
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew Ru(II) photocages operative with near-IR light: new platform for drug delivery in the PDT window†
Malik H.
Al-Afyouni
,
Thomas N.
Rohrabaugh
Jr
,
Kathlyn F.
Al-Afyouni
and
Claudia
Turro
 *
*
Department of Chemistry and Biochemistry, The Ohio State University, Columbus, Ohio 43210, USA. E-mail: turro.1@osu.edu
First published on 20th July 2018
Abstract
A series of Ru(II) complexes bearing the tridentate 2,6-di(quinolin-2-yl)pyridine (dqpy) ligand were designed to undergo photoinduced ligand dissociation with red/near-IR light. The complexes [Ru(dqpy)(L)(CH3CN)]2+, where L = 2,2′-bipyridine (bpy, 1), 4,4′dimethyl-2,2′-bipyridine (Me2bpy, 2), and 1,10-phenanthroline (phen, 3). Complexes 1–3 exhibit red-shifted lowest energy metal-to-ligand charge transfer (MLCT) absorption maxima at ∼600 nm, as compared to the corresponding tpy (2,2′;6′,2′′-terpyridine) complexes with MLCT bands at ∼565 nm which appear as shoulders to the MLCT bands at ∼455 nm. This shift is attributed to the lower energy LUMO afforded by the dqpy ligand when compared to tpy, as evidenced by the shift of the first reduction wave to ∼0.3 V more positive potentials in the former. In addition, the lowest MLCT maximum of [Ru(dqpy)(acac)(CH3CN)]+ (4; acac− = acetylacetonate) is observed at 770 nm, attributed to the additional increase in energy of the HOMO afforded by the presence of the π-donating acac− ligand and supported by calculations. Complexes 1–3 undergo ligand substitution upon irradiation with red light, λirr ≥ 610 nm, and the ligand substitution photochemistry of 4 is accessible with near-IR light, λirr ≥ 715 nm and λirr = 735 nm. Complexes 1–4 exhibit similar quantum yields of ligand exchange, ΦL, with 450 and 600 nm irradiation, however, that of 4 is 2–3 times greater than those measured for 1–3. This enhancement is explained by the difference in ligand contributions to the HOMO. Density functional theory calculations predict partial dqpy ππ* character in the MLCT states of 1–3 and a mixed Ru/acac− → dqpy metal/ligand-to-ligand charge transfer (ML-LCT) state in 4. The photoreactivity of 1–4 with tissue-penetrating red and near-IR light, together with their exceptional dark stability (>48 h), makes the new Ru(II)-dqpy platform ideal for the development of new complexes for photoinduced drug release and for other applications that require broad absorption from the ultraviolet and visible ranges into the near-IR, such as solar energy conversion.
Introduction
Conventional chemotherapeutic cancer treatments typically exhibit high systemic toxicity and poor selectivity for cancer cells, resulting in undesirable side effects and off-site activity in healthy tissue.1–5 The drawbacks to conventional cancer treatment have motivated the development of new approaches, including the use of light to attain spatiotemporal control of metallodrug activation and release of active species at the tumor site.5–11 The use of light for drug release, photochemotherapy (PCT), utilizes an agent that is nontoxic in the dark, such that cell death is only initiated through the absorption of light. An important distinction between PCT and traditional photodynamic therapy, PDT, is that the latter relies on the presence of oxygen to function, whereas the former does not. The requirement for oxygen represents a drawback, since hypoxia at the tumor site limits the action of the drug.12,13 In both PCT and PDT, the approach imparts lowered systemic toxicity and invasiveness while increasing selectivity.5–11Recent work has demonstrated the versatility and efficacy of ruthenium-based PCT agents as potential anticancer drugs.5,8–11,14 However, most of these systems utilize visible light in the 400–550 nm range, thus limiting tissue penetration since it is outside the PDT window (600–900 nm).15,16 A handful of ruthenium-based systems capable of releasing a monodentate ligand using tissue-penetrating red light in the PDT window have been recently reported, but do not absorb beyond ∼650 nm.17–20 In these systems, the metal-to-ligand charge transfer (MLCT) transitions of these complexes are stabilized through the extension of conjugation and/or the addition of steric bulk to ancillary ligands to distort the pseudo-octahedral environment around the metal.17–20 The latter approach has been most successful in complexes where the monodentate ligand is a functionalized pyridine, however, ligand photodissociation with λirr ≥ 700 nm has not been achieved to date. A red-shift in the absorption of these cages through the control of the electronic properties of the complex without the addition of steric bulk at the metal center is desirable to circumvent thermal instability in water, an important factor in the design of PCT agents.
Herein we report the synthesis of a series of Ru(II) complexes supported by the 2,6-di(quinolin-2-yl)pyridine (dqpy) tridentate ligand, [Ru(dqpy)(L)(NCCH3)]2+, where L represents 2,2′-bipyridine (bpy, 1), 4,4′-dimethyl-2,2′-bipyridine (Me2bpy, 2), 1,10-phenanthroline (phen, 3), as well as [Ru(dqpy)(acac)(NCCH3)]+ (4) featuring the anionic acac− (acetylacetonate) ligand. These model complexes are designed to probe the ability of the [Ru(dqpy)(L)] fragment to serve as a vehicle for the delivery of nitrile-containing cysteine protease inhibitors with red and near-IR light.17,18 We envisioned that the expansion of the π-system of the common tpy (2,2′:6′,2′′-terpyridine) tridentate ligand would result in a red-shift to the MLCT absorption maximum without increasing the steric bulk around the monodentate ligand, since the quinolinyl groups are positioned in a planar fashion and are perpendicular to the axis containing the Ru–NCCH3 bond. Additionally, we sought to demonstrate the facile incorporation of a number of bidentate ancillary ligands with a range of absorption energies. The present work includes the first example of photoinduced ligand release from a Ru(II) complex with λirr ≥ 715 nm and highlights the versatility of the [Ru(dqpy)(L)] fragment as a caging group for the delivery of nitrile-containing drugs and inhibitors with irradiation in the PDT window.
Experimental
Materials
All materials were used as received without further purification unless otherwise noted. Diethyl ether, acetone, acetonitrile, and methanol were procured from Fisher Scientific, and dichloromethane was obtained from Macron Fine Chemicals. N,N-Dimethylformamide was purchased from Sigma-Aldrich and was dried under nitrogen by passing through a column of activated alumina. Reineke salt, ammonium hexafluorophosphate, lithium chloride, sodium chloride, sodium acetate trihydrate, ascorbic acid, 1,3-diphenylisobenzofuran (DPBF), acetylacetonate, 1,10-phenanthroline, 4,4′-dimethylbipyridyl, acetonitrile-d3 and acetone-d6 were purchased from Sigma-Aldrich, and 2,2′-bipyridine was obtained from Alfa Aesar. Sulfuric acid and sodium bicarbonate were received from Fisher Scientific, and potassium ferrioxalate was purchased from Strem Chemicals. Neutral alumina was purchased from Fisher Scientific. The starting material [Ru(p-cymene)Cl2]2,21 and the complexes [Ru(p-cymene)(bpy)Cl]Cl, [Ru(p-cymene)(4,4′-Me2bpy)Cl]Cl, [Ru(p-cymene)(phen)Cl]Cl, and Ru(p-cymene)(acac)Cl were synthesized according to published procedures.21b2,6-Di(quinolin-2-yl)pyridine (dqpy)
The dqpy ligand was synthesized following modifications of procedures previously reported.22,23 Ethanol (20 mL) was added to Fe powder (2.259 g, 40.5 mmol) and 2-nitrobenzaldehyde (0.607 g, 4.02 mmol), followed by 8 mL of 0.1 M HCl (0.8 mmol). The mixture was heated to 95 °C in an oil bath and stirred vigorously for 3 hours; the reaction was then allowed to cool to room temperature followed by the addition of 2,6-diacetylpyridine (0.270 g, 1.65 mmol) and KOH pellets (0.725 g, 12.9 mmol). The reaction was then returned to the oil bath and stirred at 95 °C overnight. After cooling to room temperature, the reaction was diluted with 200 mL of CH2Cl2 and the suspension was filtered through a pad of celite, followed by removal of the solvent by rotary evaporation. Methanol (100 mL) was added to the residue resulting in a white precipitate. The precipitate was collected by vacuum filtration and washed with methanol (3 × 15 mL), resulting in 0.425 g (1.27 mmol, 77%) of white solid product. 1H NMR (400 MHz) in acetone-d6, δ ppm (splitting, integration, J-coupling): 8.97 (d, 2H, 2J = 8.8 Hz), 8.85 (d, 2H, 2J = 8.0 Hz), 8.56 (d, 2H, 2J = 8.8 Hz), 8.21 (m, 3H), 8.06 (d, 2H, 2J = 8.0 Hz), 7.84 (m, 2H), 7.66 (m, 2H). ESI MS: [M + H+]+ experimental m/z = 334.14 calculated m/z = 334.13.[Ru(dqpy)(bpy)Cl][PF6] (1–Cl)
In a Schlenk tube, [Ru(p-cymene)(bpy)Cl]Cl (100 mg, 0.21 mmol) was combined with dqpy (84 mg, 0.25 mmol), excess LiCl (∼100 mg), and DMF (3 mL). The mixture was allowed to reflux for 3 h during which time the color changed from yellow to dark purple; addition of the reaction mixture to 100 mL of a concentrated aqueous NH4PF6 solution resulted in the precipitation of a purple solid that was isolated on fritted glass, washed with 3 × 20 mL water and 3 × 20 mL diethyl ether. The solid was dissolved in acetone (100 mL), and filtered through celite. The acetone solution was concentrated to 10 mL and purified on an alumina column with acetone as eluent. The purple solution was concentrated to ∼5 mL under vacuum and precipitated with 100 mL diethyl ether. The purple solid was isolated by vacuum filtration (113 mg, 70% yield). 1H NMR (400 MHz) in acetone-d6, δ ppm (multiplicity, integration, J-coupling): 10.82 (d, 1H, 2J = 5.6 Hz), 9.08 (d, 2H, 2J = 8.0 Hz), 8.98 (d, 1H, 2J = 7.6 Hz), 8.79 (d, 2H, 2J = 8.4 Hz), 8.66 (t, 1H, 2J = 8.4 Hz), 8.60 (d, 2H, 2J = 8.4 Hz), 8.47 (m, 2H), 8.41 (m, 1H), 8.01 (d, 2H, 2J = 8.0 Hz), 7.61 (m, 3H), 7.31 (m, 2H), 7.24 (d, 2H, 2J = 8.8 Hz), 7.09 (m, 2H).[Ru(dqpy)(Me2bpy)Cl][PF6] (2–Cl)
[Ru(dqpy)(Me2bpy)Cl][PF6] was prepared following the same procedure for 1–Cl using dqpy (82 mg, 0.24 mmol) and substituting [Ru(p-cymene)(bpy)Cl]Cl for [Ru(p-cymene)(Me2bpy)Cl]Cl (101 mg, 0.20 mmol), resulting 110 mg (67% yield) of isolated product. 1H NMR (400 MHz) in acetone-d6, δ ppm (multiplicity, integration, J-coupling): 10.63 (d, 1H, 2J = 6.0 Hz), 9.05 (d, 2H, 2J = 8.0 Hz), 8.85 (s, 1H), 8.77 (d, 2H, 2J = 8.8 Hz), 8.57 (d, 2H, 2J = 8.8 Hz), 8.42 (t, 1H, 2J = 8.2 Hz), 8.34 (s, 1H), 8.25 (d, 1H, 2J = 6.4 Hz), 8.00 (d, 2H, 2J = 8.0 Hz), 7.61 (m, 2H), 7.38 (d, 1H, 2J = 5.6 Hz), 7.32 (m, 4H), 6.90 (d, 1H, 2J = 5.6 Hz), 3.00 (s, 3H), 2.27 (s, 3H).[Ru(dqpy)(phen)Cl][PF6] (3–Cl)
[Ru(dqpy)(phen)Cl][PF6] was prepared following the same procedure for 1–Cl using dqpy (82 mg, 0.24 mmol) and substituting [Ru(p-cymene)(bpy)Cl]Cl for [Ru(p-cymene)(phen)Cl]Cl (100 mg, 0.20 mmol), resulting in 92 mg (56% yield) of isolated product. 1H NMR (400 MHz) in acetone-d6, δ ppm (multiplicity, integration, J-coupling): 11.10 (dd, 1H, 2J = 5.6 Hz, 2J = 1.4 Hz), 9.25 (dd, 1H, 2J = 8.4 Hz, 2J = 1.2 Hz), 9.13 (d, 2H, 2J = 8.4 Hz), 8.81 (m, 3H), 8.51 (m, 4H), 8.28 (dd, 1H, 2J = 8.2 Hz, 2J = 1.4 Hz), 8.15 (d, 1H, 2J = 9.2 Hz), 7.97 (dd, 1H, 2J = 5.6 Hz, 2J = 1.2 Hz), 7.91 (dd, 2H, 2J = 8.0 Hz, 2J = 1.6 Hz), 7.46 (m, 3H), 7.20 (dq, 2H, 2J = 8.8 Hz, 4J = 0.8 Hz), 7.09 (m, 2H).[Ru(dqpy)(bpy)(CH3CN)][PF6]2 (1)
The precursor 1–Cl (50 mg, 0.065 mmol) was dissolved in 20 mL 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) CH3CN/H2O in a 50 mL round-bottom flask. The solution was allowed to reflux for 24 h, resulting in a color change from purple to dark orange. The CH3CN/H2O reaction mixture was then added to 100 mL of concentrated aqueous NH4PF6 to form an orange precipitate; the solid was isolated on fritted glass by vacuum filtration and washed with 3 × 20 mL water and 3 × 20 mL diethyl ether. The solid was then dissolved in acetone (100 mL) and filtered through celite. The orange filtrate was concentrated to ∼5 mL under reduced pressure, and precipitated with 100 mL diethyl ether. The orange solid was isolated by vacuum filtration (52 mg, 87% yield). 1H NMR (400 MHz) in acetone-d6, δ ppm (multiplicity, integration, J-coupling): 10.57 (d, 1H, 2J = 5.6 Hz), 9.22 (d, 2H, 2J = 8.4 Hz), 9.03 (d, 1H, 2J = 8.0 Hz), 8.90 (d, 2H, 2J = 8.8 Hz), 8.76 (m, 4H), 8.56 (d, 1H, 2J = 8.0 Hz), 8.46 (m, 1H), 8.11 (d, 2H, 2J = 8.0 Hz), 7.85 (td, 1H, 3J = 7.9 Hz, 2J = 1.3 Hz), 7.78 (d, 1H, 2J = 5.6 Hz), 7.70 (t, 2H, 3J = 7.6 Hz), 7.43 (m, 2H), 7.21 (m, 3H), 2.29 (s, 3H). ESI + MS: [M]2+ experimental m/z = 316.07 calcd m/z = 316.07. Elem. anal. calcd for C35H26F12N6P2Ru·2H2O·(CH3)2CO: C: 44.93; H: 3.57; N: 8.27. Found: C: 45.12; H: 3.19; N: 8.30.
:1 (v/v) CH3CN/H2O in a 50 mL round-bottom flask. The solution was allowed to reflux for 24 h, resulting in a color change from purple to dark orange. The CH3CN/H2O reaction mixture was then added to 100 mL of concentrated aqueous NH4PF6 to form an orange precipitate; the solid was isolated on fritted glass by vacuum filtration and washed with 3 × 20 mL water and 3 × 20 mL diethyl ether. The solid was then dissolved in acetone (100 mL) and filtered through celite. The orange filtrate was concentrated to ∼5 mL under reduced pressure, and precipitated with 100 mL diethyl ether. The orange solid was isolated by vacuum filtration (52 mg, 87% yield). 1H NMR (400 MHz) in acetone-d6, δ ppm (multiplicity, integration, J-coupling): 10.57 (d, 1H, 2J = 5.6 Hz), 9.22 (d, 2H, 2J = 8.4 Hz), 9.03 (d, 1H, 2J = 8.0 Hz), 8.90 (d, 2H, 2J = 8.8 Hz), 8.76 (m, 4H), 8.56 (d, 1H, 2J = 8.0 Hz), 8.46 (m, 1H), 8.11 (d, 2H, 2J = 8.0 Hz), 7.85 (td, 1H, 3J = 7.9 Hz, 2J = 1.3 Hz), 7.78 (d, 1H, 2J = 5.6 Hz), 7.70 (t, 2H, 3J = 7.6 Hz), 7.43 (m, 2H), 7.21 (m, 3H), 2.29 (s, 3H). ESI + MS: [M]2+ experimental m/z = 316.07 calcd m/z = 316.07. Elem. anal. calcd for C35H26F12N6P2Ru·2H2O·(CH3)2CO: C: 44.93; H: 3.57; N: 8.27. Found: C: 45.12; H: 3.19; N: 8.30.
[Ru(dqpy)(4,4′-Me2bpy)(CH3CN)][PF6]2 (2)
[Ru(dqpy)(4,4′-Me2bpy)(CH3CN)][PF6]2 was prepared following the same procedure for 1 starting with 2–Cl (50 mg, 0.063 mmol) instead of 1–Cl, which resulted in isolation of 55 mg of product (93% yield). Single crystals were grown from diffusion of diethyl ether into a concentrated acetonitrile solution. 1H NMR (400 MHz) in acetone-d6, δ ppm (multiplicity, integration, J-coupling): 10.38 (d, 1H, 2J = 5.6 Hz), 9.21 (d, 2H, 2J = 8.0 Hz), 8.90 (m, 3H), 8.79 (d, 2H, 2J = 8.4 Hz), 8.70 (t, 1H, 3J = 8.2 Hz), 8.42 (s, 1H), 8.32 (d, 1H, 2J = 5.6 Hz), 8.11 (d, 2H, 2J = 8.0 Hz), 7.71 (t, 2H, 2J = 7.4 Hz), 7.55 (d, 1H, 2J = 5.6 Hz), 7.47 (m, 2H), 7.26 (d, 2H, 2J = 8.8 Hz), 7.05 (d, 1H, 2J = 6.0 Hz), 2.99 (s, 3H), 2.31 (s, 3H), 2.27 (s, 3H). ESI + MS: [M]2+ experimental m/z = 330.08 calcd m/z = 330.08. Elem. anal. calcd for C38H30F12N6P2Ru·(CH3)2CO: C: 46.67; H: 3.60; N: 8.34. Found: C: 47.75; H: 3.84; N: 8.07.[Ru(dqpy)(phen)(CH3CN)][PF6]2 (3)
[Ru(dqpy)(phen)(CH3CN)][PF6]2 was prepared following the same procedure for 1, using 3–Cl (50 mg, 0.063 mmol) instead of 1–Cl, which resulted in 51 mg of isolated product (86% yield). Single crystals were grown from diffusion of diethyl ether into a concentrated acetonitrile solution. 1H NMR (400 MHz) in acetone-d6, δ ppm (multiplicity, integration, J-coupling): 11.00 (dd, 1H, 2J = 5.2 Hz, 2J = 1.2 Hz), 9.34 (dd, 1H, 2J = 8.4 Hz, 2J = 1.2 Hz), 9.26 (d, 2H, 2J = 8.0 Hz), 8.90 (d, 2H, 2J = 8.8 Hz), 8.83 (dd, 1H, 2J = 8.2 Hz, 2J = 5.4 Hz), 8.75 (m, 3H), 8.49 (d, 1H, 2J = 8.8 Hz), 8.45 (dd, 1H, 2J = 8.4 Hz, 2J = 1.2 Hz), 8.20 (d, 1H, 2J = 8.8 Hz), 8.10 (dd, 1H, 2J = 5.2 Hz, 2J = 1.2 Hz), 8.00 (dd, 2H, 2J = 8.2 Hz, 2J = 1.4 Hz), 7.57 (m, 3H), 7.23 (m, 2H), 7.15 (d, 2H, 2J = 8.8 Hz), 2.36 (s, 3H). ESI + MS: [M]2+ experimental m/z = 328.05 calcd m/z = 328.07. Elem. anal. calcd for C37H26F12N6P2Ru·(CH3CH2)2O: C: 48.29; H: 3.56; N: 8.24. Found: C: 48.01; H: 3.56; N: 7.87.[Ru(dqpy)(acac)(CH3CN)][PF6] (4)
In a Schlenk tube Ru(p-cymene)(acac)Cl (100 mg, 0.24 mmol) was combined with dqpy (96 mg, 0.29 mmol), excess LiCl (∼100 mg), and DMF (3 mL). The mixture was allowed to reflux for 3 h during which the color changed from yellow to dark green. The solution was precipitated in 100 mL of a concentrated brine solution. The resulting green solid was isolated on fritted glass, dissolved in CH2Cl2 (200 mL), and filtered through celite. The green solution was concentrated to ∼5 mL under vacuum and precipitated with 100 mL diethyl ether. The green solid 4–Cl isolated by vacuum filtration was used without further purification. In a 50 mL round-bottom flask, 4–Cl was dissolved in 30 mL 3:1 (v/v) CH3CN/H2O. The solution was allowed to reflux for 24 h, during which time the color of the solution changed from green to dark purple. The CH3CN/H2O solution was then added to 100 mL of concentrated aqueous NH4PF6 to form a purple precipitate. The solid was isolated on fritted glass by vacuum filtration and washed with 3 × 20 mL water and 3 × 20 mL diethyl ether. The solid was then dissolved in acetone (100 mL) and filtered through celite. The filtrate was concentrated to ∼5 mL under reduced pressure, and purified on an alumina column with acetone as eluent. The purple solution was concentrated to ∼5 mL under vacuum and precipitated with 100 mL diethyl ether. The purple solid was isolated by vacuum filtration (55 mg, 32% yield). 1H NMR (400 MHz) in acetone-d6, δ ppm (multiplicity, integration, J-coupling): 9.34 (d, 2H, 2J = 9.2 Hz), 8.84 (d, 2H, 2J = 8.0 Hz), 8.72 (m, 4H), 8.17 (m, 3H), 8.05 (td, 2H, 3J = 7.8 Hz, 2J = 1.6 Hz), 7.88 (td, 2H, 3J = 7.5 Hz, 2J = 1.0 Hz), 5.29 (s, 1H), 2.58 (s, 3H), 2.11 (s, 3H), 1.29 (s, 3H). ESI + MS: [M]+ experimental m/z = 575.11 calcd m/z = 575.10. Elem. anal. calcd for C30H23F6N4PO2Ru·H2O: C: 48.85; H: 3.69; N: 7.60. Found: C: 49.33; H: 4.07; N: 7.85.
Instrumentation and methods
1H NMR spectra were collected with a Bruker 400 MHz DPX spectrometer. Electrospray ionization mass spectrometry (ESI-MS) was performed with a Bruker micrOTOF instrument dissolved in methanol. Microanalysis samples were weighed with a PerkinElmer Model AD-6 Autobalance and their compositions were determined with a PerkinElmer 2400 Series II Analyzer. Steady state absorption spectra were recorded on a Hewlett-Packard 8453 diode array spectrometer. Sample irradiation was performed using a 150 W Xe arc lamp (USHIO) in a MilliArc lamp housing unit with an LPS-220 power supply and an LPS-221 igniter (PTI) or two LUXEON Far Red Rebel LEDs (λmax = 735 ± 15 nm). The desired irradiation wavelengths using the Xe arc lamp were controlled by selecting the appropriate long pass (CVI Melles Griot) or bandpass (Thorlabs) filters. In general, the samples were irradiated in water (∼5% acetone) in a 1 x 1 cm quartz cuvette. For photolysis experiments, electronic absorption spectra were recorded at various time points of irradiation. The ligand exchange quantum yields (Φ) were determined by irradiating the samples in water using the appropriate bandpass filter. Electronic absorption spectra were recorded at early irradiation times (<10% conversion) during the quantum yield determination, and the rate of consumption of the reactant was determined from the slope of the line of a plot of the moles of reactant vs. irradiation time. The photon flux of the lamp was determined using potassium ferrioxalate (λirr = 450 nm) or Reineke salt (λirr = 600 nm) as the chemical actinometer.24 The quantum yield for ligand dissociation was calculated as the rate of moles of reactant consumed divided by the photon flux and corrected for the mean fraction of light absorbed by the sample.24 Electrochemical measurements were performed under a nitrogen atmosphere using a BASi model CV-50W voltammetric analyzer (Bioanalytical Systems Inc.). Cyclic voltammagrams were obtained using a three electrode configuration with a glassy carbon (3 mm) working electrode, a Ag/AgCl (3 M NaClaq) reference electrode, and a Pt wire auxiliary electrode in a glass tube fitted with a vycor tip. Ferrocene (Fc) was added at the end of each cyclic voltammetry experiment as a secondary reference; Fc+/Fc redox couple in 1 M TBAPF6 in acetonitrile E1/2 = +0.45 V.X-ray crystallography
Single crystal X-ray diffraction data was collected on a Bruker D8 Venture with a Photon II CPAD detector. All structures were solved using ShelXT and refined using ShelXL in the Olex2 program.25 The space groups were determined based on systematic absences and intensity statistics. An intrinsic phasing solution method provided most non-hydrogen atoms from the E-map. Full-matrix least squares/difference Fourier cycles were performed which located the remaining non-hydrogen atoms. All non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were placed in ideal positions and refined as riding atoms with relative isotropic displacement parameters. For complexes 2, 3, and 4, reflection contributions from disordered solvent molecules were removed using Platon, function: Squeeze.26 A total of 57 electrons and a 255 Å3 solvent accessible void per cell, 101 electrons and a 295 Å3 solvent accessible void per cell, and 555 electrons and a 1822 Å3 solvent accessible void per cell for 2, 3, and 4, respectively. Both PF6− polyatomic ions in 2 exhibit significant positional disorder, and were modeled by dividing the disordered atoms into groups (PART instruction), and allowing the atoms to refine freely.DFT calculations
Spin restricted DFT calculations were performed with the Gaussian 09 package.27 All geometry optimization and vibrational frequency calculations were performed with the SDD28 basis set on Ru and the TZVP29 basis set on all other atoms with the PBE30 exchange–correlation functional. The geometries of all complexes were fully optimized starting from X-ray crystal structures, when available. Complex 1 was optimized starting from the optimized structure of 2, replacing the methyl groups of 4,4′-dimethylbipyridine with hydrogen atoms. All optimized geometries have positive harmonic frequencies (confirming the calculated structures as electronic energy minima). Further calculations of molecular orbitals and TD-DFT utilized the B3LYP31 hybrid functional, again with the SDD basis set on Ru and with the TZVP basis set on all other atoms with the inclusion of solvation effects using the polarized continuum model (PCM) with acetonitrile as the solvent in the TD calculations.32 TD-DFT was used to calculate the electronic transition state energies and intensities of the 75 lowest-energy states. Orbitals from the Gaussian calculations were plotted using the Chemcraft program. The analysis of the MO compositions were performed using the AOMix program.33Results and discussion
Synthesis and characterization
The synthetic steps for the preparation of 1–4 are displayed in Scheme 1 starting from the dinuclear Ru(II) η6-arene complex, [Ru(p-cymene)Cl2]2.21 The reaction of the p-cymene dimer with the desired bidentate ligand, L, in CH3CN resulted in monomeric [Ru(p-cymene)(L)Cl]n+, which was then heated to displace the arene in the presence of the dqpy ligand, affording the corresponding [Ru(dqpy)(L)Cl]+ (L = bpy, Me2bpy, phen) complex or [Ru(dqpy)(acac)Cl].20 In the final step, the desired acetonitrile complex, 1–4, was obtained by heating each chloride precursor in 3:1 (v/v) CH3CN/H2O, followed by aqueous salt metathesis with NH4PF6.
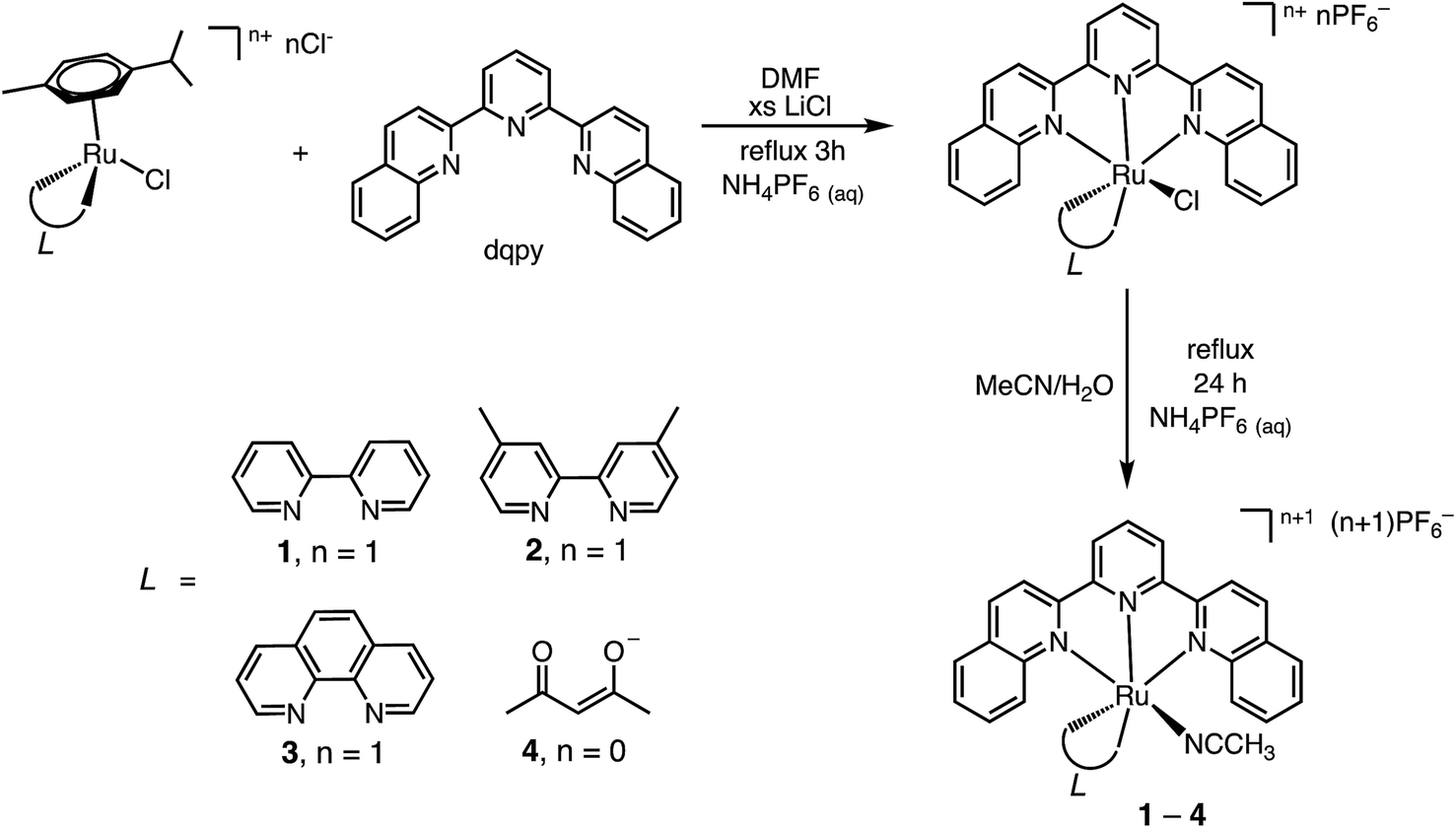 | ||
| Scheme 1 | ||
The chemical structures of 1–4 were confirmed by 1H NMR spectroscopy and positive mode electrospray ionization mass spectrometry (ESI-MS+). In general, the 1H NMR spectra of 1–4 in acetone-d6 exhibit aryl resonances in the 7.0–9.0 ppm range that are typical for polypyridyl compounds.20a,34–36 In 1–3, a distinctive resonance is observed at ∼10.5–11.0 ppm known to correspond to the aryl proton of the bidentate ligand positioned directly adjacent to the CN triple bond of the acetonitrile ligand. The deshielding of this resonance in 1–3 is a result of proximity to the nitrile group in an orientation perpendicular to the cylindrical magnetic anisotropy along the nitrile CN bond. In 4, a distinctive 1H resonance at 5.30 ppm is observed, assigned to the proton bound to the central carbon of the acac− ligand. In 1–4 a single methyl proton resonance is observed in the 2.0–2.3 ppm range corresponding to the coordinated CH3CN ligand, as previously reported for related complexes.20a,34,37
The solid-state single crystal X-ray diffraction structures of 2–4 are shown in Fig. 1 and additional information is provided in Table S1.† The bond lengths from the metal to the CH3CN ligand, Ru–N1, and to the central pyridine ring of the dqpy ligand, Ru–N3, in 2, 2.044(1) and 1.979(2) Å, respectively, are similar to those previously reported for [Ru(tpy)(4,4′-Me2bpy)(NCCH3)]2+ at 2.037 and 1.968 Å, respectively.36 The Ru–N2 and Ru–N4 bonds to the quinolinyl units of the dqpy ligand in 2 are significantly lengthened relative to Ru–N3, and are on average 0.085 Å longer than the corresponding Ru–N bonds in [Ru(tpy)(4,4′-Me2bpy)(NCCH3)]2+.36 This is a result of steric clashes between the quinolinyl protons of the dqpy ligand and the adjacent ring system of the bidentate ligand. Nevertheless, the sum of the angles around Ru formed by the three N atoms of the dqpy ligand and N5 of the bidentate ligand in 2 is 359.7°, indicating that despite the introduction of relatively bulky quinolinyl groups, dqpy binds to Ru(II) in a rigorously planar fashion analogous to terpyridine. The N1–Ru–N6 and Ru–N1–C1 angles at 171.1° and 169.7°, respectively, in the solid-state structure of 2 deviate slightly from 180° due to steric clashes with the PF6− anion and co-crystallized acetonitrile solvent (Fig. S1†). The N2–Ru–N4 angle in 2 at 156.5° is consistent with the corresponding N–Ru–N angle in [Ru(tpy)(4,4′-Me2bpy)(NCCH3)]2+, highlighting the distortion from octahedral coordination geometry imposed by both the dqpy and tpy ligands.36 The N5–Ru–N6 angle in 2 at 78.4° is also consistent with the corresponding angle in the tpy supported analog.36
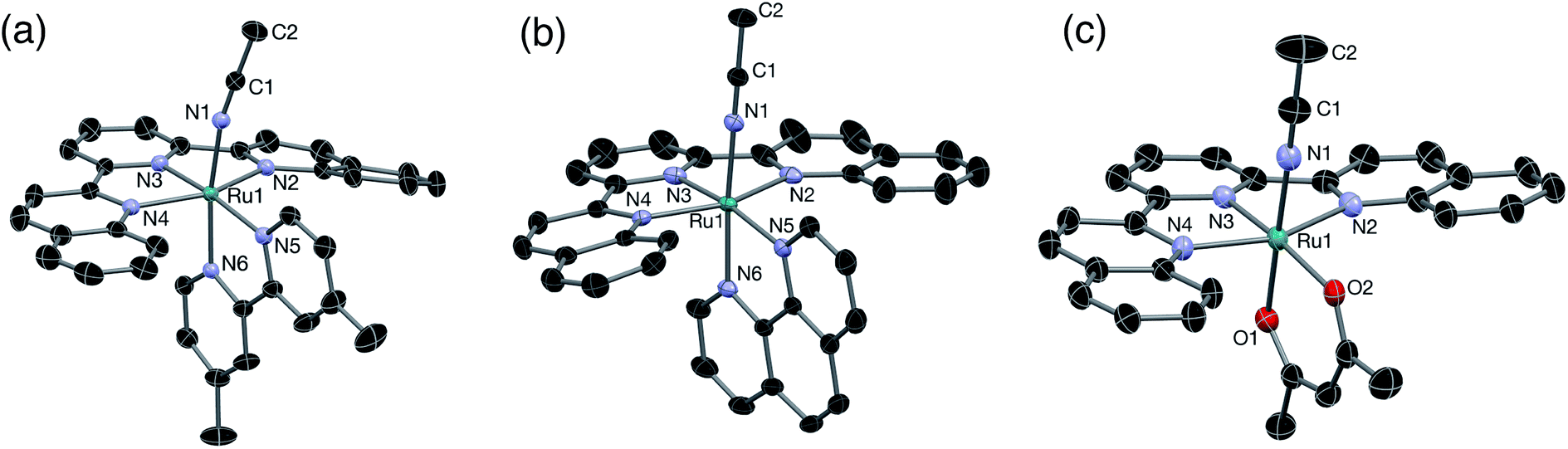 | ||
| Fig. 1 Thermal ellipsoid plots of (a) 2, (b) 3, and (c) 4 shown at 50% probability. H atoms, PF6− ions, and co-crystallized solvent molecules omitted for clarity. | ||
The Ru–N1 distance in 3, 2.036(3) Å, is in good agreement with that previously reported in related nitrile complexes,34 and the Ru–N3 distance of 1.972(3) Å is consistent with that reported for [Ru(tpy)(phen)(CH3CN)]2+.34 As expected, the structural features of 3 are similar to those of 2, where the Ru–N2 and Ru–N4 bonds in 3 are on average 0.076 Å longer than the corresponding Ru–N bonds in [Ru(tpy)(phen)(CH3CN)]2+ and the sum of the angles formed by the three N atoms of the dqpy ligand and N5 of the bidentate ligand is nearly planar at 359.9°. The N1–Ru–N6 and the Ru–N1–C1 angles in 3 are close to linear at 174.6° and 178.5°, respectively.
The structure of 4 is similar to that previously reported for [Ru(tpy)(acac)(dmso)]+ with Ru–N3, Ru–O1, and Ru–O2 distances determined to be 1.937(2) Å, 2.053(2) Å, and 2.090(2) Å, respectively.35 The Ru–N2 and Ru–N4 bond distances of 2.096(3) Å and 2.124(2) Å, respectively, are consistent with the elongation of the corresponding bonds observed in 2 and 3. The Ru–N1 bond length, 2.016(2) Å, is similar to those observed in 2 and 3. The angle in 4 formed by O1–Ru–O2 at 89.9° is large compared with the corresponding N5–Ru–N6 angles in 2 and 3 at 78.4° and 79.3°, respectively, as expected for the six-membered ring formed upon coordination of acac− in contrast to the five-membered rings in 2 and 3. Interestingly, the short Cquinoline–O2 distance observed in 4 suggests that an intramolecular H-bonding interaction may occur in the absence of an external H-bond donor. Accordingly, as discussed below, a solvochromatic shift is observed for 4 in protic solvents.
Electronic absorption and electrochemistry
The electronic absorption spectra of 1–4 are shown in Fig. 2 and relevant information is listed in Table 1. Absorption features in the ultraviolet (UV) and near-UV regions are assigned as arising from 1ππ* transitions of dqpy and the bidentate ligands, as is typical in Ru(II) polypyridyl complexes.38,39 Complex 1 exhibits an 1MLCT absorption maximum at 471 nm in acetone, which is only slightly red-shifted compared to that of [Ru(tpy)(bpy)(NCCH3)]2+ at 468 nm.40 However the 1MLCT transition of 1 also exhibits two shoulders at ∼550 (ε = 1800 M−1 cm−1) and ∼609 nm (ε = 800 M−1 cm−1), and the latter tails to ∼690 nm (Fig. 2). Complexes 2 and 3 exhibit slightly red-shifted 1MLCT transitions compared to their terpyridine analogs,34,36 and also feature low energy shoulders (Table 1 and Fig. 2). It was previously shown in an in-depth electronic structure study that the low-energy shoulders of the 1MLCT transitions in [Ru(tpy)(bpy)(L)]2+ complexes arise from Ru(II)→tpy 1MLCT transitions.41 Therefore, the low-energy 1MLCT shoulders in 1–3 are attributed to transitions from Ru(dπ) to dqpy(π*) orbitals, likely localized on the quinoline moieties based on the similar red-shift of the 1MLCT absorption observed in analogous Ru(II) complexes containing the 2,2′-biquinoline ligand.20,42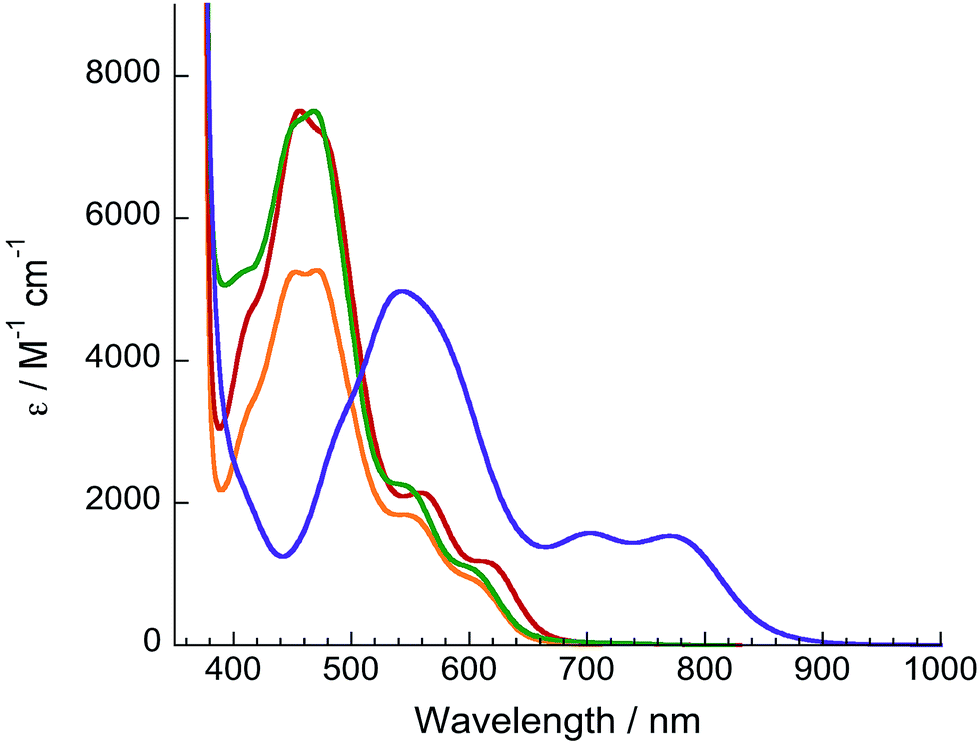 | ||
| Fig. 2 Electronic absorption spectra of 1 (orange), 2 (red), 3 (green), and 4 (purple) in acetone. | ||
| Complex | λ abs/nm (ε/×103 M−1 cm−1) | E 1/2 /V | Φ 450, Φ600 |
|---|---|---|---|
| a vs Ag/AgCl, 0.1 M TBAPF6. b Irreversible. | |||
| 1 | 352 (16), 371 (22), 471 (5.3), 550 (1.8), 609 (0.8) | +1.47, −0.93, −1.38, −1.67 | 0.0039(1), 0.0042(3) |
| 2 | 353 (22), 372 (27), 456 (7.5), 558 (2.1), 608 (1.2) | +1.43, −0.95, −1.39, −1.74 | 0.0032(3), 0.0029(2) |
| 3 | 353 (19), 372 (29), 469 (7.5), 534 (2.3), 600 (1.1) | +1.49, −0.92, −1.37, −1.65 | 0.0048(4), 0.0050(2) |
| 4 | 346 (25), 366 (16), 540 (5.0), 703 (1.6), 770 (1.5) | +0.78, −1.15, −1.67,b −1.86b | 0.0096(8), 0.010(1) |
In addition to being significantly red-shifted relative to 1–3, the 1MLCT maximum of 4 at 540 nm is accompanied by two bands with significant absorption of near-IR light at 703 nm (ε = 1600 M−1 cm−1) and 770 nm (ε = 1500 M−1 cm−1), the latter tailing to 950 nm. It is evident that the dqpy ligand provides a low-energy ligand-centered π* orbital to shift the absorption of 1–4 into the therapeutic window. In the case of 4, the electron-donating acac− ligand also raises the energy of the metal-based HOMO, further shifting the 1MLCT absorption to lower energy.
Cyclic voltammagrams of 1–4 collected in CH3CN (0.1 M TBAPF6) are shown in Fig. 3. Complexes 1–3 exhibit fully reversible oxidation events ranging from +1.43 to +1.49 V vs. Ag/AgCl assigned to the Ru(III/II) couples, similar to those for the tpy analogs [Ru(tpy)(L)(CH3CN)]2+ (L = bpy, phen, Me2bpy), reported to range from +1.32 to +1.36 V vs. Ag/AgCl in the same solvent (Table 1).34,43,44 A significant cathodic shift is observed for the reversible Ru(III/II) couple of 4 as compared to 1–3, attributed to the destabilization of filled Ru(dπ) orbitals by the strongly π-donating acac− ligand and it appears at a similar potential to that reported for [Ru(tpy)(acac)(dmso)]+, +0.83 V vs. Ag/AgCl, in the same solvent.35
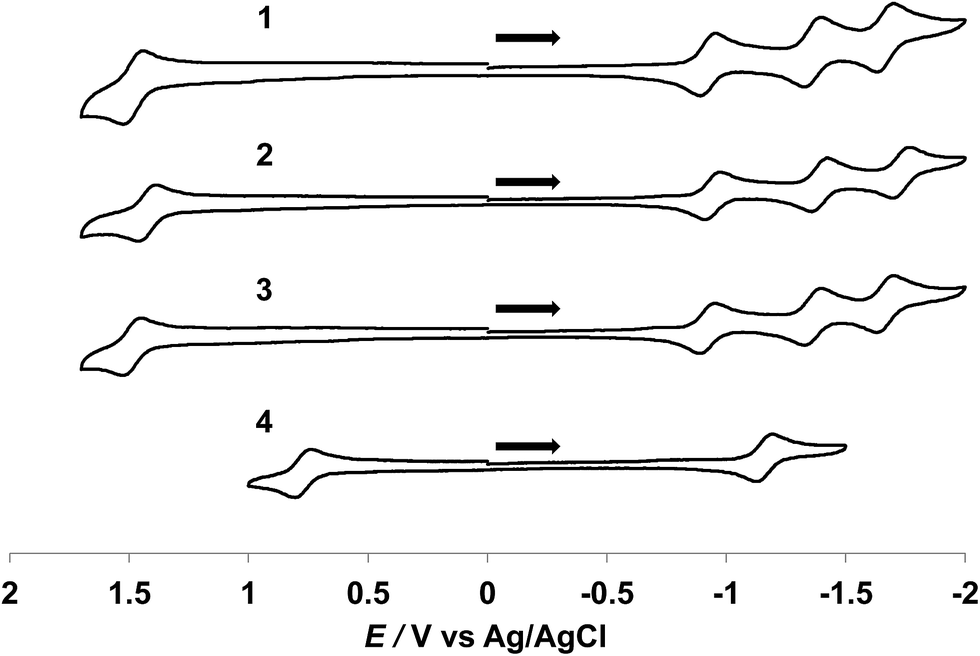 | ||
| Fig. 3 Cyclic voltammagrams of 1–4 in CH3CN (0.1 M TBAPF6, 200 mV s−1). | ||
Three reversible reduction waves are observed for 1–3 and the potentials of the first two reduction couples are independent of the bidentate ligand and appear at ∼−0.93 V and ∼−1.38 V vs. Ag/AgCl (Table 1). As such, these two couples are assigned to the placement of two successive electrons on the dqpy ligand. Two irreversible reduction waves are observed for free dqpy at ∼−2.04 V and ∼−2.56 V vs. Ag/AgCl under identical experimental conditions (Fig. S2†). A third reversible reduction wave is also present in 1–3 that is associated with the bidentate ligand. Complex 4 exhibits one reversible reduction couple at ∼−1.15 V vs. Ag/AgCl and two irreversible waves at ∼−1.86 V and at ∼−2.19 V vs. Ag/AgCl (Fig. S3†), shifted to more negative potentials relative to 1–3. As expected, the monocationic complex 4 is more difficult to reduce than the divalent 1–3 ions.
The HOMO–LUMO gaps in 1, 2, and 3 can be estimated at 2.40 eV, 2.38 eV, and 2.41 eV, respectively, from the difference in the first oxidation and first reduction couples of each complex. A value of 1.93 eV is calculated for 4, attributed to the highly cathodically shifted Ru(III/II) oxidation potential in this complex.
Electronic structure calculations
DFT calculations were performed to gain further insight into the electronic structures of 1–4. Geometry optimizations using the PBE functional, the SDD basis set for Ru, and the TZVP basis set on all other atoms gave structures in excellent agreement with experimentally determined crystal structures for 2–4 (Tables S2–S4†). The calculated molecular orbitals (MOs) and Mulliken population densities for 2 suggest that in the ground state the HOMO is of mixed Ru/dyz (58%) and dqpy/π (30%) character with minor contributions from Me2bpy/π* (7%) and CH3CN/π* (5%), while the LUMO is of almost entirely (92%) dqpy/π* in character. The rendered contour plot of the LUMO in Fig. 4a shows significant delocalization on the quinolinyl groups of the dqpy ligand. The next two higher unoccupied MOs, LUMO+1 and LUMO+2, are of dqpy/π* (96%) and Me2bpy/π* (92%) character, respectively, which supports the assignments of the second and third reduction waves in the CV of 2 as dqpy- and Me2bpy-based, respectively. As expected, the calculated MOs and Mulliken population densities of 1 and 3 show similar properties to 2. The HOMO in each is of mixed Ru/dyz and dqpy/π character, the LUMO and LUMO+1 are of predominately dqpy/π* character, and the LUMO+2 has mainly bidentate/π* character (Fig. S20 and S21†), also consistent with the electrochemical assignments for these complexes.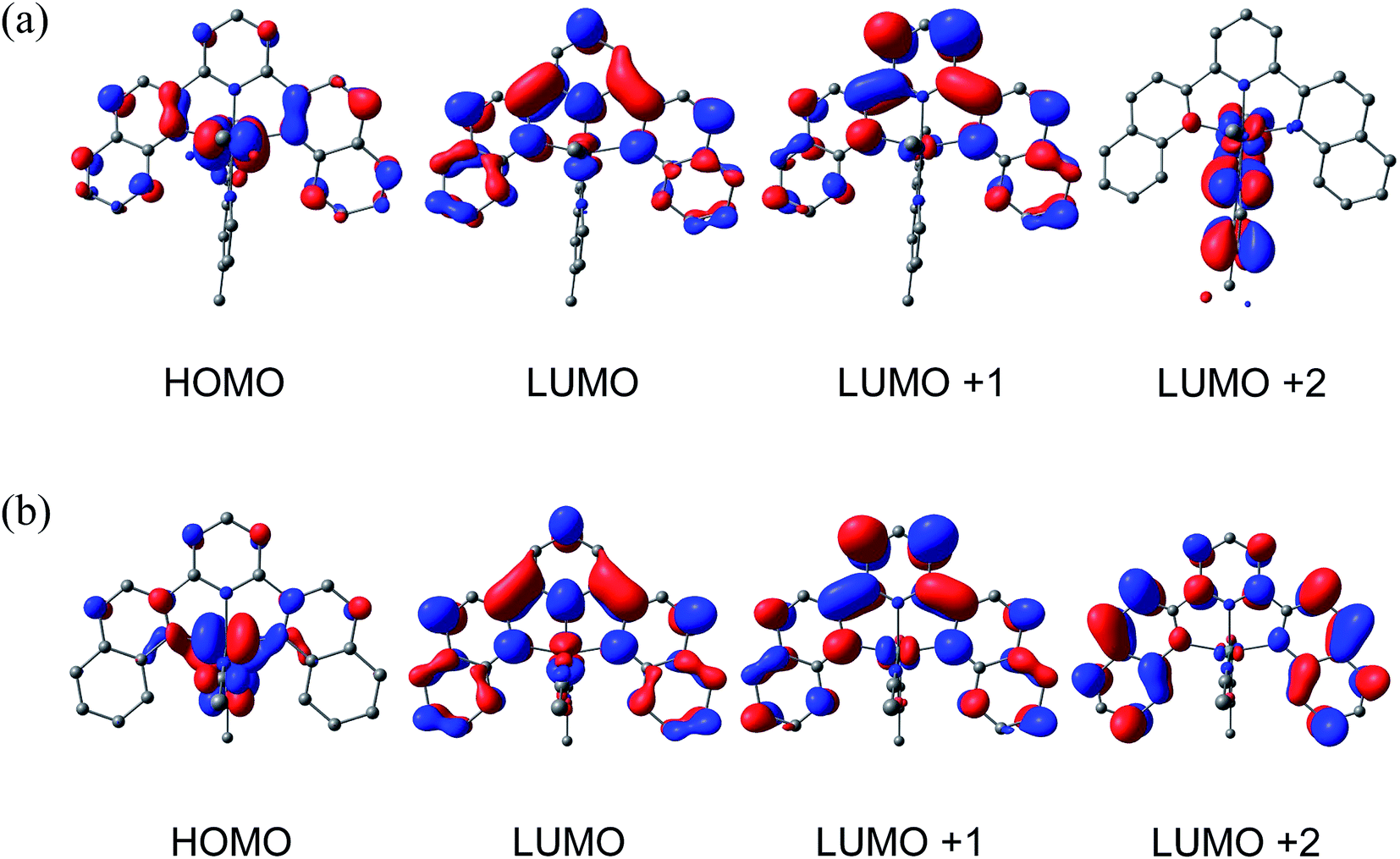 | ||
| Fig. 4 Calculated frontier molecular orbitals for (a) 2 and (b) 4. | ||
The calculated ground state HOMO of 4 shown in Fig. 4b is of mixed Ru/dyz (53%) character with considerable acac−/π (27%) character, with only 17% localization on the dqpy ligand. This result is expected based on the significant donating ability of the acac− ligand. The LUMO of 4 is calculated to be predominately dqpy/π* (90%), analogous to 1–3. It is therefore evident that the π-interaction of Ru/dyz orbitals with the acac− π-orbitals significantly destabilizes the HOMO in 4, consistent with the cathodic shift of the oxidation potential observed electrochemically. The LUMO + 1 in 4 is predominately of dqpy/π* (96%) character, but in contrast to 1–3, the LUMO + 2 is also localized on the dqpy ligand (98% dqpy/π*), reflecting the electron-rich character of the acac− ligand.
The calculated electronic absorption spectra of 1–4 using TD-DFT are in good agreement with experimental data. The calculated HOMO–LUMO energy difference in 2 is 2.17 eV, which is in good agreement with the 2.38 eV determined electrochemically. The lowest energy transition is predicted to be a 1MLCT predominately from the HOMO (Ru/dyz) to the LUMO (dqpy/π*) with a maximum at 570 nm, corresponding to the experimentally observed lowest-energy shoulder at 608 nm. The slightly higher-energy shoulder transition at 558 nm is calculated at 544 nm to be a mixed 1MLCT from HOMO-2 (Ru/dxy) and HOMO-1 (Ru/dxz) to the LUMO (dqpy/π*). Four predicted 1MLCT transitions comprise the intense features in the 420–480 nm range of the absorption spectrum of 2. The most intense transition at 456 nm, calculated at 446 nm, is of mostly HOMO (Ru/dyz) to LUMO+2 (Me2bpy/π*) character, while the other three transitions are of mixed Ru/d to dqpy/π* character. Calculated absorption energies of 1 and 3 parallel those of 2 (See Table S5†). The most intense 1MLCT transitions in 1–3 are to the bidentate ligand, whereas those at lower energies are localized on the dqpy ligand.
The HOMO–LUMO energy difference in 4 is calculated to be 1.70 eV, consistent with the electrochemical gap 1.93 eV. Using TD-DFT, a pure HOMO–LUMO transition is calculated at 730 nm, which corresponds to the absorption feature at 770 nm. A mixed HOMO and HOMO-1 (Ru/dxy) to dqpy/π* transition is calculated with a maximum at 598 nm, and four additional 1MLCT transitions of predominately Ru/d to LUMO − LUMO+2 character are calculated within the visible spectrum (Table S5†). No transitions involving the acac− ligand are predicted in the visible/near-IR range.
Photochemistry
The ligand substitution photochemistry of 1–4, depicted in Scheme 2, was monitored through the changes in the electronic absorption and 1H NMR spectra as a function of irradiation time in D2O and CD3CN (λirr ≥ 395 nm). The photolysis of 1 in D2O (5% acetone-d6) featured a decrease in the intensity of the peak at 2.07 ppm associated with the coordinated CH3CN ligand, and an increase in intensity of that corresponding to free CH3CN at 2.06 ppm. However, the ligand exchange product, [Ru(dqpy)(bpy)(D2O)]2+, generally precipitated in solution at concentrations necessary for 1H NMR characterization. Monitoring the photolysis of 1 in CD3CN was more successful. A decrease in intensity of the 1H NMR peak at 2.02 ppm associated with the coordinated CH3CN ligand was observed with the concomitant increase of the resonance at 1.96 ppm known to correspond to free CH3CN (Fig. S4†).20,45 No other spectral changes are observed, consistent with the exchange of CH3CN for solvent CD3CN during the photolysis. Similar spectral changes are observed for the photolyses of 2–4 in CD3CN (Fig. S5–S7†). | ||
| Scheme 2 | ||
The irradiation of 1 results in a decrease in the 1MLCT band at 471 nm and the appearance of a new band at 474 nm as a function of irradiation time with λirr ≥ 395 nm and λirr ≥ 610 nm (Fig. 5a). During the photolysis of 1, a single set of isosbestic points at 402, 480, and 540 nm were observed, indicative of a single transformation without secondary reactions. Similar results were observed for 2 and 3 with λirr ≥ 395 nm and λirr ≥ 610 nm (Fig. S9–S11†). It should also be noted that no changes to the electronic absorption spectra of 1–4 were observed in the absence of light for at least 48 h in H2O (5% acetone), consistent with the dark stability of the complexes (Fig. S14–S17†).
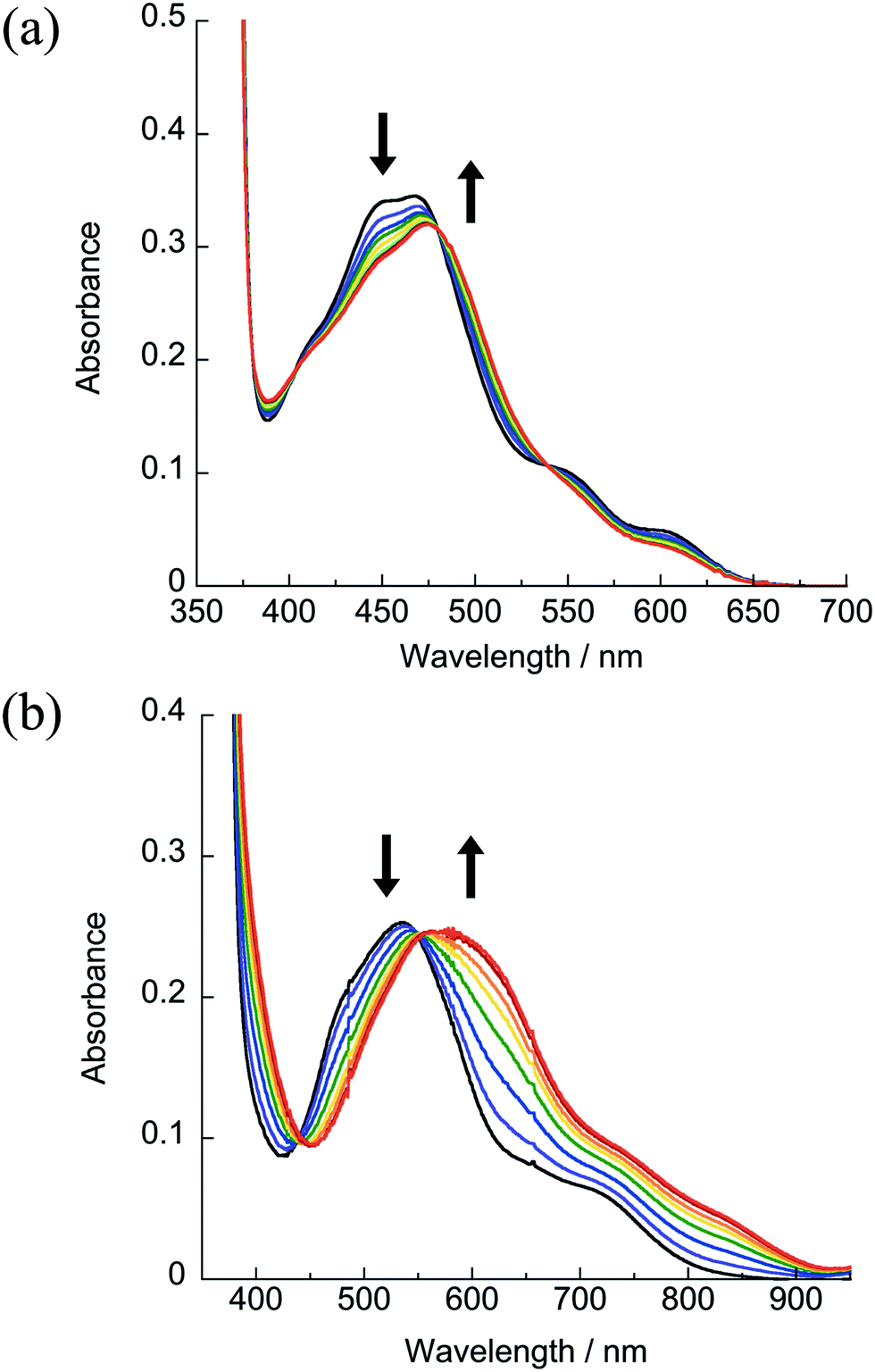 | ||
| Fig. 5 Changes in the electronic absorption spectra upon irradiation of (a) 1 (λirr ≥ 610) nm and (b) 4 (λirr ≥ 715 nm) in H2O (5% acetone), tirr = 0–2 h under a N2 atmosphere. | ||
The photoreactivity of 4 in H2O (5% acetone) with near-IR light λirr ≥ 715 nm is shown in Fig. 5b. The irradiation of nitrogen-purged samples of 4 results in similar spectral changes as those observed for 1–3, with a decrease in the 1MLCT band at 533 nm and the appearance of a new peak at 600 nm with an isosbestic point at 551 nm, confirming the release of CH3CN and the formation of a single product. Whereas the photochemistry of 1–3 is independent of oxygen, 4 undergoes a secondary photochemical reaction in water in the presence of oxygen (Fig. S12†). Given the low Ru(III/II) oxidation potential of 4, it is believed that the photochemical product, [Ru(dqpy)(acac)(OH2)]+ or its deprotonated form, Ru(dqpy)(acac)(OH), is oxidized by O2 in solution (E1/2(O20/−) = −0.82 V vs. Ag/AgCl in water) upon absorption of a photon.46 Analysis of the photochemically generated species present in aerated aqueous solutions of 4 by mass spectrometry reveals a peak with m/z = 551.13, consistent with [Ru(dqpy)(acac)(OH)]+ (Fig. S19†). However, control experiments show that this reaction requires a photon; when the photolysis of 4 is conducted in N2-purged H2O and only opened to air after the sample is removed from the light, the oxidation side-reaction is not observed (Fig. S13†). Moreover, when 4 is irradiated in water in the presence of the reducing agent ascorbic acid in air, no product oxidation is apparent, consistent with the regeneration of the Ru(II) product by ascorbate (Fig. S13†). In contrast to the reactivity in water, the photoinduced ligand exchange of 4 in aerated CD3CN is not accompanied by oxidation, as determined by following the reaction by 1H NMR spectroscopy upon irradiation of 4 in CD3CN using LEDs with λirr = 735 ± 15 nm, which results in the release of free CH3CN after 2 h with no other observable spectral changes (Fig. S8†). These results indicate that the secondary pathway likely involves photoinduced electron transfer to O2 to generate superoxide and is a topic of future work.
The quantum yields of CH3CN exchange, Φ450, for 1–4 in deaerated water with λirr = 450 nm are listed in Table 1, and those recorded using λirr = 600 nm were identical within the error of the measurement. The values of Φ450 measured for 1–3 are generally similar to each other, but the photoaquation quantum yield of 4 is greater by a factor of ∼2–3 (Table 1). In contrast to 4, photoisomerization in the related [Ru(tpy)(acac)(dmso)]+ complex is not observed (Φ < 0.0001),35 whereas that of [Ru(tpy)(bpy)(dmso)]2+ is more efficient (Φ = 0.024).47 It is therefore likely that ligand photosubstitution in 4 occurs by a different mechanism than the photoisomerization of the related dmso complexes.
It is important to note that the Ru-based HOMO of 4 possesses a significant contribution from the acac− ligand (27%), whereas the ligand contribution to the HOMOs of 1–3 are calculated to possess ∼30% dqpy character and only a small (∼7%) contribution from the bidentate ligands. The LUMO in 1–4 is predicted to be localized on the dqpy ligand and the lowest energy MLCT band is calculated to arise from a HOMO–LUMO transition. Therefore, the hole in the lowest energy MLCT states of 1–3 is expected to have significant contribution from the dqpy ligand, whereas partial oxidation of the acac− ligand is predicted for 4 in MLCT state. It is hypothesized that this difference in the localization of charge in the excited state may play a role in the ligand dissociation. In the case of 1–3, the MLCT state should possess significant dqpy ππ* character, whereas in 4 the excited state is mixed Ru/acac−→dqpy metal/ligand-to-ligand charge transfer (ML-LCT). Removal of electron density from the acac− ligand with one coordination site positioned trans to the leaving ligand may weaken the Ru–NCCH3 bond and lead to dissociation. Alternatively, the 3ML-LCT excited state in 4 may be able to mix with the dissociative metal-centered state, thus promoting ligand exchange.
Conclusion
The series of dqpy supported Ru(II) complexes presented above undergo ligand substitution upon absorption of tissue-penetrating red and near-IR light. These complexes are stable in the dark, and exhibit similar quantum yields with 450 and 600 nm irradiation. Electrochemical studies confirm that the lowered LUMO energy resulting from the extended π-system in the dqpy ligand in 1–3 relative to the terpyridine substituted analogs is responsible for the red-shifted MLCT transition energies, whereas in 4 the predominant cause of the exceptionally low energy MLCT transitions is the relatively destabilized HOMO resulting from the strongly electron-donating acac− ligand. Structural evidence reveals that dqpy imposes little or no steric hindrance on the monodentate nitrile ligand, and the influence of the dqpy ligand is primarily electronic, allowing for sterics to be easily added or removed through choice of bidentate ligand. The versatility, photoreactivity, and exceptional dark stability of dqpy supported Ru(II) complexes makes them ideal candidates for PCT agents. Studies using complexes of this type as photocages for anticancer drugs in biological systems are currently underway.Conflicts of interest
There are no conflicts of interest.Acknowledgements
The authors thank Pelotonia for financial support and the Ohio Supercomputer Center. Elemental analyses were obtained from the CENTC Elemental Analysis Facility at the University of Rochester, funded by NSF CHE-0650456. The authors are grateful to Dr Judith C. Gallucci and Dr William W. Brennessel for their guidance in crystallography.References
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436 CrossRef PubMed.
- M. Hanif and C. G. Haringer, Future Med. Chem., 2018, 10, 615 CrossRef PubMed.
- M. Yang and U. Bierbach, Eur. J. Inorg. Chem., 2017, 1561 CrossRef.
- G. Palermo, A. Magistrato, T. Riedel, T. von Erlach, C. A. Davey, P. J. Dyson and U. Rothlisberger, ChemMedChem, 2016, 11, 1199 CrossRef PubMed.
- F. Reeβsing and W. Szymanski, Curr. Med. Chem., 2017, 24, 4905 Search PubMed.
- (a) N. J. Farrer, L. Salassa and P. J. Sadler, Dalton Trans., 2009, 10690 RSC; (b) P. Zhang and P. J. Sadler, J. Organomet. Chem., 2017, 839, 5 CrossRef.
- L. C.-C. Lee, K.-K. Leung and K. K.-W. Lo, Dalton Trans., 2017, 46, 16357 RSC.
- C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660 RSC.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, Chem. Soc. Rev., 2017, 46, 5771 RSC.
- J. D. Knoll and C. Turro, Coord. Chem. Rev., 2015, 282, 110 CrossRef PubMed.
- A. Li, C. Turro and J. J. Kodanko, Chem. Commun., 2018, 54, 1280 RSC.
- Y. Mir, J. E. van Lier, B. Paquette and D. Houde, Photochem. Photobiol., 2008, 84, 1182 CrossRef PubMed.
- J. Dang, J. He, D. Chen and L. Yin, Biomater. Sci., 2017, 5, 1500 RSC.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706 RSC.
- R. R. Allison and K. Moghissi, Photodiagn. Photodyn. Ther., 2013, 10, 331 CrossRef PubMed.
- S. Mallidi, S. Anbil, A.-L. Bulin, G. Obaid, M. Ichikawa and T. Hasan, Theranostics, 2016, 6, 2458 CrossRef PubMed.
- E. Wachter, D. K. Heidary, B. S. Howerton, S. Parkin and E. C. Glazer, Chem. Commun., 2012, 48, 9649 RSC.
- (a) W. Sun, R. Thiramanas, L. D. Slep, X. Zeng, V. Mailänder and S. Wu, Chem.–Eur. J., 2017, 23, 10832 CrossRef PubMed; (b) W. Sun, S. Li, B. Häupler, J. Liu, S. Jin, W. Steffen, U. S. Schubert, H.-J. Butt, X.-J. Liang and S. Wu, Adv. Mater., 2017, 29, 1603702 CrossRef PubMed.
- L. N. Lameijer, D. Ernst, S. L. Hopkins, M. S. Meijer, S. H. C. Askes, S. E. Le Dévédec and S. Bonnet, Angew. Chem., Int. Ed., 2017, 56, 11549 CrossRef PubMed.
- (a) B. A. Albani, C. B. Durr and C. Turro, J. Phys. Chem. A, 2013, 117, 13885 CrossRef PubMed; (b) J. D. Knoll, B. A. Albani, C. B. Durr and C. Turro, J. Phys. Chem. A, 2014, 118, 10603 CrossRef PubMed; (c) L. M. Loftus, K. F. Al-Afyouni and C. Turro, Chem.–Eur. J., 2018 DOI:10.1002/chem.201802405.
- (a) A. K. Bennett and M. A. Smith, Dalton Trans., 1974, 233 RSC; (b) A. Habtemariam, M. Melchart, R. Fernández, S. Parsons, I. D. H. Oswald, A. Parkin, F. P. A. Fabbiani, J. E. Davidson, A. Dawson, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2006, 49, 6858 CrossRef PubMed.
- E. Largy, F. Hamon, F. Rosu, V. Gabelica, E. De Pauw, A. Guédin, J.-L. Mergny and M.-P. Teulade-Fichou, Chem.–Eur. J., 2011, 17, 13274 CrossRef PubMed.
- H. Bertrand, S. Bombard, D. Monchaud, E. Talbot, A. Guédin, J.-L. Mergny, R. Grünert, P. J. Bednarskid and M.-P. Teulade-Fichou, Org. Biomol. Chem., 2009, 7, 2864 RSC.
- (a) M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, Taylor & Francis Group, Boca Raton, FL, 3rd edn, 2006, p. 601 Search PubMed; (b) E. A. Wegner and A. W. Adamson, J. Am. Chem. Soc., 1966, 88, 394 CrossRef.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed; (c) O. D. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09, Revision D.01, Gaussian, 2013 Search PubMed.
- D. Andrae, U. Haussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 CrossRef.
- A. Schafer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 CrossRef.
- (a) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef PubMed; (b) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef; (b) C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef; (c) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef PubMed.
- (a) S. I. Gorelsky, Version 6.85, 2014, http://www.sg-chem.net/ Search PubMed; (b) S. I. Gorelsky and A. B. P. Lever, J. Organomet. Chem., 2001, 635, 187 CrossRef.
- S. Bonnet, J. P. Collin, N. Gruber, J. P. Sauvage and E. R. Schofield, Dalton Trans., 2003, 4654 RSC.
- A. A. Rachford, J. L. Petersen and J. J. Rack, Inorg. Chem., 2005, 44, 8065 CrossRef PubMed.
- B. A. Johnson, H. Agarwala, T. A. White, E. Mijangos, S. Maji and S. Ott, Chem.–Eur. J., 2016, 22, 14870 CrossRef PubMed.
- Y. Liu, D. B. Turner, T. N. Singh, A. M. Angeles-Boza, A. Chouai, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2009, 131, 26 CrossRef PubMed.
- A. Dovletoglou, S. A. Adeyemi and T. J. Meyer, Inorg. Chem., 1996, 35, 4120 CrossRef PubMed.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85 CrossRef.
- C. R. Hecker, P. E. Fanwick and D. R. McMillin, Inorg. Chem., 1991, 30, 659 CrossRef.
- C. Tsai, M. M. Allard, R. L. Lord, D. Luo, Y. Chen, H. B. Schlegel and J. F. Endicott, Inorg. Chem., 2011, 50, 11965 CrossRef PubMed.
- D. M. Klassen, Chem. Phys. Lett., 1982, 93, 383 CrossRef.
- T. A. White, S. Maji and S. Ott, Dalton Trans., 2014, 43, 15028 RSC.
- Z. Chen, C. R. K. Glasson, P. L. Holland and T. J. Meyer, Phys. Chem. Chem. Phys., 2013, 15, 9503 RSC.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176 CrossRef.
- D. Vasudevan and H. Wendt, J. Electroanal. Chem., 1995, 192, 69 CrossRef.
- A. A. Rachford and J. J. Rack, J. Am. Chem. Soc., 2006, 128, 14318 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1841086–1841088. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc02094a |
| This journal is © The Royal Society of Chemistry 2018 |