
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The cycloaspeptides: uncovering a new model for methylated nonribosomal peptide biosynthesis†
Kate M. J.
de Mattos-Shipley
*a,
Claudio
Greco
 a,
David M.
Heard
a,
Gemma
Hough
b,
Nicholas P.
Mulholland
b,
Jason L.
Vincent
b,
Jason
Micklefield
c,
Thomas J.
Simpson
a,
Christine L.
Willis
a,
Russell J.
Cox
de and
Andrew M.
Bailey
*f
a,
David M.
Heard
a,
Gemma
Hough
b,
Nicholas P.
Mulholland
b,
Jason L.
Vincent
b,
Jason
Micklefield
c,
Thomas J.
Simpson
a,
Christine L.
Willis
a,
Russell J.
Cox
de and
Andrew M.
Bailey
*f
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: kd4495@bris.ac.uk
bSyngenta Ltd., Jealott's Hill International Research Centre Bracknell, Berkshire, RG42 6EY, UK
cSchool of Chemistry, University of Manchester, Oxford Road, Manchester, M1 7DN, UK
dInstitute für Organsche Chemie, Leibniz Universität Hannover, Schneiderberg 1A, 30167 Hannover, Germany
eBMWZ, Leibniz Universität Hannover, Schneiderberg 38, 30167 Hannover, Germany
fSchool of Biological Sciences, University of Bristol, Life Sciences Building, 24 Tyndall Avenue, Bristol, BS8 1TQ, UK. E-mail: andy.bailey@bris.ac.uk
First published on 10th April 2018
Abstract
The cycloaspeptides are bioactive pentapeptides produced by various filamentous fungi, which have garnered interest from the agricultural industry due to the reported insecticidal activity of the minor metabolite, cycloaspeptide E. Genome sequencing, bioinformatics and heterologous expression confirmed that the cycloaspeptide gene cluster contains a minimal 5-module nonribosomal peptide synthetase (NRPS) and a new type of trans-acting N-methyltransferase (N-MeT). Deletion of the N-MeT encoding gene and subsequent feeding studies determined that two modules of the NRPS preferentially accept and incorporate N-methylated amino acids. This discovery allowed the development of a system with unprecedented control over substrate supply and thus output, both increasing yields of specific metabolites and allowing the production of novel fluorinated analogues. Furthermore, the biosynthetic pathway to ditryptophenaline, another fungal nonribosomal peptide, was shown to be similar, in that methylated phenylalanine is accepted by the ditryptophenaline NRPS. Again, this allowed the directed biosynthesis of a fluorinated analogue, through the feeding of a mutant strain. These discoveries represent a new paradigm for the production of N-methylated cyclic peptides via the selective incorporation of N-methylated free amino acids.
Introduction
Cycloaspeptides are bioactive cyclic pentapeptides which were originally identified from an unclassified Aspergillus species in 1987.1 Cycloaspeptides A–C 1–3 contain anthranilic acid (position p1), L-alanine (p2), L-phenylalanine (p3), L-leucine (p4) and L-tyrosine (p5) residues, and differ only in the N-methylation pattern of the phenylalanine and tyrosine moieties (Chart 1). A later screen of the Aspergillus genus failed to identify any cycloaspeptide producers, but multiple cycloaspeptide-producing Penicillium species have since been identified.2–4 Cycloaspeptide D 4, which contains a valine at p4, was isolated along with cycloaspeptide A 1 from Penicillium ribeum and P. algidum, in work which also identified these cycloaspeptides as being active against the malarial parasite Plasmodium falciparum.3,4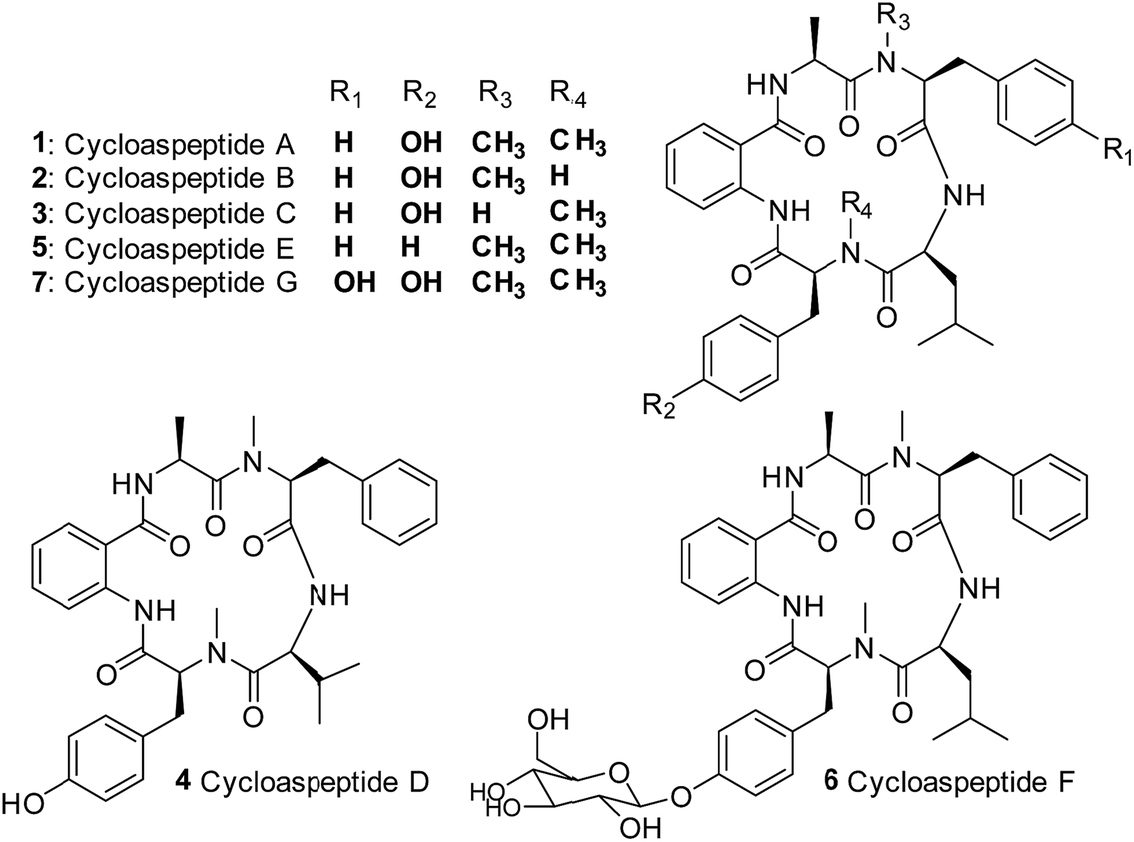 | ||
| Chart 1 Structures of the cycloaspeptides – isolated from various filamentous fungi. | ||
In 2006, cycloaspeptide E 5 was isolated as a minor metabolite from several Penicillium species and one Trichothecium species, and reported to exhibit insecticidal activity against lepidoptera.55 differs from 1 in having phenylalanine instead of tyrosine at p5 (Chart 1), and was proposed to have a neurotoxic mode of action. There is significant interest in natural products with insecticidal properties due to the huge economic and social burden created by insect damage to crop species worldwide.6
Further research into the bioactivity of cycloaspeptide E 5 has been hampered by the fact that yields are only approximately 2% of the main metabolite; cycloaspeptide A 1, meaning that purification for bioactivity screening is problematic and the natural titres are too low to be commercially viable. Most recently cycloaspeptides F 6 and G 7 from the entomopathogenic fungus Isaria farinosa, have been isolated and characterized. These compounds have cytotoxic properties against tumour cell lines.7
In order to investigate cycloaspeptide biosynthesis, with the key aim of improving the yields of 5, we set out to discover and manipulate the cycloaspeptide biosynthetic pathway, using two publicly available cycloaspeptide producers, Penicillium soppii (CBS 869.70) and Penicillium jamesonlandense (CBS 102888).2 Our original working hypothesis was that the cycloaspeptides would fit the accepted paradigm for methylated cyclic peptide biosynthesis, and as such would consist of a traditional 5-module NRPS containing the two requisite N-methyltransferase (N-MeT) domains. We reasoned that substrate promiscuity of the NRPS could account for the range of cycloaspeptides reported, whereas we considered a RiPP system unlikely due to the fidelity of ribosomally synthesised peptides to their encoded precursor peptide sequence. A combination of approaches including bioinformatic analyses, heterologous expression, gene deletions and feeding experiments have demonstrated that indeed, an NRPS is responsible for the biosynthesis of the cycloaspeptides. However, rather than containing N-Met domains, the NRPS requires a pathway-specific trans-acting methyltransferase to provide the necessary substrates and preferentially incorporates methylated amino acids at two positions. This discovery unlocked a new avenue for pathway engineering through synthetic biology, by allowing unprecedented control over substrate availability.
Results and discussion
P. soppii and P. jamesonlandense were cultured on a range of solid media and screened by LCMS for the production of cycloaspeptides. A compound with the correct mass for cycloaspeptide A 1 could be reliably detected by LCMS, from both species and on all media after 2 weeks. This was confirmed by purification and NMR analysis (Table S8†). P. soppii was also grown in three different liquid media (CYB, MEB and YEB), and again 1 could be detected in all, with the highest concentrations being achieved with CYB (Fig. S21†). A compound with the correct mass for cycloaspeptide E 5 could be detected in certain cultures, but only in trace amounts and not reliably. Analysis using the highly sensitive UPLC-Orbitrap mass analyser system confirmed that the accurate masses matched the chemical formulae of the predicted compounds (Fig. S22 and Table S6†). The sensitivity of this system also allowed the detection of a compound with the correct predicted formula to be cycloaspeptide G 7, a compound which has not been detected in a Penicillium species before.Cycloaspeptide A 1 could also be easily detected by LCMS in MEB cultures of P. jamesonlandense (Fig. S23, S26 and Table S7†). There were also compounds present with the correct masses to be cycloaspeptides D 4, E 5 and G 7 (Table S7†). Unfortunately, the yields of 5 were too low to purify for NMR and structural validation.
Paired-end genomic sequence data was generated for both species and assembled to produce draft genomes (Table S2†), which were analysed using AntiSMASH8 to identify putative biosynthetic gene clusters (BGCs). A total of 82 and 83 BGCs were detected for P. soppii and P. jamesonlandense respectively, demonstrating the rich metabolic potential of these species (Table S3†).
P. soppii contains 20 NRPS clusters whereas P. jamesonlandense contains 17, with 8 such clusters being common to both species. The domain architecture of each predicted NRPS was analysed using the NCBI conserved domain database (CDD) (Table S4†). Only one was found to contain the five modules that would be expected for pentapeptides such as the cycloaspeptides (Fig. S7†). Unexpectedly this NRPS lacked any integral N-methyltransferase (N-MeT) domains, which appeared inconsistent with cycloaspeptide biosynthesis, as N-methylation, if present, is normally performed by such domains embedded within the NRPS. However, an adjacent gene present in both species is predicted to encode a methyltransferase, so might perform this activity in trans. Additionally, the NRPS appears to contain a final condensation-like (CT) domain, where rather than having the highly conserved HHXXXDXXS/T motif found in condensation domains, it has the modified motif SHXXXDXXS/T (5377SHAQYDGVS5385). CT domains are known to catalyse the macrocyclisation of cyclic peptides in filamentous fungi9 – a role analogous to that of the final thiolesterase in bacterial cyclic peptide biosynthesis.
In P. jamesonlandense, adjacent to the NRPS and putative N-MeT encoding genes are various additional predicted genes that could potentially be involved in cycloaspeptide production. These encode an amino acid transporter, a Zn2Cys6 transcription factor and an aminotransferase. In P. soppii however, these genes are translocated on another scaffold (sc038, Fig. 1). It is still possible that they are involved in cycloaspeptide biosynthesis, as split secondary metabolite clusters have been observed in fungi multiple times.10 However, it is also possible that the NRPS and N-MeT are the only proteins required for the production of cycloaspeptides.
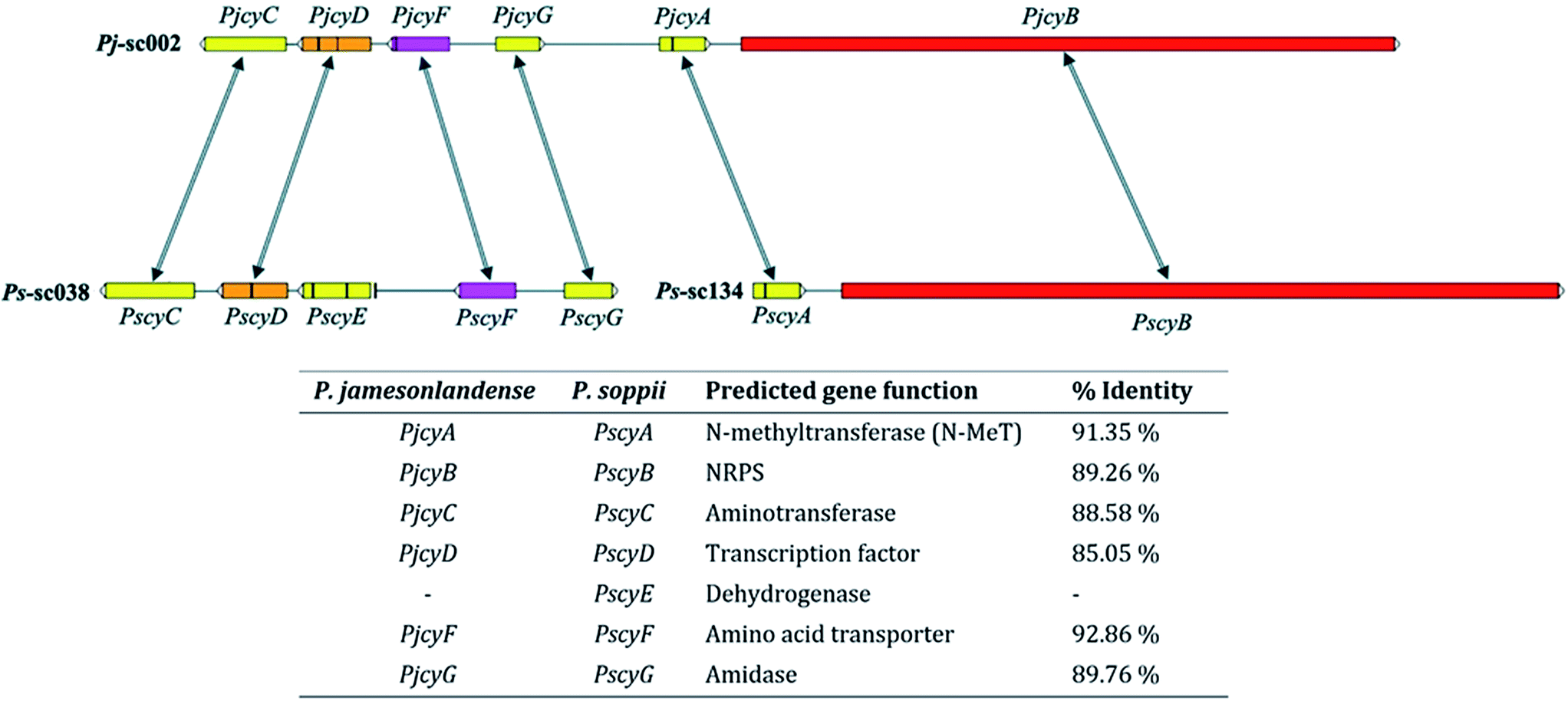 | ||
| Fig. 1 Cycloaspeptide gene clusters from P. jamesonlandense (top) and P. soppii (bottom). The N-methyltransferase (PjcyA and PscyA) and NRPS (PjcyB and PscyB) genes are adjacent to one another in both species. Additional genes identified at the same locus in P. jamesonlandense, which could potentially be involved in secondary metabolite biosynthesis, such as an amino acid transporter and transcription factor were identified on a separate scaffold in P. soppii. Arrows denote homologous genes; vertical black bars within genes denote introns. | ||
Bioinformatic analysis was conducted for the adenylation (A) domains of the NRPS. This focused on the predicted presence of an anthranilic acid adenylating domain, due to their distinct nature.11 The online NRPS predictor tool12 was used to generate the 10 amino acid code for each domain (Table 1), and these were compared with those of known fungal NRPS enzymes.11 The presence of a glycine in the first position of the 10 amino acid code for domain 1, rather than the aspartic acid typically seen, is indicative of domain 1 being an anthranilic acid adenylating domain. This is consistent with the fact that all known fungal anthranilic acid A-domains are positioned within the first module of the synthetase.11 It then follows that the second module incorporates an alanine, the third a phenylalanine, the fourth a leucine, and the fifth, either a tyrosine (to produce 1) or a phenylalanine (to produce 5). Evidence supporting this assignment is the similarity of the third and fifth modules, which are predicted to incorporate Phe and Phe/Tyr respectively. In P. soppii these domains have 80% sequence similarity when comparing the 10 amino acid code, and 85% sequence similarity when comparing the 34 extracted residues. Interestingly, it was not possible to identify homologous gene clusters in publicly available fungal genomes, even when specifically searching Aspergillus, Penicillium, Trichothecium or Isaria genomes for homologues of PscyA and PscyB. This suggests that although the cycloaspeptides have been reported from various fungal genera, the gene cluster is not particularly wide-spread and cycloaspeptide biosynthesis may be limited to a fairly small fungal clade, or clades.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12
12
| Module | Pos1 (235) | Pos2 (236) | Pos3 (239) | Pos4 (278) | Pos5 (299) | Pos6 (301) | Pos7 (322) | Pos8 (330) | Pos9 (331) | Pos10 (517) | Predicted substrate |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | G | V | I | F | I | A | A | G | I | K | Ant |
| 2 | D | V | F | F | V | V | G | V | L | K | Ala |
| 3 | D | A | Y | A | V | G | G | I | C | K | Phe |
| 4 | D | L | M | L | V | G | A | V | I | K | Leu |
| 5 | D | A | Y | T | S | G | G | I | C | K | Tyr/Phe |
To allow an investigation of gene function in P. soppii, a protoplast-based transformation system was developed, using either hygromycin or geneticin resistance as selectable markers. This transformation system was optimized using eGFP as a reporter gene (Fig. S9†) and a bipartite knock-out strategy13 was then used to disrupt specific genes. The disruption of either the NRPS (PscyB) or the N-MeT (PscyA) led to a complete loss of production of both cycloaspeptide A 1 and E 5, (Fig. 2A–C) confirming that both genes are required for cycloaspeptide biosynthesis and that 1 and 5 are synthesized by the same pathway.
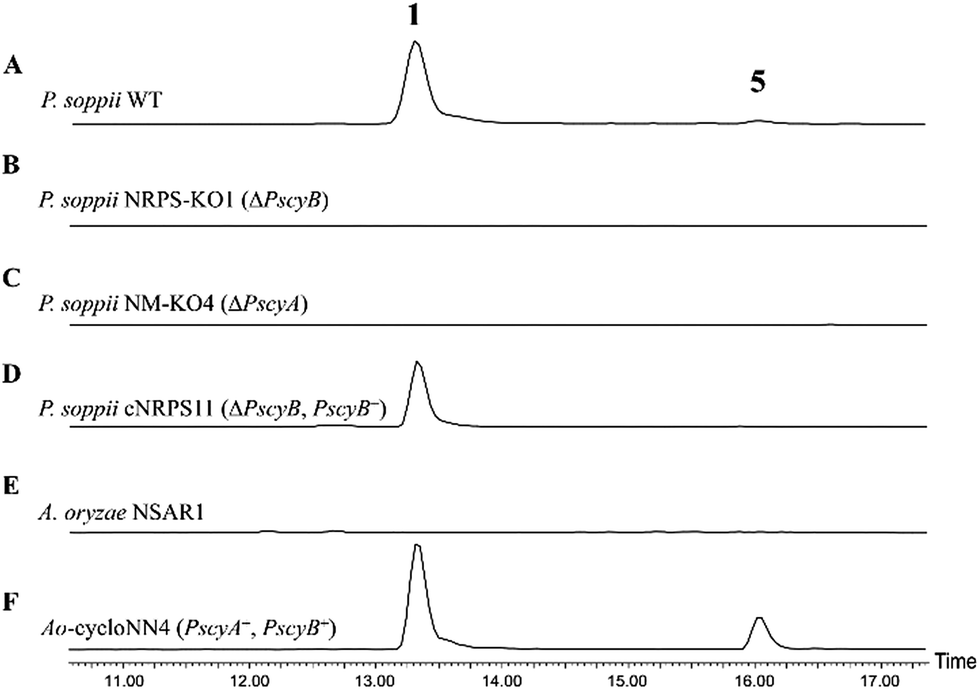 | ||
| Fig. 2 ES+ extracted ion chromatograms for 1 ([M + H]+: 642.7) and 5 ([M + H]+: 627.7) in various fungal cultures: (A) wild-type P. soppii produces both 1 and 5; (B) P. soppii ΔPscyB strain, NRPS-KO1, produces no cycloaspeptides; (C) P. soppii ΔPscyA strain, NM-KO1, produces no cycloaspeptides; (D) NRPS-KO1 complemented with a wild-type copy of the cycloaspeptide NRPS (ΔPscyB, PscyB+), demonstrates a restoration in cycloaspeptide biosynthesis; (E) wild-type A. oryzae NSAR1; (F) strain Ao-cycloasNN4, which is an A. oryzae NSAR1 transformant co-expressing PscyA and PscyB, produces both cycloaspeptide A and cycloaspeptide E. | ||
The disruption of the transcription factor (PscyD) led to a marked reduction in cycloaspeptide yields, suggesting that although it is translocated in P. soppii, it is still involved in the regulation of cycloaspeptide biosynthesis (Fig. S24†).
To further confirm the identity of the gene cluster and conclusively demonstrate that the loss of production was not an indirect consequence of the transformation system, strain NRPS-KO1 (ΔPscyB) was complemented by transformation with the plasmid pTYGen–N-MeT–NRPS which contains PscyA and PscyB under the control of Padh (the alcohol dehydrogenase promoter from A. oryzae) and Ptub (the tubulin promoter from P. soppii) respectively. Selection of geneticin resistant transformants led to the restoration of cycloaspeptide production (Fig. 2D).
Heterologous production of both 1 and 5 was achieved in Aspergillus oryzae NSAR1 via the co-expression of PscyA and PscyB from a multi-gene expression vector with arginine prototrophy selection (Fig. 2E and F).14 This demonstrates that PscyA and PscyB are the only two structural genes required for cycloaspeptide biosynthesis. Interestingly, the ratio of cycloaspeptide A 1 to E 5 appeared to differ in A. oryzae when compared to the natural producers, with cycloaspeptide E 5 being easily detectable in initial screens of the A. oryzae transformants. An apparent decrease in fitness and growth rate was observed in A. oryzae transformants producing cycloaspeptides, and production was not stable. A loss of production occurring after subculturing prevented quantification of titres. In an attempt to push the ratio of 5:1 further in favour of cycloaspeptide E 5 biosynthesis, a full A domain swap was conducted. The seemingly promiscuous A domain from module 5 (Tyr/Phe) was replaced with a second copy of the A domain from module 3 (Phe). This engineered NRPS was expressed both in A. oryzae and the P. soppii ΔPscyB strain but the NRPS was dysfunctional, with no cycloaspeptides being detected in either strain (data not shown).
To ascertain the order of events in the cycloaspeptide biosynthetic pathway, LCMS chromatograms for the N-MeT knock-out strains were searched for any putative un-methylated intermediates. The absence of any such compounds suggested that rather than the N-methylations serving to decorate the product of the NRPS, the N-MeT may act first, providing methylated amino acids for incorporation by the NRPS. To test this, N-MeT knock-out strain 4 (NM-KO4) was fed with 1 mM of both N-methylated tyrosine (N-mTyr 8) and N-methylated phenylalanine (N-mPhe 9). This fully restored cycloaspeptide biosynthesis (Fig. 3B), demonstrating that the NRPS accepts free methylated amino acids, a feature not previously observed in non-ribosomal peptide systems in either fungi or bacteria.
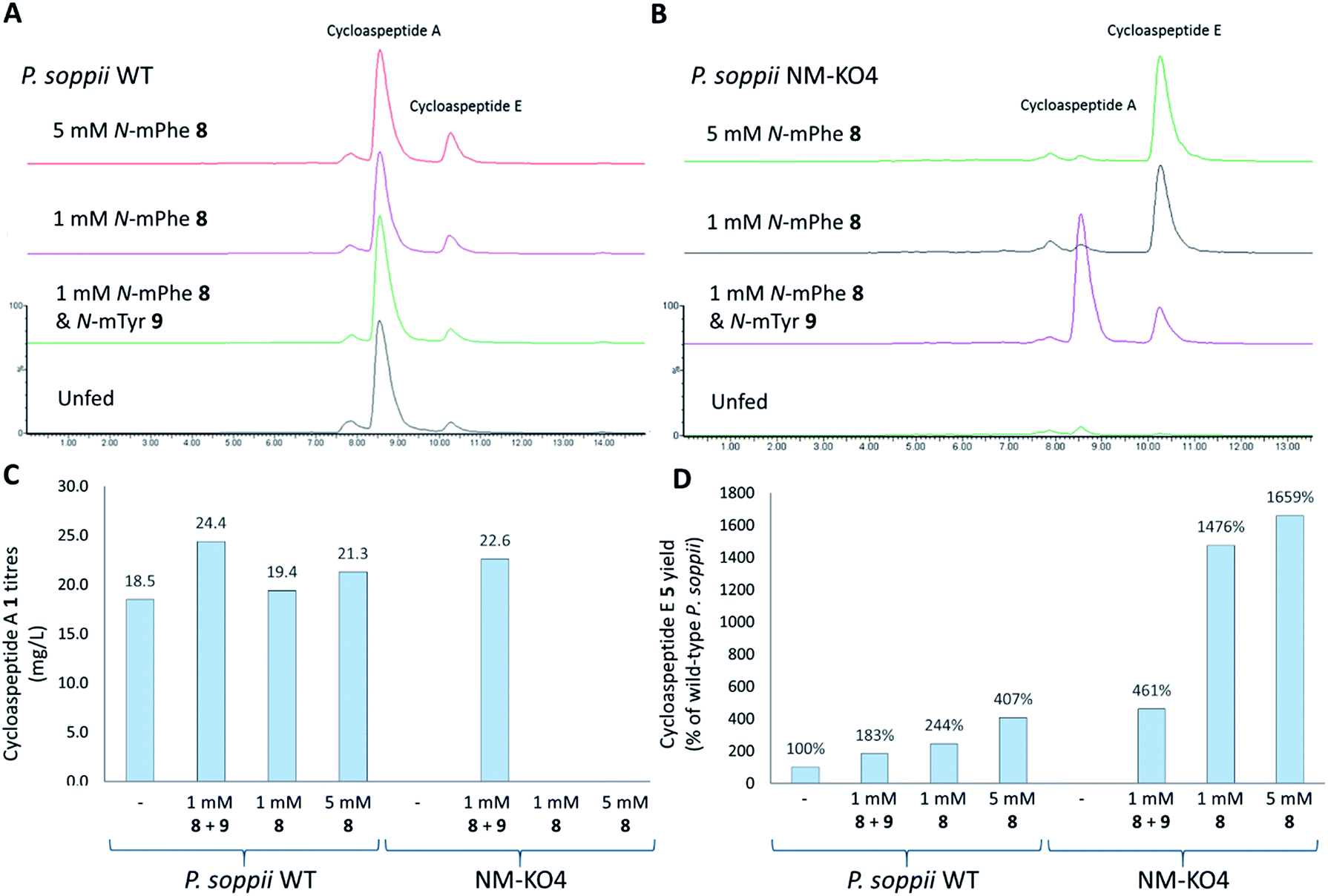 | ||
| Fig. 3 Feeding P. soppii cultures with N-methylated phenylalanine 8 and N-methylated tyrosine 9 was shown to impact cycloaspeptide biosynthesis, increasing titres in wild-type P. soppii and restoring cycloaspeptide production in an N-MeT knock-out strain (NMKO4: ΔPscyA). (A) ES+ extracted ion chromatograms for cycloaspeptide A 1 ([M + H]+: 642.7) and cycloaspeptide E 5 ([M + H]+: 627.7). Wild-type cultures, from bottom to top: unfed control. Fed with 1 mM each of 8 and 9. Fed with 1 mM 8. Fed with 5 mM 8. (B) NMKO4 (ΔPscyA) cultures, from bottom to top: unfed control. Fed with 1 mM each of 8 and 5. Fed with 1 mM 8. Fed with 5 mM 8. (C) Absolute titres of 1 (mg L−1) in various fed cultures. Some differences were observed for the fed wild-type cultures and complete restoration was seen for the N-MeT knock-out strain. (D) Relative titres of 5 (% of wild-type) in the various cultures demonstrating an increase in 5 with feeding, particularly in the knock-out strain NM-KO4. Quantification was not possible for 5 due to a lack of cycloaspeptide E standard, so relative titres were calculated. | ||
In further experiments, wild-type P. soppii and NM-KO4 cultures were fed with either a 1:1 mixture of N-mPhe 8 and N-mTyr 9 (1 mM each), or with N-mPhe alone (either 1 mM or 5 mM final culture concentration) (Fig. 3). Increases in the cycloaspeptide yields when the WT strain was fed with the N-methylated amino acids suggests that substrate availability is a limiting factor in this system (Fig. 3C). The yields of the cycloaspeptides in the fed NM-KOS cultures were most striking. In addition to producing wild-type yields of cycloaspeptide A 1, cultures fed with both N-mPhe 8 and N-mTyr 9 produced over four times more cycloaspeptide E 5 than the unfed wild-type strain (Fig. 3D). When supplemented with either 1 mM or 5 mM N-mPhe 8 cycloaspeptide E 5 titres were increased further, to approximately 14.5 and 16.5 times respectively (Fig. 3D).
Such increased yields allowed purification of cycloaspeptide E 5 for structural confirmation by NMR (Table S9 and Fig. S23 and S24†). In addition to 5, a compound could be detected in NM-KO4 cultures fed with N-mPhe 8 that has the expected mass and UV to be cycloaspeptide B 2 (Fig. 4). 2 was identified from the original Aspergillus sp. NE-45,1 and has a methylated phenylalanine at p3, but an unmethylated tyrosine at p5. This compound has not been reported in Penicillium species before.
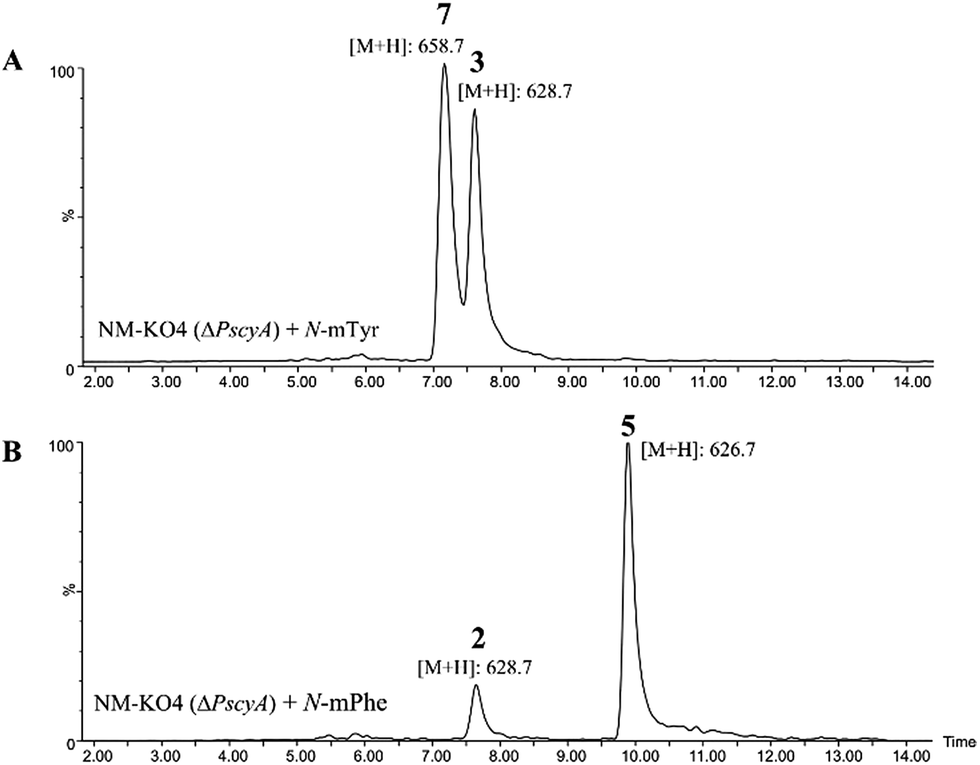 | ||
| Fig. 4 (A) ES+ extracted ion chromatogram for masses of 628.7, which corresponds to cycloaspeptide C 3 ([M + H]+) and 658.7 which corresponds to cycloaspeptide G 7 ([M + H]+); (B) ES+ extracted ion chromatogram for masses of 628.7 which corresponds to cycloaspeptide B 2 ([M + H]+) and 626.7 which corresponds to compound cycloaspeptide E 5 ([M + H]+). These compounds are not present in NM-KO4 (ΔPscyA), unless cultures are fed with methylated amino acids. | ||
A minor metabolite with the correct accurate mass to be cycloaspeptide G 7, which has methylated tyrosine at both p3 and p5, was detected in wild-type P. soppii cultures using an UPLC-Orbitrap mass analyser. To determine whether the titres of this compound could be increased, NM-KO4 cultures were fed with N-mTyr alone.
This resulted in the easy detection of 7 in all fed cultures using standard LC-MS methods (Fig. 4). Feeding with N-mTyr also led to the production of a compound not detected in the wild-type cultures which has the expected mass to be cycloaspeptide C 3. 3 has un-methylated phenylalanine at p3 and N-mTyr at p5. As with 5, this compound has not been previously observed in Penicillium strains. These results indicate that the third and fifth modules of the NRPS have a strong preference for methylated amino acids, but also the ability to accept and incorporate un-methylated amino acids.
To further investigate the substrate selectivity of the NRPS, a range of alternative amino acids were fed to strain NM-KO4. N-Methyl-leucine 10, N-methyl-isoleucine 11, N-methyl-tryptophan 12, α-methyl-phenylalanine 13 and N-methyl-D-phenylalanine 14 were obtained commercially (Chart 2). Racemic p-methyl-N-mPhe 15, and a range of fluorinated N-mPhe analogues (with the fluorine at the ortho, meta and para positions 18–20) were synthesised by alkylation of the Boc–N-methylglycine dianion with the appropriate benzyl bromides, followed by Boc deprotection with trifluoroacetic acid (TFA) (Scheme 1A). N,O-Dimethyl-L-Tyr 17 was synthesised by double deprotonation of Boc-L-tyrosine with sodium hydride, followed by dimethylation with methyl iodide (Scheme 1C). Boc deprotection was again performed with TFA. N-Ethyl-L-Phe 16 was synthesised by reductive amination of L-phenylalanine with acetaldehyde and sodium cyanoborohydride (Scheme 1D).
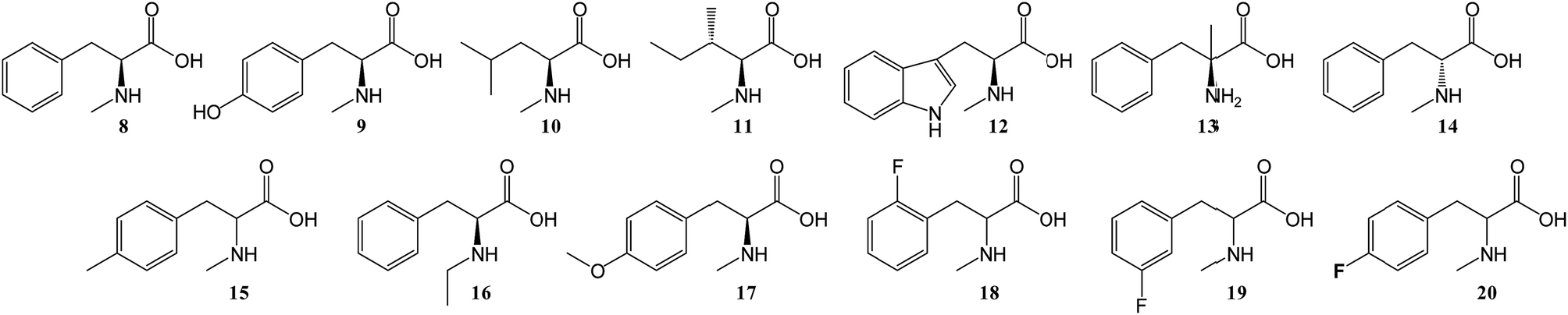 | ||
| Chart 2 Various amino acid analogues fed to P. soppii NM-KO4 (ΔPscyA). | ||
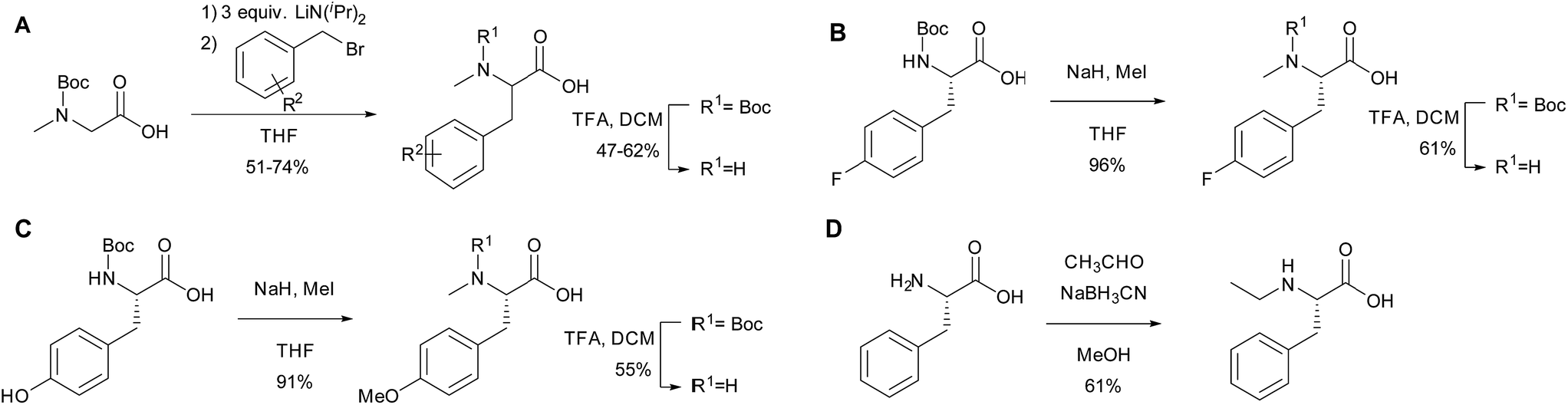 | ||
| Scheme 1 The synthesis of amino acid analogues. | ||
These compounds were either fed alone (5 mM) or in combination with N-mPhe 8 (at a ratio of 5:1–5 mM:1 mM). The only cultures to produce novel compounds in the initial screens were those supplemented with fluorinated phenylalanine analogues. The synthesis of 4F–N-mPhe 20 was therefore scaled up (see ESI†) and an LCMS analysis identified compounds with the correct masses to be fluorinated analogues of cycloaspeptide E 5, and cycloaspeptide B 2 (Fig. 5). 4F–cycloaspeptide E 22 was purified and characterized using proton, carbon and fluorine NMR (Table S10 and Fig. S28–S30†). Purified 5 and 22 were screened for anti-lepidopteran activity using the tobacco budworm (H. virescens) as an industrially relavent target organism, but no activity was observed against this species following injection with either compound (Table S12†).
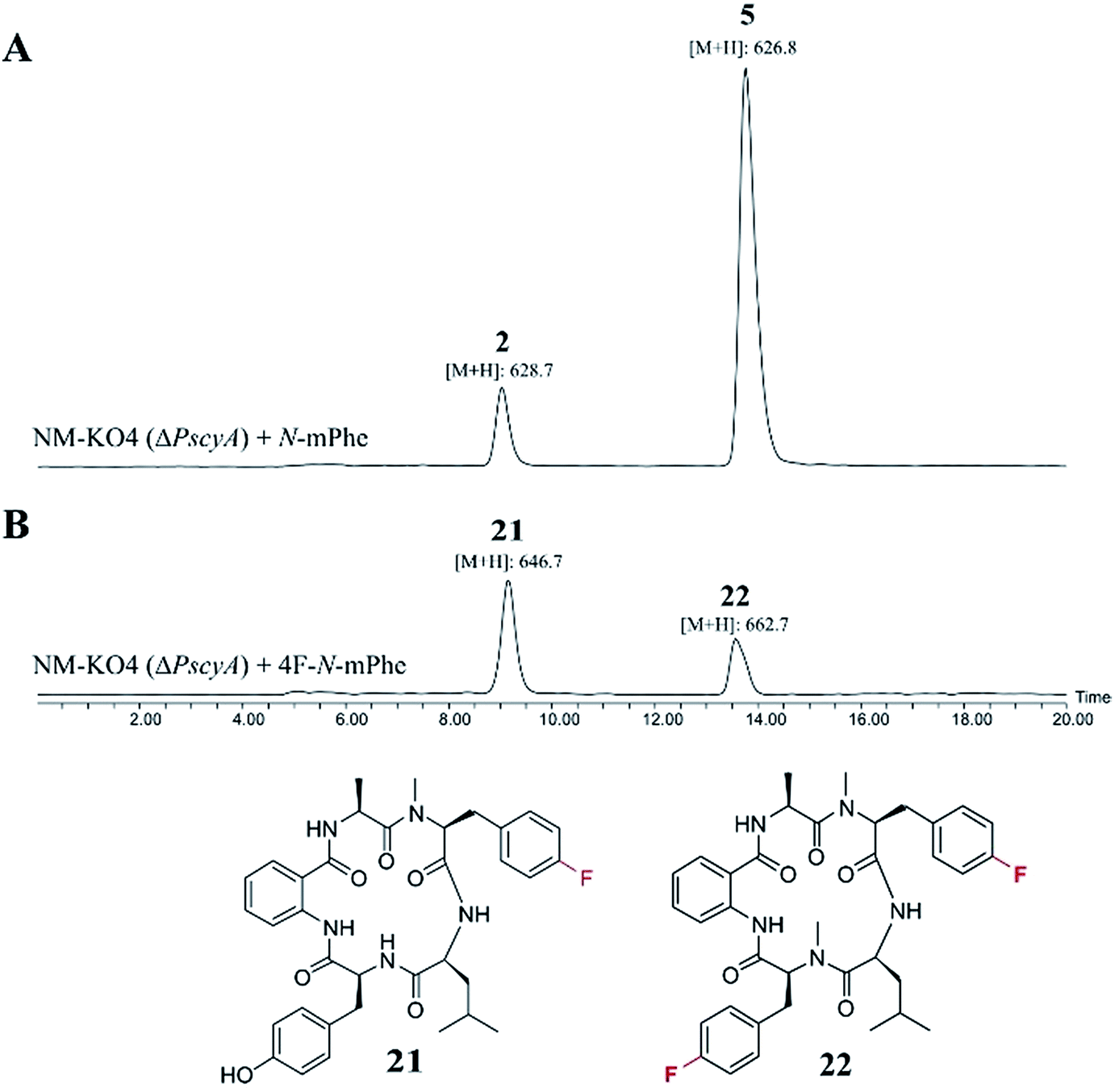 | ||
| Fig. 5 ES+ extracted ion chromatograms ([M + H]+) for cycloaspeptides B 2 (m/z: 628.7) and E 5 (m/z: 626.7), and their fluorinated counterparts 21 (m/z: 646.7) and 22 (m/z: 662.7). (A) NM-KO4 (ΔPscyA) cultures fed with N-mPhe 8, produced 2 and 5. (B) Cultures fed with 4F–N-Met–Phe 20, produced 21 and 22. | ||
A search of the NCBI database for any potential PscyA homologues identified an N-MeT from A. flavus with 39% sequence identity to PscyA at the protein level (Table S5†). Investigating the genomic context of this gene determined that it is part of a BGC containing a 2 module NRPS and cytochrome P450, which has been identified as the ditryptophenaline 23 gene cluster by Watanabe and coworkers.1523 is a dimeric diketopiperazine consisting of two cyclic Trp:N-Met-Phe dipeptides.
An A. flavus strain with the ditryptophenaline N-MeT disrupted (ΔdtpB) was kindly supplied by Prof. Kenji Watanabe of the University of Shizuoka, and feeding studies were conducted. Analogous to the cycloaspeptide system, feeding cultures with N-mPhe 8 fully restored ditryptophenaline 23 production, demonstrating the acceptance of 8 by the ditryptophenaline NPRS (dtpA). Furthermore, the production of a fluorinated ditryptophenylene analogue 24 was again achieved by feeding cultures with 4F–N-mPhe 20 (Fig. 6). Interestingly, feeding with N-mTyr 9 did not result in the production of hydroxylated analogues, demonstrating that the ditryptophenaline NRPS exhibits lower promiscuity than the cycloaspeptide NRPS. The rapid discovery of a second NRPS which accepts methylated amino acids suggests that these systems represent a new fungal route to methylated peptide natural products, rather than the cycloaspeptide system being unique.
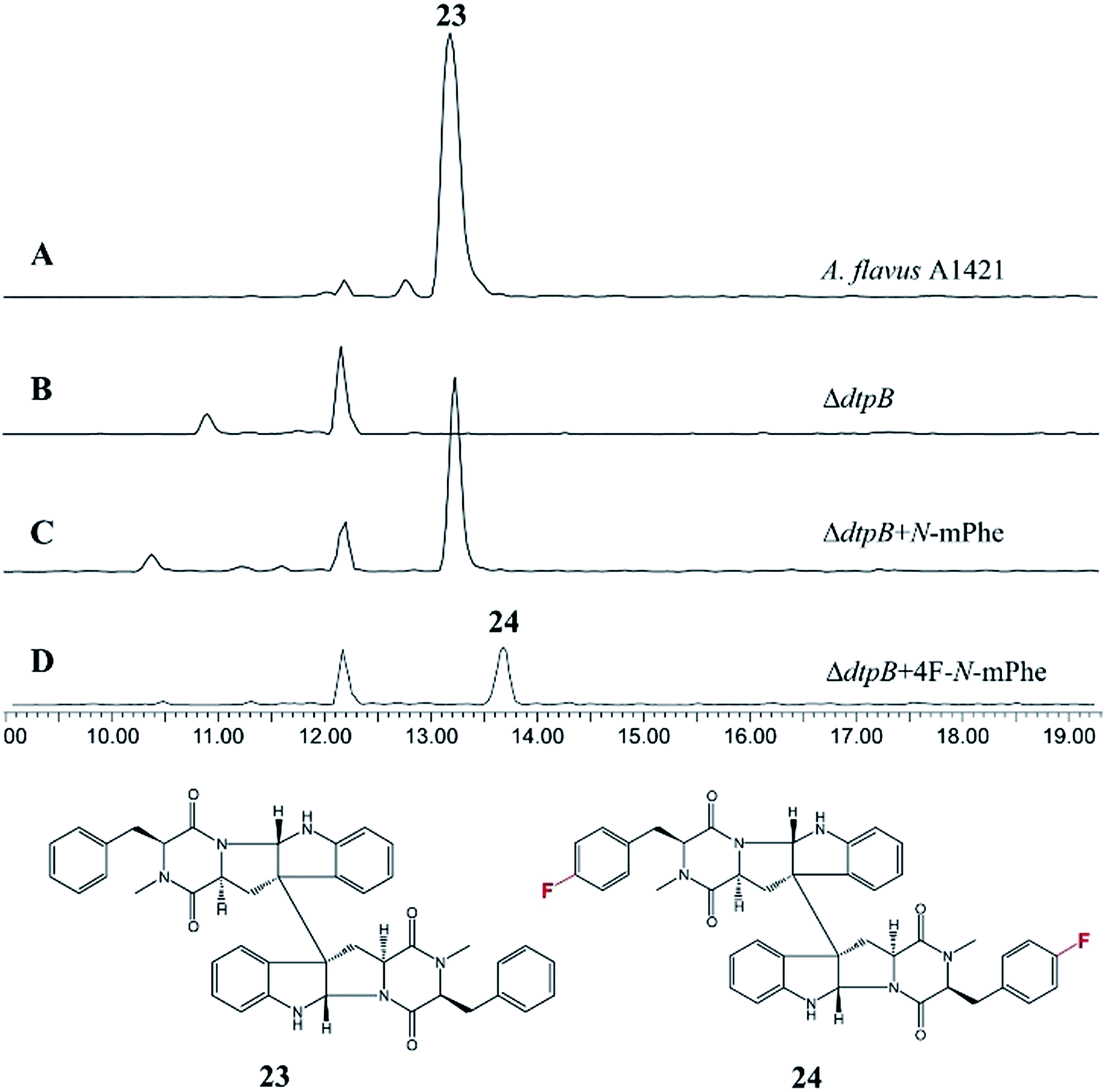 | ||
| Fig. 6 HPLC ES+ spectrum for the crude extracts of various A. flavus cultures: (A) A. flavus strain A1421 produces ditryptophenaline 23; (B) a ΔdtpB strain can no longer produce 23; (C) feeding the ΔdtpB strain with N-methylated phenylalanine restores the production of ditryptophenaline 23. (D) Feeding the ΔdtpB strain with a fluorinated phenylalanine analogue (4F–N-mPhe 20) led to the production of a fluorinated ditryptophenaline analogue 24. | ||
Conclusions
The gene cluster responsible for the biosynthesis of the cycloaspeptides has been identified in two psychrotolerant Penicillium species: P. soppii and P. jamesonlandense. Heterologous expression in A. oryzae has demonstrated that the minimal gene set required to produce both cycloaspeptide A 1 and cycloaspeptide E 2 is a 5-module NRPS and a new type of pathway specific N-methyltransferase (N-MeT). Gene knock-outs and feeding studies have demonstrated that two modules of the NRPS preferentially accept and incorporate N-methylated amino acids, which are provided by the pathway specific N-MeT, a system not previously seen in secondary metabolism. Selective amino acid N-methyltransferases are very rare in either primary or secondary metabolism. The only known enzyme to catalyse a single N-methylation of a proteogenic amino acid is glycine N-methyltransferase from primary metabolism. The most similar known examples from fungal secondary metabolism are 4-dimethylallyltryptophan N-methyltransferase, which is the second pathway-specific enzyme in the production of ergot alkaloids,16 and a methyltransferase from the ergothioneine pathway which trimethylates histidine.17Deletion of the cycloaspeptide N-Met allowed the supply of methylated amino acids to the NRPS to be controlled, and this was exploited to direct biosynthesis towards specific minor metabolites, accomplishing the original aim of increasing cycloaspeptide E 5 yields. Feeding cultures with a fluorinated phenylalanine analogue also led to the production of novel fluorinated cycloaspeptides. The ditryptophenaline pathway from Aspergillus flavus was quickly confirmed as being a second example of such a system, suggesting that as more NRPS gene clusters are identified and characterised, further examples will be uncovered. Again, feeding a N-Met knock-out strain allowed the production of an unnatural ditryptophenaline analogue.
The preferential acceptance of methylated amino acids by an NRPS combined with the ability to remove the natural substrate supply provides a unique biotransformation opportunity over traditional feeding of modified amino acids. Firstly, methylated amino acids fed into the system have little competition from unmethylated cytoplasmic amino acids. Secondly, the incorporation will be more efficient because synthetic methylated amino acids are not at risk of being consumed by other cellular processes such as protein production.
The ability to produce natural product analogues is valuable due to the potential for altered bioactivities or pharmacokinetic properties. N-Methylated peptides are particularly desirable as they are known to often display improved stability over their non-methylated counterparts. Currently medicinal chemistry employs expensive and toxic reagents to synthesise N-methylated peptides. Consequently, new approaches for de novo biosynthesis of N-methylated peptides could be an attractive alternative to chemical synthesis. The ability to generate fluorinated natural product analogues is particularly relevant for natural product research due to their extreme rarity in nature (less than 0.005% of identified natural products contain fluorine18) combined with the various benefits observed with compounds containing one or more fluorine atom,19,20 demonstrated by the fact that 15–20% of pharmaceuticals now contain at least one fluorine atom.18 Such compounds could also be more amenable to semi-synthetic derivatization, which is a major route to drug development.
Using A-domains which accept methylated amino acids in domain swaps could also be used to introduce methylated amino acids, or unnatural analogues such as the fluorinated amino acids into other NRP natural products of agricultural of pharmaceutical interest. Indeed, there have been some major advances in NRPS structural biology and engineering recently,21–23 which means that the prospect of swapping in domains/modules to introduce N-methylated amino acids into existing NRPs could become feasible. Detailed in vitro studies of the N-MeT enzyme and the N–Me amino acid activating A-domains and modules, including crystallography, could help to elucidate where the selectivity and specificity of such systems lie, and the roles that the individual NRPS domains play in controlling the output of such systems. In the longer term, such an understanding could help guide engineering to alter the substrate specificity of these systems, enabling methylation and incorporation of a wider range of N-methylated amino acids. Also, further genome sequencing and mining could lead to the discovery of other N-MeT and NRPS that incorporate different N-methylated amino acids in nature. Taken together, such studies could provide a set of N-Met enzymes and NRPS domains/modules for engineering the de novo biosynthesis of N-methylated peptide ‘non-natural’ products through the assembly of novel chimeric NRPS.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by funding from BBSRC and Syngenta (BB/K002341/1), BBSRC and EPSRC through BrisSynBio, the Bristol Centre for Synthetic Biology (BB/L01386X/1) and the Bristol Chemical Synthesis Centre for Doctoral Training which provided a PhD studentship for DMH (EP/L015366/1). Genome sequencing and the production of assembled draft genomes was carried out at the DNA sequencing facility in the Biochemistry Department of Cambridge University. Bioactivity screening was conducted at Syngenta with the assistance of Emily Aldridge. Thanks go to all members of the natural product sLola consortium for discussions and input to experimental design.References
- R. Kobayashi, Y. Samejima, S. Nakajima, K. Kawai and S. Udagawa, Chem. Pharm. Bull., 1987, 35, 1347–1352 CrossRef CAS PubMed
.
- J. C. Frisvad, T. O. Larsen, P. W. Dalsgaard, K. A. Seifert, G. Louis-Seize, E. K. Lyhne, B. B. Jarvis, J. C. Fettinger and D. P. Overy, Int. J. Syst. Evol. Microbiol., 2006, 56, 1427–1437 CrossRef CAS PubMed
- P. W. Dalsgaard, T. O. Larsen and C. Christophersen, J. Antibiot., 2005, 58, 141–144 CrossRef CAS PubMed
- P. W. Dalsgaard, T. O. Larsen, K. Frydenvang and C. Christophersen, J. Nat. Prod., 2004, 67, 878–881 CrossRef CAS PubMed
- P. Lewer, P. R. Graupner, D. R. Hahn, L. L. Karr, D. O. Duebelbeis, J. M. Lira, P. B. Anzeveno, S. C. Fields, J. R. Gilbert and C. Pearce, J. Nat. Prod., 2006, 69, 1506–1510 CrossRef CAS PubMed
- M. P. Zalucki, A. Shabbir, R. Silva, D. Adamson, L. Shu-Sheng and M. J. Furlong, J. Econ. Entomol., 2012, 105, 1115–1129 CrossRef PubMed
- Y. Zhang, S. Liu, H. Liu, X. Liu and Y. Che, J. Nat. Prod., 2009, 72, 1364–1367 CrossRef CAS PubMed
- M. H. Medema, K. Blin, P. Cimermancic, V. de Jager, P. Zakrzewski, M. A. Fischbach, T. Weber, E. Takano and R. Breitling, Nucleic Acids Res., 2011, 39, W339–W346 CrossRef CAS PubMed
- X. Gao, S. W. Haynes, B. D. Ames, P. Wang, L. P. Vien, C. T. Walsh and Y. Tang, Nat. Chem. Biol., 2012, 8, 823–830 CrossRef CAS PubMed
- H.-C. Lo, R. Entwistle, C.-J. Guo, M. Ahuja, E. Szewczyk, J.-H. Hung, Y.-M. Chiang, B. R. Oakley and C. C. C. Wang, J. Am. Chem. Soc., 2012, 134, 4709–4720 CrossRef CAS PubMed
- B. D. Ames and C. T. Walsh, Biochemistry, 2010, 49, 3351–3365 CrossRef CAS PubMed
- M. Röttig, M. H. Medema, K. Blin, T. Weber, C. Rausch and O. Kohlbacher, Nucleic Acids Res., 2011, 39, W362–W367 CrossRef PubMed
- M. L. Nielsen, L. Albertsen, G. Lettier, J. B. Nielsen and U. H. Mortensen, Fungal Genet. Biol., 2006, 43, 54–64 CrossRef CAS PubMed
- K. A. K. Pahirulzaman, K. Williams and C. M. Lazarus, Methods Enzymol., 2012, 517, 241–260 CAS
- T. Saruwatari, F. Yagishita, T. Mino, H. Noguchi, K. Hotta and K. Watanabe, ChemBioChem, 2014, 15, 656–659 CrossRef CAS PubMed
- O. Rigbers and S.-M. Li, J. Biol. Chem., 2008, 283, 26859–26868 CrossRef CAS PubMed
- A. Vit, L. Misson, W. Blankenfeldt and F. P. Seebeck, ChemBioChem, 2015, 16, 119–125 CrossRef CAS PubMed
- K. Chan and D. O'Hagan, Methods Enzymol., 2012, 516, 219–235 CAS
- M. C. Walker and M. C. Chang, Chem. Soc. Rev., 2014, 43, 6527–6536 RSC
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2013, 114, 2432–2506 CrossRef PubMed
- C. Steiniger, S. Hoffmann, A. Mainz, M. Kaiser, K. Voigt, V. Meyer and R. D. Süssmuth, Chem. Sci., 2017, 8, 7834–7843 RSC
- E. J. Drake, B. R. Miller, C. Shi, J. T. Tarrasch, J. A. Sundlov, C. L. Allen, G. Skiniotis, C. C. Aldrich and A. M. Gulick, Nature, 2016, 529, 235–238 CrossRef CAS PubMed
- J. G. Owen, M. J. Calcott, K. J. Robins and D. F. Ackerley, Cell Chem. Biol., 2016, 23, 1395–1406 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc00717a |
| This journal is © The Royal Society of Chemistry 2018 |