynamic stabilities (weaker linked gO/CFs vs. strongly bonded RFs), the distinct reaction orders of their formation (as reported here), their dependence on various solution parameters such as pH, ionic strength, specific ion effects, and of course, their dependence on the primary sequence of the polypeptide chain itself.
Recently, it has been proposed that the switch from RFs to gO/CFs in β-lactoglobulin at strongly hydrolyzing conditions (90 °C, pH 2) results from concentration-dependent changes to the distributions of hydrolyzed fragments.81 It is possible that different peptides do undergo either gO/CF or RF formation at identical solution conditions. For our hewL assembly conditions (52 °C, pH 2) we have shown that the switch from RF to gO/CF formation can be readily induced by increasing salt at fixed protein concentrations (see horizontal lines in Fig. 1B),46 while hydrolysis rates across the COC were unchanged (Fig. 4 in ref. 54). Here we show that pre-hydrolyzing hewL for multiple days has no discernible effect on RF kinetics which, due to its long lag periods, would be most susceptible to hydrolysis (see Fig. S5†). Hence, we believe hydrolysis is not fundamental to the switch from RF to gO/CF formation.
The sharp transition from “oligomer-free” to “oligomeric” RF growth upon crossing the COC allowed us to address a long-standing question: what role do metastable gO/CFs play in the nucleation–polymerization of RFs? Qualitatively, NCC fits well with the lag-free emergence of gO/CFs and the biphasic kinetics for gO/CF growth with late-stage RF nucleation and growth. However, the inability of gO/CFs to seed RF growth below the COC (Fig. 3A) and their inhibitory effects on RF formation above the COC (Fig. 5E and F) are inconsistent with NCC. We note, however, that protein-specific factors such as the structure of the protein undergoing assembly or the size and structure of its oligomers might affect whether NCC is a feasible RF nucleation mechanism under other conditions. Large liquid-like droplets have been reported to arise during polyQ fibril assembly or β2 microglobulin disassembly.82,83 Hence, there remains uncertainty whether there are multiple, potentially distinct metastable states, and how those might be related to each other and to late-stage fibrils.
Extrapolating NP scaling laws, established below the COC, to monomer concentrations above the COC allowed us to quantify how gO/CF formation altered NP. Crossing the COC, the scaling law for lag periods with monomer concentration initially developed a kink. Kinks in the scaling behavior of RF lag periods, albeit to a lesser degree, have been reported for insulin, bovine serum albumin and lysozyme.39,41,84 Even inverse relationships between half-time and/or lag-time of amyloid fibril growth have been observed before for the light chain variable domain LEN,85 ribosomal protein S6,86 glucagon-like peptide-1,31 and Aβ66 and have been explained with competing off-pathway aggregation. A model for competing off-pathway aggregation by Powers and Powers75 did generate an inverse relationship between half-time for reaction completion (but not lag-time) of amyloid formation at high protein concentration, due to monomer depletion by off-pathway precipitates. Importantly, though, COC-limited monomer depletion can only explain an arrest but not the increase in the RF lag-times reported here. The identified active inhibition of RF formation by gO/CFs necessitates modification of the standard cNP model (Fig. 7).
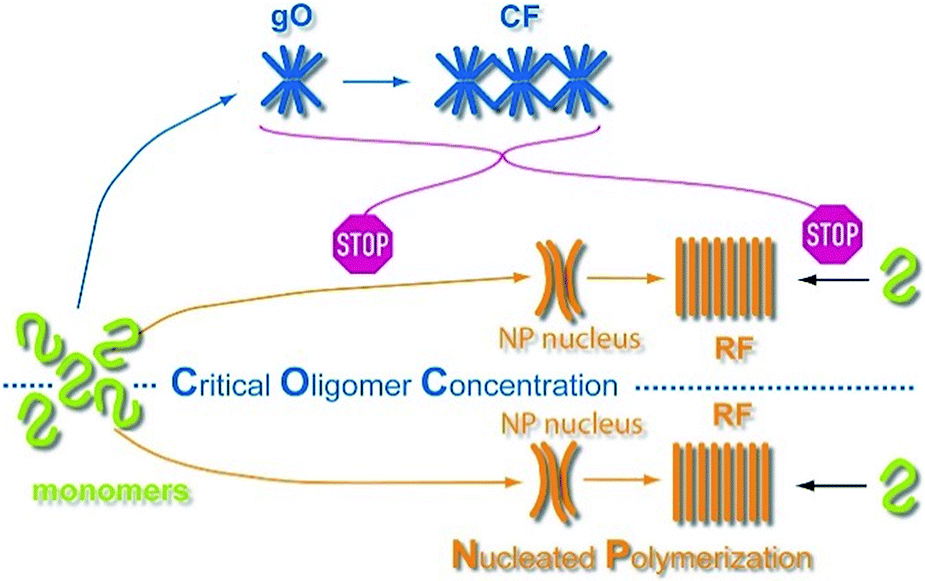 | ||
| Fig. 7 Scheme of amyloid oligomer and fibril formation. GO/CFs are metastable off-pathway assemblies formed to a significant extent above the COC. GO/CFs inhibit RF formation not only by buffering monomer concentration to the COC level, thereby slowing fibril nucleation and elongation, but also by actively disrupting either process (STOP signs). The precise mechanism of this active inhibition remains to be elucidated. | ||
The COCs observed here are relatively high compared to typical in vivo protein concentrations. For example, dimAβ, although showing increased oligomerization propensity due to covalent linkage of two Aβ units, has a COC of the order of 1 μM (in low salt buffer at neutral pH), whereas physiological Aβ concentrations are in the nanomolar range.87 Similar discrepancies, though, exist for Aβ fibril solubilities, with the latter ranging from hundreds of nanomolar to millimolars. It is important to realize in this context that there are multiple factors that can significantly reduce COCs or increase local protein concentrations in vivo. These include salt concentration and pH (see Fig. 1B) (e.g., accumulation of micromolar concentrations of Aβ in acidic vesicles87); interfaces, such as membrane or fibril surfaces;88 post-translational modifications such as cross-links;30 macromolecular crowding;89 and disease-related mutations, some of which strongly promote gO/CF formation.17,90,91 Moreover, interactions with other cellular components may affect gO/CF formation. For example, interaction with another aggregation-prone protein, TDP-43, was shown to strongly promote gO/CF formation of Aβ at the expense of RF formation.92 The relatively high COCs observed for the two model systems therefore likely result from the present experimental conditions. Interestingly, reduced expression of APP in mouse models that did not affect plaque load led to reduced formation of plaque-unrelated oligomers, suggesting that the concentration dependence of gO/CF formation has a correlate in vivo.93,94
The above results paint a complex picture of how RF formation proceeds in the presence of gO/CFs. The mutual interactions among gO/CFs and RFs, either direct or via the monomer pool, are likely to feature prominently in the temporal evolution of gO/CFs vs. RF populations in vivo, as well. There are solid indications that gO/CFs and RFs have distinct biological/pathological activity profiles that might vary with protein identity, its growth conditions, and its cellular environment.20,27,57,95–98 The retarding effects of early-stage gOs on the nucleation and growth of late-stage RFs might result in extended exposure to toxic oligomers and low rates of RF formation and gO/CF depletion in vivo. This provides one possible explanation for the paradoxical observations that post-mortem RF loads correlate poorly with the severity of clinical symptoms.99,100 Overall, systematic investigation of the thermodynamic and kinetic factors regulating the assembly of distinct amyloid species in vitro and their mutual interplay can provide important insights into the mechanisms regulating amyloid assembly in vivo.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NIH grant 2R15GM097723-02 (M. M.), NIH grant R01AG053988 (G. U.), ERC Consolidator grant 726368 (W. H.), Alzheimer Forschungs Initiative (W. H.), and Sidney Sussex College Cambridge (G. Me.). Access to the Jülich-Düsseldorf Biomolecular NMR Center is acknowledged.Notes and references
- E. H. Koo, J. Lansbury, P. T. Lansbury Jr and J. W. Kelly, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9989–9990 CrossRef.
- C. A. Ross and M. A. Poirier, Nat. Med., 2004, 10, S10–S17 CrossRef PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef PubMed.
- M. Stefani, Biochim. Biophys. Acta, 2004, 1739, 5–25 CrossRef PubMed.
- M. Sunde and C. F. F. Blake, Q. Rev. Biophys., 1998, 31, 1–39 CrossRef PubMed.
- J. N. Buxbaum, Curr. Opin. Rheumatol., 2004, 16, 67–75 CrossRef PubMed.
- L. M. Blancas-Mejia and M. Ramirez-Alvarado, Annu. Rev. Biochem., 2013, 82, 745–774 CrossRef PubMed.
- D. Eisenberg and M. Jucker, Cell, 2012, 148, 1188–1203 CrossRef PubMed.
- M. B. Pepys, P. N. Hawkins, D. R. Booth, D. M. Vigushin, G. A. Tennent, A. K. Soutar, N. Totty, O. Nguyen, C. C. F. Blake, C. J. Terry, T. G. Feest, A. M. Zalin and J. J. Hsuan, Nature, 1993, 362, 553–557 CrossRef PubMed.
- D. M. Fowler, A. V. Koulov, W. E. Balch and J. W. Kelly, Trends Biochem. Sci., 2007, 32, 217–224 CrossRef PubMed.
- I. Benilova, E. Karran and B. De Strooper, Nat. Neurosci., 2012, 15, 349–357 CrossRef PubMed.
- R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman and C. G. Glabe, Science, 2003, 300, 486–489 CrossRef PubMed.
- R. Kodali and R. Wetzel, Curr. Opin. Struct. Biol., 2007, 17, 48–57 CrossRef PubMed.
- D. M. Walsh, A. Lomakin, G. B. Benedek, M. M. Condron and D. B. Teplow, J. Biol. Chem., 1997, 272, 22364–22372 CrossRef PubMed.
- H. A. Lashuel, C. Wurth, L. Woo and J. W. Kelly, Biochemistry, 1999, 38, 13560–13573 CrossRef PubMed.
- Y. E. Kim, F. Hosp, F. Frottin, H. Ge, M. Mann, M. Hayer-Hartl and F. U. Hartl, Mol. Cell, 2016, 63, 951–964 CrossRef PubMed.
- K. A. Conway, S.-J. Lee, J.-C. Rochet, T. T. Ding, R. E. Williamson and P. T. Lansbury, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 571–576 CrossRef.
- C. A. Lasagna-Reeves, D. L. Castillo-Carranza, M. J. Guerrero-Muñoz, G. R. Jackson and R. Kayed, Biochemistry, 2010, 49, 10039–10041 CrossRef PubMed.
- V. Novitskaya, O. V. Bocharova, I. Bronstein and I. V. Baskakov, J. Biol. Chem., 2006, 281, 13828–13836 CrossRef PubMed.
- W. B. Stine Jr, K. N. Dahlgren, G. A. Krafft and M. J. LaDu, J. Biol. Chem., 2003, 278, 11612–11622 CrossRef PubMed.
- W. S. Gosal, I. J. Morten, E. W. Hewitt, D. A. Smith, N. H. Thompson and S. E. Radford, J. Mol. Biol., 2005, 351, 850–864 CrossRef PubMed.
- R. Kayed, Y. Sokolov, B. Edmonds, T. M. McIntire, S. C. Milton, J. E. Hall and C. G. Glabe, J. Biol. Chem., 2004, 279, 46363–46366 CrossRef PubMed.
- M. N. N. Vieira, L. Forny-Germano, L. M. Saraiva, A. Sebollela, A. M. B. Martinez, J.-C. Houzel, F. G. De Felice and S. T. Ferreira, J. Neurochem., 2007, 103, 736–748 CrossRef PubMed.
- K. Ono, M. M. Condron and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14745–14750 CrossRef PubMed.
- L. V. Kalia, S. K. Kalia, P. J. McLean, A. M. Lozano and A. E. Lang, Ann. Neurol., 2013, 73, 155–169 CrossRef PubMed.
- K. N. Dahlgren, A. M. Manelli, W. B. Stine, J. Baker, K. Lorinda, G. A. Krafft and M. J. LaDu, J. Biol. Chem., 2002, 277, 36046–36053 CrossRef PubMed.
- M. Malisauskas, A. Darinskas, V. Zamotin, A. Gharibyan, I. Kostanyan and L. Morozova-Roche, Biochemistry, 2006, 71, 505–512 Search PubMed.
- M. P. Lambert, A. K. Barlow, B. A. Chromy, C. Edwards, R. Freed, M. Liosatos, T. E. Morgan, I. Rozovsky, B. Trommer, K. L. Viola, P. Wals, C. Zhang, C. E. Finch, G. A. Krafft and W. L. Klein, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6448–6453 CrossRef.
- R. Kayed and C. A. Lasagna-Reeves, J. Alzheimer's Dis., 2013, 33, S67–S78 Search PubMed.
- B. O'Nuallain, D. B. Freir, A. J. Nicoll, E. Risse, N. Ferguson, C. E. Herron, J. Collinge and D. M. Walsh, J. Neurosci., 2010, 30, 14411–14419 CrossRef PubMed.
- K. L. Zapadka, F. J. Becher, S. Uddin, P. G. Varley, S. Bishop, A. L. Gomes dos Santos and S. E. Jackson, J. Am. Chem. Soc., 2016, 138, 16259–16265 CrossRef PubMed.
- A. L. Fink, Folding Des., 1998, 3, R9–R23 CrossRef PubMed.
- M. Muschol and F. Rosenberger, J. Chem. Phys., 1997, 107, 1953–1962 CrossRef.
- S. M. Vaiana, M. B. Palma-Vittorelli and M. U. Palma, Proteins: Struct., Funct., Bioinf., 2003, 51, 147–153 CrossRef PubMed.
- A. Patel, H. O. Lee, L. Jawerth, S. Maharana, M. Jahnel, M. Y. Hein, S. Stoynov, J. Mahamid, S. Saha, T. M. Franzmann, A. Pozniakovski, I. Poser, N. Maghelli, L. A. Royer, M. Weigert, E. W. Myers, S. Grill, D. Drechsel, A. A. Hyman and S. Alberti, Cell, 2015, 162, 1066–1077 CrossRef PubMed.
- L. Wei, P. Jiang, W. Xu, H. Li, H. Zhang, L. Yan, M. B. Chan-Park, X.-W. Liu, K. Tang, Y. Mu and K. Pervushin, J. Biol. Chem., 2011, 286, 6291–6300 CrossRef PubMed.
- C. Bleiholder, N. F. Dupuis, T. Wyttenbach and M. T. Bowers, Nat. Chem., 2010, 3, 172 CrossRef PubMed.
- T. P. J. Knowles, C. A. Waudby, G. L. Devlin, S. I. A. Cohen, A. Aguzzi, M. Vendruscolo, E. M. Terentjev, M. E. Welland and C. M. Dobson, Science, 2009, 326, 1533–1537 CrossRef PubMed.
- G. Meisl, J. B. Kirkegaard, P. Arosio, T. C. T. Michaels, M. Vendruscolo, C. M. Dobson, S. Linse and T. P. J. Knowles, Nat. Protoc., 2016, 11, 252–272 CrossRef PubMed.
- G. Meisl, X. Yang, B. Frohm, T. P. J. Knowles and S. Linse, Sci. Rep., 2016, 6, 18728 CrossRef PubMed.
- G. Meisl, L. Rajah, S. A. I. Cohen, M. Pfammatter, A. Saric, E. Hellstrand, A. K. Buell, A. Aguzzi, S. Linse, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Chem. Sci., 2017, 8, 7087–7097 RSC.
- T. R. Serio, A. G. Cashikar, A. S. Kowal, G. J. Sawicki, J. J. Moslehi, L. Serpell, M. F. Arnsdorf and S. L. Lindquist, Science, 2000, 289, 1317–1321 CrossRef PubMed.
- J. Lee, E. K. Culyba, E. T. Powers and J. W. Kelly, Nat. Chem. Biol., 2011, 7, 602–609 CrossRef PubMed.
- M. Necula, R. Kayed, S. Milton and C. G. Glabe, J. Biol. Chem., 2007, 282, 10311–10324 CrossRef PubMed.
- S. Matsumura, K. Shinoda, M. Yamada, S. Yokojima, M. Inoue, T. Ohnishi, T. Shimada, K. Kikuchi, D. Masui, S. Hashimoto, M. Sato, A. Ito, M. Akioka, S. Takagi, Y. Nakamura, K. Nemoto, Y. Hasegawa, H. Takamoto, H. Inoue, S. Nakamura, Y.-i. Nabeshima, D. B. Teplow, M. Kinjo and M. Hoshi, J. Biol. Chem., 2011, 286, 11555–11562 CrossRef PubMed.
- T. Miti, M. Mulaj, J. D. Schmit and M. Muschol, Biomacromolecules, 2015, 16, 326–335 CrossRef PubMed.
- J. N. Israelachvili, in Intermolecular and Surface Forces, Academic Press, Burlington, MA, 3rd edn, 2011, ch. 19 Search PubMed.
- M. Adachi, M. So, K. Sakurai, J. Kardos and Y. Goto, J. Biol. Chem., 2015, 290, 18134–18145 CrossRef PubMed.
- T. R. Jahn and S. E. Radford, Arch. Biochem. Biophys., 2008, 469, 100–117 CrossRef PubMed.
- S. E. Hill, T. Miti, T. Richmond and M. Muschol, PLoS One, 2011, 6, e18171 CrossRef PubMed.
- S. E. Hill, J. Robinson, G. Matthews and M. Muschol, Biophys. J., 2009, 96, 3781–3790 CrossRef PubMed.
- D.-P. Hong, A. L. Fink and V. N. Uversky, J. Mol. Biol., 2008, 383, 214–223 CrossRef PubMed.
- R. H. Pires, Á. Karsai, M. J. Saraiva, A. M. Damas and M. S. Z. Kellermayer, PLoS One, 2012, 7, e44992 CrossRef PubMed.
- J. Foley, S. E. Hill, T. Miti, M. Mulaj, M. Ciesla, R. Robeel, C. Persichilli, R. Raynes, S. Westerheide and M. Muschol, J. Chem. Phys., 2013, 139, 121901–121912 CrossRef PubMed.
- G. Zandomeneghi, M. R. H. Krebs, M. G. McCammon and M. Fändrich, Protein Sci., 2004, 13, 3314–3321 CrossRef PubMed.
- E. Cerf, R. Sarroukh, S. Tamamizu-Kato, L. Breydo, S. Derclaye, Y. F. Dufrêne, V. Narayanaswami, E. Goormaghtigh, J.-M. Ruysschaert and V. Raussens, Biochem. J., 2009, 421, 415–423 CrossRef PubMed.
- A. Laganowsky, C. Liu, M. R. Sawaya, J. P. Whitelegge, J. Park, M. Zhao, A. Pensalfini, A. B. Soriaga, M. Landau, P. K. Teng, D. Cascio, C. Glabe and D. Eisenberg, Science, 2012, 335, 1228–1231 CrossRef PubMed.
- M. I. Apostol, K. Perry and W. K. Surewicz, J. Am. Chem. Soc., 2013, 135, 10202–10205 CrossRef PubMed.
- U. Sengupta, A. N. Nilson and R. Kayed, Exp. Biol. Med., 2016, 6, 42–49 Search PubMed.
- T. T. O'Malley, N. A. Oktaviani, D. Zhang, A. Lomakin, B. O'Nuallain, S. Linse, G. B. Benedek, M. J. Rowan, F. A. A. Mulder and D. M. Walsh, Biochem. J., 2014, 461, 413–426 CrossRef PubMed.
- T. T. O'Malley, W. M. Witbold, S. Linse and D. M. Walsh, Biochemistry, 2016, 55, 6150–6161 CrossRef PubMed.
- T. Yamaguchi, H. Yagi, Y. Goto, K. Matsuzaki and M. Hoshino, Biochemistry, 2010, 49, 7100–7107 CrossRef PubMed.
- Z. Fu, D. Aucoin, J. Davis, W. E. Van Nostrand and S. O. Smith, Biochemistry, 2015, 54, 4197–4207 CrossRef PubMed.
- A. Lomakin, D. S. Chung, G. B. Benedek, D. A. Kirschner and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 1125–1129 CrossRef.
- L. M. Luheshi, W. Hoyer, T. P. de Barros, I. van Dijk Härd, A.-C. Brorsson, B. Macao, C. Persson, D. C. Crowther, D. A. Lomas, S. Ståhl, C. M. Dobson and T. Härd, PLoS Biol., 2010, 8, e1000334 CrossRef PubMed.
- M. Nick, Y. Wu, N. W. Schmidt, S. B. Prusiner, J. Stöhr and W. F. DeGrado, Biopolymers, 2018, e23096 CrossRef PubMed.
- C. S. R. Grüning, S. Klinker, M. Wolff, M. Schneider, K. Toksöz, A. N. Klein, L. Nagel-Steger, D. Willbold and W. Hoyer, J. Biol. Chem., 2013, 288, 37104–37111 CrossRef PubMed.
- S. I. A. Cohen, M. Vendruscolo, M. E. Welland, C. M. Dobson, E. M. Terentjev and T. P. J. Knowles, J. Chem. Phys., 2011, 135, 065105 CrossRef PubMed.
- R. Roychaudhuri, M. Yang, M. M. Hoshi and D. B. Teplow, J. Biol. Chem., 2009, 284, 4749–4753 CrossRef PubMed.
- S. L. Bernstein, N. F. Dupuis, N. D. Lazo, T. Wyttenbach, M. M. Condron, G. Bitan, D. B. Teplow, J.-E. Shea, B. T. Ruotolo, C. V. Robinson and M. T. Bowers, Nat. Chem., 2009, 1, 326–331 CrossRef PubMed.
- M. Wolff, B. Zhang-Haagen, C. Decker, B. Barz, M. Schneider, R. Biehl, A. Radulescu, B. Strodel, D. Willbold and L. Nagel-Steger, Sci. Rep., 2017, 7, 2493 CrossRef PubMed.
- M. Cernescu, T. Stark, E. Kalden, C. Kurz, K. Leuner, T. Deller, M. Göbel, G. P. Eckert and B. Brutschy, Anal. Chem., 2012, 84, 5276–5284 CrossRef PubMed.
- G. Meisl, X. Yang, C. M. Dobson, S. Linse and T. P. J. Knowles, Chem. Sci., 2017, 8, 4352–4362 RSC.
- S. I. A. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9758–9763 CrossRef PubMed.
- E. T. Powers and D. L. Powers, Biophys. J., 2008, 94, 379–391 CrossRef PubMed.
- R. Sabaté and J. Estelrich, J. Phys. Chem. B, 2005, 109, 11027–11032 CrossRef PubMed.
- B. Soreghan, J. Kosmoski and C. Glabe, J. Biol. Chem., 1994, 269, 28551–28554 Search PubMed.
- E. Rhoades and A. Gafni, Biophys. J., 2003, 84, 3480–3487 CrossRef PubMed.
- M. Fändrich and C. M. Dobson, EMBO J., 2002, 21, 5682–5690 CrossRef.
- J. C. Stroud, C. Liu, P. K. Teng and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7717–7722 CrossRef PubMed.
- X. Ye, M. S. Hedenqvist, M. Langton and C. Lendel, RSC Adv., 2018, 8, 6915–6924 RSC.
- S. L. Crick, K. M. Ruff, K. Garai, C. Frieden and R. V. Pappu, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20075–20080 CrossRef PubMed.
- K. W. Tipping, T. K. Karamanos, T. Jakhria, M. G. Iadanza, S. C. Goodchild, R. Tuma, N. A. Ranson, E. W. Hewitt and S. E. Radford, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5691–5696 CrossRef PubMed.
- K. Eden, R. Morris, J. Gillam, C. E. MacPhee and R. J. Allen, Biophys. J., 2015, 108, 632–643 CrossRef PubMed.
- P. O. Souillac, V. N. Uversky, I. S. Millett, R. Khurana, S. Doniach and A. L. Fink, J. Biol. Chem., 2002, 277, 12666–12679 CrossRef PubMed.
- T. Deva, N. Lorenzen, B. S. Vad, S. V. Petersen, I. Thørgersen, J. J. Enghild, T. Kristensen and D. E. Otzen, Biochim. Biophys. Acta, Proteins Proteomics, 2013, 1834, 677–687 CrossRef PubMed.
- X. Hu, S. L. Crick, G. Bu, C. Frieden, R. V. Pappu and J.-M. Lee, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20324–20329 CrossRef PubMed.
- A. Quist, I. Doudevski, H. Lin, R. Azimova, D. Ng, B. Frangione, B. Kagan, J. Ghiso and R. Lal, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10427–10432 CrossRef PubMed.
- L. Huang, R. Jin, J. Li, K. Luo, T. Huang, D. Wu, W. Wang, R. Chen and G. Xiao, FASEB J., 2010, 24, 3536–3543 CrossRef PubMed.
- C. Nilsberth, A. Westlind-Danielsson, C. B. Eckman, M. M. Condron, K. Axelman, C. Forsell, C. Stenh, J. Luthman, D. B. Teplow, S. G. Younkin, J. Naslund and L. Lannfelt, Nat. Neurosci., 2001, 4, 887–893 CrossRef PubMed.
- T. Tomiyama, T. Nagata, H. Shimada, R. Teraoka, A. Fukushima, H. Kanemitsu, H. Takuma, R. Kuwano, M. Imagawa, S. Ataka, Y. Wada, E. Yoshioka, T. Nishizaki, Y. Watanabe and H. Mori, Ann. Neurol., 2008, 63, 377–387 CrossRef PubMed.
- Y.-S. Fang, K.-J. Tsai, Y.-J. Chang, P. Kao, R. Woods, P.-H. Kuo, C.-C. Wu, J.-Y. Liao, S.-C. Chou, V. Lin, L.-W. Jin, H. S. Yuan, I. H. Cheng, P.-H. Tu and Y.-R. Chen, Nat. Commun., 2014, 5, 4824 CrossRef PubMed.
- S. W. Fowler, A. C. A. Chiang, R. R. Savjani, M. E. Larson, M. A. Sherman, D. R. Schuler, J. R. Cirrito, S. E. Lesné and J. L. Jankowsky, J. Neurosci., 2014, 34, 7871–7885 CrossRef PubMed.
- P. Liu, M. N. Reed, L. A. Kotilinek, M. K. O. Grant, C. L. Forster, W. Qiang, S. L. Shapiro, J. H. Reichl, A. C. A. Chiang, J. L. Jankowsky, C. M. Wilmot, J. P. Cleary, K. R. Zahs and K. H. Ashe, Cell Rep., 2015, 11, 1760–1771 CrossRef PubMed.
- W. L. Klein, G. A. Krafft and C. E. Finch, Trends Neurosci., 2001, 24, 219–224 CrossRef PubMed.
- M. F. Mossuto, A. Dhulesia, G. Devlin, E. Frare, J. R. Kumita, P. P. de Laureto, M. Dumoulin, A. Fontana, C. M. Dobson and X. Salvatella, J. Mol. Biol., 2010, 402, 783–796 CrossRef PubMed.
- N. Reixach, S. Deechongkit, X. Jiang, J. W. Kelly and J. N. Buxbaum, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 2817–2822 CrossRef PubMed.
- W.-F. Xue, A. L. Hellewell, W. S. Gosal, S. W. Homans, E. W. Hewitt and S. E. Radford, J. Biol. Chem., 2009, 284, 34272–34282 CrossRef PubMed.
- D. W. Dickson, Neurobiol. Aging, 1997, 18, S21–S26 CrossRef PubMed.
- B. T. Hyman, Neurobiol. Aging, 1997, 18, S27–S32 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01479e |
| ‡ Equal contributions, joint first authors. |
| This journal is © The Royal Society of Chemistry 2018 |