
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReductive coupling of benzyl oxalates with highly functionalized alkyl bromides by nickel catalysis†
Xiao-Biao
Yan
,
Chun-Ling
Li
,
Wen-Jie
Jin
,
Peng
Guo
and
Xing-Zhong
Shu
 *
*
State Key Laboratory of Applied Organic Chemistry (SKLAOC), College of Chemistry and Chemical Engineering, Lanzhou University, 222 South Tianshui Road, Lanzhou, 730000, China. E-mail: shuxingzh@lzu.edu.cn
First published on 20th April 2018
Abstract
Coupling reactions involving non-sulfonated C–O electrophiles provide a promising method for forming C–C bonds, but the incorporation of functionalized or secondary alkyl groups remains a challenge due to the requirement for well-defined alkylmetal species. In this study, we report a reductive nickel-catalyzed cross-coupling of benzyl oxalates with alkyl bromides, using oxalate as a new leaving group. A broad range of highly functionalized alkyl units (such as functional groups: alkyl chloride, alcohol, aldehyde, amine, amide, boronate ester, ether, ester, heterocycle, phosphonate, strained ring) were efficiently incorporated at the benzylic position. The utility of this synthetic method was further demonstrated by late-stage modification of complex bioactive compounds. Preliminary mechanistic experiments revealed that a radical process might be involved in the reaction.
Introduction
Transition metal-catalyzed cross-coupling reactions have become one of the most important tools in organic synthesis.1 Recent efforts have been focused on the use of non-sulfonated C–O electrophiles (e.g., carboxylates, ethers) as coupling partners versus organic halides.2 Their advantages involve low toxicity, low cost and ready availability, and abundant C–O bonds in a wide range of natural and artificial compounds. The high activation barrier for C–O cleavage and selectivity challenge in the presence of multiple C–O bonds3 mean that their coupling reactions have been realized only recently by using organometallic species (e.g., Grignards, organozincs, and boronic acids) as coupling partners (Scheme 1, path (a)).2 In contrast to the major advances achieved in the field of arylation reactions,4 the development of alkylation reactions has proved more difficult.5 To date, only a few elegant studies have demonstrated the Csp3–Csp3 coupling of relatively unreactive C–O electrophiles. The incorporation of functionalized or secondary alkyl groups remains a particular challenge,6 which can partially be ascribed to the low availability and high reactivity profiles of alkylmetal reagents. The development of protocols using electrophiles instead of organometallic reagents to couple with C–O electrophiles may provide a solution to these problems, offering a unique opportunity to discover new reactivities within this field (Scheme 1, path (b)).7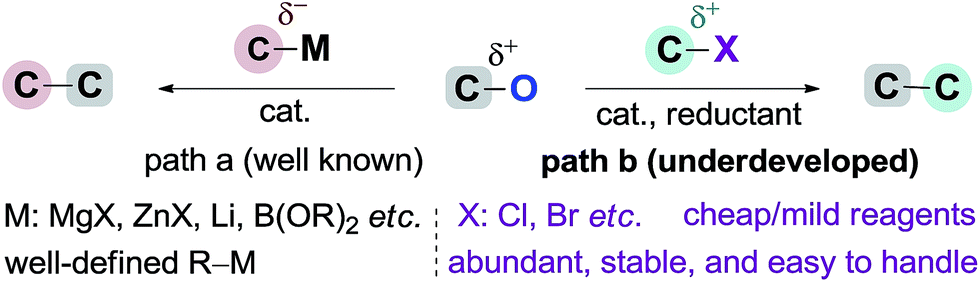 | ||
| Scheme 1 Catalytic cross-coupling reactions via C–O bond cleavage. | ||
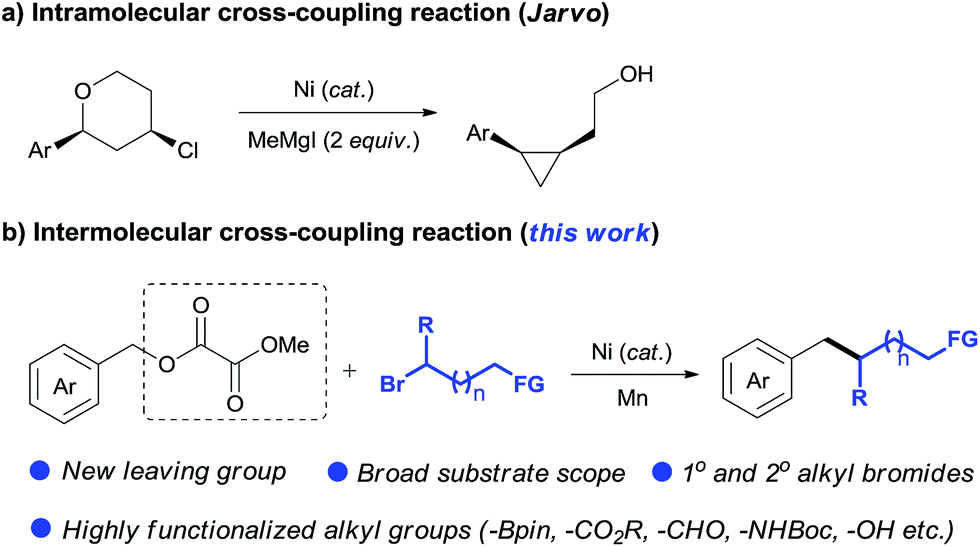
The reductive cross-coupling of two electrophiles has emerged as an increasingly popular approach for constructing the C–C bond.8 One of the major challenges in this field is to expand the scope of electrophiles. Encouraged by the pioneer work of Weix,8e the groups of Martin, Jarvo, Shi, Molander, and Shu have launched a program to disclose the potential of nickel-catalyzed reductive cross-coupling of relatively unreactive C–O electrophiles.7d–l Notably Jarvo's group has disclosed intramolecular Csp3–Csp3 coupling of benzyl ethers with alkyl halides (Scheme 2a).7k However, intermolecular Csp3–Csp3 coupling is still unresolved because of difficulty and complexity with controlling selectivity for the cross-product.9 Herein, we demonstrate the use of oxalate10 as a leaving group to allow an intermolecular Csp3–Csp3 coupling between benzyl carboxylates and alkyl bromides (Scheme 2b). This method accomplishes the incorporation of a wide range of highly functionalized alkyl groups at the benzylic position and tolerates both primary and secondary alkyl bromides. Our study demonstrates this method is an attractive alternative for the alkylation of benzylic derivatives using alkyl nucleophiles.6,11
 | ||
| Scheme 2 Reductive Csp3–Csp3 cross-coupling reactions via benzylic C–O bond cleavage. | ||
Results and discussion
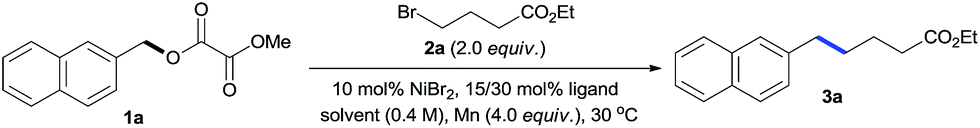
We started our investigations by exploring the reaction of oxalate 1a with alkyl bromide 2a (Table 1, see Table S1 and S2† for more details). Initial experiments revealed that reactions under the conditions of NiBr2 (10 mol%), Mn (4.0 equiv.) in DMF using nitrogen ligands gave no or low yields of the desired product 3a (entries 1–4), along with large amounts of ArCH3 (2-methylnaphthalene) and ArCH2–OH (2-naphthalenemethanol) side products. A review of the literature revealed that phosphine ligands were mostly ineffective for reductive cross-coupling reactions.8 However, the use of PPh3 gave 3a with an unexpected 34% yield (entry 5), encouraging us to study the electronic and steric effects of the ligands. We found that P(4-CF3Ph)3 was the most effective (entries 6–9). Screening of solvents revealed that DMSO was crucial to increasing reaction efficiency, as it significantly inhibited formation of the ArCH2–OH side product (entries 9–13). No reaction was observed in the absence of the Ni catalyst or Mn reductant (entries 14–15). Due to the solubility problem, reactions using non-polar alkyl bromides for the preparation of 3b–3e and 3y–3aa were sluggish in DMSO. Thus, the conditions of entry 13 using DMSO/DMF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were used as standard conditions under which the gram-scale reaction of 1a gave 3a with 82% yield.
:1) were used as standard conditions under which the gram-scale reaction of 1a gave 3a with 82% yield.
|
|
|||
|---|---|---|---|
| Entry | Ligand | Solvent | Yield (%) |
| a Substrates 1a (0.2 mmol), monodentate ligand (30 mol%), or bidentate ligand (15 mol%) were used and reacted for 24 h; yields were determined by 1H NMR using anisole as an internal standard. b Yields are isolated yields. c 1a (4 mmol, 0.976 g) was used; isolated yield. d No Ni catalyst. e No Mn. | |||
| 1 | L1 | DMF | 20 |
| 2 | L2 | DMF | 18 |
| 3 | L3 | DMF | 32 |
| 4 | L4 | DMF | 0 |
| 5 | PPh3 | DMF | 34 |
| 6 | P(4-MePh)3 | DMF | 16 |
| 7 | P(2-MePh)3 | DMF | 0 |
| 8 | P(4-FPh)3 | DMF | 50 |
| 9 | P(4-CF3Ph)3 | DMF | 58 |
| 10 | P(4-CF3Ph)3 | DMA | 52 |
| 11 | P(4-CF3Ph)3 | DMSO | 73 (79)b |
| 12 | P(4-CF3Ph)3 | DMSO/DMA 1:1 |
68 (73)b |
| 13 | P(4-CF3Ph)3 | DMSO/DMF 1:1 |
70 (75)b, (82)c |
| 14d | P(4-CF3Ph)3 | DMSO/DMF 1:1 |
0 |
| 15e | P(4-CF3Ph)3 | DMSO/DMF 1:1 |
0 |
|
|||
The effects of various leaving groups were then studied (Table 2). Simple benzyl ether (4) was unreactive. While carboxylates were more reactive than methyl ether, reactions of commonly used carboxylates gave no or very low yields of the desired product (5–9). In these cases, most starting materials of 4–9 remained unchanged. By increasing the leaving ability of carboxylate,12 a full conversion of trifluoroacetate 10 was observed, affording 3a with 19% yield along with large amounts of ArCH2–CH2Ar (30%), ArCH2–OH (15%), and ArCH3 (11%) side products. While 3-pyridyl ester 11 was totally unreactive under standard conditions, the reaction of 2-pyridyl ester 12 gave 3a with 22% yield. This result indicated that use of the bidentate leaving group was beneficial to this reaction,13 prompting us to examine the effects of several others. Ether 13 and acetate 14 were found to be unreactive, even though they were active leaving groups for the benzylic C–O cleavage.7e,13 The use of a stronger coordinating group, 2-(methylthio)acetate (15), led to trace amounts of 3a. The reaction of ethyl oxalate 16 gave a comparable result to that of methyl oxalate 1a, affording 3a with 72% NMR yield.14
| a Substrates 4–16 (0.2 mmol) were used and reacted for 24 h; yields were determined by 1H NMR using anisole as an internal standard. |
|---|
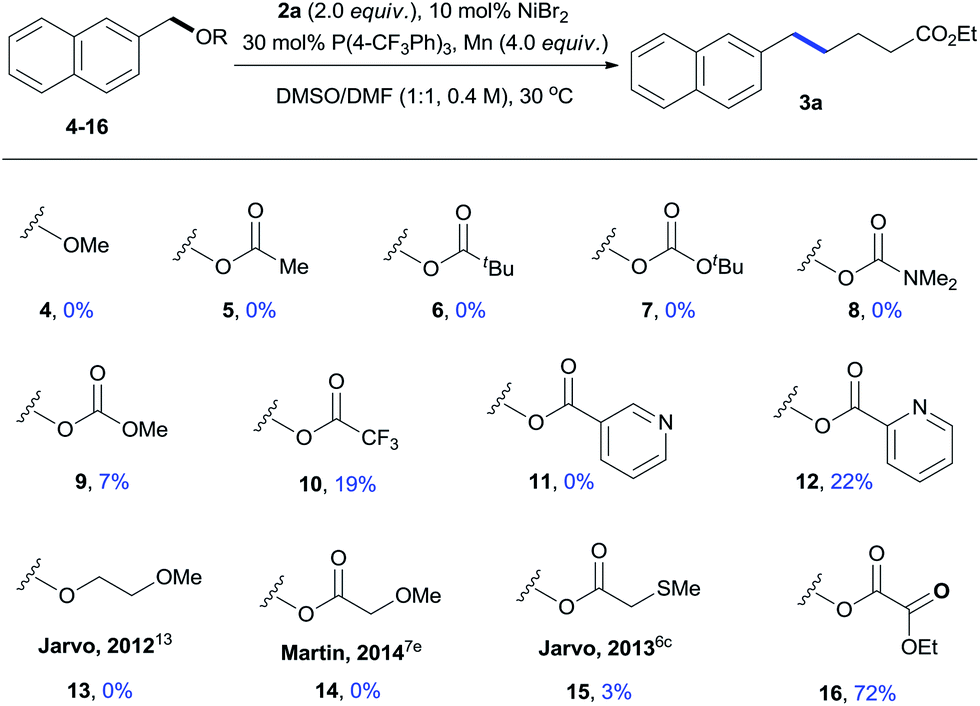
|
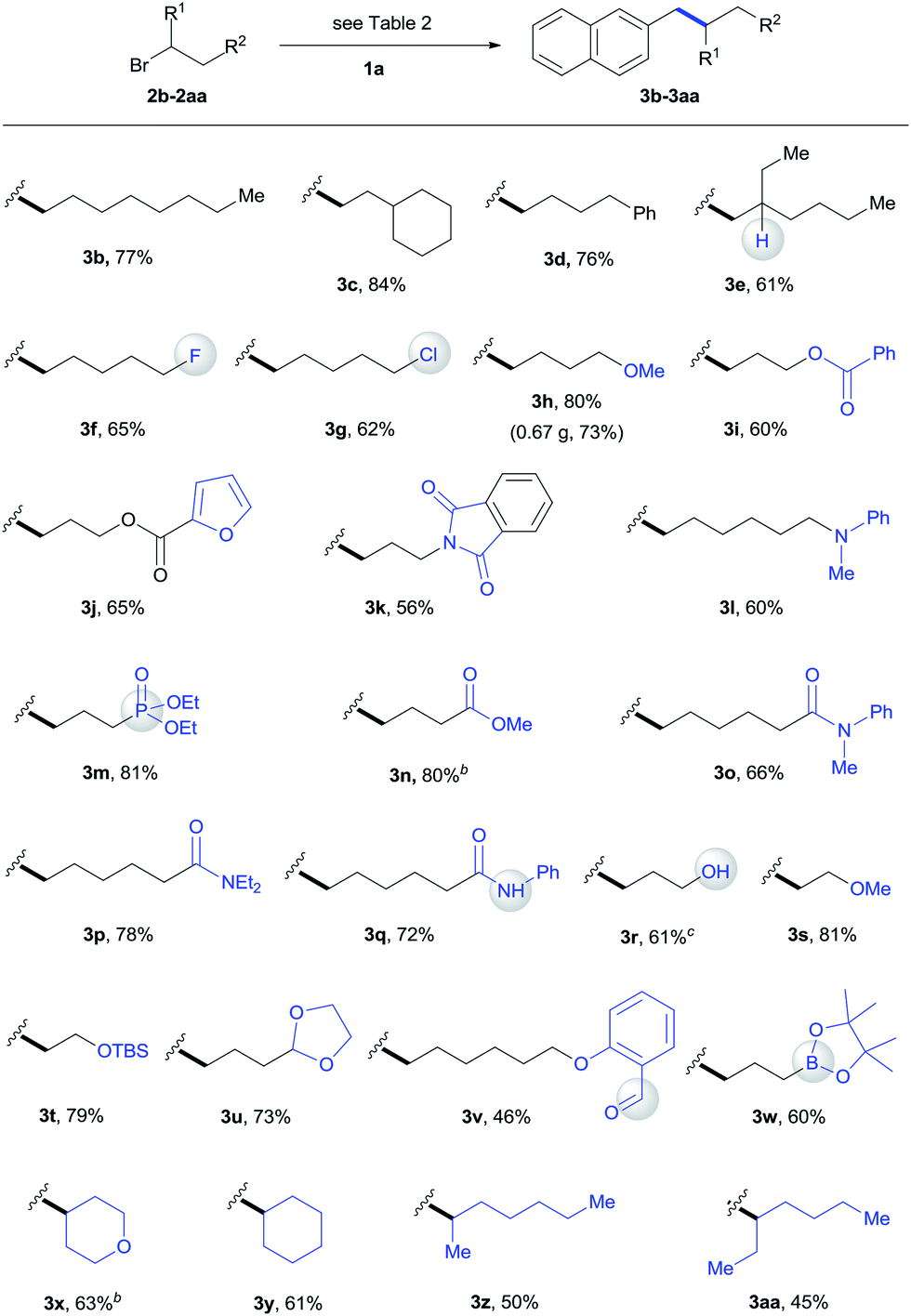
We subsequently focused on examining the generality of this protocol. The incorporation of functionalized alkyl units is important in the synthesis of complex molecules. The use of functionalized alkylmetal reagents is severely restricted because of limited availability and high reactivity profiles of the reagent itself.5b The use of alkyl halides would circumvent these problems. In this work, both simple long-chain alkyl substrates (3b–3d) and a wide range of functionalized alkyl bromides coupled with 1a efficiently under standard conditions (Table 3). The incorporation of β-substituted alkyl unit represents a challenge due to the fast β-H elimination of alkylmetal intermediates.6e Product 3e was formed in a moderate yield under our conditions. The reaction was highly chemoselective for functionalization of the C–Br bond over C–F (3f) and C–Cl (3g) bonds. Moreover, a gamut of functionalities such as methyl and silyl ethers (3h, 3s, 3t), esters (3i, 3j, 3n), heterocycle (3j), tertiary amides (3k, 3o, 3p), amine (3l), phosphonate (3m), acidic amide (3q), alcohol (3r), aldehyde (3v), and boronate ester (3w) were accommodated. Although the direct coupling of aldehyde substrate (3v) was less effective, the use of alkyl bromides bearing protected aldehyde function gave a good yield of product 3u. The reaction was selective for functionalization of the C–Br bond, leaving the nucleophilic C–B bond for additional transformation (3w). Furthermore, the scope of this alkylation protocol could be extended to secondary alkyl bromides to give cyclic (3x, 3y) and acyclic (3z, 3aa) products. The reaction could be scaled up to gram-scale and produced 3h with 73% yield.
A wide range of benzyl oxalates coupled with functionalized alkyl bromide 2n efficiently under standard conditions (Table 4). The reaction was highly chemoselective for alkylation of benzyl esters, leaving a number of aryl esters intact (3ab–3af).2 Functional units such as strained rings, amino acid and α-oxy acid derivatives, as well as acidic carbamates were tolerated under reductive conditions (3ac–3ag). No erosion in enantioselectivity was found en route to either 3ae or 3af. The reaction of 1-naphthyl oxalate (3ai) gave a comparable result to that for 3ah. Nitrogen and sulfur heterocycles are prevalent in pharmaceuticals but always represent a challenge for metal catalysis. The expected products 3aj–3an were formed with moderate yields under standard conditions. Unfortunately, our method did not allow the coupling reaction of secondary benzyl electrophiles (3ap).
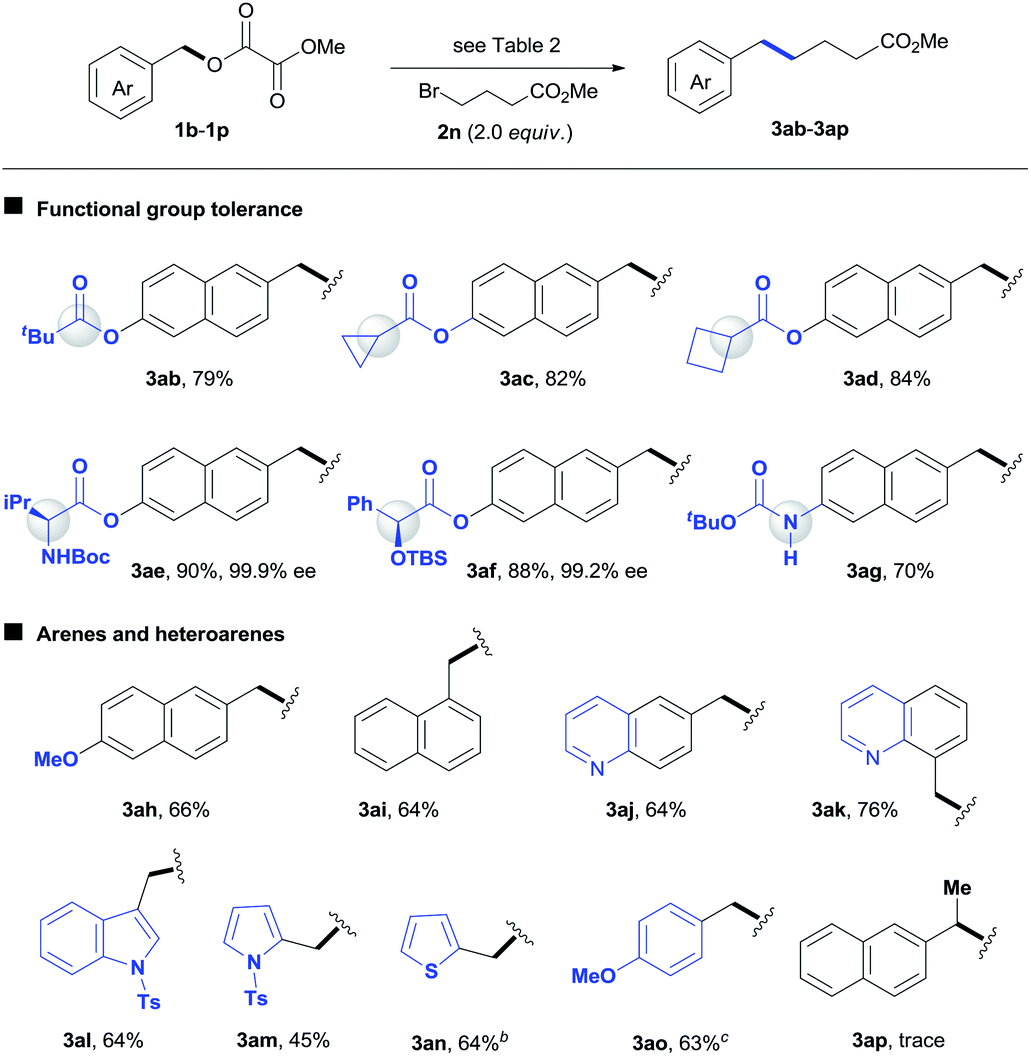
To date, most Ni-catalyzed cross-coupling reactions via inert C–O bond cleavage are limited to substrates with π-extended systems like naphthalene.2 One possible explanation is that, unlike a regular arene, the π-extended system can be coordinated to Ni(0) efficiently,15 because the binding complex might retain a certain aromaticity and might still be partially stable.7e Indeed, under standard conditions the reaction of a regular benzyl oxalate only gave trace amounts of product 3ao. The vast majority of substrate was converted to benzyl alcohol. This prompted us to study the reaction conditions again. Finally, we found that by changing the ligand to dppb and using MgBr2 (1.5 equiv.) and 3-CF3-Py (5 mol%) as additives, the reaction afforded a useful 63% yield of 3ao. Although the role of additives is unclear, the use of MgBr2 significantly improved selectivity for 3ao over benzyl alcohol, and the addition of 3-CF3-Py inhibited the formation of benzyl dimer.
The late-stage modification of complex molecules provides a promising approach to altering the pharmacological profiles of natural products. To further demonstrate the synthetic utility of our method, we studied the alkylation reactions of several complex bioactive compounds (Scheme 3). The reaction of α-D-(+)-glucose derivate 17 with oxalate 1a gave product 18 with 54% yield. The presence of highly coordinative thioether and amide groups makes functionalization of D-biotin a challenge for metal catalysis. The expected product 20 was formed with 63% yield under standard conditions. Lithocholic acid derivate 21 selectively coupled with oxalate 1a, leaving a free alcohol group intact. In addition, this approach allows for incorporation of a naphthyl group at the secondary alkyl carbon, affording 24 with moderate yield.
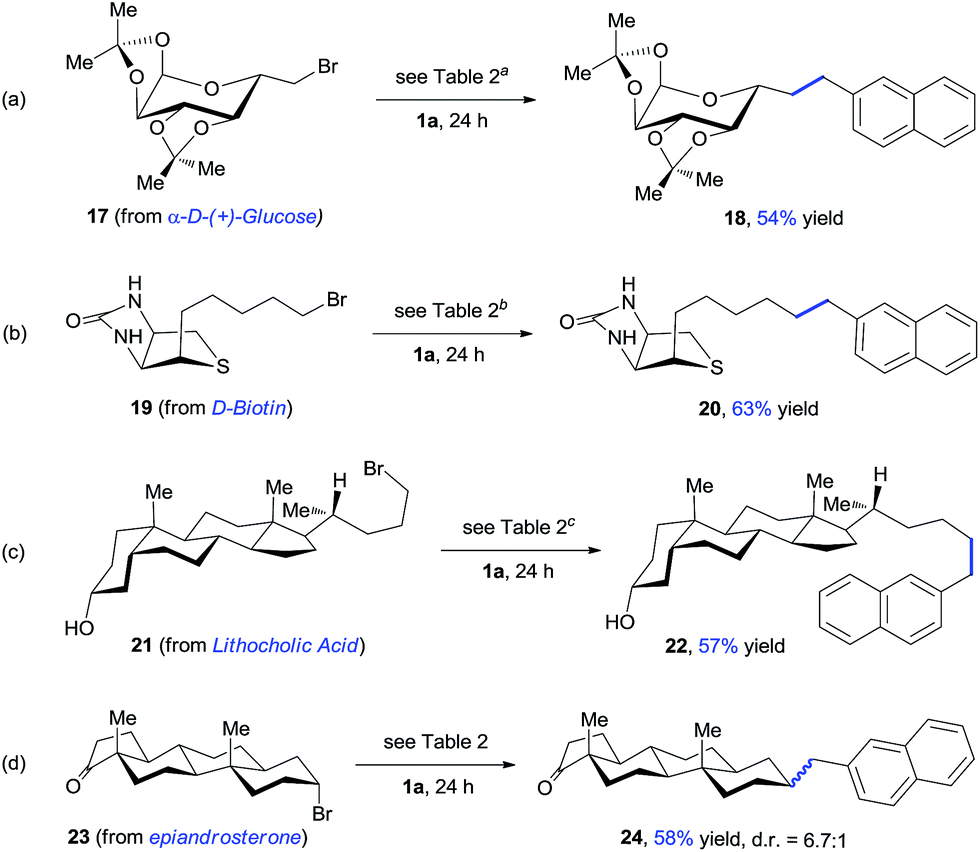 | ||
| Scheme 3 Late-stage modification of biologically active molecules. aOxalate 1a (2.0 equiv.), 20 mol% NiBr2, 45 °C. b20 mol% NiBr2, 45 °C. c20 mol% NiBr2. | ||
Use of the optically pure alkyl bromide 23 gave 24 with a dr of 6.7:1, indicating a potential radical mechanism (Scheme 3d). The reaction of 1a and 2a was significantly inhibited in the presence of a radical scavenger such as TEMPO and 1,1-diphenylethylene (Scheme 4a). The radical trapping product 25 was obtained with 50% NMR yield. Further, radical clock experiments revealed that the reaction of cyclopropylmethyl bromide 26 only produced the ring opening product 28 (Scheme 4b), which is consistent with the process of rearrangement of the cyclopropylmethyl radical to the homoallyl radical.16 These results suggest the presence of a C-centered radical in the reaction pathway.
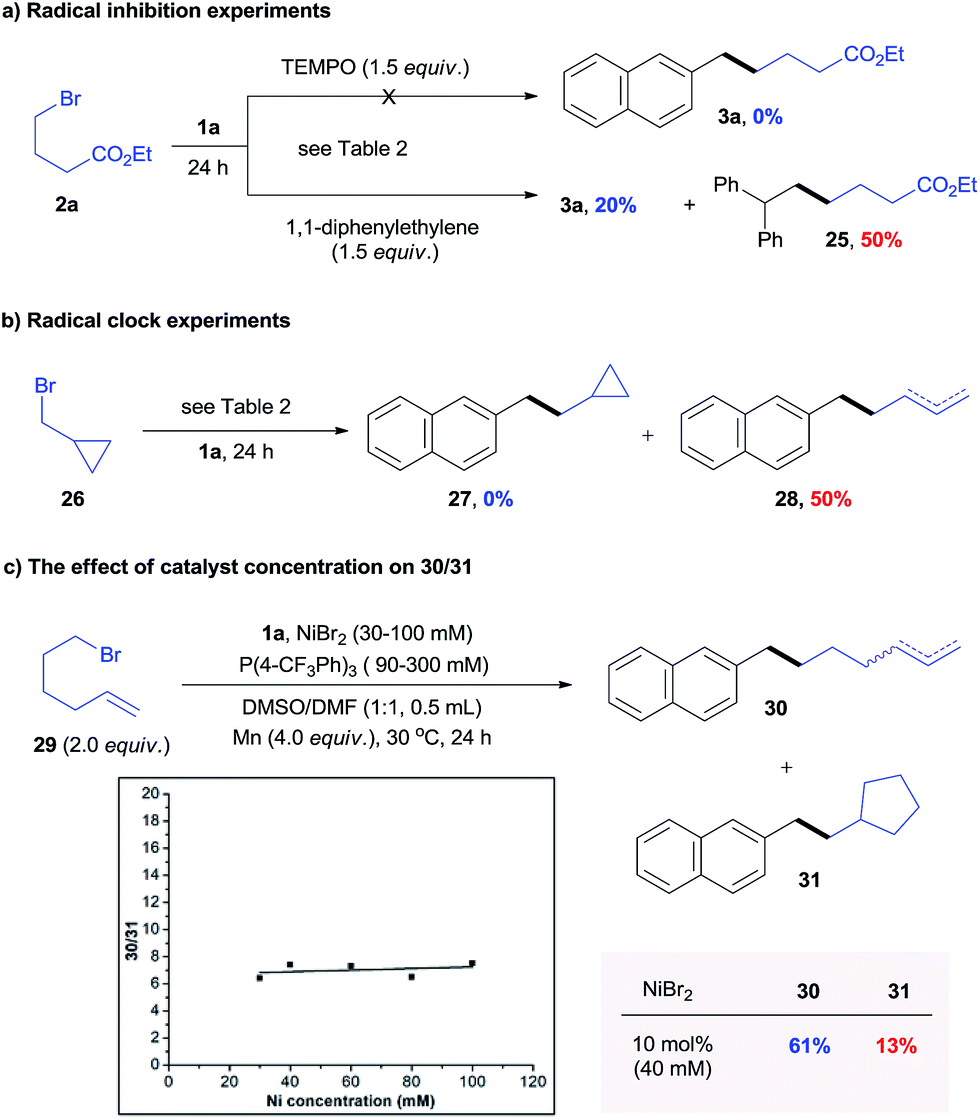 | ||
| Scheme 4 Mechanistic studies. | ||
To verify a potential radical chain mechanism, the effect of catalyst concentrations on products of reaction 1a with 6-bromo-1-hexene 29 was then studied (Scheme 4c).17 Under higher catalyst concentrations, the C-centered radical was more easily trapped by catalysts before cyclization.17 We expected that if a radical chain mechanism was to apply, the ratio of un-rearranged to rearranged products (30/31) would increase with increasing catalyst concentration. However, our result in Scheme 4c shows that 30/31 was not dependent upon catalyst concentrations. This result indicates that the oxidative addition of the alkyl halide appears to occur via a non-chain process.18
Conclusions
In summary, we have demonstrated a nickel-catalyzed cross-coupling of benzyl carboxylate with alkyl bromide by using oxalate as a leaving group. This study suggests that the reductive cross-coupling of electrophiles might be the foundation for new discoveries in the field of metal-catalyzed cleavages of the C–O bond. This method's excellent functional group compatibility suggests that it can be a powerful alternative to established protocols using alkyl nucleophiles for the alkylation of benzylic derivatives. Further mechanistic investigation and extension to other electrophiles are ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support from NSFC (21502078, 21772072), FRFCU (lzujbky-2016-ct09), and 1000 Talents Plan Program.Notes and references
- Recent reviews: (a) F. Diederich and P. J. Stang, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, Germany, 1998 Search PubMed; (b) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587 CrossRef CAS PubMed.
- Selected reviews: (a) D.-G. Yu, B.-J. Li and Z.-J. Shi, Acc. Chem. Res., 2010, 43, 1486 CrossRef CAS PubMed; (b) B. M. Rosen, K. W. Quasdorf, D. A. Wilkson, N. Zhang, A.-M. Resmerita, N. K. Garg and B. Percec, Chem. Rev., 2011, 111, 1346 CrossRef CAS PubMed; (c) J. Yamaguchi, K. Muto and K. Itami, Eur. J. Org. Chem., 2013, 19 CrossRef CAS; (d) J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081 RSC; (e) M. Tobisu and N. Chatani, Acc. Chem. Res., 2015, 48, 1717 CrossRef CAS PubMed; (f) E. J. Tollefson, L. E. Hanna and E. R. Jarvo, Acc. Chem. Res., 2015, 48, 2344 CrossRef CAS PubMed.
- (a) Z. Li, S.-L. Zhang, Y. Fu, Q.-X. Guo and L. Liu, J. Am. Chem. Soc., 2009, 131, 8815 CrossRef CAS PubMed; (b) X. Hong, Y. Liang and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 2017 CrossRef CAS PubMed.
- Selected references: (a) E. Wenkert, E. L. Michelotti and C. S. Swindell, J. Am. Chem. Soc., 1979, 101, 2246 CrossRef CAS; (b) B.-T. Guan, Y. Wang, B.-J. Li, D.-G. Yu and Z.-J. Shi, J. Am. Chem. Soc., 2008, 130, 14468 CrossRef CAS PubMed; (c) K. W. Quasdorf, X. Tian and N. K. Garg, J. Am. Chem. Soc., 2008, 130, 14422 CrossRef CAS PubMed; (d) H. Duan, L. Meng, D. Bao, H. Zhang, Y. Li and A. Lei, Angew. Chem., Int. Ed., 2010, 49, 6387 CrossRef CAS PubMed; (e) K. Muto, J. Yamaguchi and K. Itami, J. Am. Chem. Soc., 2012, 134, 169 CrossRef CAS PubMed; (f) Y. Zhao and V. Snieckus, J. Am. Chem. Soc., 2014, 136, 11224 CrossRef CAS PubMed; (g) T. Iwasaki, Y. Miyata, R. Akimoto, Y. Fujii, H. Kuniyasu and N. Kambe, J. Am. Chem. Soc., 2014, 136, 9260 CrossRef CAS PubMed; (h) Q. Zhou, K. M. Cobb, T. Tan and M. P. Watson, J. Am. Chem. Soc., 2016, 138, 12057 CrossRef CAS PubMed.
- Selected reviews on catalytic alkylation reactions with alkylmetal reagents: (a) J. Choi and G. C. Fu, Science, 2017, 356, 152 CrossRef CAS PubMed; (b) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed. Recent elegant works on the alkylation of unreactive C–O electrophiles: (c) M. Leiendecker, C.-C. Hsiao, L. Guo, N. Alandini and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 12912 CrossRef CAS PubMed; (d) D. Gärtner, A. L. Stein, S. Grupe, J. Arp and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2015, 54, 10545 CrossRef PubMed; (e) M. Tobisu, T. Takahira, T. Morioka and N. Chatani, J. Am. Chem. Soc., 2016, 138, 6711 CrossRef CAS PubMed.
- Selected elegant works: (a) B.-T. Guan, S.-K. Xiang, B.-Q. Wang, Z.-P. Sun, Y. Wang, K.-Q. Zhao and Z.-J. Shi, J. Am. Chem. Soc., 2008, 130, 3268 CrossRef CAS PubMed; (b) B. L. H. Taylor, E. C. Swift, J. D. Waetzig and E. R. Jarvo, J. Am. Chem. Soc., 2011, 133, 389 CrossRef CAS PubMed; (c) H. M. Wisniewska, E. C. Swift and E. R. Jarvo, J. Am. Chem. Soc., 2013, 135, 9083 CrossRef CAS PubMed; (d) E. J. Tollefson, D. D. Dawson, C. A. Osborne and E. R. Jarvo, J. Am. Chem. Soc., 2014, 136, 14951 CrossRef CAS PubMed; (e) I. M. Yonova, A. G. Johnson, C. A. Osborne, C. E. Moore, N. S. Morrissette and E. R. Jarvo, Angew. Chem., Int. Ed., 2014, 53, 2422 CrossRef CAS PubMed.
- Selected elegant works on the coupling of activated allylic carboxylates with alkyl electrophiles, see: (a) X. Qian, A. Auffrant, A. Felouat and C. Gosmini, Angew. Chem., Int. Ed., 2011, 50, 10402 CrossRef CAS PubMed; (b) L. L. Anka-Lufford, M. R. Prinsell and D. J. Weix, J. Org. Chem., 2012, 77, 9989 CrossRef CAS PubMed; (c) H. Chen, X. Jia, Y. Yu, Q. Qian and H. Gong, Angew. Chem., Int. Ed., 2017, 56, 13103 CrossRef CAS PubMed. Limited reports on reductive coupling of unreactive C–O electrophiles, see: homo-coupling: (d) Z.-C. Cao and Z.-J. Shi, J. Am. Chem. Soc., 2017, 139, 6546 CrossRef CAS PubMed. With π-electrophiles: (e) A. Correa, T. León and R. Martin, J. Am. Chem. Soc., 2014, 136, 1062 CrossRef CAS PubMed; (f) A. Correa and R. Martin, J. Am. Chem. Soc., 2014, 136, 7253 CrossRef CAS PubMed. With aryl electrophiles: (g) M. O. Konev, L. E. Hanna and E. R. Jarvo, Angew. Chem., Int. Ed., 2016, 55, 6730 CrossRef CAS PubMed; (h) Z.-C. Cao, Q.-Y. Luo and Z.-J. Shi, Org. Lett., 2016, 18, 5978 CrossRef CAS PubMed; (i) B. A. Vara, N. R. Patel and G. A. Molander, ACS Catal., 2017, 7, 3955 CrossRef CAS PubMed; (j) X.-G. Jia, P. Guo, J.-C. Duan and X.-Z. Shu, Chem. Sci., 2018, 9, 640 RSC. With alkyl electrophiles (intramolecular): (k) E. J. Tollefson, L. W. Erickson and E. R. Jarvo, J. Am. Chem. Soc., 2015, 137, 9760 CrossRef CAS PubMed; (l) L. W. Erickson, E. L. Lucas, E. J. Tollefson and E. R. Jarvo, J. Am. Chem. Soc., 2016, 138, 14006 CrossRef CAS PubMed.
- Recent reviews: (a) C. E. I. Knappke, S. Grupe, D. Gärtner, M. Corpet, C. Gosmini and A. Jacobi von Wangelin, Chem.–Eur. J., 2014, 20, 682 CrossRef PubMed; (b) T. Moragas, A. Correa and R. Martin, Chem.–Eur. J., 2014, 20, 8242 CrossRef CAS PubMed; (c) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767 CrossRef CAS PubMed; (d) J. Gu, X. Wang, W. Xue and H. Gong, Org. Chem. Front., 2015, 2, 1411 RSC. Selected references on nickel catalysis: (e) D. A. Everson, R. Shrestha and D. J. Weix, J. Am. Chem. Soc., 2010, 132, 920 CrossRef CAS PubMed; (f) H. Xu, C. Zhao, Q. Qian, W. Deng and H. Gong, Chem. Sci., 2013, 4, 4022 RSC; (g) Y. Zhao and D. J. Weix, J. Am. Chem. Soc., 2014, 136, 48 CrossRef CAS PubMed; (h) C. Zhao, X. Jia, X. Wang and H. Gong, J. Am. Chem. Soc., 2014, 136, 17645 CrossRef CAS PubMed; (i) A. H. Cherney and S. E. Reisman, J. Am. Chem. Soc., 2014, 136, 14365 CrossRef CAS PubMed; (j) K. M. Arendt and A. G. Doyle, Angew. Chem., Int. Ed., 2015, 54, 9876 CrossRef CAS PubMed; (k) L. K. G. Ackerman, L. L. Anka-Lufford, M. Naodovic and D. J. Weix, Chem. Sci., 2015, 6, 1115 RSC; (l) L. Hu, X. Liu and X. Liao, Angew. Chem., Int. Ed., 2016, 55, 9743 CrossRef CAS PubMed; (m) A. García-Domínguez, Z. Li and C. Nevado, J. Am. Chem. Soc., 2017, 139, 6835 CrossRef PubMed; (n) X. Lu, Y. Wang, B. Zhang, J.-J. Pi, X.-X. Wang, T.-J. Gong, B. Xiao and Y. Fu, J. Am. Chem. Soc., 2017, 139, 12632 CrossRef CAS PubMed.
- (a) T.-Y. Luh, M.-K. Leung and K.-T. Wong, Chem. Rev., 2000, 100, 3187 CrossRef CAS PubMed; (b) D. A. Everson and D. J. Weix, J. Org. Chem., 2014, 79, 4793 CrossRef CAS PubMed.
- Oxalate substrates are readily available from methyl chlorooxoacetate (0.56$ per g, HEOWNS). For elegant works using oxalate acids as radical precursors to couple with aryl halides and activated alkenes by metallophotoredox catalysis, see: (a) C. C. Nawrat, C. R. Jamison, Y. Slutskyy, D. W. C. MacMillan and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 11270 CrossRef CAS PubMed; (b) X. Zhang and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 13862 CrossRef CAS PubMed.
- Selected reviews: (a) B. Liégault, J.-L. Renaud and C. Bruneau, Chem. Soc. Rev., 2008, 37, 290 RSC; (b) J. L. Bras and J. Muzart, Eur. J. Org. Chem., 2016, 2565 CrossRef. Selected examples: (c) R. Kuwano, Y. Kondo and Y. Matsuyama, J. Am. Chem. Soc., 2003, 125, 12104 CrossRef CAS PubMed; (d) B. M. Trost and L. C. Czabaniuk, J. Am. Chem. Soc., 2012, 134, 5778 CrossRef CAS PubMed.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, Sausalito, CA (USA), 2006 Search PubMed.
- The chelation of in situ formed Mn2+ to bidentate leaving groups might weaken the C–O bond, thus accelerating the rate of oxidative addition. For related references, see: B. L. Taylor, M. R. Harris and E. R. Jarvo, Angew. Chem., Int. Ed., 2012, 51, 7790 CrossRef CAS PubMed . Also see: ref. 6c and ref. 7e. We also observed that the use of extra Lewis acid significantly accelerated the conversion of oxalate (see Fig. S1 and S2†). Unfortunately, those reactions using Lewis acids did not improve the yields of the desired product.
- At present, the reasons for the success of the oxalate is still not clear. We tentatively suggested that both the high leaving ability (pKa: oxalic acid 1.27, CF3CO2H 0.52, CH3CO2H 4.76) and bidentate nature of oxalate might be important for the reaction.
- D. J. Brauer and C. Krueger, Inorg. Chem., 1977, 16, 884 CrossRef CAS.
- J. P. Stevenson, W. F. Jackson and J. M. Tanko, J. Am. Chem. Soc., 2002, 124, 4271 CrossRef CAS PubMed.
- (a) S. Biswas and D. J. Weix, J. Am. Chem. Soc., 2013, 135, 16192 CrossRef CAS PubMed; (b) J. Breitenfeld, J. Ruiz, M. D. Wodrich and X. Hu, J. Am. Chem. Soc., 2013, 135, 12004 CrossRef CAS PubMed.
- (a) G. D. Jones, J. L. Martin, C. McFarland, O. R. Allen, R. E. Hall, A. D. Haley, R. J. Brandon, T. Konovalova, P. J. Desrochers, P. Pulay and D. A. Vicic, J. Am. Chem. Soc., 2006, 128, 13175 CrossRef CAS PubMed; (b) X. Lin and D. L. Phillips, J. Org. Chem., 2008, 73, 3680 CrossRef CAS PubMed; (c) A. Wilsily, F. Tramutola, N. A. Owston and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 5794 CrossRef CAS PubMed; (d) A. S. Dudnik and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 10693 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc00609a |
| This journal is © The Royal Society of Chemistry 2018 |