
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structurally characterized terminal manganese(IV) oxo tris(alkoxide) complex†
Robert L.
Halbach
,
David
Gygi
,
Eric D.
Bloch‡
,
Bryce L.
Anderson
and
Daniel G.
Nocera
 *
*
Department of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, USA. E-mail: dnocera@fas.harvard.edu
First published on 26th April 2018
Abstract
A Mn(IV) complex featuring a terminal oxo ligand, [MnIV(O)(ditox)3][K(15-C-5)2] (3; ditox = tBu2MeCO−, 15-C-5 = 15-crown-5-ether) has been isolated and structurally characterized. Treatment of the colorless precursor [MnII(ditox)3][K(15-C-5)2] (2) with iodosobenzene affords 3 as a green free-flowing powder in high yields. The X-ray crystal structure of 3 reveals a pseudotetrahedral geometry about the central Mn, which features a terminal oxo (d(Mn–Oterm = 1.628(2) Å)). EPR spectroscopy, SQUID magnetometry, and Evans method magnetic susceptibility indicate that 3 consists of a high-spin S = 3/2 Mn(IV) metal center. 3 promotes C–H bond activation by a hydrogen atom abstraction. The [MnIV(O)(ditox)3]− furnishes a model for the proposed terminal oxo of the unique manganese of the oxygen evolving complex of photosystem II.
High valent metal oxos are a linchpin for manipulating the O–O bond in energy conversion transformations involving the oxygen reduction reaction (ORR) and its reverse process, the oxygen evolution reaction (OER).1,2 For metals left of the oxo wall,3 a terminal metal oxo is often invoked as the primary product of O–O bond cleavage of the ORR, and conversely, O–O bond formation of the OER is often proposed to proceed from a high-valent metal oxo. In the OER conversion of photosystem II, the “dangler” manganese,4 of the manganese oxygen evolving complex (OEC) is proposed to support a terminal metal oxo in the high valent S4 state of the Kok cycle.5–12 Most high valent manganese oxos are proposed as transient intermediates in oxidation reactions.13–16 In this regard, the stabilization of the terminal Mn-oxo moiety by hydrogen bonding within the trigonal ligand field of [buea]3− (tris[(N′-tert-butylureaylato)-N-ethylene]aminato) is notable for its elegant and concise design17,18 as is the stabilization of a terminal oxo in tetragonal ligand fields of nitrogen-donating macrocycles.19 Nonetheless, in any ligand field, isolation and structural characterization of high valent Mn oxos remain elusive with the peculiarity that oxos in their higher formal oxidation state of Mn(V) are better structurally characterized19–22 than their Mn(IV) counterparts.
The reactivity of the metal-oxo functional group is largely governed by the coordination geometry about the metal center. The preponderance of metal-oxo complexes, including that for Mn,19–23 feature a pseudo-octahedral or tetragonal ligand field,24 beginning with the early description of the electronic structure of the vanadyl oxo.25 When metals have higher d electron counts, the oxo is better stabilized in the more uncommon trigonal ligand field,26,27 as demonstrated by the C3 symmetry of the [H3buea]3− ligand.28 We have explored the exceptionally weak trigonal ligand field engendered by the tris(alkoxide) platform of ditox (Hditox = tBu2MeCOH)29 to support the metal-oxo unit.30,31 The steric bulk of the ditox ligand is less than that of the related tritox32,33 ligand as a result of the replacement of one tert-butyl group for one methyl group. The ditox ligand allows for the preferential formation of tris(alkoxide) metal complexes, which readily accommodate a terminal-oxo in a pseudotetrahedral ligand field upon oxygen-atom transfer, as has been demonstrated for a series of 3d transition metals (M = V, Cr, Fe).30,31 Herein, we show that a trigonal ligand field of the ditox ligand can support a Mn(IV) terminal oxo. The isolation, structural characterization and reactivity of [Mn(O)(ditox)3][K(15-C-5)2] (3) provides a benchmark for Mn(IV) in an exclusive oxygen ligand field, as is the case for the Mn(IV) centers of the oxygen evolving complex (OEC) of photosystem II.
The preparative reaction chemistry utilized to obtain Mn(II) and Mn(IV)-oxo compounds in the tris(ditox) environment is outlined in Fig. 1. The addition of three equivalents of K(ditox) to MnCl2 in THF at ambient temperature readily furnishes a colorless solid and a faint yellow solution of MnII(ditox)3K(THF)2 (1). Filtration, removal of solvent in vacuo, and crystallization from pentane at −40 °C gives analytically pure colorless crystals of 1 in 54% yield. The potassium salt, [MnII(ditox)3][K(15-C-5)2] (2), is prepared by treatment of a pentane solution of 1 with two equivalents of 15-C-5 at room temperature, quantitatively furnishing 2 as a colorless fine powder. Crystals of 2 suitable for X-ray diffraction can be obtained from a concentrated Et2O solution cooled at −40 °C overnight. Treating 2 with one equivalent of PhIO in THF at room temperature rapidly generates a dark green solution of [MnIV(O)(ditox)3][K(15-C-5)2] (3). Filtration, removal of solvent in vacuo, and multiple pentane washings of the resultant solid furnishes 3 as an intensely dark green powder in 79% yield following drying in vacuo. Solutions of 3 stored under an inert atmosphere at ambient temperature change from dark green to brown over the course of a few hours, indicating the decomposition of 3, which can be stored indefinitely as a solid in the dark at −40 °C. The negative mode ESI-MS spectrum of 3 (Fig. S1†) exhibits a prominent ion peak at m/z 542.4 with mass and isotopic distribution patterns corresponding to [MnIV(O)(diox)3]− (calcd m/z 542.4). A mass shift from m/z 542.4 to 544.4 was observed when 3 was generated using 18O-enriched PhIO (46% 16O: 54% 18O). Crystals of 3 suitable for X-ray diffraction can be obtained from a concentrated Et2O solution of 3 cooled overnight at −40 °C.
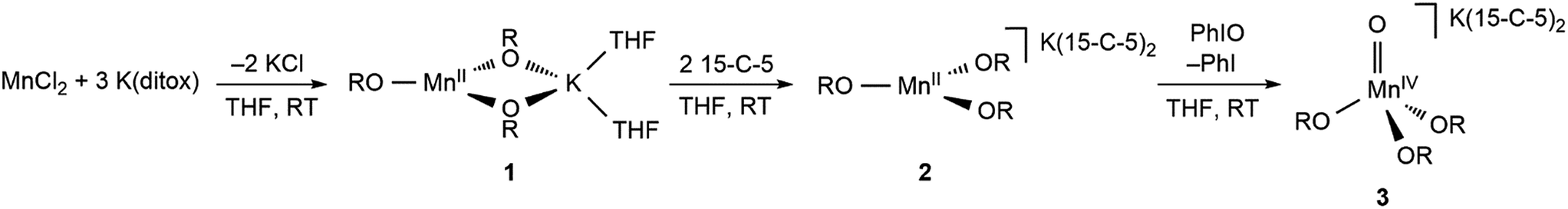 | ||
| Fig. 1 Preparative reactions of Mn ditox compounds, where OR = ditox and 15-C-5 = 15-crown-5-ether. | ||
Single crystal X-ray diffraction studies were performed on compounds 1–3. Details of the data collection are provided in Table S1† and X-ray crystal structures are shown in Fig. 2 and in Fig. S2–S4,† which also provide selected bond lengths and angles for the complexes. Compound 1 crystallizes in the monoclinic space group C2c and exhibits a Y-shaped geometry about manganese with the potassium cation coordinated directly to two ditox ligands (Fig. 2). The average Mn–Oalk bond distance is 1.941 ± 0.052 Å. The sum of the Oalk–Mn–Oalk bond angles of 1 is 359.2°, highlighting the planar nature of the compound and indicating that the manganese center resides in the plane of the alkoxides. However, the O(1)–Mn(1)–O(1A) bond angle of 97.64(8)° in 1 is significantly contracted relative to the two other O–Mn–O angles in the trigonal plane. One ditox ligand of 1 was disordered over two positions in a 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :47 ratio, and each THF molecule was disordered over two positions in a 50:50 ratio.
:47 ratio, and each THF molecule was disordered over two positions in a 50:50 ratio.
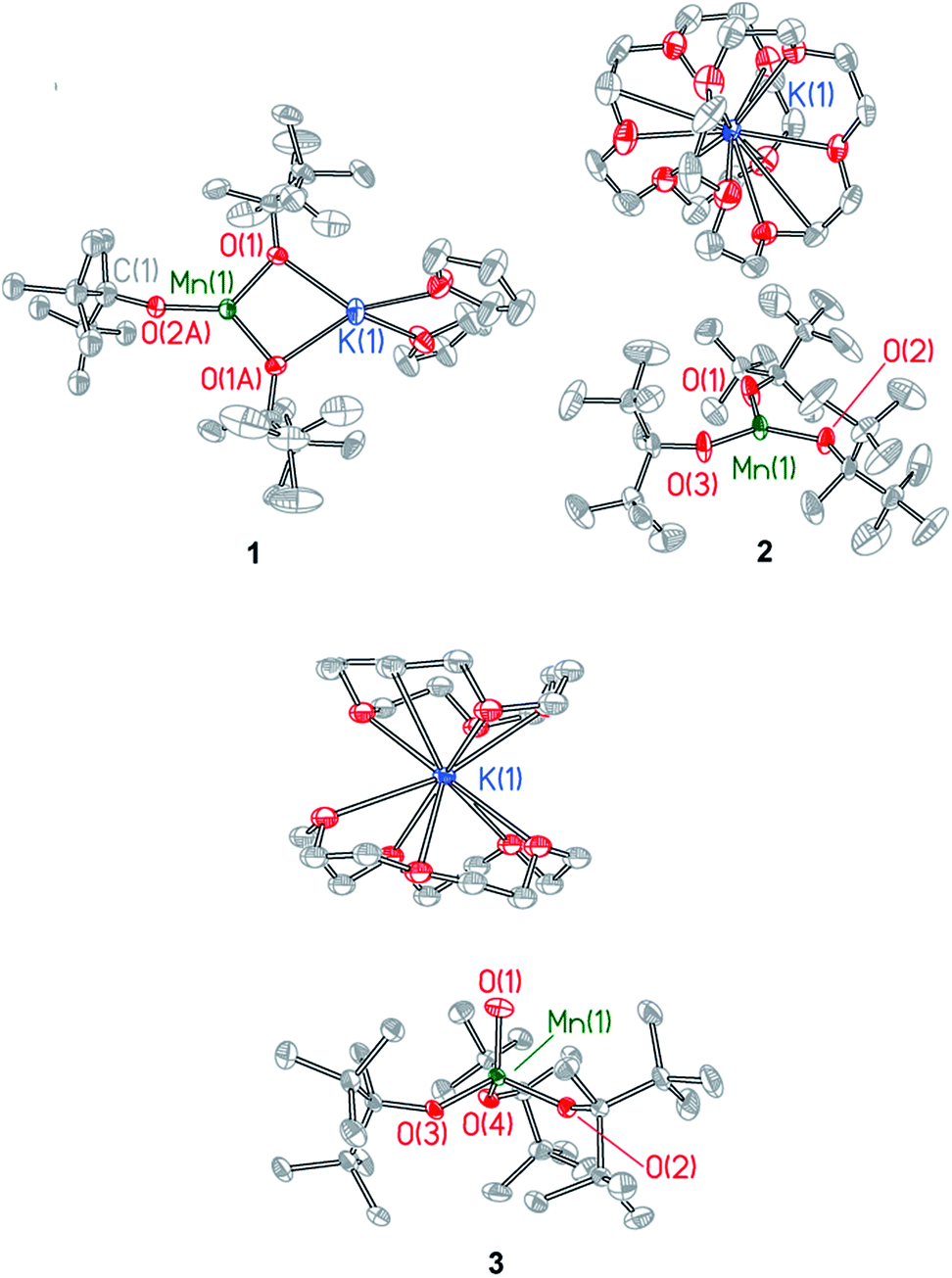 | ||
| Fig. 2 X-ray structures of Mn(II) complexes 1 and 2 and Mn(IV)-oxo complex 3. Selected bond distances and angles are provided in the ESI.† | ||
Compound 2 crystallizes in the monoclinic space group P21/c as a discrete anion/cation pair (Fig. 2). The average Mn–Oalk bond distance is 1.925 ± 0.003 Å, and the sum of the Oalk–Mn–Oalk bond angles is 359.8°, indicating the trigonal planar nature of 2. The sequestration of the potassium countercation by two crown ether molecules prevents distortion of the Oalk–Mn–Oalk bond angles and renders a pseudo-D3h symmetry about manganese in which each angle is very nearly 120°.
Compound 3 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) as a discrete anion/cation pair (Fig. 2). The manganese center exhibits a slightly axially distorted pseudotetrahedral geometry with approximate C3v symmetry. The terminal Mn–Oterm function has a bond length of 1.628(2) Å and is bent at an angle of 85.86(8)° relative to the plane defined by the alkoxide oxygen atoms. The average Mn–Oalk bond distance is 1.849 ± 0.024 Å, which is notably shorter than the average Mn–Oalk bond distance of 1 and 2, consistent with the higher oxidation state of manganese in 3. The average Oterm–Mn–Oalk bond angle is observed to be 111.23 ± 4.52° while the average Oalk–Mn–Oalk bond angle is 107.71 ± 3.69°, an angle that is consistent with a pseudotetrahedral geometry. One of the crown ether molecules is disordered over two positions in a 50:50 ratio.
as a discrete anion/cation pair (Fig. 2). The manganese center exhibits a slightly axially distorted pseudotetrahedral geometry with approximate C3v symmetry. The terminal Mn–Oterm function has a bond length of 1.628(2) Å and is bent at an angle of 85.86(8)° relative to the plane defined by the alkoxide oxygen atoms. The average Mn–Oalk bond distance is 1.849 ± 0.024 Å, which is notably shorter than the average Mn–Oalk bond distance of 1 and 2, consistent with the higher oxidation state of manganese in 3. The average Oterm–Mn–Oalk bond angle is observed to be 111.23 ± 4.52° while the average Oalk–Mn–Oalk bond angle is 107.71 ± 3.69°, an angle that is consistent with a pseudotetrahedral geometry. One of the crown ether molecules is disordered over two positions in a 50:50 ratio.
The nature of the Mn(IV)–Oterm is in line with a wide range of metal oxo complexes. Table 1 lists structural metrics for a series of related compounds to 3. Beginning with a trigonal ditox ligand platform, the Mn–Oterm bond distance in 3 is similar to its V and Cr counterparts of d0 and d1 electron counts. Inasmuch as the d0–d4 systems occupy the same e-orbital set of primarily dxz/yz character, the similarity of the d(Mn–Oterm) bond distances for M = V, Cr and Mn is unsurprising. The d(Mn–Oterm) bond distance, which is much shorter than that of hydroxides bound to metals in a ditox trigonal field as reflected by Fe(III)(OH)(ditox)3−, lies between the Mn(IV)-oxygen single bond distance and the Mn(V)-oxygen triple bond distance of structurally characterized manganese complexes with a terminal oxygen ligand (Table 1). Additionally, though not crystallographically characterized, Mn(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond lengths have been determined by EXAFS for two Mn(IV) oxo compounds (see Table 1), and they are in line with that observed for 3.
O bond lengths have been determined by EXAFS for two Mn(IV) oxo compounds (see Table 1), and they are in line with that observed for 3.
| M–O moiety | Complex | d(M–O)/Å | Reference |
|---|---|---|---|
| a [H3buea] = tris[(N′-tert-butylureaylato)-N-ethylene]amide. b Distance determined from EXAFS. c H4TAML = 6,6-diethyl-3,3,9, tetramethyl-1,8,9,11-tetrahydro-4λ2-pyrido[2,3-e][1,4,7,10]tetraazacyclo-tridecine-2,5,7,10(3H,6H)-tetraone. d H4TAML′ = 3,4,8,9-tetrahydro3,3,6,6,9,9-hexamethyl-1H-1,4,8,11-benzotetraazocyclotridecane-2,5,7,10(6H,11H)tetrone. e H4PHAB = 1,2-bis(2,2-diphenyl-2-hydroxyethanamido)benzene. f H3TBP8Cz = octakis(p-tert-butylphenyl)corrolazine. g BnTPEN = N-benzyl-N,N′,N′-tris(2-pyridylmethyl)-1,2-diaminoethane. h Me2EBC = 4,11-dimethyl-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane. | |||
| Mn(IV)–Oterm | MnO(ditox)3− | 1.628(2) | This paper |
| Mn(IV)–Oterm | MnO(BnTPEN)2+g | 1.69b | 43 |
| Mn(IV)–Oterm | MnO(OH)(Me2EBC)2+h | 1.71b | 34 |
| Cr(V)–Oterm | CrO(ditox)3 | 1.649(2) | 31 |
| V(V)–Oterm | VO(ditox)3 | 1.605(1) | 31 |
| Fe(III)–OHterm | Fe(III)(OH)(ditox)3− | 1.890(1) | 30 |
| Mn(IV)–OHterm | Mn(IV)(OH)(buea)−a | 1.83b | 35 |
| Mn(V)–Oterm | MnO(TAML)−c | 1.549(3) | 20 |
| Mn(V)–Oterm | MnO(TAML′)−d | 1.5555(12) | 19 |
| Mn(V)–Oterm | MnO(PHAB)−e | 1.558(4) | 21 |
| Mn(V)–Oterm | MnO(TBP8Cz)f | 1.5455(18) | 22 |
The FTIR spectra of 1–3 are reproduced in Fig. S5.† The M–OR stretching region of 1 shows a strong absorption at 575 cm−1 as well as two weak absorptions at 637 cm−1 and 659 cm−1. The addition of 15-C-5 to 1 to furnish 2 results in the lowest-energy band of 1 shifting from 575 cm−1 to 583 cm−1 and the two higher-energy bands of 1 coalescing into a single band at 646 cm−1. The expansion of one of the Oalk–Mn–Oalk bond angles of 2 relative to 1 appears to be correlated with a higher-energy M–OR feature in 2. The lower-energy band of 2 shifts from 583 cm−1 to 591 cm−1 and the higher-energy band shifts from 646 cm−1 to 649 cm−1 upon its reaction with PhIO to give 3. These shifts to higher energy are consistent with the higher oxidation state of 3 and the attendant decrease in the Mn–Oalk bond distance. The FTIR spectra of 3 and its 18Ooxo-isotopomer (Fig. S6†) display Mn–Ooxo stretching frequencies at 845 cm−1 and 809 cm−1 (ν(Mn16O)/ν(Mn18O) = 1.04; calcd 1.06), respectively.
Solution susceptibility measurements of 1 and 2 at 298 K yield μeff values of 6.03 μB and 5.77 μB, respectively, which is consistent with both complexes possessing a high-spin S = 5/2 spin state (μspin-only = 5.92 μB). The solution susceptibility measurement of 3 at 298 K yields a μeff value of 4.54 μB, which is comparable to values for other observed Mn(IV)–oxo complexes possessing a spin state of S = 3/2 (μspin-only = 3.87 μB).23,36 SQUID DC magnetic susceptibility of powdered 3 from 2–298 K confirms a high-spin configuration, which is consistent with the weak-field nature of the ditox ligand.29 SQUID magnetometry at 20 °C provides a solid-state susceptibility of 4.75 μB for 3, which is significantly greater than the spin-only susceptibility of 3.87 μB expected for an isotropic S = 3/2 system (Fig. S7†) and is more in-line with a highly anisotropic species. Indeed, X-band EPR spectroscopy performed in perpendicular mode on 3 shows a pseudo-axial signal at gx = 4.135, gy = 3.860, and gz = 1.980 (Fig. 3). Sharp hyperfine coupling can be observed at gz yielding |Az| = 299 MHz. A plot of χT versus T for 3 shows antiferromagnetic behavior with χT decreasing steadily as temperature decreases to ∼20 K, after which χT drops quickly to a minimal value of 0.35 cm3 K mol−1 at 2 K (Fig. S7†). The large temperature dependence of χT suggests the presence of significant magnetic anisotropy, as anisotropic spins possess energy separations between their MS levels that are often within an order of magnitude of kBT, leading to temperature dependent behavior. Attempts to model the χT versus T data without considering magnetic anisotropy were unsuccessful. In order to attempt to quantify the axial zero-field splitting parameter D, the χT versus T data (Fig. S8†) were fit to the Hamiltonian: Ĥ = DŜz2 + μBgS·H, where D is the zero-field splitting, Ŝz is the spin operator, μB is the Bohr magneton, g is the Landé g-factor, S is the spin, and H is the magnetic field. The model employs only axial D and g-tensors, ignoring any transverse anisotropy (E) since including this term did not improve the fit to the data and resulted in g-factors that varied significantly from those determined via EPR spectroscopy. The best fit to the data produced gx, gy, and gz values of 4.15, 3.57, and 1.92, respectively, and a value of D of −12.31 cm−1, all of which are in good agreement with the values obtained from EPR. To more accurately determine the magnitude of D and E, we collected low-temperature magnetization data at various applied DC fields. The resulting plot of reduced magnetization for 3 is shown in Fig. S8.† To quantify the effect of strong magnetic anisotropy, the data were modeled according to the Hamiltonian: Ĥ = DŜz2 + E(Ŝx2 − Ŝy2) + μBgS·H which is similar to the above mentioned model but contains an additional transverse zero-field splitting parameter, E. Fits to the data using PHI37 and an average g-factor of 3.13 give axial and transverse zero-field splitting parameters of D = −23.4 cm−1 and |E| = 6. This D-value is several orders of magnitude larger than those reported for a suite Mn(IV)(OH) compounds.38,39
 | ||
Fig. 3 Experimental (green line  ) and simulated (black dots ) and simulated (black dots  ) X-band CW-EPR spectrum of [MnIV(O)(ditox)3][K(15-C-5)2] (3) at 6 K in butyronitrile glass. See text for simulation parameters. ) X-band CW-EPR spectrum of [MnIV(O)(ditox)3][K(15-C-5)2] (3) at 6 K in butyronitrile glass. See text for simulation parameters. | ||
Compound 3 can be oxidized under mild conditions. Cyclic voltammograms of 3 in a 0.2 M [TBA][PF6]/THF solution show an electrochemically reversible oxidation wave with an E1/2 value of −0.65 V versus Fc+/0 that we attribute to the Mn(V/IV) couple (Fig. 4). A cathodic sweep to negative potentials engenders an irreversible reduction wave with a peak current at −2.27 V versus Fc+/0. This irreversible reduction wave is accompanied by the visible formation of a brown insoluble solid.
 | ||
| Fig. 4 Cyclic voltammogram of [MnIV(O)(ditox)3][K(15-C-5)2] (3) in THF/[TBA][PF6]. | ||
Complex 3 reacts with the relatively weak C–H bonds of 9,10-dihydroanthracene (76.2 kcal mol−1)40 and 1,4-cyclohexadiene (CHD) (76.0 kcal mol−1)40 and produces the Mn(III) hydroxide, as the properties of the product in color and solubility are similar with the previously reported FeIII–OH ditox compound.30 The reaction between 3 and excess CHD generates nearly half of an equivalent of benzene and trace amounts of 1,3-cyclohexadiene (Fig. S10†). Attempts to oxidize stronger C–H bonds, such as those of ethyl benzene (85.4 kcal mol−1),40 show no reaction by 1H NMR. Notably 3 also does not react with oxygen-atom acceptors. For instance, treatment of 3 with a ten-fold excess of PPh3 shows no evidence of the oxygen-atom transfer product, OPPh3, or any other phosphorous-containing products, as indicated by 31P{1H} NMR. This lack of oxygen-atom transfer reactivity is similar to that observed for the nucleophilic Cr d1 complex [CoCp2][O2Cr(NRAr)2] prepared by Cummins and coworkers,41 who contrast the reactivity of their nucleophilic Cr d1 complex against that of the electrophilic Re d1 complex [HB(pz)3]ReO2Cl prepared by Mayer and coworkers42 who concluded that the highly electrophilic nature of the oxidized complex [HB(pz)3]ReO2Cl+ was mainly a result of its cationic charge. In turn, Cummins and coworkers attribute the lack of oxidizing ability of their complex to its negative charge in conjunction with the strongly electron-donating amido ligands. Similarly, we believe that the lack of oxygen atom transfer chemistry of 3 is the result of its anionic charge and strong electron-donating properties of the ditox ligand set. For example, in a neutral all N-donor field, a similar manganese oxo prepared by the group of Nam is able to attack the much stronger bonds of cyclohexane (99.5 kcal mol−1).43
The ability of the all oxygen anionic ligand field of ditox to stabilize a Mn(IV) oxo may shed light on the ability of the Oxygen Evolving Complex (OEC) to protect itself in the S3 state proposed by Lubitz in which all manganese centers are in the +4 oxidation state.6 As for [MnIV(O)(ditox)3]−, the OEC has an anionic all oxygen ligand field, suggesting that a muted reactivity of the Mn(IV)-oxo in a donating oxygen ligand field may be a strategy for the OEC to circumvent the oxidation of the amino acid environment in lower S states of the Kok cycle such as S3, thus preserving the OEC to perform water splitting in the higher and more reactive S4 oxidation state. In this regard, the Mn(IV) oxo ditox system suggests a mechanism by which the reactivity of traditionally high energy reactive intermediates may be attenuated in a pre-catalytic resting state, a property that protein cofactors such as OEC have long mastered.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSF grant CHE-1464232. D. G. acknowledges the National Science Foundation (NSF) for a Graduate Research Fellowship.Notes and references
- D. G. Nocera, Inorg. Chem., 2009, 48, 10001 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS PubMed.
- J. R. Winkler and H. B. Gray, in Structure and Bonding, ed. D. M. P. Mingos, P. Day and J. P. Dahl, Springer, Berlin Heidelberg, 2012, vol. 142, p. 17 Search PubMed.
- J. M. Peloquin, K. A. Campbell, D. W. Randall, M. A. Evanchik, V. L. Pecoraro, W. H. Armstrong and R. D. Britt, J. Am. Chem. Soc., 2000, 122, 10926 CrossRef CAS.
- R. Gupta, T. Taguchi, B. Lassalle-Kaiser, E. L. Bominaar, J. Yano, M. P. Hendrich and A. S. Borovik, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5319 CrossRef CAS PubMed.
- N. Cox, M. Retegan, F. Neese, D. A. Pantazis, A. Boussac and W. Lubitz, Science, 2014, 345, 804 CrossRef CAS PubMed.
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Acc. Chem. Res., 2013, 46, 1588 CrossRef CAS PubMed.
- A. Grundmeier and H. Dau, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 88 CrossRef CAS PubMed.
- J. Barber, Chem. Soc. Rev., 2009, 38, 185 RSC.
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455 CrossRef CAS PubMed.
- T. J. Meyer, M. H. V. Huynh and H. H. Thorp, Angew. Chem., Int. Ed., 2007, 46, 5284 CrossRef CAS PubMed.
- V. L. Pecoraro, M. J. Baldwin, M. T. Caudle, W.-Y. Hsieh and N. A. Law, Pure Appl. Chem., 1998, 70, 925 CrossRef CAS.
- E. N. Jacobsen, in Comprehensive Organometallic Chemistry II, ed. G. Wilkinson, F. G. A. Stone, E. W. Abel and L. S. Hegedus, Pergamon, New York, 1995, vol. 12, p. 1097 Search PubMed.
- W. J. Song, M. S. Seo, S. DeBeer George, T. Ohta, R. Song, M.-J. Kang, T. Tosha, T. Kitagawa, E. I. Solomon and W. Nam, J. Am. Chem. Soc., 2007, 129, 1268 CrossRef CAS PubMed.
- H. M. Neu, R. A. Baglia and D. P. Goldberg, Acc. Chem. Res., 2015, 48, 2754 CrossRef CAS PubMed.
- W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727 CrossRef CAS PubMed.
- T. Taguchi, R. Gupta, B. Lassalle-Kaiser, D. W. Boyce, V. K. Yachandra, W. B. Tolman, J. Yano, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2012, 134, 1996 CrossRef CAS PubMed.
- T. Taguchi, K. L. Stone, R. Gupta, B. Lassalle-Kaiser, J. Yano, M. P. Hendrich and A. S. Borovik, Chem. Sci., 2014, 5, 3064 RSC.
- S. Hong, Y.-M. Lee, M. Sankaralingam, A. K. Vardhaman, Y. J. Park, K.-B. Cho, T. Ogura, R. Sarangi, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2016, 138, 8523 CrossRef CAS PubMed.
- D.-L. Popescu, A. Chanda, M. Stadler, F. T. de Oliveira, A. D. Ryabov, E. Munck, E. L. Bominaar and T. J. Collins, Coord. Chem. Rev., 2008, 252, 2050 CrossRef CAS.
- F. M. MacDonnell, N. L. P. Fackler, C. Stern and T. V. O'Halloran, J. Am. Chem. Soc., 1994, 116, 7431 CrossRef CAS.
- R. A. Baglia, K. A. Prokop-Prigge, H. M. Neu, M. A. Siegler and D. P. Goldberg, J. Am. Chem. Soc., 2015, 137, 10874 CrossRef CAS PubMed.
- J. Chen, Y.-M. Lee, K. M. Davis, X. Wu, M. S. Seo, K.-B. Cho, H. Yoon, Y. Jun Park, S. Fukuzumi, Y. N. Pushkar and W. Nam, J. Am. Chem. Soc., 2013, 135, 6388 CrossRef CAS PubMed.
- R. H. Holm, Chem. Rev., 1987, 87, 1401 CrossRef CAS.
- C. J. Ballhausen and H. B. Gray, Inorg. Chem., 1961, 1, 111 CrossRef.
- T. A. Betley, Q. Wu, T. V. Voorhis and D. G. Nocera, Inorg. Chem., 2008, 47, 1849 CrossRef CAS PubMed.
- T. A. Betley, Y. Surendranath, M. V. Childress, G. E. Alliger, R. Fu, C. C. Cummins and D. G. Nocera, Philos. Trans. R. Soc., B, 2008, 363, 1293 CrossRef CAS PubMed.
- S. A. Cook and A. S. Borovik, Acc. Chem. Res., 2015, 48, 2407 CrossRef CAS PubMed.
- M. P. Marshak and D. G. Nocera, Inorg. Chem., 2013, 52, 1173 CrossRef CAS PubMed.
- M. B. Chambers, S. Groysman, D. Villagrán and D. G. Nocera, Inorg. Chem., 2013, 52, 3159 CrossRef CAS PubMed.
- S. Groysman, D. Villagrán and D. G. Nocera, Inorg. Chem., 2010, 49, 10759 CrossRef CAS PubMed.
- T. V. Lubben and P. T. Wolczanski, J. Am. Chem. Soc., 1987, 109, 424 CrossRef CAS.
- P. T. Wolczanski, Polyhedron, 1995, 14, 3335 CrossRef CAS.
- D. F. Leto and T. A. Jackson, Inorg. Chem., 2014, 53, 6179 CrossRef CAS PubMed.
- T. Taguchi, K. L. Stone, R. Gupta, B. Kaiser-Lassalle, J. Yano, M. P. Hendrich and A. S. Borovik, Chem. Sci., 2014, 5, 3064 RSC.
- W. Zhang, J. L. Loebach, S. R. Wilson and E. N. Jacobsen, J. Am. Chem. Soc., 1990, 112, 2801 CrossRef CAS.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164 CrossRef CAS PubMed.
- R. Gupta, T. Taguchi, A. S. Borovik and M. P. Hendrich, Inorg. Chem., 2013, 52, 12568 CrossRef CAS PubMed.
- D. F. Leto, A. A. Massie, H. E. Colmer and T. A. Jackson, Inorg. Chem., 2016, 55, 3272 CrossRef CAS PubMed.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Taylor & Francis Group, Boca Raton, 2007 Search PubMed.
- A. L. Odom, D. J. Mindiola and C. C. Cummins, Inorg. Chem., 1999, 38, 3290 CrossRef CAS PubMed.
- D. D. DuMez and J. M. Mayer, Inorg. Chem., 1998, 37, 445 CrossRef CAS PubMed.
- X. Wu, M. S. Seo, K. M. Davis, Y.-M. Lee, J. Chen, K.-B. Cho, Y. N. Pushkar and W. Nam, J. Am. Chem. Soc., 2013, 133, 20088 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Single crystal X-ray diffraction data; ESI-MS spectra and data: IR spectra; and magnetic data. CCDC 1829119–1829121. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01164h |
| ‡ Present address: Department of Chemistry and Biochemistry, University of Delaware, Newark, Delaware 19716, United States. E-mail: E-mail: edb@udel.edu |
| This journal is © The Royal Society of Chemistry 2018 |