
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into the mechanism and aging of a noble-metal free H2-evolving dye-sensitized photocathode†
Nicolas
Kaeffer‡§
 a,
Christopher D.
Windle‡¶
a,
Romain
Brisse||
b,
Corinne
Gablin
c,
Didier
Leonard
c,
Bruno
Jousselme
b,
Murielle
Chavarot-Kerlidou
a and
Vincent
Artero
*a
a,
Christopher D.
Windle‡¶
a,
Romain
Brisse||
b,
Corinne
Gablin
c,
Didier
Leonard
c,
Bruno
Jousselme
b,
Murielle
Chavarot-Kerlidou
a and
Vincent
Artero
*a
aLaboratoire de Chimie et Biologie des Métaux, Université Grenoble Alpes, CNRS UMR 5249, CEA, 17 rue des Martyrs, F-38054 Grenoble, Cedex, France. E-mail: vincent.artero@cea.fr; Web: http://www.solhycat.com
bLaboratory of Innovation in Surface Chemistry and Nanosciences (LICSEN), NIMBE, CEA, CNRS, Université Paris-Saclay, CEA Saclay, 91191 Gif-sur-Yvette, Cedex, France
cUniv Lyon, CNRS, Université Claude Bernard Lyon 1, ENS de Lyon, Institut des Sciences Analytiques, UMR 5280, 5, rue de la Doua, F-69100 Villeurbanne, France
First published on 10th July 2018
Abstract
Dye-sensitized photo-electrochemical cells (DS-PECs) form an emerging technology for the large-scale storage of solar energy in the form of (solar) fuels because of the low cost and ease of processing of their constitutive photoelectrode materials. Preparing such molecular photocathodes requires a well-controlled co-immobilization of molecular dyes and catalysts onto transparent semiconducting materials. Here we used a series of surface analysis techniques to describe the molecular assembly of a push–pull organic dye and a cobalt diimine–dioxime catalyst co-grafted on a p-type NiO electrode substrate. (Photo)electrochemical measurements allowed characterization of electron transfer processes within such an assembly and to demonstrate for the first time that a CoI species is formed as the entry into the light-driven H2 evolution mechanism of a dye-sensitized photocathode. This co-grafted noble-metal free H2-evolving photocathode architecture displays similar performances to its covalent dye–catalyst counterpart based on the same catalytic moiety. Post-operando time-of-flight secondary ion mass spectrometry (ToF-SIMS) analysis of these photoelectrodes after extensive photoelectrochemical operation suggested decomposition pathways of the dye and triazole linkage used to graft the catalyst onto NiO, providing grounds for the design of optimized molecular DS-PEC components with increased robustness upon turnover.
Introduction
The production of fuels via artificial photosynthesis is a promising solution for the storage of solar energy.1–3 Among other solar fuel technologies, photoelectrochemical cells (PECs) couple light harvesting with fuel-forming catalysis in a single device.4 Dye-sensitized PECs (DS-PECs) are a particular class of such devices that relies on molecular active components – a catalyst and a photosensitizer – immobilized at the surface of transparent conductive electrode substrates, extending the concept of (tandem) dye-sensitized solar cell (DSSC) technology.5–10 In that frame, significant achievements have been reported in the last four years regarding the preparation of dye-sensitized photoanodes achieving water oxidation8,9,11–15 as well as photocathodes active for H2 production13,16–28 or CO2 reduction,29,30 including their implementation in operating tandem DS-PECs.20,30–32 The development of dye-sensitized photoanodes has reached milestones owing to surface engineering approaches14,15 leading to improved light-harvesting and catalytic efficiencies, coupled to the comprehension of degradative mechanisms33 and their circumvention. In contrast, H2-evolving photocathodes, which only came to light recently, are lagging behind in terms of understanding and triggering factors that govern their activity and are thus the weak part in the construction of tandem DS-PECs. The challenging development of these photocathodes has explored several architectures that use either layer-by-layer,21 supramolecular,18 covalent22,25 assemblies or co-grafting19,20,26,34–36 to couple a catalyst and a dye on a suitable cathode substrate, typically p-type semiconducting nickel oxide (NiO).37,38 Among them, the co-grafting strategy has proven effective to prepare photocathodes implemented in the first operating tandem DS-PECs.19,20 However, direct comparison with other architectures has never been addressed in the literature to date, and while detailed mechanistic information becomes available for co-grafted photocathodes based on FeFe H2-evolving catalysts,26,34,36 such information is still scarce on these best performing architectures exploiting the popular cobaloxime and diimine–dioxime cobalt catalysts.18–20,22,25 Finally, in-depth, including post-operando, molecular characterization of such architectures is also lacking, although they are direct prerequisites to further improve performance.In this study, we developed and characterized a novel co-grafted photocathode combining the organic push–pull dye RBG-174 (Fig. 1), previously reported for p-DSSC applications,39,40 with a novel derivative of the cobalt diimine–dioxime hydrogen-evolving catalyst [Co(DO)(DOH)pnBr2],41CoC11P, bearing a terminal phosphonic acid anchoring group (Fig. 1). Full characterization at the molecular level of the co-sensitized NiO photocathode was achieved using a combination of X-ray photoelectron spectroscopy (XPS), time of flight-secondary ion mass spectrometry (ToF-SIMS), and UV-visible absorption spectroscopy. Importantly, we use electrochemistry to describe electron transfer processes and catalyst activation within mesoporous NiO films. Under visible light irradiation, the photo-electrochemical generation of the reduced Co(I) state of CoC11P was demonstrated, as an entry point into H2 evolution catalysis. This construction proved operational for photoelectrochemical H2 evolution under fully aqueous conditions with performances similar to the previously reported covalent dye–catalyst assembly design based on the same molecular catalyst.22 Finally, we show here that ToF-SIMS surface analysis is instrumental in identifying decomposition pathways of the grafted molecular components.
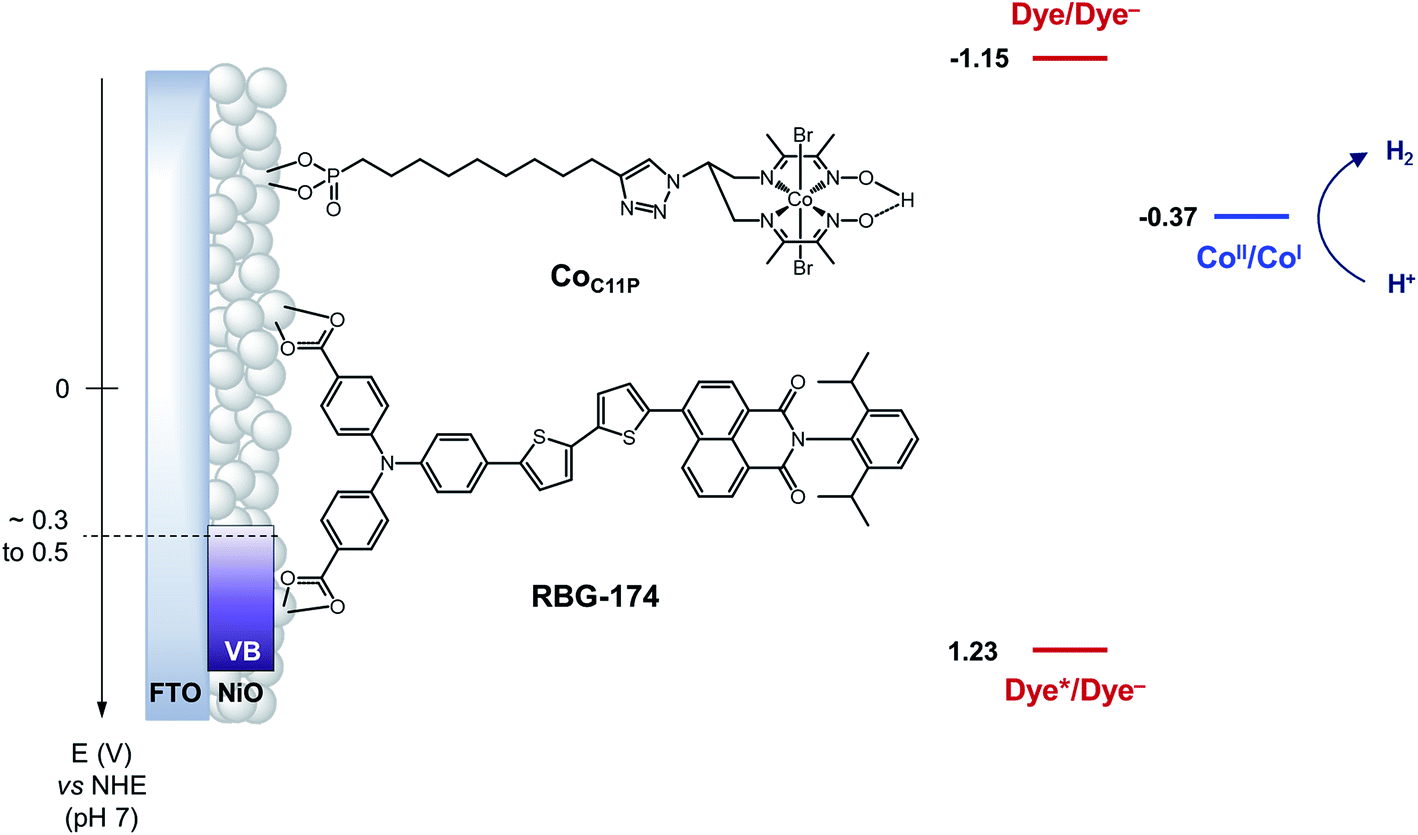 | ||
| Fig. 1 Schematic energy diagram of the NiOǀRBG-174ǀCoC11P photocathode. See text for the estimation of energy levels at pH = 7. Potentials for the redox couples of RBG-174 have been determined as described in ref. 48 from data found in ref. 40. | ||
Experimental part
Reagents and materials
Solvents, starting materials, and supporting electrolyte salts were purchased from Sigma-Aldrich and used without further purification, unless otherwise stated. F108 polymer and decylphosphonic acid (C10P) were purchased from Sigma-Aldrich. Anhydrous NiCl2 was purchased from Strem and stored in a glovebox. Diethyl undec-10-yn-1-ylphosphonate (PEtC9C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH) was purchased from Sikemia. N2-N2′-(2-Azidopropane-1,3-diyl)bis(butan-2-imine-3-oxime) ((DOH)2N3pn) was custom-synthesized by Provence Technologies according to our previously reported procedure.42 [Co(DO)(DOH)pn]Br2 and [Cu(DO)(DOH)N3pn(OH2)](ClO4) were prepared following reported procedures.42,43 The synthesis of RBG-174 has been described elsewhere.39 Dry dichloromethane and dry acetonitrile were obtained by distillation on CaH2 and kept under Ar. Unless otherwise stated, all reactions in solution were performed at room temperature under atmospheric conditions. Two types of NiO substrates were used. Screen-printed NiO films (two layers, ∼1.7 μm thick) were purchased from Dyenamo AB, Stockholm, Sweden.38 Alternatively, spin-coated NiO films (∼0.5 μm thick) were prepared using F108-templated sol–gel chemistry as previously described.22
CH) was purchased from Sikemia. N2-N2′-(2-Azidopropane-1,3-diyl)bis(butan-2-imine-3-oxime) ((DOH)2N3pn) was custom-synthesized by Provence Technologies according to our previously reported procedure.42 [Co(DO)(DOH)pn]Br2 and [Cu(DO)(DOH)N3pn(OH2)](ClO4) were prepared following reported procedures.42,43 The synthesis of RBG-174 has been described elsewhere.39 Dry dichloromethane and dry acetonitrile were obtained by distillation on CaH2 and kept under Ar. Unless otherwise stated, all reactions in solution were performed at room temperature under atmospheric conditions. Two types of NiO substrates were used. Screen-printed NiO films (two layers, ∼1.7 μm thick) were purchased from Dyenamo AB, Stockholm, Sweden.38 Alternatively, spin-coated NiO films (∼0.5 μm thick) were prepared using F108-templated sol–gel chemistry as previously described.22
Synthesis
CH (108 mg, 0.24 mmol, 1.0 equiv.; Fig. 2) were introduced in 6 mL of a methanol/water (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture, without complete dissolution of the starting materials. The mixture was flushed with Ar, and sodium ascorbate (43 mg, 0.22 mmol, 0.9 equiv.) and CuSO4·5H2O (18 mg, 0.07 mmol, 0.3 equiv.) were added. The mixture was stirred at room temperature for 2 days. The reaction mixture was concentrated. The crude mixture (containing the perchlorate salt of CuC11PEt) was purified by flash chromatography on silica gel using a gradient (39:1 to 8:2) of MeCN/aqueous KNO3 solution (saturated solution diluted at 10%).22,43 The purified fraction contains CuC11PEt as a nitrate salt. MS (ESI+): m/z calcd for C26H47CuN7O5P 631.3; found 631.3 [M − OH2 − NO3−]+. It was dissolved in the minimum volume of MeOH and precipitated by addition of an aqueous KPF6-saturated solution to yield CuC11PEt (hexafluorophosphate salt) (86 mg, 48% yield; Fig. 2). HR-MS (ESI+): m/z calcd for C26H47CuN7O5P 631.2667; found 631.2668 [M − OH2 − PF6−]+.
:1) mixture, without complete dissolution of the starting materials. The mixture was flushed with Ar, and sodium ascorbate (43 mg, 0.22 mmol, 0.9 equiv.) and CuSO4·5H2O (18 mg, 0.07 mmol, 0.3 equiv.) were added. The mixture was stirred at room temperature for 2 days. The reaction mixture was concentrated. The crude mixture (containing the perchlorate salt of CuC11PEt) was purified by flash chromatography on silica gel using a gradient (39:1 to 8:2) of MeCN/aqueous KNO3 solution (saturated solution diluted at 10%).22,43 The purified fraction contains CuC11PEt as a nitrate salt. MS (ESI+): m/z calcd for C26H47CuN7O5P 631.3; found 631.3 [M − OH2 − NO3−]+. It was dissolved in the minimum volume of MeOH and precipitated by addition of an aqueous KPF6-saturated solution to yield CuC11PEt (hexafluorophosphate salt) (86 mg, 48% yield; Fig. 2). HR-MS (ESI+): m/z calcd for C26H47CuN7O5P 631.2667; found 631.2668 [M − OH2 − PF6−]+.
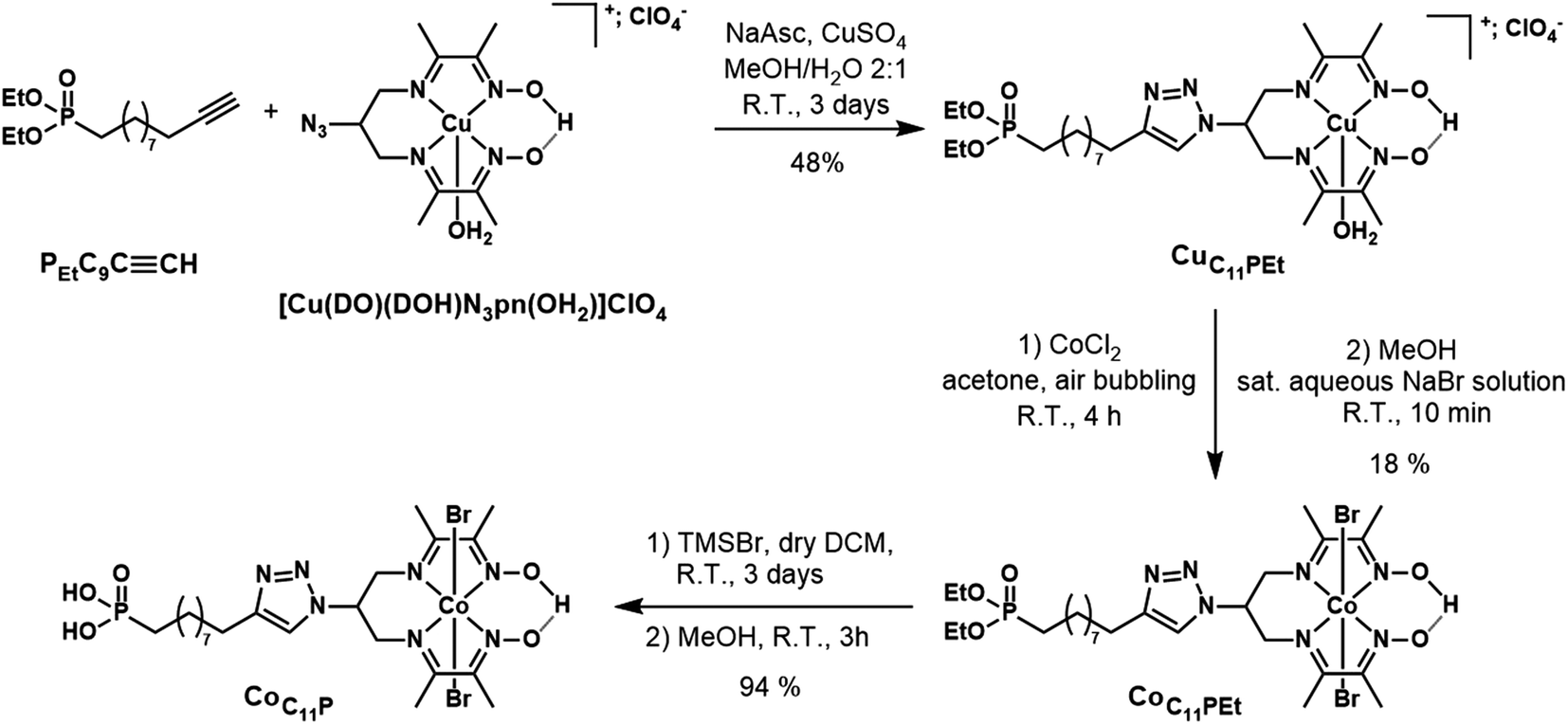 | ||
| Fig. 2 Synthetic pathway to the CoC11P phosphonic acid derivative. | ||
:0 to 8:2 gradient of MeCN/aqueous KNO3 solution (saturated solution diluted at 10%). The solid obtained was dissolved in a minimal amount of MeOH and an aqueous NaBr-saturated solution was added until a green compound precipitated. After concentration to remove MeOH, the precipitate was filtered and washed with several fractions of water before drying under vacuum to yield the expected CoC11PEt compound as a green powder (42 mg, 18% yield; Fig. 2). 1H NMR (CD3CN, 300 MHz): δ (ppm) 19.28 (s, 1H, bridging H), 7.85 (broad s, 1H), 5.76–5.50 (m, 1H), 4.67–4.31 (m, 4H), 4.10–3.84 (m, 4H), 2.79–2.62 (m, 2H), 2.59 (s, 6H), 2.54 (s, 6H) 1.78–1.61 (m, 4H), 1.60–1.44 (m, 2H), 1.45–1.29 (m, 10H), 1.26 (t, 6H, J = 7.1 Hz). 13C NMR (CD3CN, 75 MHz): δ (ppm) 177.9, 157.9, 148.8, 122.4, 62.1, 62.0, 58.6, 31.2, 31.0, 30.2, 30.0, 29.8, 29.8, 26.8, 26.2, 25.0, 23.3, 23.2, 18.6, 16.9, 16.8, 14.1. 31P NMR (CD3CN, 121 MHz): δ (ppm) 33.30. MS (ESI+): m/z calcd for C26H47BrCoN7O5P 706.2; found 706.1 [M − Br−]+. HR-MS (ESI+): m/z calcd for C26H47BrCoN7O5P 706.1886; found 706.1887 [M − Br−]+.
Molecular grafting onto NiO films
Freshly prepared spin-coated NiO substrates were treated in an ozone cleaner (Ossila E511) for 10 minutes just before dipping into the sensitizing bath. Commercial Dyenamo substrates were sintered under atmospheric conditions in a Harry Gestigkeit flat titanium furnace operated with a Detflef Gestigkeit-Programmer PR5 control board with the following temperature program: R.T. to 450 °C (30 min), 30 min at 450 °C. The substrates were cooled to ca. 90 °C before dipping into the sensitizing bath. Sensitization was performed in PTFE boxes containing 10 mL of a CoC11P or a decylphosphonic acid (C10P) methanolic solution (0.5 mM; 5 μmol of compound). Between 3 and 6 electrodes were introduced in the same PTFE box. The boxes were closed and the electrodes were left for 24 h under orbital stirring. The electrodes were removed from the solution, washed by dipping into pristine methanol for 10 min under orbital stirring and dried with N2, to yield the CoC11P-modified (or C10P-modified) electrodes.For co-grafting, Dyenamo films were first sintered at 450 °C, then cooled to ca. 90 °C and a first UV-visible spectrum of the NiO electrodes (blank) was recorded. Then, the films were heated again (ca. 90 °C) for 5 min. The electrodes were then dipped in the grafting solution for 66 h under orbital stirring. The films were removed from the solution, washed by dipping into pristine methanol for 5–10 min under orbital stirring and dried with N2 to give the NiOǀCoC11P electrodes. A second UV-visible spectrum of the sensitized NiO electrodes was recorded at this stage. The plates were further dipped in PTFE boxes containing 10 mL of a 0.5 mM RBG-174 (5 μmol) methanolic solution. The boxes were closed and the electrodes allowed to sensitize for 2 h under orbital stirring. The films were removed from the solution, washed by dipping for 10 min in methanol under orbital stirring and dried with N2 to yield the NiOǀRBG-174ǀCoC11P electrode. A final UV-visible spectrum of the co-sensitized NiO electrodes was recorded. NiOǀCoC11P control electrodes were kept after the first grafting stage. A distinct procedure was used to co-sensitize spin-coated NiO substrates. These substrates require ozone treatment before soaking overnight in CoC11P or decylphosphonic acid (0.5 mM, CH3OH) solution, rinsed with CH3OH for 5 min and then soaked in RBG-174 (0.5 mM, CH2Cl2) solution for 15 min. Reference NiOǀRBG-174 electrodes were prepared from Dyenamo NiO films sintered with the temperature program described above, but allowed to cool down to R.T. before sensitization. A first UV-visible spectrum of the pristine NiO electrodes was recorded. Then, the plates were dipped into a 0.5 mM RBG-174 methanolic solution (10 mL). The boxes were closed and the electrodes allowed to sensitize for 2 h under orbital stirring. The films were removed from the solution, washed by dipping into pristine methanol for 10 min (orbital stirring) and dried with N2 to yield the NiOǀRBG-174 control electrodes. Finally, a UV-visible spectrum of the sensitized NiO electrodes was recorded.
Physical measurements
NMR spectra were recorded at 298 K in 5 mm o.d. tubes on a Bruker 300 spectrometer equipped with a QNP probehead operating at 300.0 MHz for 1H, 75.5 MHz for 13C and 121.5 MHz for 31P. Chemical shifts reported in ppm are given relative to TMS. 1H and 13C NMR spectra were referenced internally to the residual solvent resonance. 31P NMR spectra were calibrated against an external 85% H3PO4 reference or an internal H3PO4 85% reference by inserting a glass-sealed pipette tip filled with 85% H3PO4 standard in the 5 mm o.d. tube containing a solution of the compound to be analyzed. Mass spectra were acquired on a Thermo Scientific LXQ mass spectrometer equipped with an electrospray source, at the SYMMES laboratory of INAC (CEA-Grenoble). Accurate mass measurements (HRMS) were performed on a Bruker maXis mass spectrometer by the “Fédération de Recherche” ICOA/CBM (FR2708) platform and elemental analysis on a Thermofisher Scientific “Flash 2000” by the “Plateforme d'analyse pour la chimie” (GDS 3648, Strasbourg). UV-visible absorption spectra of the plates were recorded with an Agilent Technologies Cary 60 UV-Vis spectrometer. For each spectrum, a baseline corresponding to the related underlying glass|FTO support was recorded and automatically subtracted from the spectrum of the electrode. Differential spectra were obtained by subtraction of spectra prior and after sensitization with RBG-174. 2 m-long optic fibers and coupling unit (Hellma) were used to measure spectra in the glovebox. XPS analyses were performed with a VersaProbe II spectrometer from Physical Electronics using a high-resolution monochromatic Al-Kα line X-ray source at 1486.7 eV. A fixed analyzer pass energy of 23 eV was used for core level scans leading to an overall energy resolution of 0.6 eV. Survey spectra were captured at pass energy of 117 eV. The photoelectron take-off angle was 45°, which provided an integrated sampling depth of approximately 5 nm. All spectra were referenced against an internal signature, typically by adjusting the C 1s (C–C/C–H) level peak at a binding energy of 284.8 eV. When necessary, the background was fitted by a 4th order polynomial contribution that was then subtracted to obtain background-subtracted spectra. Time-of-Flight Secondary Ion Mass Spectrometry (ToF-SIMS) were recorded on a Physical Electronics TRIFT III ToF-SIMS instrument operated with a pulsed 22 keV Au+ ion gun (ion current of 2 nA) rastered over a 300 μm × 300 μm area. An electron gun was operated in pulsed mode at low electron energy for charge compensation. Ion dose was kept below the static conditions limit. Data were analyzed using the WinCadence™ software. Mass calibration was performed on hydrocarbon secondary ions.Electrochemistry
Electrochemical data were acquired with a Biologic VSP 300 or a Princeton Applied Research Ametek PARSTAT 4000 potentiostat for measurements in the glovebox. Electrochemical measurements were conducted in 3-electrode cells made of two compartments separated by a porous glass frit or a Nafion® N-117 (180 μm, 0.90 meq g−1 exchanging capacity) membrane in the case of aqueous electrolytes. The working electrode was a decorated or a pristine NiO electrode (Dyenamo). In one case, the NiO electrode was covered by a galvanoplastic scotch tape bearing a hole of 8 mm diameter to determine the electroactive area (S = 0.5 cm2) and clamped against a 9 mm diameter hole in the electrochemical cell, with an intercalary O-ring seal to ensure tightness at the junction. In the other case, the electroactive area was determined by applying a galvanoplastic tape at the top of the mesoporous NiO part, which was dipped in the electrolyte. The electrical contact was taken at the FTO part of the electrodes. The reference electrode was made of a Ag/AgCl wire dipped into a KCl 3 M aqueous solution, separated from the supporting electrolyte by a Vycor® frit, and denoted below as Ag/AgCl. The counter electrode was a titanium wire. The working and reference electrodes were placed in the same compartment and the counter electrode in the other one. A NaCl 0.1 M aqueous solution (approx. pH 6.4) was used as the supporting electrolyte for the electrochemical characterization of the plates. The supporting electrolyte was degassed with a flow of water-saturated N2 for at least 10 min before the measurements. The N2 flow was removed from the solution but left in the headspace of the cell for the duration of the experiment. Cyclic voltammograms were typically conducted at a scan rate of 10 mV s−1. The potential of the reference electrode was calibrated at the beginning and the end of each day in an external electrochemical setup, using a glassy carbon electrode (S = 0.196 cm2) and a platinum wire as working and auxiliary electrodes, respectively. The calibration was realized with [K4Fe(CN)6] in either 0.1 M or 0.9 M potassium phosphate buffer at pH 7 (ref. 44) and conversion of potentials against the Normal Hydrogen Electrode (NHE) and the Reversible Hydrogen Electrode (RHE) was performed as previously described.22 Spectro-electrochemistry measurements were performed in a 1 cm × 1 cm × 4 cm quartz cell as the electrochemical cell, placed in the beam of an Agilent Technologies Cary 60 UV-Vis spectrometer. The working electrode was a pristine or a decorated NiO electrode and was set perpendicular to the analytical beam. A Pt wire and a Ag/AgCl electrode served as counter and reference electrodes, respectively. The NaCl 0.1 M aqueous solution was degassed with Ar prior to the measurements and a constant flow of Ar was let in the headspace of the cell during the acquisition. Chronoamperometric measurements at different applied potentials were performed. UV-visible spectra were acquired after 30 s of equilibration at the considered potential. Spectro-photoelectrochemistry measurements were achieved on functionalized spin-coated NiO electrodes in a glovebox. The sensitized electrode was taken into the glovebox and measurements were made in dry CH3CN with 0.1 M nBu4NPF6 (dried overnight under high vacuum). The counter electrode was a Pt wire and the reference was a leak-free Ag/AgCl (Multichannel Systems). Potentials were referenced to NHE as previously reported.45 Each potential was held for 5 min under irradiation. Then the electrode was disconnected, irradiation was shut down and a UV-visible spectrum was taken.Photoelectrochemistry
The light beam was generated by a Newport ozone-free xenon lamp operated at 280 W mounted with a Spectra-Physics 6123NS liquid filter for elimination of IR radiation and a Spectra-Physics 59472 UV cut-off filter (λ > 400 nm). Incident light flux on the working electrode was calibrated at 50 mW cm−2 (nearly equivalent to 1 sun) with a Newport PM1918-R power-meter coupled to a Newport 919-020-12 photodiode or with a Newport Control 91150V photodiode/power-meter setup. A Jeulin frequency generator was used to drive an automatic shutter via a Newport electronic shutter controller to shine light at regular intervals in the case of chopped-light measurements. Photoelectrochemical measurements were conducted in a specifically designed three-electrode cell made of two compartments separated by a porous glass frit. The working electrode was a functionalized or pristine spin-coated NiO substrate dipped in the electrolyte. The electroactive area was determined by applying galvanoplastic tape at the top of the mesoporous NiO film. The working electrodes were irradiated from the front. A stream of N2 was bubbled in the electrolyte for at least 20 min prior to the measurement, under stirring. Linear sweep voltammograms were recorded at a scan rate of 10 mV s−1, sweeping from anodic to cathodic potentials. Before starting the experiment, the cathodic compartment of the electrochemical cell was tightly closed and the N2 stream was stopped. The volumes of the electrolyte and headspace in the cathodic part were typically ca. 4.0 mL and 1.3 mL, respectively. In a typical experiment, the electrode was poised at the desired potential in the dark for 60 s prior to irradiation. The electrolyte used was a MES (2-(N-morpholino)ethanesulfonic acid) 0.1 M/NaCl 0.1 M aqueous buffer at pH 5.5. GC analysis of the headspace was realized by sampling 50 μL of the headspace with a gas tight syringe through a silicone septum. The content of the syringe was introduced into the injector of a Perkin-Elmer Clarus 500 gas chromatograph equipped with a Porapack Q 80/100 column (6′ 1/8′′) thermostatted at 40 °C and a TCD detector thermostatted at 100 °C. The area under the H2 peak relates to the concentration of hydrogen in the headspace using a calibration curve plotted from known concentrations of H2.Results
Synthesis of a phosphonate-derivatized cobalt diimine–dioxime complex
The phosphonate-derivatized CoC11P complex was prepared using the copper-catalyzed azide–alkyne cycloaddition (CuAAC) of [CuII(DO)(DOH)N3pn(OH2)](ClO4)43 with the commercially available alkyne linker PEtC9CCH featuring a terminal diethyl ester phosphonate anchoring group. The resulting CuC11PEt complex (Fig. 2) was characterized by mass spectrometry before cobalt substitution for copper in the diimine–dioxime coordination sphere.22 The crude mixture was purified by flash chromatography on silica gel using a mixed organic/aqueous KNO3 eluent. This procedure was previously employed for positively charged substituted copper diimine–dioxime complexes.22,43 Under these conditions, anation partially occurs and precipitation with a saturated aqueous NaBr solution yielded the pure CoC11PEt derivative containing axial bromide ligands. Final deprotection of the diethyl ester phosphonate with TMSBr afforded the phosphonic acid derivative CoC11P with good yields.
Preparation of functionalized NiO electrodes
Mesoporous NiO films were soaked in a 0.5 mM CoC11P methanolic solution to yield the NiOǀCoC11P sensitized electrodes (Fig. 3) as described in detail in the experimental part. Co-sensitized NiOǀRBG-174ǀCoC11P electrodes were obtained using a two-step procedure. Alternatively, decylphosphonic acid (C10P) was used as a cobalt-free CoC11P mimic to obtain NiOǀRBG-174ǀC10P control electrodes with similar surface organization but no catalytic core (Fig. 3). For the sake of comparison, we also prepared NiOǀRBG-174 electrodes by sensitization of the NiO substrates with RBG-174 only (Fig. 3).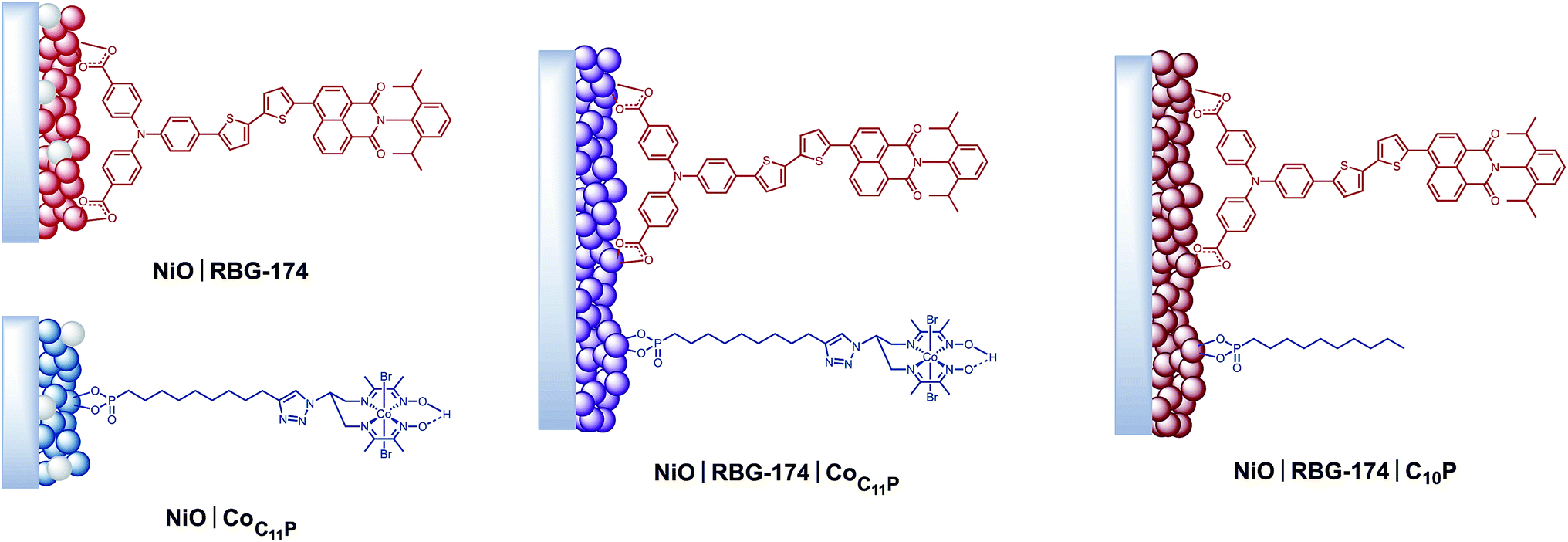 | ||
| Fig. 3 Architectures of the different sensitized electrodes studied in this work. | ||
XPS analyses of the electrodes
The XPS spectrum of NiOǀCoC11P displays two peaks in the Co 2p core level region (Fig. 4 and S4, S5†). These peaks superimpose on the top of a broad and intense signal at 765–790 eV present in the pristine NiO spectrum (Fig. S3–S5†) and likely assigned to some of the LMM Auger lines of Ni.46 A polynomial background subtraction was used to identify the contribution of the Co 2p3/2 and Co 2p1/2 signatures (Fig. 4) of grafted CoC11P centered at 780.8 and 795.6 eV, respectively. A signal in the 398–402 eV range of the N 1s core level region (Fig. S6†) corresponds to N atoms present in grafted CoC11P.47 The P 2p core level region evidences a peak centered at 131.8 eV (Fig. S6†), which was assigned to the phosphonate anchoring group.48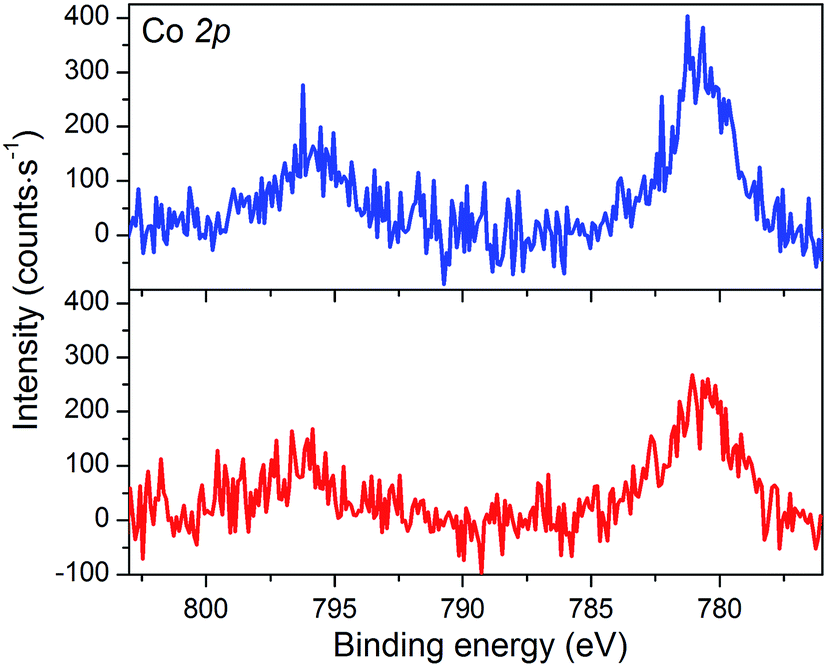 | ||
| Fig. 4 XPS analyses of the NiOǀCoC11P (blue line) and NiOǀRBG-174ǀCoC11P (red line) electrodes at the baseline-corrected Co 2p core level. | ||
The NiOǀRBG-174 electrode displays a broad signal around 400 eV in the N 1s core level region (Fig. S6†) which likely originates from the nitrogen atoms of the triphenylamine49,50 and naphthalene monoimide moieties in RBG-174. The peak centered at 163.8 eV in the S 2p core level region correlates quite well with the one evidenced for thiophene moieties in similar TiO2-immobilized organic backbones.49 This signal is hence ascribed to the two sulfur atoms in RBG-174.
The NiO|RBG-174|CoC11P electrodes display an X-ray photoelectron spectrum in the Co 2p core level region (Fig. 4) similar to that of the NiOǀCoC11P electrodes (Fig. 4) with two peaks at 780.6 and 796.6 eV. Also, the signature of the phosphonic acid anchor of CoC11P is observed (Fig. S6†) with a peak centered around 132 eV in the P 2p core level region. The S 2p signal assigned to the sulfur atoms of RBG-174 is also seen (Fig. S6†). The N 1s core level region displays a large peak at 398–402 eV (Fig. S6†). This peak is likely a combination of the signatures of the N atoms in CoC11P and in RBG-174.
ToF-SIMS analyses of the electrodes
The electrodes were further analyzed by Time-of-Flight Secondary Ion Mass Spectrometry (ToF-SIMS). This technique probes the extreme surface of films to provide mass-related molecular information at the monolayer level, and is thus extremely relevant in the context of co-grafted electrodes. For the NiOǀCoC11P electrode, parts of the spectrum obtained in the positive mode are shown in Fig. 5a and b. Even though no molecular peak was detected, several characteristic secondary ions were identified for the different parts of the molecule, giving a trustful indication of the detection of the complete CoC11P after grafting. Possible fragmentation pathways leading to the main high mass characteristic peaks detected in the positive mode are summarized in Fig. S7†. A set of peaks of interest is located around m/z = 650 (Fig. 5a). In this set, a contribution from CoC11P minus its phosphonate group can be identified, with a theoretical isotopic pattern exhibiting a major peak at m/z = 650.068 (the peak is detected at m/z = 650.09) and secondary ones at m/z = 648.070 (the peak is detected at m/z = 648.07) and 652.066 (the peak is detected at m/z = 652.09) expected to be in the 1/2/1 intensity ratio given the two main isotopes of Br and a two Br-based composition (Fig. S7†). Please note that for the sake of clarity, ToF-SIMS detected m/z are given with two decimal places while the exact m/z of the proposed fragments for peak attributions are given with three decimal places. By the subsequent loss of a Br− ligand, joined contributions can be detected in the set of peaks around m/z = 571, more precisely at m/z = 569.14 and 568.12, respectively (Fig. 5b). These contributions either imply deprotonation or an in-source reduction of CoIII to CoII during MS data collection. Interestingly, a peak at m/z = 627.14 (Fig. 5a) was also observed and was ascribed to the molecule without any Br− ligand but containing Ni and corresponding to the exact mass of 627.129 (Fig. S7†). This signature emphasizes a grafting on the NiO surface via the phosphonate binding, which is also confirmed by the observation of fragments containing Ni, O and P in the negative mode spectra ([NiPO4H]− and [NiPO5H2]− with exact mass 153.897, resp. 170.899, detected at m/z = 153.88, resp. 170.90; Fig. S8†).48 On the other hand, the set of peaks around m/z = 650 (Fig. 5a) clearly exhibits intensity ratios still different from the ones expected for the contribution proposed above (CoC11P minus phosphonate). Another contribution can indeed be CoC11P minus one Br− ligand that should display two major signatures with exact mass of 650.127 and 652.124 in similar intensities (due to the two main isotopes of Br and a one Br-based composition) (Fig. S7†). This fragment can further lose a second Br− ligand with deprotonation or in-source reduction to CoII leading to peaks with exact mass of 570.201 and 571.208, being detected at m/z = 570.13 and 571.17, respectively (Fig. S7†). These signatures are actually observed in the set of peaks around m/z = 571 (Fig. 5b). This second fragmentation pathway might indicate a non-covalent grafting mode (H-bonding of phosphonic acid groups or physisorption) for a certain amount of CoC11P molecules. Of note, both fragmentation pathways described above further lead to the same lower mass fragment (Fig. S7†) corresponding to the molecule after loss of one phosphonate and two Br− ligands with exact mass of 489.227 and detected at m/z = 489.21 (Fig. S9†). This fragment is additionally split into a series of secondary ions by the stepwise losses of CH2 fragments, well correlated with the set of peaks with a m/z = 14 difference (peaks detected at 475.22/461.19/447.19/433.18/419.15/405.15 in Fig. S9†, exact masses are given in Fig. S7†).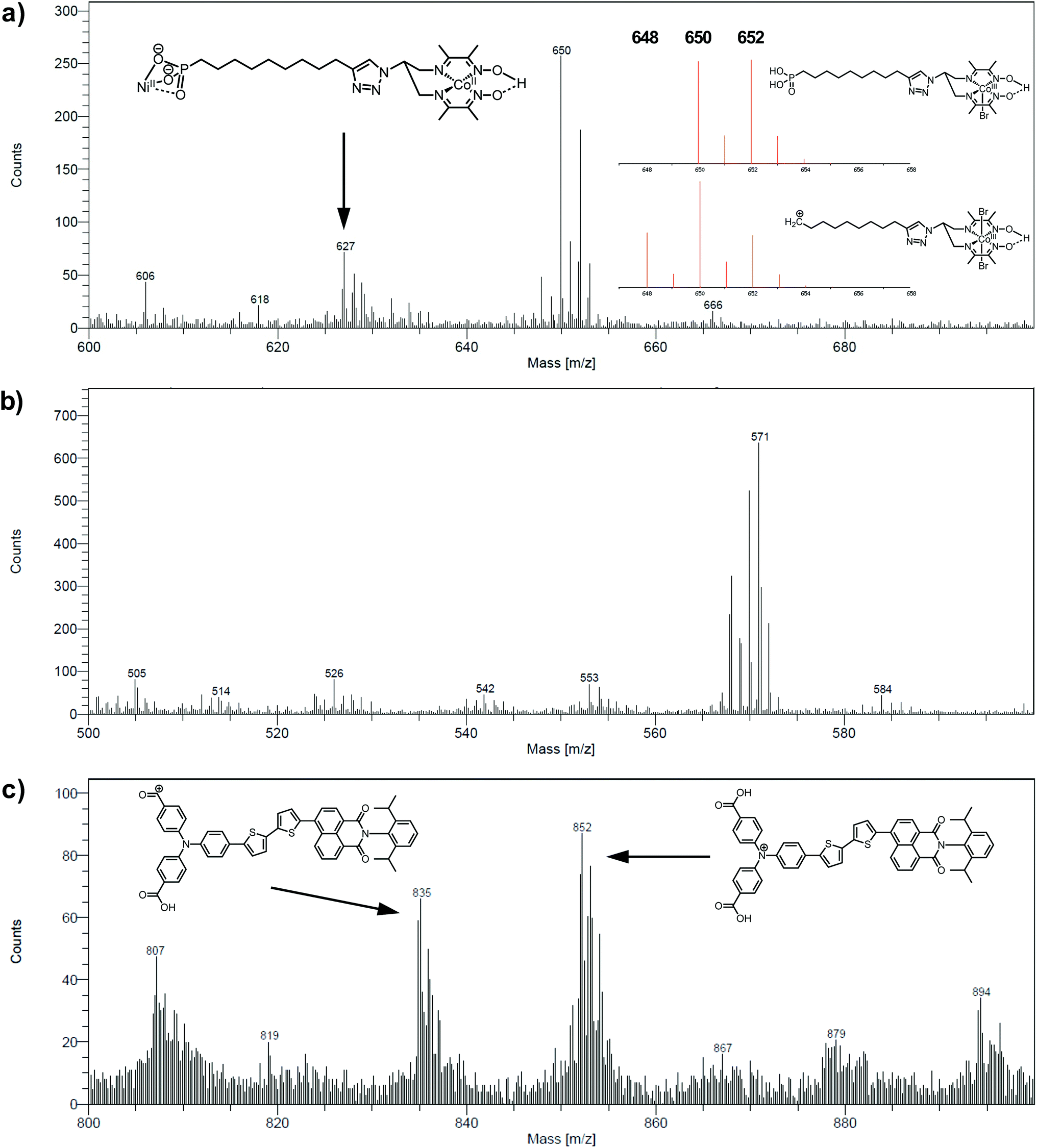 | ||
| Fig. 5 Positive mode ToF-SIMS spectra of the NiOǀCoC11P electrode in the m/z range (a) 600–700 and (b) 500–600 and (c) of the NiOǀRBG-174 electrode in the m/z range 800–900. Insets in (a) and (c) show proposed fragments and (a) theoretical isotopic patterns for fragments corresponding to the peak around m/z = 650. | ||
The analysis at the NiOǀRBG-174 electrode in the positive mode displays noteworthy fragments at high molecular mass (Fig. 5c). A set of peaks in the m/z = 850–855 range (amongst others a peak detected at m/z = 852.26) is a clear indication of the molecular ion [RBG-174]+ of exact mass = 852.233 (Fig. 5c). This signature is direct evidence that the molecule is intact after grafting. Additional peaks detected at m/z = 835.21 and 807.25 respectively correspond to the loss of a hydroxyl group from a carboxylic moiety yielding the [RBG-174 – OH]+ fragment (exact m/z = 835.230) and to a further decarbonylation to give [RBG-174 – OH – CO]+ (exact m/z = m/z = 807.235), as shown in Fig. S10†.
The NiOǀRBG-174ǀCoC11P co-grafted electrode displays the expected mixed set of peaks. First, the set of peaks detected at m/z = 852.21 and 835.24 (Fig. 6a) are in line with the ones observed on NiOǀRBG-174 electrodes (Fig. 5c). On the other hand, the peaks detected at m/z = 650.07, 627.11 and 569.14 (Fig. 6b and c) correspond to CoC11P fragments (Fig. 5a and b). Thus, the signatures of both RBG-174 and CoC11P are detected at the surface of the NiOǀRBG-174ǀCoC11P electrode.
 | ||
| Fig. 6 Positive mode ToF-SIMS spectra of the NiOǀRBG-174ǀCoC11P electrode in the m/z ranges (a) 800–900, (b) 600–700 and (c) 500–600. | ||
UV-visible spectroscopy
The NiO background-subtracted spectrum of NiOǀRBG-174 is displayed as an orange line in Fig. 7. The signature of immobilized RBG-174 dye at the surface of NiO is a broad band rising from ca. 580 nm, with a maximum at 360 nm and a marked shoulder at 460 nm. Indeed, this feature fits well with the solution spectrum of the tert-butoxide protected analogue RBG-174tBu40 (Fig. 7), taking into account the broadening of the UV-visible signature commonly observed for organic photosensitizers upon immobilization on transparent conducting oxides.49–53 From the absorbance measured at the shoulder Aelectrode(442 nm) = 0.654 and the molar absorptivity of RBG-174tBu at 442 nm (εsolution = 27100 M−1 cm−1), we estimated the grafting density of RBG-174 to be ca. 25 nmol cm−2 using eqn (1):54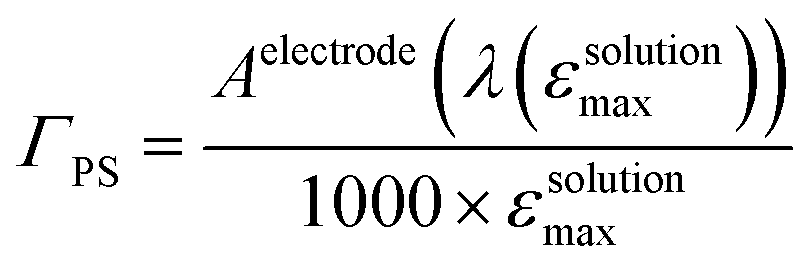 | (1) |
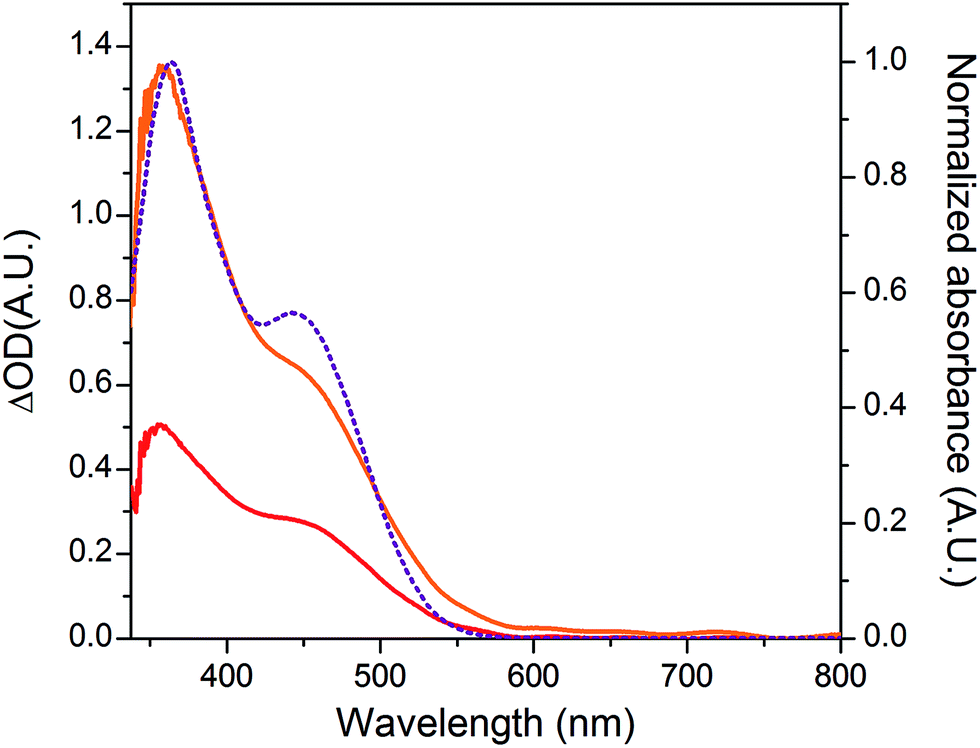 | ||
| Fig. 7 UV-visible absorption spectra of the NiOǀRBG-174ǀCoC11P electrode after subtraction of the spectrum of the NiOǀCoC11P electrode (red line), of the NiOǀRBG-174 electrode after subtraction of the spectrum of the pristine NiO electrode (orange line; left scale) and of a solution of RBG-174tBu in CH2Cl2 (purple dotted line; right scale). | ||
The UV-visible spectrum of NiOǀRBG-174ǀCoC11P is shown as a red line in Fig. 7 after subtraction of the NiOǀCoC11P spectrum recorded on the same electrode prior to dying with RBG-174. In that way, we picture the sole contribution of RBG-174. Accordingly, a signature similar to NiOǀRBG-174 is observed, although with a 43% lower intensity at the shoulder, corresponding to a grafting density estimated at ΓRBG−174 = 10.4 nmol cm−2.
(Spectro)electrochemistry of NiOǀCoC11P electrodes
The cyclic voltammograms recorded at NiOǀCoC11P and NiOǀRBG-174ǀCoC11P in NaCl 0.1 M aqueous electrolyte are shown in Fig. 8. A reversible wave at ½ (Epa + Epc) = −0.37 V vs. NHE is observed on the NiOǀCoC11P electrode (Fig. 8a and S11†). This wave is not displayed on the blank electrode (Fig. S11†) and is hence attributed to a redox couple introduced after sensitization with CoC11P. No major modification of the wave is observed upon cycling, which points to a surface-immobilized redox species.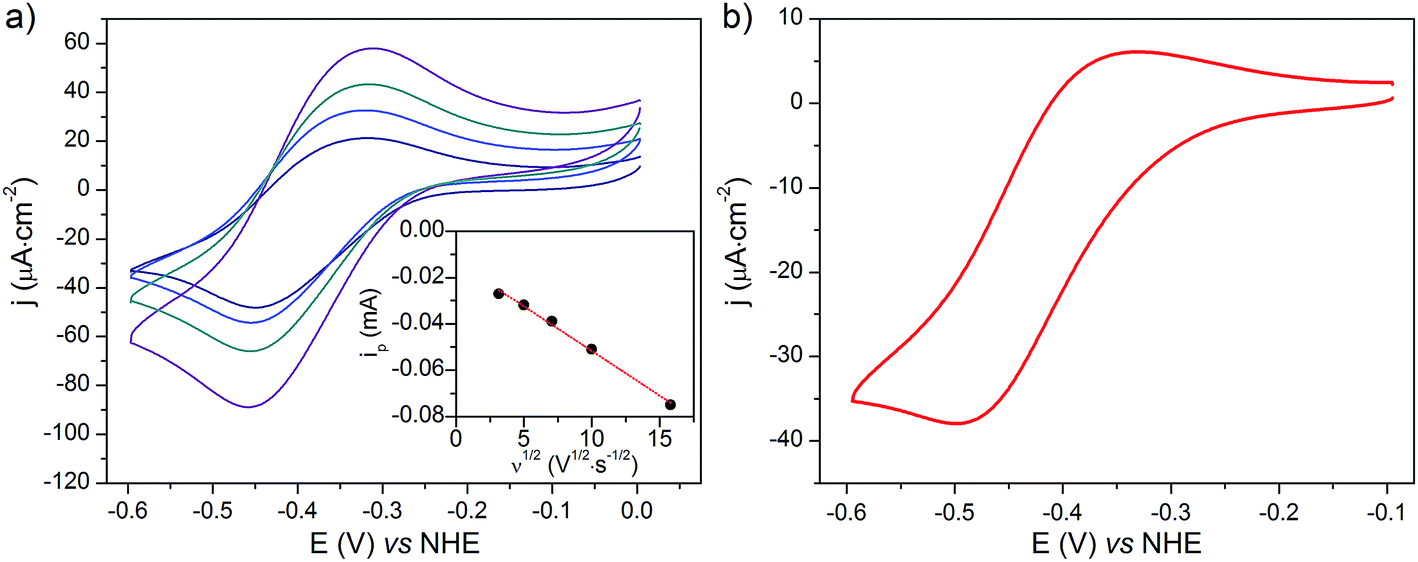 | ||
| Fig. 8 CVs recorded in a NaCl 0.1 M aqueous electrolyte at (a) a NiO/CoC11P electrode at scan rates of 10 (navy line), 25 (blue line), 50 (cyan line) and 100 (purple line) mV s−1 with (inset) the dependency of baseline-corrected cathodic peak current on the square root of the scan rate (dots) and its linear fit (dashed lines) and (b) a NiOǀRBG-174ǀCoC11P electrode at a scan rate of 10 mV s−1. | ||
UV-visible spectroelectrochemistry allowed this redox process to be assigned to the CoII/I transition in [Co(DO)(DOH)pnBr2]. The UV-visible spectrum recorded at the NiOǀCoC11P electrode poised at a potential more cathodic than this process (−0.84 V vs. NHE) indeed evidences a signature of the CoI state of [Co(DO)(DOH)pnBr2],55 with two broad peaks at ca. 573 and 708 nm (Fig. S12†). This signal is not observed for the same electrode poised at a potential anodic to the redox process (−0.24 V vs. NHE) nor for the blank electrode (Fig. S12†).
When the applied potential is cathodically swept, progressive building of the CoI species is clearly monitored on the difference UV/Vis spectra depicted in Fig. 9. The signature of the formed surface-immobilized species is in very good agreement with the one of the [CoI(DO)(DOH)pnL] complex55 and compares well with that previously reported for an ITO-grafted similar cobalt diimine–dioxime complex.56 Assuming that the molar absorptivity coefficient (εCo(I)601 nm ≈ 8300 L mol−1 cm−1) of [CoI(DO)(DOH)pn(CH3CN)] (Fig. S13†) at peak maximum does not vary significantly when shifting from MeCN to water, this signature allowed us to evaluate the surface coverage in CoC11P; we determined the surface loading ΓCoC11P ≈ 4.5 nmol cm−2 from the photoelectrochemically measured absorbance value ANiOǀCoC11P(601 nm) = 0.037 (Fig. 9) using eqn (1).54
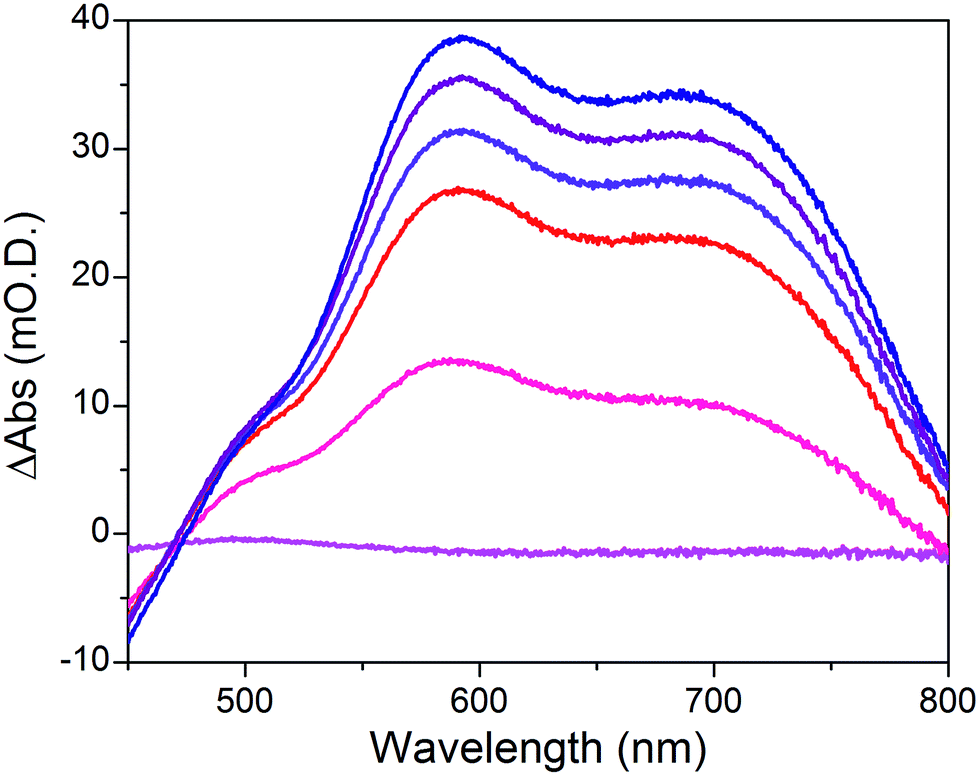 | ||
| Fig. 9 Differential UV-visible spectra of a NiOǀCoC11P electrode poised in a NaCl 0.1 M aqueous electrolyte at −0.44 (magenta line), −0.54 (pink line), −0.64 (red line), −0.74 (violet line), −0.84 (purple line) and −0.94 (navy blue line) V vs. NHE, with subtraction of the spectrum of the same electrode poised at −0.34 V vs. NHE under identical conditions. | ||
NiO is a p-type semiconductor and should behave as an insulator at applied potentials negative to its valence band edge, usually reported at +0.3 to +0.5 V vs. NHE at pH 7.57 To better understand how CoI species can be electrochemically generated below −0.37 V vs. NHE, the scan rate dependence of the signal was studied (Fig. 8a) between 10 and 100 mV s−1. Actually, the peak current linearly depends on the square root of the scan rate (Fig. 8a, inset). This observation clearly rules out the occurrence of an interfacial electron transfer process (for which the current would linearly depend on the scan rate) and is indicative of a diffusion-limited process likely occurring within the thickness of the mesoporous electrode. Modelling the process by a single electron transfer at the interface under diffusion-limited conditions, the peak current might be related to the scan rate ν through the following equation:58,59
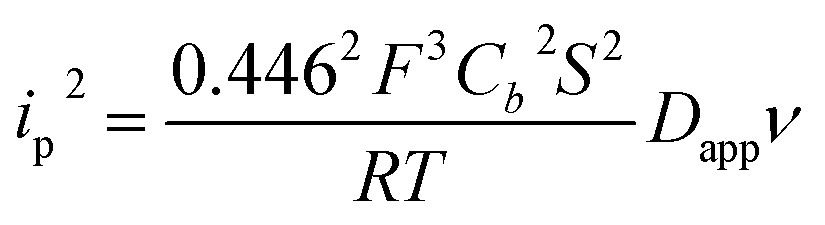 | (2) |
The experimental data could be fitted (inset in Fig. 8a) with a linear function of slope: a = 2.0 × 10−8 A2 s V−1. The apparent diffusion coefficient may thus be recovered as such:
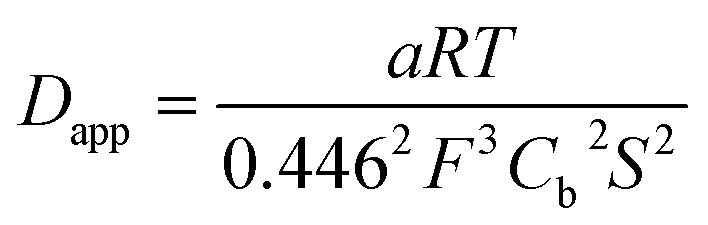 | (3) |
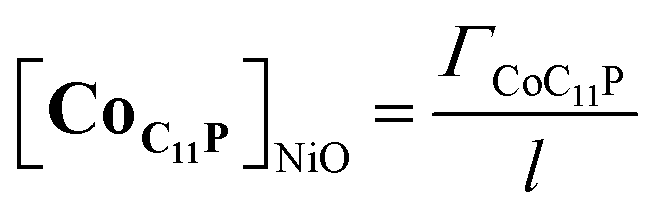 | (4) |
We thus estimated an apparent diffusion coefficient of Dapp = 4.4 × 10−8 cm2 s−1. This value ranges two orders of magnitude higher than that expected for a charge transport limited by the diffusion of counter-ions to species grafted on mesoporous semi-conducting films.60 We will propose an alternative interpretation of this value in the Discussion section (vide infra).
(Spectrophoto)electrochemistry at the dye–catalyst co-grafted NiOǀRBG-174ǀCoC11P electrode
The cyclic voltammogram recorded at a NiOǀRBG-174ǀCoC11P electrode in the cathodic region is shown in Fig. 8b. An electrochemical signature at ca. −0.40 V vs. NHE fits well with that observed for the CoII/I couple of CoC11P (−0.37 V vs. NHE) at NiOǀCoC11P electrodes. The electrochemical addressability of the attached cobalt diimine–dioxime complex is thus retained in the presence of RBG-174. We further investigated if the presence of the photosensitizer could enable light-driven electrochemical reduction of the CoC11P to the CoI state when the electrode is poised at a potential anodic to the CoII/I couple. The NiOǀRBG-174ǀCoC11P electrode poised at −0.4 V vs. Ag/AgCl (−0.3 V vs. NHE)61 in MeCN features a band centered at 475 nm on the UV-visible spectrum (Fig. 10) indicating the presence of a CoII complex immobilized onto the electrode surface.55 When the potential is set further downward to −0.7 V vs. Ag/AgCl (−0.6 V vs. NHE) the band centered at 475 nm vanishes and the broad signal characteristic of the CoI state appears between 500 and 880 nm (Fig. 10) as shown above for the NiOǀCoC11P electrode. Finally, we illuminated a NiOǀRBG-174/CoC11P electrode poised at −0.3 V vs. NHE. The UV-visible spectrum of the electrode clearly displayed the signature of the CoI state of immobilized CoC11P (Fig. 10). This observation demonstrates that the NiOǀRBG-174ǀCoC11P electrode can be used as a photoelectrode converting photon energy into electrochemical potential and enabling electron transfer between the NiO support and the catalyst uphill to the (dark) electrochemical equilibrium.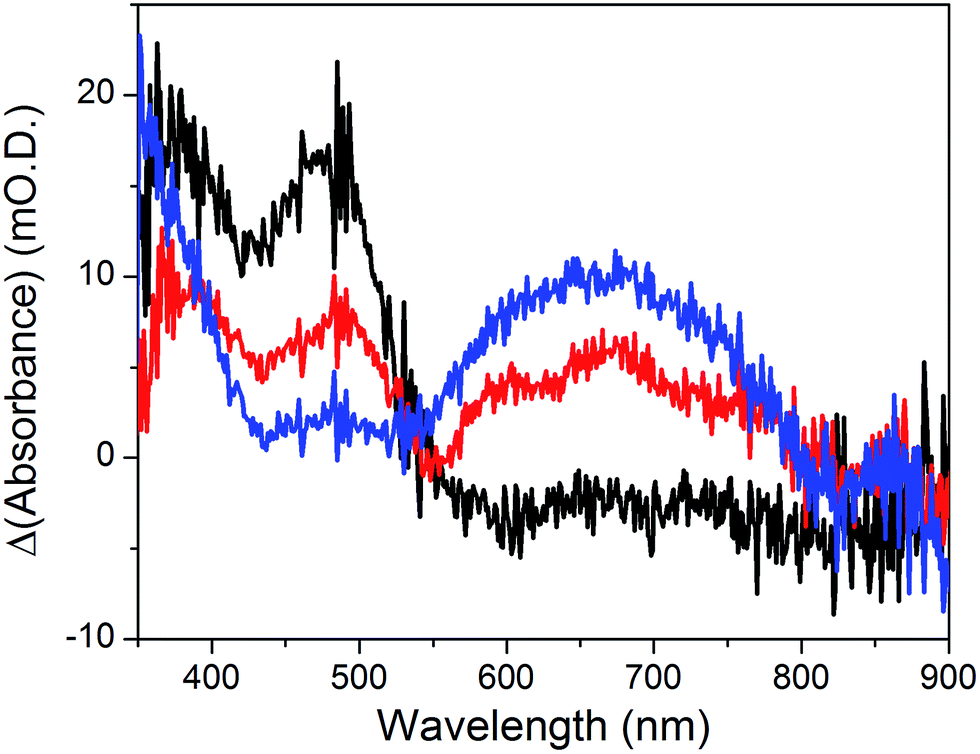 | ||
| Fig. 10 Differential UV-visible spectra recorded at a NiOǀRBG-174ǀCoC11P electrode poised at −0.4 V vs. Ag/AgCl (−0.3 V vs. NHE, black line) with subtraction of the spectrum of the same electrode poised at 0 V vs. Ag/AgCl (+0.1 V vs. NHE taking into account interliquid junction potential45), −0.7 V vs. Ag/AgCl (−0.6 V vs. NHE, blue line) with subtraction of the spectrum of the same electrode poised at −0.4 V vs. Ag/AgCl and −0.4 V vs. Ag/AgCl (−0.3 V vs. NHE) under light irradiation (red line) with subtraction of the spectrum of the same electrode poised at −0.4 V vs. Ag/AgCl in the dark, in CH3CN nBu4NPF6 0.1 M electrolyte. | ||
Photo-electrocatalytic activity of NiOǀRBG-174ǀCoC11P photocathode
The NiOǀRBG-174ǀCoC11P electrode was analyzed by linear sweep voltammetry (LSV) performed in MES 0.1 M/NaCl 0.1 M aqueous electrolyte at pH 5 without and with chopped visible light irradiation (λ > 400 nm). A photocurrent was observed throughout the potential range from ca. 0.65 to −0.05 V vs. RHE (Fig. 11a) with maximum photoresponse at 0.1 V vs. RHE. Chronoamperometry was performed to ascertain the magnitude and stability of the photocurrent at applied potentials relevant for solar-assisted H2 generation (Fig. 11b and S14†). At 0.54 V and 0.34 V vs. RHE, stable but low cathodic photocurrents of 0.1 and 1.0 μA cm−2 were observed, respectively (Fig. S14†). At 0.14 V vs. RHE the photocurrent increases significantly to give −8 μA cm−2 and then decays slightly over 5 minutes (Fig. 11b). The control NiOǀRBG-174/C10P electrode without the catalytic cobalt center showed almost no photocurrent under identical conditions (Fig. 11b and S15†).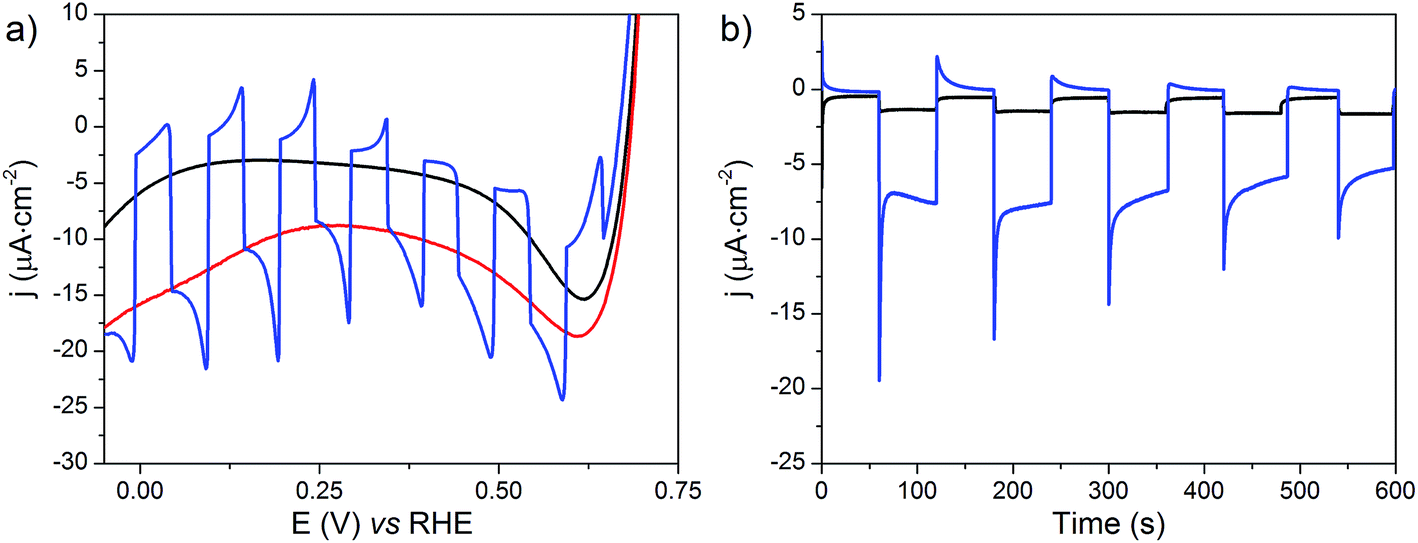 | ||
| Fig. 11 (a) LSVs at the NiOǀRBG-174ǀCoC11P electrode with (red line), without (black line) or with chopped (blue line) light irradiation recorded at a scan rate of 10 mV s−1 and (b) current densities vs. time at NiOǀRBG-174ǀCoC11P (blue line) and NiOǀRBG-174ǀC10P (black line) electrodes poised at 0.14 V vs. RHE under chopped light irradiation, in MES 0.1 M/NaCl 0.1 M aqueous electrolyte at pH 5.5. | ||
Chronoamperometry at 0.14 V vs. RHE was performed under continuous irradiation for 2 hours at the NiOǀRBG-174ǀCoC11P electrode (Fig. S16†). The electrode produced H2 with a faradaic efficiency (FE) of 9.3% ± 1.5. By contrast, the NiOǀRBG-174ǀC10P control electrode produced only a trace of H2 (FE 0.7% ± 1.2).
Discussion
We recently reported on the activity of a dye-sensitized photocathode built by immobilization of a molecular dyad on mesoporous NiO. The dyad was composed of a push–pull organic photosensitizer and the [Co(DO)(DOH)pnBr2] catalytic core.22 This photocathode was proven operative to generate H2 under photoelectrochemical conditions in aqueous media. Charge transfer properties within this dyad are of great interest to understand how such architecture enables H2 evolution and are being currently investigated. However, the preparation of such dyads is synthetically challenging. In an alternative approach to decrease synthetic complexity and deconvolute effects of the photosensitizer and the catalyst on the activity, we turned our attention to the design of a co-grafted electrode using the same cobalt diimine–dioxime catalyst. Phosphonate moieties are efficient anchors on transparent conducting oxides (TiO2, ITO)62,63 and their utilization to attach molecular catalysts onto NiO has been recently reported.20,34 As such, we prepared CoC11P, a phosphonate derivative of the [Co(DO)(DOH)pnBr2] complex, starting from commercial PEtC9CCH phosphonate (Fig. 2) and the previously reported [Cu(DO)(DOH)N3pn(OH2)]ClO4 complex using Cu-catalyzed alkyne–azide coupling.22,43 Further exchanging copper(II) for cobalt(II) coupled to air oxidation to the Co(III) state22 allowed us to isolate CoC11P after deprotection of the phosphonic esters.
CoC11P readily grafts onto mesoporous NiO electrodes, as demonstrated by XPS analysis. The recorded Co 2p3/2 signature (Fig. 4) is in good agreement with those reported for similar cobalt diimine–dioxime complexes grafted on ITO or MWCNTs.42,47 The presence of N and P in the backbone of CoC11P is also observed by XPS (Fig. S6†) and partially matches XPS characterization made on a different phosphonated [Co(DO)(DOH)pn] complex reported by Reisner and coworkers.47 Overall, the XPS analysis is well correlated with a surface of NiO decorated with CoC11P.
To give stronger evidence for the structural integrity of the molecules upon grafting, functionalized electrodes were also characterized with a surface-specific molecular mass spectrometric technique, namely ToF-SIMS.64 Although the CoC11P molecular peak was not detected (ToF-SIMS does not systematically give the detection of the molecular fragment), high mass specific signatures of all functions of the molecule are strong indicators for the grafting of intact CoC11P. In addition, the observation of secondary ions containing Ni, O and P (Fig. S8†) supports that a fraction of CoC11P is grafted on the NiO surface through stable Ni–O–P bonds, as previously reported for model phosphonic acid derivatives at TiO2 surfaces65 and for Ru-trisdiimine photosensitizers onto NiO films.48 Moreover, the detection of the preserved high mass molecular ions of both the dye and the catalyst at the co-grafted electrode indicates that the co-sensitization process alters neither the chemical structure of the catalyst nor that of the dye. This observation is additional evidence for a successful co-immobilization process.
Phosphonate grafting proved stable enough to allow electrochemical investigations of the interface in near-neutral aqueous electrolytes. Cyclic voltammetry revealed a reversible system at −0.37 V vs. NHE (Fig. 8a and S11†), which was correlated with the CoII/I event by UV-visible spectroelectrochemistry (Fig. 9). The UV-visible signature of the reduced species of this couple matches well with that observed for cobalt(I) diimine–dioxime complexes in solution (Fig. S13†) or on ITO electrodes.56 The reversibility of the CoII/I wave and the observation of the CoI state further indicates poor intrinsic catalytic activity of the NiOǀCoC11P electrode for H2 evolution at neutral pH. This is in agreement with cobalt diimine–dioxime complexes, which are active at mildly acidic pH values but not at pH 7.42,47 Nevertheless, a closer examination of the conduction process within the NiOǀCoC11P architecture showed that the CoII/I process is diffusion-limited with an apparent diffusion coefficient of 4.4 × 10−8 cm2 s−1. Such a value is about two orders of magnitude higher than that observed for species adsorbed within mesoporous semi-conducting films for which the charge transport is limited by the physical diffusion of ions from the electrolyte throughout the film to ensure electroneutrality.60 This value thus rules out a conduction process through the NiO electrode, in line with the insulating behavior of this substrate in the electrochemical potential window (NiO valence band edge around 0.37 V vs. NHE at pH 7 (ref. 57) and band gap energy of ca. 4 eV (ref. 66)). By contrast, apparent diffusion coefficients in the same order of magnitude have been reported for charges hopping at the surface of n-type (TiO2) or insulating (ZrO2, Al2O3) mesoporous films67 or metal organic frameworks (MOFs)68,69 deposited onto conductive glass and to which redox probes were attached. We postulate that a similar process of electron hopping between Co centers immobilized along the insulating NiO layer is responsible for the conduction observed at NiOǀCoC11P under our electrochemical conditions. To the best of our knowledge, this behavior has not been characterized for metal-based complexes grafted at the surface of NiO, although a self-exchange mechanism between neighboring C343 molecules grafted on NiO was proposed by Hammarström and coworkers to explain the kinetics of the photo-electrochemical processes in another series of co-grafted H2-evolving photocathodes.26,34,70 Taken together, these results confirmed that the CoI state, which is the entry point to the HER catalytic cycle under appropriate conditions, could be reached at NiOǀCoC11P, thus paving the way to photoelectrocatalytic studies.
With that aim, we co-grafted CoC11P and the RBG-174 organic photosensitizer onto NiO. XPS (Fig. 4 and S6†) and ToF-SIMS (Fig. 6) analyses of NiOǀRBG-174ǀCoC11P clearly support the presence of both entities on the co-grafted electrode. In particular, fragments assigned to grafted CoC11P (m/z = 650.07, Fig. 6b) and RBG-174 (m/z = 852.21 and 835.24, Fig. 6a) demonstrate that their molecular structures are retained on NiOǀRBG-174ǀCoC11P electrodes. These results hence validate the straightforward co-grafting approach to obtain dye–catalyst hybrid electrodes, which was also used by others,20 but for which in-depth molecular characterization of the final architecture was lacking. Under the grafting conditions used, the dye loading of NiO|RBG-174ǀCoC11P electrodes was estimated from UV-visible spectroscopy to be 43% of that observed at model NiOǀRBG-174 electrodes (Fig. 7), confirming that a substantial fraction of surface anchoring sites is occupied by CoC11P moieties.
The photoelectrochemical activity of NiOǀRBG-174ǀCoC11P electrodes was assessed in aqueous buffer (pH 5.5) under conditions similar to those used previously to characterize our covalent dye–catalyst construct.22 The onset of cathodic photocurrents is ca. +0.65 V vs. RHE, this reflects the potential from which light-driven hole injection into the NiO can occur. This potential lies within the NiO valence band, the edge of which is estimated at ca. 0.46 V vs. RHE at pH 5.5 by a Nernstian pH correction of the potential determined at pH 7.57 However, the maximum steady-state photocurrents are only reached at lower potential (0.14 V vs. RHE, Fig. S14a†) and chopped-light photoelectrochemical measurements at 0.54 and 0.34 V vs. RHE display strong negative and positive transient current spikes when the light is switched on and off, respectively. We note that such transient current spikes are not observed on control NiOǀRBG-174ǀC10P electrodes suggesting that these peaks arise from the possibility of accumulating electrons on the cobalt centers. Although a detailed study of the dynamics of such a light-driven process is beyond the scope of this study, the observation of distinct behaviors depending on the applied potential is likely in line with the existence of two populations of holes71 with distinct dynamics72 at the surface of NiO nanoparticles. In terms of photocurrent densities, the performance of co-grafted NiOǀRBG-174ǀCoC11P electrodes is close to those of reported covalent constructs based on the same catalytic centers22,25 and compares well to other NiO-based molecular photocathodes.18,21,35 Long-term chronoamperometry measurements under irradiation allowed us to correlate the photocurrent with H2 evolution with an overall faradaic yield of 10%, a value in line with previous reports for other molecular photocathodes.8,9,21,22 Reasons for such a low faradaic efficiency are related to the reduction of traces of O2 trapped in the mesoporous NiO material as well as to the partial reduction of the NiO support itself. The latter hypothesis was previously assessed by us22 and was recently confirmed by Simonov and coll.73
The operating mechanism of the NiOǀRBG-174ǀCoC11P photocathode is shown in Fig. 1 with a CoI intermediate being the entry into the H2 evolution catalytic cycle.41 To verify that such an intermediate could be formed under photo-electrochemical conditions, we performed spectro-photoelectrochemical measurements at various applied potentials (Fig. 10) and in the absence of available protons that would drive the system into catalysis. First, spectroelectrochemical measurements in the dark showed that the broad absorption band between 550 and 800 nm (Fig. 10) corresponding to the CoI form of the catalyst is only generated at a quite negative potential (−0.7 V vs. NHE) and that the CoII form dominates at −0.3 V vs. NHE (i.e. ∼0 V vs. RHE under the conditions used during photoelectrochemical measurements). Second, we observed the same UV-visible signature after 5 min of irradiation with visible light with the electrode held at the same potential (−0.3 V vs. NHE), confirming the generation of a CoI intermediate. Remarkably, this signal has a lifetime of several minutes at least under the open-circuit conditions used for the UV-visible measurements. Such a lifetime for the reduced intermediate should likely enable it to undergo protonation and experience further reduction so as to produce the CoII hydride species required for H2 evolution to proceed.41
Overall, these data demonstrate that photocathode architectures based on a co-grafted dye and catalyst can be as effective as those based on covalent dye–catalyst dyads. Whatever the architecture, all molecular photocathodes reported so far suffer from stability issues and display significant loss in activity over prolonged photoelectrolysis operation. To gain more insights into the degradation mechanisms responsible for the loss in activity we undertook a post-operando ToF-SIMS characterization. We took care here to make a fair but cautious interpretation of data as there are no reference ToF-SIMS spectra for the chemical structures related to possible degradation routes.
We first observed that the relative intensity of all signals attributed to CoC11P and RBG-174 fragments in the positive mode was lower on post-electrolysis electrodes (Fig. S17†) than on pristine ones, which points to partial detachment of dyes and catalysts from the surface, as previously noted for similar photocathodes in the literature.31,35 Still, after photoelectrocatalytic activity we observed the signals corresponding to intact RBG-174 (m/z = 852.19; Fig. S17a†) and a dichloride derivative of CoC11P (Fig. S18a†) at m/z = 642.19 (Fig. S17b†). Secondary ions containing Br− ligands are also significantly less observed in the negative mode spectrum recorded post-electrolysis (see for example the group of peaks at m/z = 79–81 in Fig. S19† at significantly lower intensity, with the peak detected at 78.96 being furthermore related to PO3− of exact mass 78.958), in line with previous studies indicating displacement of axial halide ligands in the lower oxidation states of cobalt diimine–dioxime complexes and replacement by chloride anions after completion of the catalytic turnover.41,74
Concerning CoC11P, comparing positive mode spectra (Fig. S20†) shows that the relative intensity between the peak detected at m/z = 489.22 (cleavage in α of the phosphonate moiety) and those at lower masses characterized by the successive losses of CH2 fragments (a series of peaks at m/z from 475 to 377 described in Fig. S7†) changes upon activity. As an example, the [m/z = 489.22]/[m/z = 447.10] intensity ratio is going from ∼4.3 (±0.4) before operation to ∼0.9 (±0.2) in the post-operando characterization. This observation points that the cleavage of the P–C bond becomes less favored compared to that of C–C bonds in the aliphatic chain, for CoC11P retained on the post-activity electrode. Such a result can be tentatively explained by the preferable retention upon activity of CoC11P being tightly bound to NiO and the leaching of physisorbed CoC11P species. Another significant point is the apparition of several new fragments in the negative mode spectrum (m/z = 250.05 and 277.09; Fig. S21†). These fragments were tentatively assigned to structures for which the triazole ring of CoC11P underwent reductive hydrogenations and further cleavage (Fig. S18b†). Although the 1,2,3-triazole ring is reported to be resistant to hydrolysis, oxidation, reduction, or other modes of cleavage,75 these observations could indicate that it might be prone to degradation under the photoreductive conditions generated here, leading to a loss of active grafted CoC11P complexes. Although the extent of the degradation cannot be easily quantified with this technique, we speculate that such loss of catalytic cores probably plays a substantial role in the decrease of activity during catalysis. Given the versatility of the alkyne–azide coupling click reaction for the construction of photoactive systems,25,43,76–80 this observation also calls for a dedicated study elucidating the factors and mechanism governing such a photoreductive degradation of triazole rings. Obviously, this will require the design and synthesis of model systems that can be studied in homogeneous solution under visible-light irradiation with a range of spectroscopic techniques.81
Regarding RBG-174, compared to the molecular peak, lower mass fragments (at m/z = 835.20 and 807.18) are less detected after activity (Fig. S17a†). As an example, the [m/z = 852.19]/[m/z = 835.20] intensity ratio increases from ∼1.2 (±0.1) before operation to ∼2.1 (±0.2) in the post-operando characterization. As for CoC11P, this observation might indicate that the retained RBG-174 underwent a modification of its binding mode upon turnover, which further influences the bond strength within the carboxylic acid anchoring group, and leaching of physisorbed RBG-174 entities. Some degradation of the RBG-174 dye structure is also observed post-operando. Namely, peaks detected at m/z = 240.03 and 242.01 (Fig. S22†) could illustrate a possible cleavage at the triphenylamine moiety while a peak at m/z = 398.06 (Fig. S23†) was attributed to a fragment of the dye cleaved at the dithiophene unit (Fig. S18c†). The observation of such degradative pathways is of main note given the widespread use of organic dyes based on triphenylamine and thiophene units in DS-PEC applications.8–10
In a broader context, photoelectrode materials for direct sunlight-driven water splitting in photoelectrochemical cells need to fulfil three main characteristics: efficiency, stability and cost-effectiveness.82 Many options are available regarding the light-harvesting material: extended inorganic phases, molecular species or a hybrid combination of both as investigated here with dye-sensitized photoelectrodes. The same applies to catalysts, which can be purely inorganic materials or molecular entities, including enzymes.
As far as photosensitive units are concerned, crystalline inorganic semi-conductors, such as Si83–86 and group III–V semi-conductors (InP, GaP or GaInP2 to quote only a few),7,87 generate very high photocurrent densities and can be used in photoanodes or photocathodes. However, these highly efficient photovoltaic materials are generally expensive and all are quite unstable when operated in the presence of aqueous electrolytes. These materials therefore require efficient protection against corrosion, which comes with a cost.88–90 Metal oxides like α-Fe2O3, WO3 or BiVO4 are also widely considered as visible light-harvesting materials for photoanodes91–94 because of their low cost and good stability in aqueous electrolytes and despite the comparatively lower photocurrent densities they can reach. By contrast, this class of light-harvesters is less developed for photocathode materials. Indeed, the heavily investigated visible-light absorbing p-type metal oxides Cu2O and CuO suffer from photocorrosion issues, unless properly protected.95–98 Alternative options are thus explored today, for example with tungsten selenide.99,100 The approach consisting of sensitizing semi-conductors with organic dyes – the so-called dye-sensitized photoelectrode – combines the low-cost of the latter approach with the tunability of molecular compounds to enhance the light-harvesting properties of the material. A fourth approach relies only on organic semi-conductors that are cost-effective, produce high photocurrents7,101–105 but also display limited stability during photoelectrocatalysis. Clearly, it is too early to pick a winner within these four approaches towards photoelectrochemical water splitting. Moreover, other effective solutions can emerge from efforts directed to solving current limitations of these approaches, just as the photovoltaic perovskite technology originated from attempts to develop more stable dye-sensitized solar cells.106,107
The development of dye-sensitized photoelectrodes for water splitting is without doubt the youngest of these approaches. Capitalizing on the DSSC technology that exploits ultra-fast charge injection from molecular dyes into the n-type transparent semi-conducting TiO2, dye-sensitized photoanodes for O2 evolution rapidly reached photocurrent densities at the mA cm−2 level.8,9,11–15 By contrast, dye-sensitized photocathodes were first reported in 2012 (ref. 16 and 17) and only a few operative systems have been described so far,10,108,109 which still display low photocurrent densities (tens of μA cm−2), limited faradaic yields and short-term stability. Indeed, such photocathodes have been mostly constructed on a NiO support. The low conductivity of NiO and the presence of intra-bandgap states that favour recombination processes are intrinsic limitations for photocurrent generation in such architectures.10,110,111 Indium tin oxide (ITO), a transparent conducting oxide (TCO) with improved charge-transport properties, can be used to solve this issue.27,112,113 Also, novel p-type transparent materials are currently investigated. Namely delafossites CuGaO2 (ref. 32) and CuCrO2 (ref. 35) already proved promising to increase photocurrent densities as well as to shift the onset photocurrent potential by a few hundreds of millivolts. Surface treatments are also instrumental in efficiently passivating surface trap states and enhancing photocurrent generation.18,114 With such knowledge, the design of novel dyes absorbing a larger portion of the solar spectrum and of faster, more stable, catalysts will eventually allow progress towards higher performances for dye-sensitized photocathodes.
These strategies are also supported by a clearer view on the mechanisms and limitations of the approach, as addressed in this work, and proposals to circumvent them. In particular, the origin of the low faradaic efficiency towards HER observed here and in previous studies engaging NiO-based photocathodes21,22,73 is related to the reduction of traces of O2 trapped in the mesoporous NiO film and to partial reduction of NiO itself. The latter hypothesis, initially proposed by us,22 was recently confirmed by Simonov and colleagues.73 If higher photocurrent densities are reached, these competitive processes may become minor phenomena. However, solutions may as well come from smart coverage of the NiO substrate using atomic layer deposition (ALD) of passivating oxides or alternative surface treatments, including non-hydrolytic sol–gel deposition,115 already exploited to enhance performances of other metal oxide-based photoelectrodes.116 Interestingly, ALD was proven successful to stabilize the grafting of molecular dyes and catalysts onto TCO.23,117 ToF-SIMS data gained in this study actually demonstrate that the phosphonate moiety of CoC11P is firmly bound to the NiO support, confirming data gained from the grafting of Ru-based dyes by phosphonate anchors on the same support.48 Nevertheless, post-operando measurements indicate partial leaching of phosphonate-attached species, which calls for additional stabilization of this specific linkage. So far, TiO2 and Al2O3 are the most widely used passivation/stabilization layers deposited by ALD, although the former has n-type character118 and the latter displays poor stability in basic aqueous media.119 Research in this area should thus focus on the atomically precise deposition of materials that will be redox-neutral, or of p-type nature and stable under aqueous conditions on the long term.120 A final point regards the intrinsic durability of the molecular components themselves during turnover. In this study, we highlight for the first time the degradation of the triazole ring under photoreductive aqueous conditions as well as possible cleavages of the push–pull organic dye. Here again, ALD coating can provide a solution, namely through the “mummy” strategy developed by T. J. Meyer and coworkers121 in which the whole molecular components are embedded into the deposited layer. In addition, our findings may also guide the development of more stable molecular dyes and catalysts by introducing a requirement for resistance to reductive conditions in the early design of chemical structures for dye-sensitized photocathodes.
Conclusion
Two main types of architectures for H2-evolving dye-sensitized photocathodes have been described so far, depending on whether the catalytic moiety is directly grafted on the transparent conducting oxide substrates or not. This study allows for the first time to benchmark these two architectures based on the same catalytic moiety under similar conditions. At this stage, we cannot conclude on the superiority of one or the other strategy as far as the photocathode performance is concerned. Clearly, the co-grafting strategy offers more flexibility regarding the possibility to tune surface ratios between the dye and catalyst and is much less demanding in terms of synthesis allowing the easy combination of various dye and catalyst structures. In that respect, we add with this study a new phosphonate derivative of a cobalt diimine–dioxime catalyst, CoC11P, that can be grafted on the surface of transparent conducting oxides and extend the short list of such graftable H2-evolution catalyst derivatives.19,20,26,47,122 Our study also evidences the prominent role of the cobalt catalytic core in H2 evolution catalyzed under photoelectrochemical reduction. We have shown that under applied potentials typically used for molecular photocathodes the cobalt catalyst is reduced from CoIII to CoII without light. However, at potentials positive of 0 V vs. RHE, irradiation is essential to produce the Co(I) state and enter the catalytic cycle. In addition, we present the first use of ToF-SIMS to characterize a co-grafted molecular photocathode and diagnose the fate of the molecular component after extended turnover. Importantly, a significant fraction of both dye and catalyst were found to be present at the surface of the electrode after 2 h photoelectrolysis, consistent with the observation that the photoelectrode was still active after such a period of operation. ToF-SIMS was also key to identifying possible decomposition pathways of the dye and the catalyst, providing rationale for the observed decay in activity and now allowing for the design of new molecular components with increased robustness upon turnover.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the French National Research Agency (PhotoCAT project – ANR-14-JTIC-0004-01 and the Labex program ARCANE – ANR-11-LABX-0003-01). We thank E. Martinez from the Nanocharacterization platform at MINATEC campus (Univ Grenoble Alpes, CEA-Leti) for XPS measurements and Colette Lebrun from SyMMES (Univ Grenoble Alpes, CNRS, CEA-INAC) for ESI-MS measurements.References
- T. A. Faunce, W. Lubitz, A. W. Rutherford, D. MacFarlane, G. F. Moore, P. Yang, D. G. Nocera, T. A. Moore, D. H. Gregory, S. Fukuzumi, K. B. Yoon, F. A. Armstrong, M. R. Wasielewski and S. Styring, Energy Environ. Sci., 2013, 6, 695–698 RSC.
- T. Faunce, S. Styring, M. R. Wasielewski, G. W. Brudvig, A. W. Rutherford, J. Messinger, A. F. Lee, C. L. Hill, H. deGroot, M. Fontecave, D. R. MacFarlane, B. Hankamer, D. G. Nocera, D. M. Tiede, H. Dau, W. Hillier, L. Wang and R. Amal, Energy Environ. Sci., 2013, 6, 1074–1076 RSC.
- A. Thapper, S. Styring, G. Saracco, A. W. Rutherford, B. Robert, A. Magnuson, W. Lubitz, A. Llobet, P. Kurz, A. Holzwarth, S. Fiechter, H. de Groot, S. Campagna, A. Braun, H. Bercegol and V. Artero, Green, 2013, 3, 43–57 Search PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef PubMed.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 CrossRef PubMed.
- V. Nikolaou, A. Charisiadis, G. Charalambidis, A. G. Coutsolelos and F. Odobel, J. Mater. Chem. A, 2017, 5, 21077–21113 RSC.
- N. Queyriaux, N. Kaeffer, A. Morozan, M. Chavarot-Kerlidou and V. Artero, J. Photochem. Photobiol., C, 2015, 25, 90–105 CrossRef.
- Z. Yu, F. Li and L. Sun, Energy Environ. Sci., 2015, 8, 760–775 RSC.
- P. T. Xu, N. S. McCool and T. E. Mallouk, Nano Today, 2017, 14, 42–58 CrossRef.
- E. A. Gibson, Chem. Soc. Rev., 2017, 46, 6194–6209 RSC.
- L. L. Duan, L. P. Tong, Y. H. Xu and L. C. Sun, Energy Environ. Sci., 2011, 4, 3296–3313 RSC.
- D. L. Ashford, M. K. Gish, A. K. Vannucci, M. K. Brennaman, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, Chem. Rev., 2015, 115, 13006–13049 CrossRef PubMed.
- D. Wang, M. V. Sheridan, B. Shan, B. H. Farnum, S. L. Marquard, B. D. Sherman, M. S. Eberhart, A. Nayak, C. J. Dares, A. K. Das, R. M. Bullock and T. J. Meyer, J. Am. Chem. Soc., 2017, 139, 14518–14525 CrossRef PubMed.
- M. S. Eberhart, D. Wang, R. N. Sampaio, S. L. Marquard, B. Shan, M. K. Brennaman, G. J. Meyer, C. Dares and T. J. Meyer, J. Am. Chem. Soc., 2017, 139, 16248–16255 CrossRef PubMed.
- J. T. Kirner and R. G. Finke, J. Mater. Chem. A, 2017, 5, 19560–19592 RSC.
- L. Li, L. L. Duan, F. Y. Wen, C. Li, M. Wang, A. Hagfeld and L. C. Sun, Chem. Commun., 2012, 48, 988–990 RSC.
- L. Tong, A. Iwase, A. Nattestad, U. Bach, M. Weidelener, G. Gotz, A. Mishra, P. Bauerle, R. Amal, G. G. Wallace and A. J. Mozer, Energy Environ. Sci., 2012, 5, 9472–9475 RSC.
- Z. Ji, M. He, Z. Huang, U. Ozkan and Y. Wu, J. Am. Chem. Soc., 2013, 135, 11696–11699 CrossRef PubMed.
- K. Fan, F. Li, L. Wang, Q. Daniel, E. Gabrielsson and L. Sun, Phys. Chem. Chem. Phys., 2014, 16, 25234–25240 RSC.
- F. Li, K. Fan, B. Xu, E. Gabrielsson, Q. Daniel, L. Li and L. Sun, J. Am. Chem. Soc., 2015, 137, 9153–9159 CrossRef PubMed.
- M. A. Gross, C. E. Creissen, K. L. Orchard and E. Reisner, Chem. Sci., 2016, 7, 5537–5546 RSC.
- N. Kaeffer, J. Massin, C. Lebrun, O. Renault, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2016, 138, 12308–12311 CrossRef PubMed.
- R. J. Kamire, M. B. Majewski, W. L. Hoffeditz, B. T. Phelan, O. K. Farha, J. T. Hupp and M. R. Wasielewski, Chem. Sci., 2017, 8, 541–549 RSC.
- K. A. Click, D. R. Beauchamp, Z. J. Huang, W. L. Chen and Y. Y. Wu, J. Am. Chem. Soc., 2016, 138, 1174–1179 CrossRef PubMed.
- P. B. Pati, L. Zhang, B. Philippe, R. Fernández-Terán, S. Ahmadi, L. Tian, H. Rensmo, L. Hammarström and H. Tian, ChemSusChem, 2017, 10, 2480–2495 CrossRef PubMed.
- A. M. Brown, L. J. Antila, M. Mirmohades, S. Pullen, S. Ott and L. Hammarström, J. Am. Chem. Soc., 2016, 138, 8060–8063 CrossRef PubMed.
- B. Shan, A. K. Das, S. Marquard, B. H. Farnum, D. Wang, R. M. Bullock and T. J. Meyer, Energy Environ. Sci., 2016, 9, 3693–3697 RSC.
- B. Shan, B. D. Sherman, C. M. Klug, A. Nayak, S. L. Marquard, Q. Liu, R. M. Bullock and T. J. Meyer, J. Phys. Chem. Lett., 2017, 8, 4374–4379 CrossRef PubMed.
- Y. Kou, S. Nakatani, G. Sunagawa, Y. Tachikawa, D. Masui, T. Shimada, S. Takagi, D. A. Tryk, Y. Nabetani, H. Tachibana and H. Inoue, J. Catal., 2014, 310, 57–66 CrossRef.
- G. Sahara, R. Abe, M. Higashi, T. Morikawa, K. Maeda, Y. Ueda and O. Ishitani, Chem. Commun., 2015, 51, 10722–10725 RSC.
- G. Sahara, H. Kumagai, K. Maeda, N. Kaeffer, V. Artero, M. Higashi, R. Abe and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 14152–14158 CrossRef PubMed.
- H. Kumagai, G. Sahara, K. Maeda, M. Higashi, R. Abe and O. Ishitani, Chem. Sci., 2017, 8, 4242–4249 RSC.
- J. T. Hyde, K. Hanson, A. K. Vannucci, A. M. Lapides, L. Alibabaei, M. R. Norris, T. J. Meyer and D. P. Harrison, ACS Appl. Mater. Interfaces, 2015, 7, 9554–9562 CrossRef PubMed.
- L. J. Antila, P. Ghamgosar, S. Maji, H. N. Tian, S. Ott and L. Hammarstrom, ACS Energy Lett., 2016, 1, 1106–1111 CrossRef.
- C. E. Creissen, J. Warnan and E. Reisner, Chem. Sci., 2018, 9, 1439–1447 RSC.
- M. G. Gatty, S. Pullen, E. Sheibani, H. Tian, S. Ott and L. Hammarstrom, Chem. Sci., 2018, 9, 4983–4991 RSC.
- F. Odobel and Y. Pellegrin, J. Phys. Chem. Lett., 2013, 4, 2551–2564 CrossRef.
- C. J. Wood, G. H. Summers, C. A. Clark, N. Kaeffer, M. Braeutigam, L. R. Carbone, L. D'Amario, K. Fan, Y. Farre, S. Narbey, F. Oswald, L. A. Stevens, C. D. J. Parmenter, M. W. Fay, A. La Torre, C. E. Snape, B. Dietzek, D. Dini, L. Hammarstrom, Y. Pellegrin, F. Odobel, L. Sun, V. Artero and E. A. Gibson, Phys. Chem. Chem. Phys., 2016, 18, 10727–10738 RSC.
- R. Brisse, R. Faddoul, T. Bourgeteau, D. Tondelier, J. Leroy, S. Campidelli, T. Berthelot, B. Geffroy and B. Jousselme, ACS Appl. Mater. Interfaces, 2017, 9, 2369–2377 CrossRef PubMed.
- R. Brisse, C. Praveen, V. Maffeis, T. Bourgeteau, D. Tondelier, T. Berthelot, B. Geffroy, T. Gustavsson, J. M. Raimundo and B. Jousselme, Sustainable Energy Fuels, 2018, 2, 648–654 RSC.
- N. Kaeffer, M. Chavarot-Kerlidou and V. Artero, Acc. Chem. Res., 2015, 48, 1286–1295 CrossRef PubMed.
- E. S. Andreiadis, P. A. Jacques, P. D. Tran, A. Leyris, M. Chavarot-Kerlidou, B. Jousselme, M. Matheron, J. Pecaut, S. Palacin, M. Fontecave and V. Artero, Nat. Chem., 2013, 5, 48–53 CrossRef PubMed.
- N. Queyriaux, E. S. Andreiadis, S. Torelli, J. Pecaut, B. S. Veldkamp, E. A. Margulies, M. R. Wasielewski, M. Chavarot-Kerlidou and V. Artero, Faraday Discuss., 2017, 198, 251–261 RSC.
- J. E. O'Reilly, Biochim. Biophys. Acta, 1973, 292, 509–515 CrossRef.
- T. N. Huan, P. Simon, G. Rousse, I. Genois, V. Artero and M. Fontecave, Chem. Sci., 2017, 8, 742–747 RSC.
- K. S. Kim and N. Winograd, Surf. Sci., 1974, 43, 625–643 CrossRef.
- N. M. Muresan, J. Willkomm, D. Mersch, Y. Vaynzof and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 12749–12753 CrossRef PubMed.
- N. Queyriaux, R. A. Wahyuono, J. Fize, C. Gablin, M. Wachtler, E. Martinez, D. Leonard, B. Dietzek, V. Artero and M. Chavarot-Kerlidou, J. Phys. Chem. C, 2017, 121, 5891–5904 CrossRef PubMed.
- M. Hahlin, E. M. J. Johansson, S. Plogmaker, M. Odelius, D. P. Hagberg, L. Sun, H. Siegbahn and H. Rensmo, Phys. Chem. Chem. Phys., 2010, 12, 1507–1517 RSC.
- H. Ellis, S. K. Eriksson, S. M. Feldt, E. Gabrielsson, P. W. Lohse, R. Lindblad, L. C. Sun, H. Rensmo, G. Boschloo and A. Hagfeldt, J. Phys. Chem. C, 2013, 117, 21029–21036 CrossRef.
- G. Boschloo and A. Hagfeldt, Acc. Chem. Res., 2009, 42, 1819–1826 CrossRef PubMed.
- Y. Pellegrin, L. Le Pleux, E. Blart, A. Renaud, B. Chavillon, N. Szuwarski, M. Boujtita, L. Cario, S. Jobic, D. Jacquemin and F. Odobel, J. Photochem. Photobiol., A, 2011, 219, 235–242 CrossRef.
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, in Photochemistry and Photophysics of Coordination Compounds I, ed. V. Balzani and S. Campagna, Springer, Berlin Heidelberg, 2007, vol. 280, pp. 117–214 Search PubMed.
- J. Massin, M. Bräutigam, N. Kaeffer, N. Queyriaux, M. J. Field, F. H. Schacher, J. Popp, M. Chavarot-Kerlidou, B. Dietzek and V. Artero, Interface Focus, 2015, 5, 20140083 CrossRef PubMed.
- P. Zhang, P.-A. Jacques, M. Chavarot-Kerlidou, M. Wang, L. Sun, M. Fontecave and V. Artero, Inorg. Chem., 2012, 51, 2115–2120 CrossRef PubMed.
- M. R. J. Scherer, N. M. Muresan, U. Steiner and E. Reisner, Chem. Commun., 2013, 49, 10453–10455 RSC.
- G. Natu, P. Hasin, Z. Huang, Z. Ji, M. He and Y. Wu, ACS Appl. Mater. Interfaces, 2012, 4, 5922–5929 CrossRef PubMed.
- J. M. Savéant, Elements of Molecular and Biomolecular Electrochemistry, Wiley, 2006 Search PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical methods: fundamentals and applications, 2nd edn, Wiley, 2001 Search PubMed.
- C. Renault, L. Nicole, C. Sanchez, C. Costentin, V. Balland and B. Limoges, Phys. Chem. Chem. Phys., 2015, 17, 10592–10607 RSC.
- C. Costentin, S. Drouet, M. Robert and J. M. Saveant, Science, 2012, 338, 90–94 CrossRef PubMed.
- C. Queffélec, M. Petit, P. Janvier, D. A. Knight and B. Bujoli, Chem. Rev., 2012, 112, 3777–3807 CrossRef PubMed.
- S. P. Pujari, L. Scheres, A. T. M. Marcelis and H. Zuilhof, Angew. Chem., Int. Ed., 2014, 53, 6322–6356 CrossRef PubMed.
- A. Benninghoven, Angew. Chem., Int. Ed. Engl., 1994, 33, 1023–1043 CrossRef.
- N. Adden, L. J. Gamble, D. G. Castner, A. Hoffmann, G. Gross and H. Menzel, Langmuir, 2006, 22, 8197–8204 CrossRef PubMed.
- G. A. Niklasson and C. G. Granqvist, J. Mater. Chem., 2007, 17, 127–156 RSC.
- P. Bonhôte, E. Gogniat, S. Tingry, C. Barbé, N. Vlachopoulos, F. Lenzmann, P. Comte and M. Grätzel, J. Phys. Chem. B, 1998, 102, 1498–1507 CrossRef PubMed.
- I. Hod, P. Deria, W. Bury, J. E. Mondloch, C. W. Kung, M. So, M. D. Sampson, A. W. Peters, C. P. Kubiak, O. K. Farha and J. T. Hupp, Nat. Commun., 2015, 6, 8304 CrossRef PubMed.
- N. Kornienko, Y. Zhao, C. S. Kley, C. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. Yang, J. Am. Chem. Soc., 2015, 137, 14129–14135 CrossRef PubMed.
- J. M. Gardner, M. Beyler, M. Karnahl, S. Tschierlei, S. Ott and L. Hammarstrom, J. Am. Chem. Soc., 2012, 134, 19322–19325 CrossRef PubMed.
- L. D'Amario, R. Jiang, U. B. Cappel, E. A. Gibson, G. Boschloo, H. Rensmo, L. Sun, L. Hammarström and H. Tian, ACS Appl. Mater. Interfaces, 2017, 9, 33470–33477 CrossRef PubMed.
- L. D'Amario, J. Fohlinger, G. Boschloo and L. Hammarstrom, Chem. Sci., 2018, 9, 223–230 RSC.
- D. A. Hoogeveen, M. Fournier, S. A. Bonke, A. Nattestad, A. Mishra, P. Bäuerle, L. Spiccia, A. J. Mozer and A. N. Simonov, J. Phys. Chem. C, 2017, 121, 25836–25846 CrossRef.
- M. Giorgetti, M. Berrettoni, I. Ascone, S. Zamponi, R. Seeber and R. Marassi, Electrochim. Acta, 2000, 45, 4475–4482 CrossRef.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef PubMed.
- K. Ladomenou, V. Nikolaou, G. Charalambidis and A. G. Coutsolelos, Coord. Chem. Rev., 2016, 306(Part 1), 1–42 CrossRef.
- I. Nierengarten and J.-F. Nierengarten, Chem. Rec., 2015, 15, 31–51 CrossRef PubMed.
- F. Odobel, M. Séverac, Y. Pellegrin, E. Blart, C. Fosse, C. Cannizzo, C. R. Mayer, K. J. Elliott and A. Harriman, Chem.–Eur. J., 2009, 15, 3130–3138 CrossRef PubMed.
- S. M. Marinho, M. H. Ha-Thi, V. T. Pham, A. Quaranta, T. Pino, C. Lefumeux, T. Chamaille, W. Leibl and A. Aukauloo, Angew. Chem., Int. Ed., 2017, 56, 15936–15940 CrossRef PubMed.
- Y. Chen, H. Chen and H. Tian, Chem. Commun., 2015, 51, 11508–11511 RSC.
- G. Smolentsev, B. Cecconi, A. Guda, M. Chavarot-Kerlidou, J. A. van Bokhoven, M. Nachtegaal and V. Artero, Chem.–Eur. J., 2015, 43, 15158–15162 CrossRef PubMed.
- J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407–414 CrossRef.
- J. R. McKone, E. L. Warren, M. J. Bierman, S. W. Boettcher, B. S. Brunschwig, N. S. Lewis and H. B. Gray, Energy Environ. Sci., 2011, 4, 3573–3583 RSC.
- S. W. Boettcher, E. L. Warren, M. C. Putnam, E. A. Santori, D. Turner-Evans, M. D. Kelzenberg, M. G. Walter, J. R. McKone, B. S. Brunschwig, H. A. Atwater and N. S. Lewis, J. Am. Chem. Soc., 2011, 133, 1216–1219 CrossRef PubMed.
- J. J. Leung, J. Warnan, D. H. Nam, J. Z. Zhang, J. Willkomm and E. Reisner, Chem. Sci., 2017, 8, 5172–5180 RSC.
- T. R. Hellstern, J. D. Benck, J. Kibsgaard, C. Hahn and T. F. Jaramillo, Adv. Energy Mater., 2016, 6, 1501758 CrossRef.
- Q. Huang, Z. Ye and X. Xiao, J. Mater. Chem. A, 2015, 3, 15824–15837 RSC.
- M. J. Kenney, M. Gong, Y. G. Li, J. Z. Wu, J. Feng, M. Lanza and H. J. Dai, Science, 2013, 342, 836–840 CrossRef PubMed.
- X. Zhou, R. Liu, K. Sun, K. M. Papadantonakis, B. S. Brunschwig and N. S. Lewis, Energy Environ. Sci., 2016, 9, 892–897 RSC.
- C. Ros, T. Andreu, M. D. Hernández-Alonso, G. Penelas-Pérez, J. Arbiol and J. R. Morante, ACS Appl. Mater. Interfaces, 2017, 9, 17932–17941 CrossRef PubMed.
- B. D. Alexander, P. J. Kulesza, I. Rutkowska, R. Solarska and J. Augustynski, J. Mater. Chem., 2008, 18, 2298–2303 RSC.
- S. Hilliard, D. Friedrich, S. Kressman, H. Strub, V. Artero and C. Laberty-Robert, ChemPhotoChem, 2017, 1, 273–280 CrossRef.
- W. Hamd, S. Cobo, J. Fize, G. Baldinozzi, W. Schwartz, M. Reymermier, A. Pereira, M. Fontecave, V. Artero, C. Laberty-Robert and C. Sanchez, Phys. Chem. Chem. Phys., 2012, 14, 13224–13232 RSC.
- S. Hilliard, G. Baldinozzi, D. Friedrich, S. Kressman, H. Strub, V. Artero and C. Laberty-Robert, Sustainable Energy Fuels, 2017, 1, 145–153 RSC.
- J. Toupin, H. Strub, S. Kressmann, M. Boudot, V. Artero and C. Laberty-Robert, Phys. Chem. Chem. Phys., 2017, 19, 30675–30682 RSC.
- K. Sekizawa, S. Sato, T. Arai and T. Morikawa, ACS Catal., 2018, 8, 1405–1416 CrossRef.
- M. Schreier, J. S. Luo, P. Gao, T. Moehl, M. T. Mayer and M. Gratzel, J. Am. Chem. Soc., 2016, 138, 1938–1946 CrossRef PubMed.
- A. Paracchino, V. Laporte, K. Sivula, M. Gratzel and E. Thimsen, Nat. Mater., 2011, 10, 456–461 CrossRef PubMed.
- J. R. McKone, A. P. Pieterick, H. B. Gray and N. S. Lewis, J. Am. Chem. Soc., 2012, 135, 223–231 CrossRef PubMed.
- X. Y. Yu, M. S. Prevot, N. Guijarro and K. Sivula, Nat. Commun., 2015, 6, 7596 CrossRef PubMed.
- T. Bourgeteau, D. Tondelier, B. Geffroy, R. Brisse, S. Campidelli, R. Cornut and B. Jousselme, J. Mater. Chem. A, 2016, 4, 4831–4839 RSC.
- T. Bourgeteau, D. Tondelier, B. Geffroy, R. Brisse, R. Cornut, V. Artero and B. Jousselme, ACS Appl. Mater. Interfaces, 2015, 7, 16395–16403 CrossRef PubMed.
- T. Bourgeteau, D. Tondelier, B. Geffroy, R. Brisse, C. Laberty-Robert, S. Campidelli, R. de Bettignies, V. Artero, S. Palacin and B. Jousselme, Energy Environ. Sci., 2013, 6, 2706–2713 RSC.
- A. Morozan, T. Bourgeteau, D. Tondelier, B. Geffroy, B. Jousselme and V. Artero, Nanotechnology, 2016, 27, 355401 CrossRef PubMed.
- L. Steier, S. Bellani, H. C. Rojas, L. Pan, M. Laitinen, T. Sajavaara, F. Di Fonzo, M. Gratzel, M. R. Antognazza and M. T. Mayer, Sustainable Energy Fuels, 2017, 1, 1915–1920 RSC.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef PubMed.
- N.-G. Park, Mater. Today, 2015, 18, 65–72 CrossRef.
- B. Shan, A. Nayak, M. K. Brennaman, M. Liu, S. L. Marquard, M. S. Eberhart and T. J. Meyer, J. Am. Chem. Soc., 2018, 140, 6493–6500 CrossRef PubMed.
- B. Shan, A. Nayak, R. N. Sampaio, M. S. Eberhart, L. Troian-Gautier, M. K. Brennaman, G. J. Meyer and T. J. Meyer, Energy Environ. Sci., 2018, 11, 447–455 RSC.
- L. D'Amario, L. J. Antila, B. Pettersson Rimgard, G. Boschloo and L. Hammarström, J. Phys. Chem. Lett., 2015, 6, 779–783 CrossRef PubMed.
- R. J. Dillon, L. Alibabaei, T. J. Meyer and J. M. Papanikolas, ACS Appl. Mater. Interfaces, 2017, 9, 26786–26796 CrossRef PubMed.
- Z. Huang, M. He, M. Yu, K. Click, D. Beauchamp and Y. Wu, Angew. Chem., Int. Ed., 2015, 54, 6857–6861 CrossRef PubMed.
- B. H. Farnum, K.-R. Wee and T. J. Meyer, Nat. Chem., 2016, 8, 845 CrossRef PubMed.
- C. J. Flynn, S. M. McCullough, E. Oh, L. Li, C. C. Mercado, B. H. Farnum, W. Li, C. L. Donley, W. You, A. J. Nozik, J. R. McBride, T. J. Meyer, Y. Kanai and J. F. Cahoon, ACS Appl. Mater. Interfaces, 2016, 8, 4754–4761 CrossRef PubMed.
- F. Héroguel, L. Silvioli, Y.-P. Du and J. S. Luterbacher, J. Catal., 2018, 358, 50–61 CrossRef.
- K. Sivula, J. Phys. Chem. Lett., 2013, 4, 1624–1633 CrossRef PubMed.
- K. Hanson, M. D. Losego, B. Kalanyan, G. N. Parsons and T. J. Meyer, Nano Lett., 2013, 13, 4802–4809 CrossRef PubMed.
- B. Seger, S. D. Tilley, T. Pedersen, P. C. K. Vesborg, O. Hansen, M. Gratzel and I. Chorkendorff, J. Mater. Chem. A, 2013, 1, 15089–15094 RSC.
- G. C. Correa, B. Bao and N. C. Strandwitz, ACS Appl. Mater. Interfaces, 2015, 7, 14816–14821 CrossRef PubMed.
- M. Schreier, F. Héroguel, L. Steier, S. Ahmad, J. S. Luterbacher, M. T. Mayer, J. Luo and M. Grätzel, Nat. Energy, 2017, 2, 17087 CrossRef.
- A. M. Lapides, B. D. Sherman, M. K. Brennaman, C. J. Dares, K. R. Skinner, J. L. Templeton and T. J. Meyer, Chem. Sci., 2015, 6, 6398–6406 RSC.
- C. E. Creissen, J. Warnan and E. Reisner, Chem. Sci., 2017, 9, 1439–1447 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc00899j |
| ‡ These authors contributed equally. |
| § Present address: Department of Chemistry and Applied Biosciences, Vladimir Prelog Weg 1-5, ETH Zürich, CH-8093 Zürich, Switzerland. |
| ¶ Present address: Department of Chemical Engineering, University College London, Torrington Place, London, WC1E 7JE, UK. |
| || Present address: Sorbonne Université, MONARIS, 4 Place Jussieu, 75252 Paris cedex 05, France. |
| This journal is © The Royal Society of Chemistry 2018 |