
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct catalytic enantioselective amination of ketones for the formation of tri- and tetrasubstituted stereocenters†
B. M.
Trost
 *,
J. S.
Tracy
and
T.
Saget
*,
J. S.
Tracy
and
T.
Saget
Department of Chemistry, Stanford University, Stanford, CA 94305-5080, USA. E-mail: bmtrost@stanford.edu
First published on 14th February 2018
Abstract
Herein, we report a Zn–ProPhenol catalyzed direct asymmetric amination reaction of unactivated aryl and vinyl ketones using di-tert-butyl azodicarboxylate as a cheap and practical electrophilic nitrogen source. Importantly, this methodology works with both α-branched and unbranched ketones for the construction of tri- and tetrasubstituted N-containing stereocenters. The reaction can be run at gram-scale with low catalyst loadings and features a recoverable and reusable ligand. Finally, the enantioenriched hydrazine products can be readily converted into versatile building blocks such as α-amino carbonyl compounds and β-amino alcohols.
Introduction
Nitrogen-containing molecules are ubiquitous among natural products and pharmaceuticals. In this regard, the stereoselective formation and functionalization of amines has been an area of continuous interest in synthetic organic chemistry.1 Despite considerable advances in this field, the enantioselective synthesis of α-tertiary amines, a common motif in naturally occurring and synthetic bioactive molecules,2 still represents a synthetic challenge.3 In this context, we report a direct catalytic enantioselective amination of cyclic ketones for the construction of tri- and tetrasubstituted N-containing stereogenic centers. This transformation allows efficient and stereocontrolled access to a broad array of α-amino carbonyl and β-amino alcohol motifs starting from widely available ketone precursors (Scheme 1).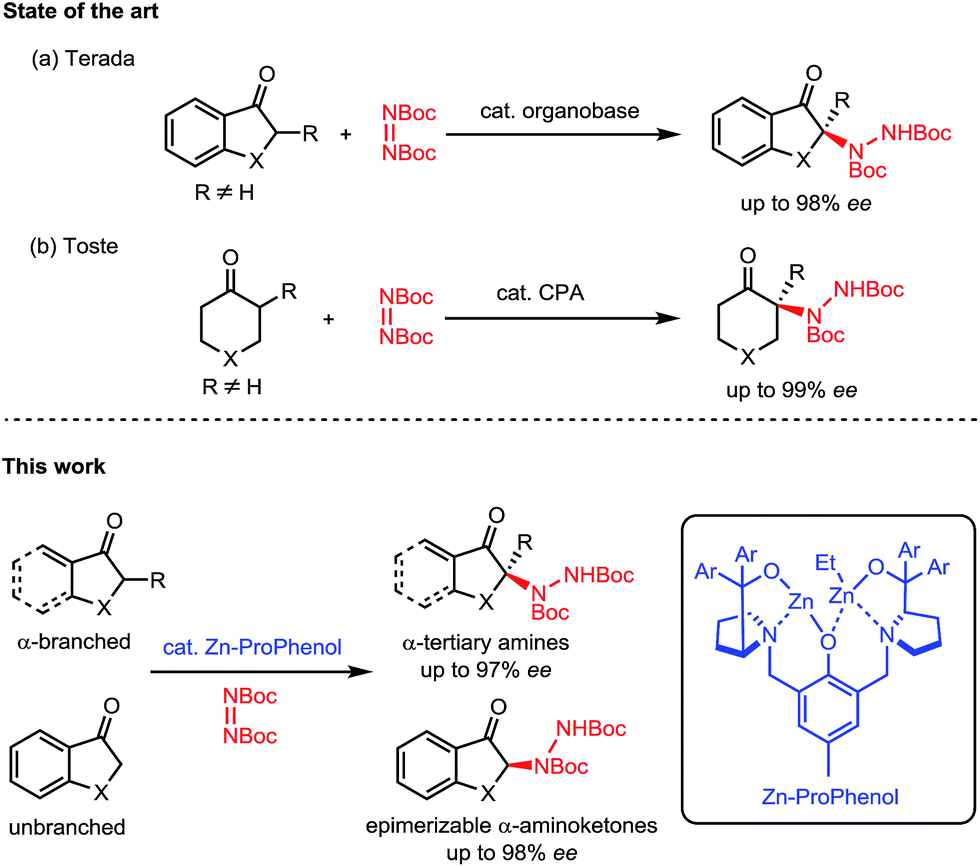 | ||
| Scheme 1 Direct catalytic enantioselective amination of α-branched ketones. | ||
Nitrogen atoms are usually incorporated within organic scaffolds by the addition of a nitrogen-based nucleophile to a carbon-based electrophile, often with the intermediacy of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond which can undergo further reactivity to afford amines. In comparison, the electrophilic amination of carbon-based nucleophiles has received much less attention.4–6 This latter strategy is particularly relevant for the enantioselective synthesis of α-tertiary amines, since the catalytic asymmetric addition of nucleophiles to ketimines is an extremely challenging process.5 Indeed, the direct catalytic enantioselective α-amination of carbonyl compounds6 has proven to be a useful strategy to access molecules decorated with tetrasubstituted N-containing stereogenic centers. However, these reactions rely on the use of aldehydes7 or activated carbonyl compounds such as 1,3-dicarbonyls,8 α-cyanocarbonyls,9 α-keto esters,10 and 2-oxindoles.11 There are only a few reports of direct enantioselective amination of unactivated ketones12,13 and only two examples describe the use of α-branched ketones for the synthesis of α-tertiary amines. Terada's initial report utilizing a chiral organosuperbase with cyclic aromatic ketones14 (Scheme 1a) was followed by Toste's chiral phosphoric acid methodology with cyclic aliphatic ketones15 (Scheme 1b). Both methods require the formation of a tetrasubstituted carbon alpha to the carbonyl to avoid epimerization of the final product. To date, there is no general method allowing for the direct catalytic asymmetric amination of both α-branched and unbranched ketones.
N bond which can undergo further reactivity to afford amines. In comparison, the electrophilic amination of carbon-based nucleophiles has received much less attention.4–6 This latter strategy is particularly relevant for the enantioselective synthesis of α-tertiary amines, since the catalytic asymmetric addition of nucleophiles to ketimines is an extremely challenging process.5 Indeed, the direct catalytic enantioselective α-amination of carbonyl compounds6 has proven to be a useful strategy to access molecules decorated with tetrasubstituted N-containing stereogenic centers. However, these reactions rely on the use of aldehydes7 or activated carbonyl compounds such as 1,3-dicarbonyls,8 α-cyanocarbonyls,9 α-keto esters,10 and 2-oxindoles.11 There are only a few reports of direct enantioselective amination of unactivated ketones12,13 and only two examples describe the use of α-branched ketones for the synthesis of α-tertiary amines. Terada's initial report utilizing a chiral organosuperbase with cyclic aromatic ketones14 (Scheme 1a) was followed by Toste's chiral phosphoric acid methodology with cyclic aliphatic ketones15 (Scheme 1b). Both methods require the formation of a tetrasubstituted carbon alpha to the carbonyl to avoid epimerization of the final product. To date, there is no general method allowing for the direct catalytic asymmetric amination of both α-branched and unbranched ketones.
Our group's aza-hemicrown family of ProPhenol ligands form dinuclear main group metal catalysts when treated with alkyl metal reagents such as Et2Zn.16 The ability of the Zn–ProPhenol catalytic system to promote a dual-activation of both nucleophiles and electrophiles has allowed for the development of a wide range of enantioselective addition reactions, but the use of heteroatom electrophiles has so far not been achieved.17 In this context, we wished to evaluate the potential of our Zn–ProPhenol catalysts for amination reactions of unactivated ketones using azodicarboxylates.
Results and discussion
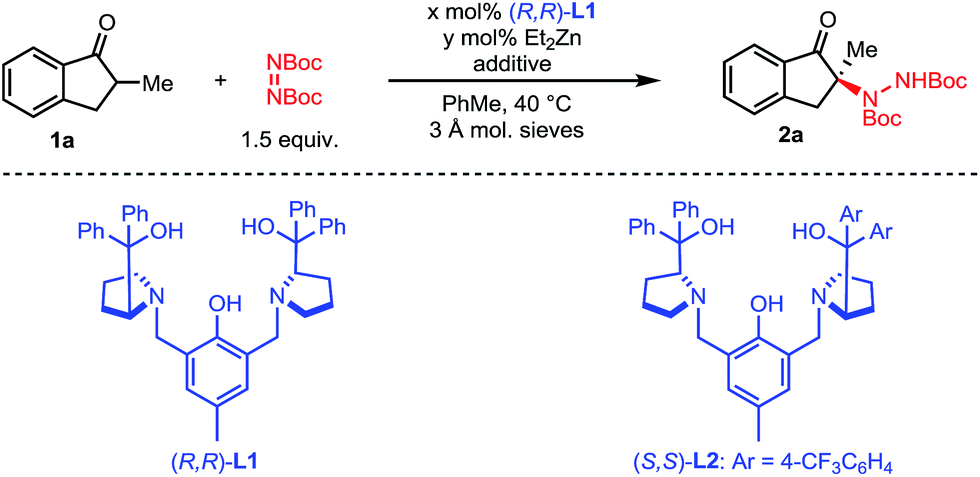
We initiated our studies with 2-methyl-1-indanone 1a and di-tert-butyl azodicarboxylate as a cheap and practical electrophilic nitrogen source (Table 1). Pleasingly, our standard Zn–ProPhenol catalyst promoted the desired amination in toluene at 40 °C to afford 2a with promising yield and selectivity (entry 1). Following this initial hit, a screen of Lewis-basic additives was undertaken based upon the previous observation that such additives can significantly impact the catalyst turnover and resulting enantioselectivity of the Zn–ProPhenol catalyst.17 The use of triphenylphosphine oxide (TPPO) or tert-butyl-N-methylcarbamate (3) as additives resulted in modest increases in ee (entries 2 and 3). However, bis-phosphine oxide (R)-BINAPO provided a significant boost in both yield (97%) and ee (89%) (entry 4). A small but measurable mismatched effect was observed when (S)-BINAPO was used, since the same enantiomer of 2a was obtained albeit with lower selectivities (entry 5). Keeping with bidentate phosphine oxides, additives 4 and 5 were screened, but both resulted in a significant reduction in ee (entries 6 and 7). Finally, while the bis-phosphine oxide of 1,4-bis(diphenylphosphino)butane (dppbO2) proved no better than TPPO, the use of the bis-phosphine oxide of 1,2-bis(diphenylphosphino)ethane (dppeO2) restored the selectivity and reactivity of the additive (R)-BINAPO with the added benefit of being achiral (entries 8 and 9). Switching ligands from (R,R)-L1 to the non-C2 symmetric (S,S)-L2 resulted in an improved enantioselectivity at the expense of yield (entry 10). However, a slight increase of catalyst loading to 15 mol% gave 2a in high yield and selectivity (entry 11).|
|
|||||
|---|---|---|---|---|---|
| Entry | x | y | Additive [mol%] | Yieldb [%] | eec [%] |
| a 0.20 mmol 1a, 0.30 mmol di-tert-butyl azodicarboxylate, x mol% L1, y mol% ZnEt2, 10 mg 3 Å mol. sieves, in 1.0 mL of toluene at 40 °C. b Yield of the isolated product. c Determined by HPLC on a chiral stationary phase. d (S,S)-L2 utilized. | |||||
| 1 | 11 | 20 | None | 77 | 70 |
| 2 | 11 | 20 | TPPO [10] | 79 | 79 |
| 3 | 11 | 20 | 3 [10] | 72 | 78 |
| 4 | 11 | 20 | (R)-BINAPO [10] | 97 | 89 |
| 5 | 11 | 20 | (S)-BINAPO [10] | 73 | 75 |
| 6 | 11 | 20 | 4 [10] | 69 | 11 |
| 7 | 11 | 20 | 5 [10] | 72 | 51 |
| 8 | 11 | 20 | dppbO2 [10] | 79 | 83 |
| 9 | 11 | 20 | dppeO2 [10] | 93 | 89 |
| 10d | 11 | 20 | dppeO2 [10] | 66 | −91 |
| 11d | 16 | 30 | dppeO2 [15] | 98 | −97 |
|
|||||
With optimized conditions in hand (Table 1, entry 11), the scope of the process was explored (Scheme 2). Indanones having a linear alkyl chain at the 2-position all gave excellent results (2a–2f). Of note, functional groups such as methyl esters, alkenes and terminal alkynes are well tolerated (2d–2f). Introduction of a bulkier isopropyl group at the 2-position of indanone still afforded product 2g with good enantioselectivity, albeit with a lower yield. Substrates were not limited to 5-membered rings, as 6-membered aryl and even vinyl ketones showed excellent reactivity and selectivity (2h–2i). To further highlight the broad scope of our catalytic amination process, two 3-alkyl-2-oxindole substrates were aminated using a reduced catalyst loading at room temperature (2j–2k).
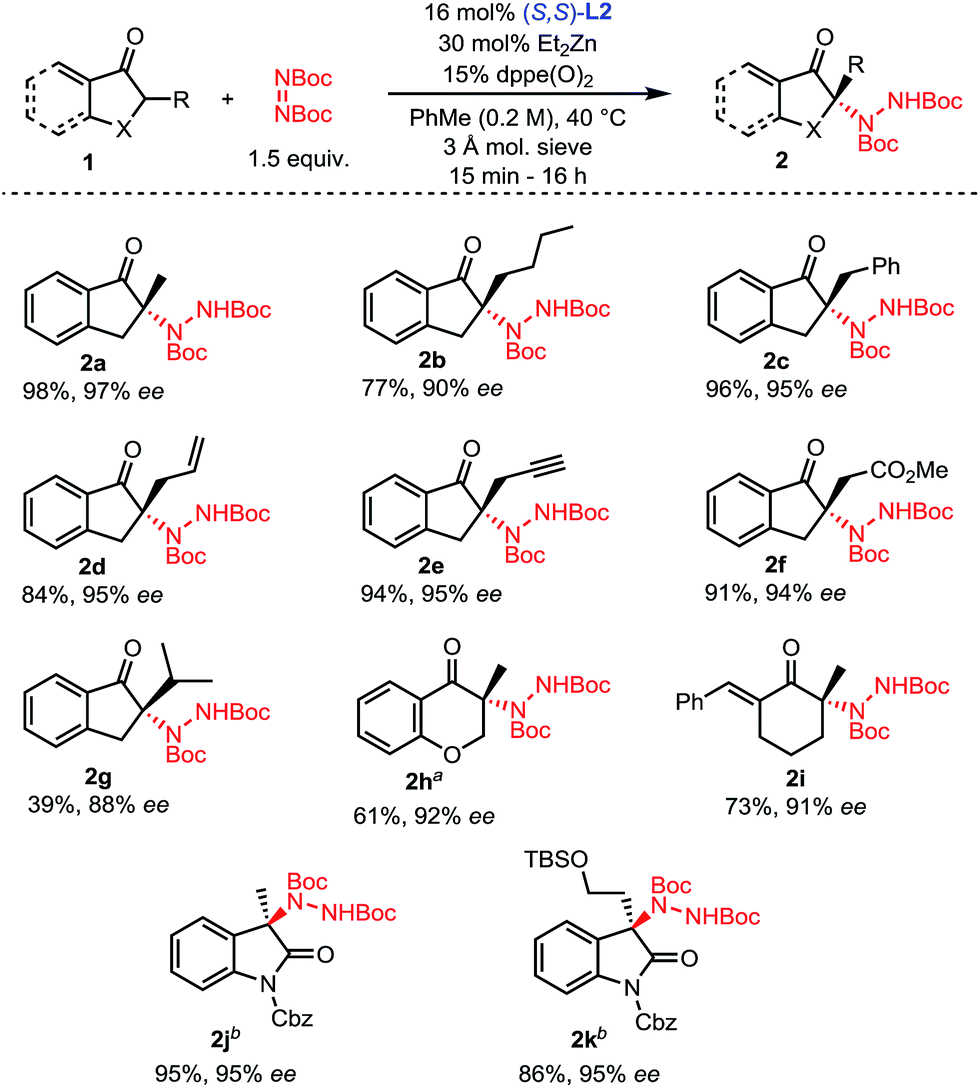 | ||
| Scheme 2 Scope of the amination of α-branched ketones. aReaction performed at 60 °C. bReaction performed with 5.5 mol% L2, 10 mol% ZnEt2, at 23 °C without any phosphine oxide additive. | ||
We next turned our attention to the use of unbranched aromatic ketones as nucleophiles in our amination reaction. Importantly, these substrates are not compatible with either Toste's or Terada's protocols due to product epimerization from the strongly acidic or strongly basic catalysts required. We felt the mild reaction conditions provided by our bifunctional Zn–ProPhenol catalyst should be perfectly suited for such challenging substrates.
When 1-tetralone 1l was reacted with 2.0 equivalents of di-tert-butyl azodicarboxylate in THF (0.4 M) at room temperature using 11 mol% of (R,R)-L1 and 20 mol% of diethylzinc, the desired amination product 2l was isolated in a promising 66% yield and 88% ee (Table 2, entry 1). Lowering the equivalents of electrophile to 1.1 resulted in an increase in yield without impacting the ee (entry 2). The molecular sieves were found to be important to catalyst turnover but not selectivity (entry 3). Decreasing the reaction concentration to 0.25 M improved the selectivity to 94% ee without sacrificing catalyst activity, while performing the reaction at 4 °C significantly impacted catalyst turnover (entries 4 and 5).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | x | y | z | Conc. | Yieldb [%] | eec [%] |
| a 0.20 mmol 1l, z equiv di-tert-butyl azodicarboxylate, x mol% L1, y mol% ZnEt2, 10 mg 3 Å mol. sieves, in 0.8 mL of THF at rt. b Yield of the isolated product. c Determined by HPLC on a chiral stationary phase. d No 3 Å mol. sieves. e Reaction performed at 4 °C. | ||||||
| 1 | 11 | 20 | 2.0 | 0.4 M | 66 | 88 |
| 2 | 11 | 20 | 1.1 | 0.4 M | 86 | 86 |
| 3d | 11 | 20 | 1.1 | 0.4 M | 49 | 90 |
| 4 | 11 | 20 | 1.1 | 0.25 M | 86 | 94 |
| 5e | 11 | 20 | 1.1 | 0.25 M | 28 | 87 |
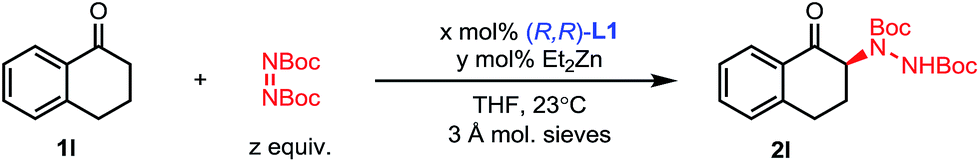
The scope of this amination process was then explored (Scheme 3). A range of 1-tetralones substituted at the 5-, 6- and 7-positions were successfully aminated in high yields and selectivities (2l–2q). Of note, tetralones 1o, 1p and 1q substituted with strongly electron-withdrawing groups showed no evidence of epimerization under the reaction conditions, showcasing the mild activation provided by the Zn–ProPhenol bifunctional catalyst. Importantly, such electron deficient tetralones were poor substrates for previously reported amination methods. The use of 1-chromanone 1r was challenging and lower temperatures as well as partial conversion were required to prevent significant epimerization of 2r. On the other hand, 1-benzosuberone 1s gave excellent results using our protocol. Finally, the 1-indanone class of substrates was studied. This class of substrates proved to be a significant challenge for previous amination reactions because they afford highly epimerizable products which were isolated with poor enantioselectivities.13c,e Using our mild Zn–ProPhenol catalyst and stopping the reaction at partial conversion led to the isolation of products 2t and 2u with great enantioselectivities and good yields. In the case of substrate 1u, the use of L2 was beneficial.
 | ||
| Scheme 3 Scope of the amination of unbranched ketones. aReaction performed at 4 °C. bReaction performed with 16 mol% L1 and 30 mol% ZnEt2 at 0.4 M. cReaction performed with 5.5 mol% (S,S)-L2 and 10 mol% ZnEt2. dBased on recovered starting material (brsm). | ||
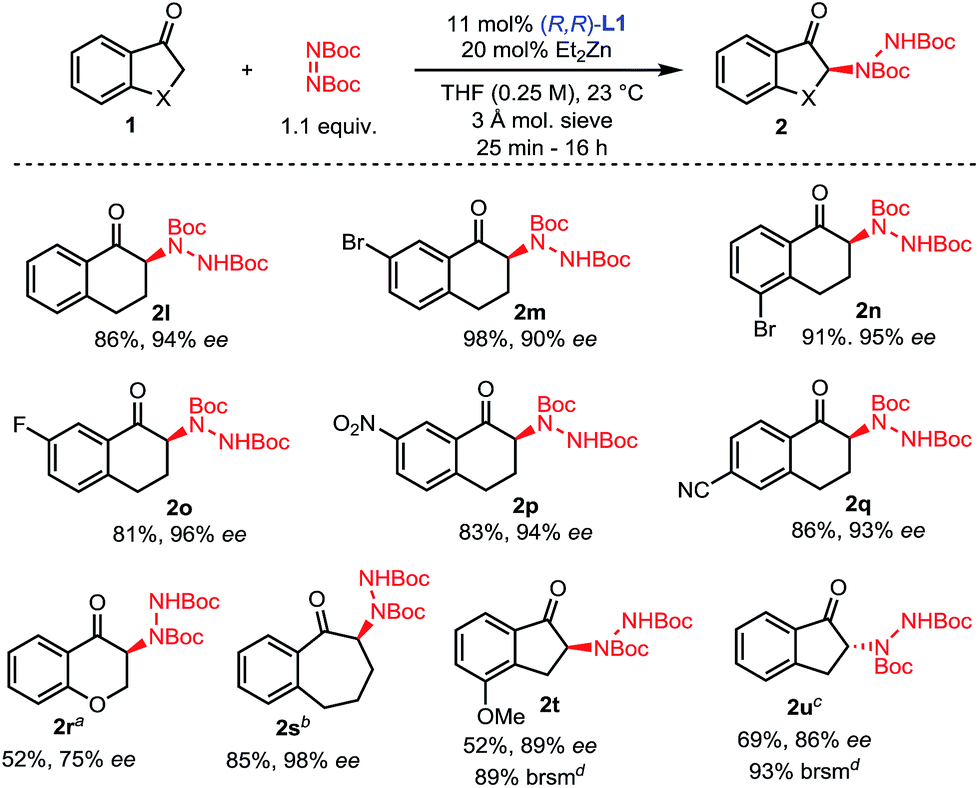
To showcase the scalability and practicality of the process, we performed some amination reactions on a 1 to 2 mmol scale with reduced catalyst loadings (Scheme 4). Amination of oxindole 1j was scaled to 2.1 mmol using 2 mol% catalyst without any significant change in outcome. Similarly, the amination of 2-methyl-1-indanone 1a could be run on a 1 mmol scale using 10 mol% of catalyst without a significant impact on the efficiency of the reaction. Finally, the amination of 7-fluorotetralone 1o was performed on a 1 mmol scale with 3 mol% catalyst and 2o was obtained with comparably excellent yield and enantioselectivity.
 | ||
| Scheme 4 Large scale reactions. | ||
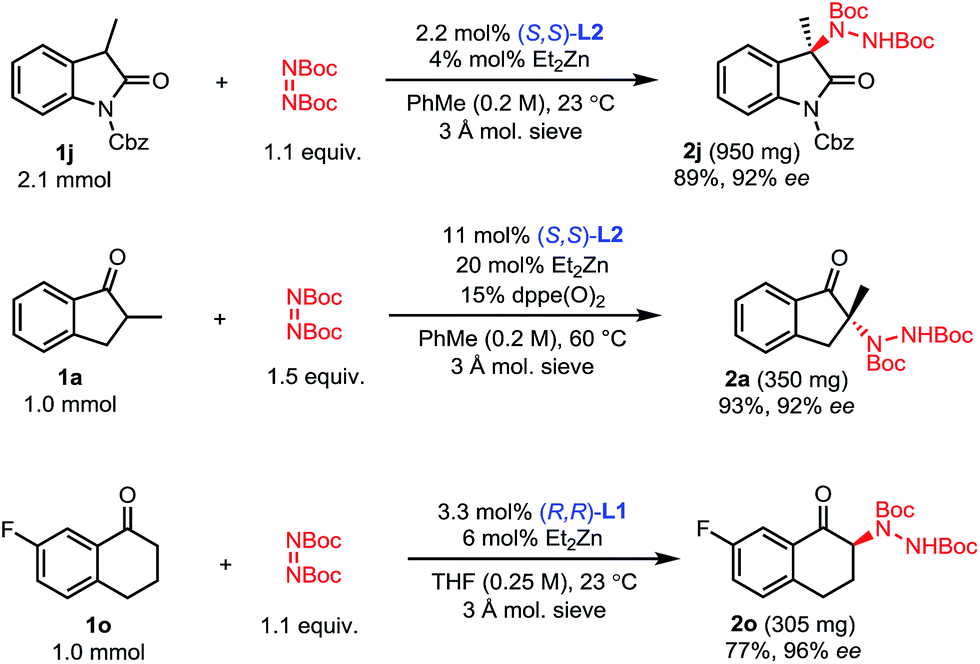
While some of these amination processes require relatively high catalyst loadings, these loadings can be mitigated by the easy recovery of the ProPhenol ligand through column chromatography and a subsequent acid–base extraction. To showcase this feature, the amination of 1-benzosuberone 1s was performed on a 0.6 mmol scale using our conditions from Scheme 3 (Scheme 5). After workup and purification, 83% of ligand L1 was recovered. This recovered ligand was then reused in a subsequent reaction with no loss of reactivity or selectivity.
 | ||
| Scheme 5 Recovery of ligand. | ||
A requirement for any useful amination technique utilizing dialkyl azodicarboxylate electrophiles is to show that the resulting hydrazines can be readily transformed into synthetically desirable amino products. In this respect, the use of di-tert-butyl azodicarboxylate represents a significant improvement over related methods requiring use of the less readily deprotected dimethyl azodicarboxylate (DMAD) or diethyl azodicarboxylate (DEAD).13a,c,e To highlight the utility of our products, oxindole 2j was converted to Boc-amine 6 in a two-step sequence involving an alkylation with methyl bromoacetate followed by an E1cB elimination promoted by cesium carbonate (Scheme 6a).18 In addition, both syn and anti amino alcohol derivatives could be accessed from ketone 2s with no erosion of ee (Scheme 6b). Bulky L-selectride delivers hydride from the more sterically accessible face to form the syn-product while the use of lithium aluminium hydride results in an internal delivery of the hydride to form the anti-product.19 We then developed a one-pot procedure for the subsequent alkylation–elimination cleavage of the N–N bond to access compounds syn-7 and anti-8 in 3 and 2 steps respectively. Overall, this approach allows a stereodivergent synthesis of cyclic compounds incorporating a phenylethanolamine motif. Such a motif can be found in many bioactive natural products, including the Ephedra alkaloids.20 It is also a common motif in marketed and clinical drugs as exemplified by salbutamol, a bronchodilator used to treat asthma, and naxagolide, Merck's antiparkinson compound.21,22
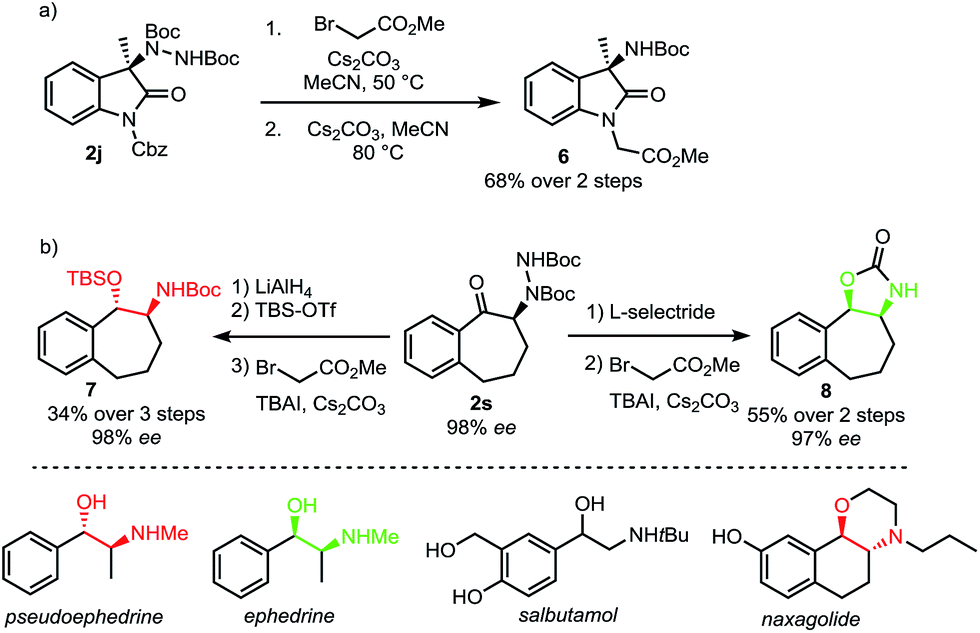 | ||
| Scheme 6 Cleavage of N–N bond to form amino products. | ||
A proposed catalytic cycle is depicted in Scheme 7 using (S,S)-L1 instead of the non-C2 symmetric (S,S)-L2 for purposes of simplicity. The initially formed dinuclear Zn–ProPhenol catalyst enters the catalytic cycle through a deprotonation of the ketone to form complex I. Bi-dentate coordination of the electrophile produces complex II which sets the electrophile up for attack from the re-face of the enolate. The resulting zinc–hydrazine product (III) serves as a source of base to deprotonate another equivalent of ketone while simultaneously releasing the product. Importantly, the stereochemical outcome of the reaction is in perfect agreement with our recent work on related Mannich reactions.23 The source of selectivity in complex II is made clearer when viewed down the axis of the phenol (IIa, bottom of Scheme 7). From this orientation, steric interactions are minimized when the bulk of the reactants is directed, as shown in IIa, towards the western pseudo-equatorial phenyl group (green) and would be maximized if the bulk of the reactants is instead positioned towards the eastern pseudo-axial phenyl group (purple).
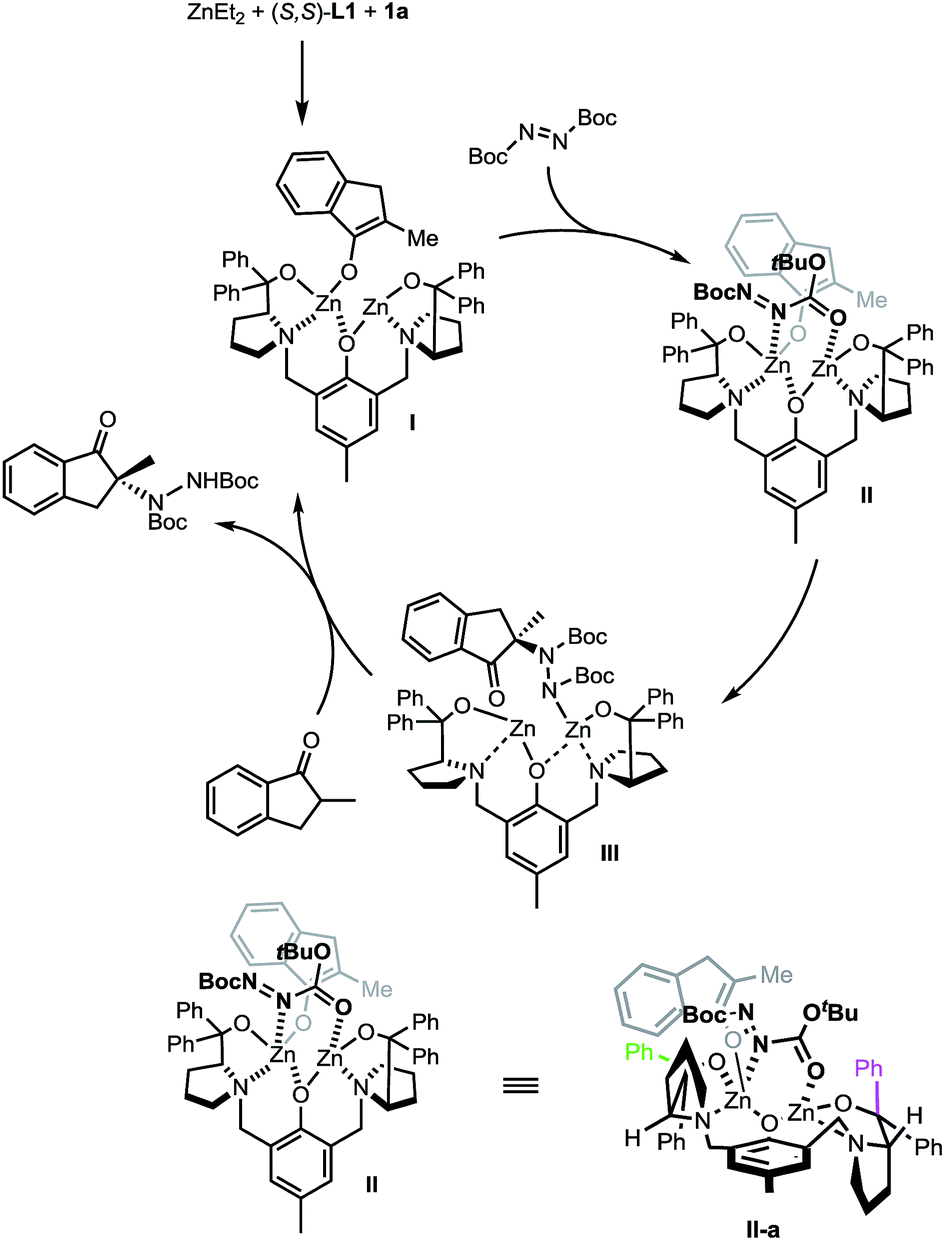 | ||
| Scheme 7 Proposed catalytic cycle. | ||
Conclusion
In conclusion, we have developed a mild catalytic method for the direct amination of aromatic and vinyl ketones as well as of oxindoles. The procedure is unique in its ability for a single catalyst to form tri- and tetrasubstituted N-containing stereocenters with high enantioselectivities, even those whose products contain readily epimerizable indanone or electron-withdrawing tetralone scaffolds that do not fare well under previously reported conditions.13 This method negates the need for cryogenic temperatures and multi-day reaction times characteristic of previous reports.14,15 The reactions can be scaled up with reduced catalyst loadings without impacting the efficiency of the process and the ligand can be recovered and reused, making the effective catalyst loadings very low. Finally, the resulting hydrazine products are readily converted into valuable amines utilizing a simplified one-pot alkylation–elimination procedure.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the NIH (GM-033049) and the NSF (CHE-1360634) for their generous support of our program. JST acknowledges support from the John Stauffer Memorial Fellowship. TS acknowledges support from the Swiss National Science Foundation for a postdoctoral fellowship.Notes and references
- (a) A. Ricci, Modern Amination Methods, Wiley-VCH, Weinheim, Germany, 2000 Search PubMed; (b) R. Hili and A. K. Yudin, Nat. Chem. Biol., 2006, 2, 284–287 CrossRef CAS PubMed; (c) T. C. Nugent, Chiral Amine Synthesis: Methods, Developments and Applications, Wiley-VCH, Weinheim, Germany, 2010 Search PubMed.
- For selected reviews, see: (a) J. Kobayashi and H. Morita, Alkaloids, 2003, 60, 165–205 CAS; (b) D. J. Ramon and M. Yus, Curr. Org. Chem., 2004, 8, 149–183 CrossRef CAS; (c) I. Moldvai, E. Temesvari-Major, M. Incze, G. Doernyei, E. Szentirmay and C. Szantay, Helv. Chim. Acta, 2005, 88, 1344–1356 CrossRef CAS; (d) T. Reynolds, Phytochemistry, 2005, 66, 1399–1406 CrossRef CAS PubMed.
- J. Clayden, M. Donnard, J. Lefranc and D. J. Tetlow, Chem. Commun., 2011, 47, 4624–4639 RSC.
- C. Greck and J.-P. Genet, Synlett, 1997, 741–748 CrossRef CAS.
- (a) R. Bloch, Chem. Rev., 1998, 98, 1407–1438 CrossRef CAS PubMed; (b) S. Kobayashi, Y. Mori, J. S. Fossey and M. M. Salter, Chem. Rev., 2011, 111, 2626–2704 CrossRef CAS PubMed.
- For recent reviews on general amination of carbonyl compounds see: (a) V. Nair, A. T. Biju, S. C. Mathew and B. P. Babu, Chem.–Asian J., 2008, 3, 810–820 CrossRef CAS PubMed; (b) T. Vilaivan and W. Bhanthumnavin, Molecules, 2010, 15, 917–958 CrossRef CAS PubMed; (c) A. M. R. Smith and K. K. Hii, Chem. Rev., 2011, 111, 1637–1656 CrossRef CAS PubMed; (d) A. Vallribera, R. M. Sebastian and A. Shafir, Curr. Org. Chem., 2011, 15, 1539–1577 CrossRef CAS; (e) T. A. Ramirez, B. Zhao and Y. Shi, Chem. Soc. Rev., 2012, 41, 931–942 RSC; (f) F. Zhou, F.-M. Liao, J.-S. Yu and J. Zhou, Synthesis, 2014, 46, 2983–3003 CrossRef CAS.
- For a review on the amination of aldehydes, see: (a) A. Desmarchelier, V. Coeffard, X. Moreau and C. Greck, Tetrahedron, 2014, 70, 2491–2513 CrossRef CAS. For selected references on the amination of aldehydes, see: (b) A. Bøgevig, K. Johl, N. Kumaragurubaran, W. Zhuang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2002, 41, 1790–1793 CrossRef; (c) B. List, J. Am. Chem. Soc., 2002, 124, 5656–5657 CrossRef CAS PubMed; (d) N. S. Chowdari, D. B. Ramachary and C. F. Barbas III, Org. Lett., 2003, 5, 1685–1688 CrossRef CAS PubMed; (e) S. Bertelsen, M. Marigo, S. Brandes, P. Dinér and K. A. Jørgensen, J. Am. Chem. Soc., 2006, 128, 12973–12980 CrossRef CAS PubMed; (f) G. Cecere, C. M. König, J. L. Alleva and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 11521–11524 CrossRef CAS PubMed.
- For selected references on the amination of 1,3-carbonyl compounds see: (a) M. Marigo, K. Juhl and K. A. Jørgenson, Angew. Chem., Int. Ed., 2003, 42, 1367–1369 CrossRef CAS PubMed; (b) X. Xu, T. Yabuta, P. Yuan and Y. Takemoto, Synlett, 2006, 1, 137–140 Search PubMed; (c) T. Mashiko, K. Hara, D. Tanaka, Y. Fujiwara, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2007, 129, 11342–11343 CrossRef CAS PubMed; (d) T. Mashiko, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 14990–14999 CrossRef CAS PubMed; (e) H. Konishi, T. Y. Lam, J. P. Malerich and W. H. Rawal, Org. Lett., 2010, 12, 2028–2031 CrossRef CAS PubMed; (f) Z. Yang, Z. Weng, S. Bai, X. Liu, L. Lin and X. Feng, Org. Lett., 2011, 13, 596–599 CrossRef CAS PubMed; (g) Q.-H. Deng, T. Bleith, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2013, 135, 5356–5359 CrossRef CAS PubMed; (h) C. Xu, L. Zhang and S. Luo, Angew. Chem., Int. Ed., 2014, 53, 4149–4152 CrossRef CAS PubMed; (i) H. M. Nelson, J. S. Patel, H. P. Shunatona and F. D. Toste, Chem. Sci., 2015, 6, 170–173 RSC; (j) K. Ohmatsu, Y. Ando, T. Nakashima and T. Ooi, Chem, 2016, 1, 802–810 CrossRef CAS.
- For selected references on the amination of α-cyanocarbonyl compounds, see: (a) S. Saaby, M. Bella and K. A. Jørgensen, J. Am. Chem. Soc., 2004, 126, 8120–8121 CrossRef CAS PubMed; (b) X. Liu, H. Li and L. Deng, Org. Lett., 2005, 7, 167–169 CrossRef CAS PubMed; (c) Y. Hasegawa, M. Watanabe, I. D. Gridnev and T. Ikariya, J. Am. Chem. Soc., 2008, 130, 2158–2159 CrossRef CAS PubMed; (d) H. Yasuharu, I. D. Gridnev and T. Ikariya, Bull. Chem. Soc. Jpn., 2012, 85, 316–334 CrossRef.
- For the amination of α-keto esters, see: (a) K. Juhl and K. A. Jørgensen, J. Am. Chem. Soc., 2002, 124, 6254–6255 CrossRef; (b) M. Terada, K. Amagai, K. Ando, E. Kwon and H. Ube, Chem.–Eur. J., 2011, 17, 9037–9041 CrossRef CAS PubMed.
- For selected references on the amination of 2-oxindoles see: (a) L. Cheng, L. Liu, D. Wang and Y.-J. Chen, Org. Lett., 2009, 11, 3874–3877 CrossRef CAS PubMed; (b) T. Bui, M. Borregan and C. F. Barbas III, J. Org. Chem., 2009, 74, 8935–8938 CrossRef CAS PubMed; (c) Z.-Q. Qian, F. Zhou, T.-P. Du, B.-L. Wang, M. Ding, X.-L. Wang, M. Ding, X.-L. Zhao and J. Zhou, Chem. Commun., 2009, 6753–6755 RSC; (d) Z. Yang, Z. Wang, S. Bai, K. Shen, D. Chen, X. Liu, L. Lin and S. Feng, Chem.–Eur. J., 2010, 16, 6632–6637 CrossRef CAS PubMed; (e) S. Mouri, Z. Chen, H. Mitsunuma, M. Furutachi, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 1255–1257 CrossRef CAS PubMed.
- For methods involving enamine catalysis, see: (a) N. Kumaragurubaran, K. Juhl, W. Zhuang, A. Bøgevig and K. A. Jørgensen, J. Am. Chem. Soc., 2002, 124, 2420–2421 CrossRef; (b) T.-Y. Liu, H.-L. Cui, Y. Zhang, K. Jiang, W. Du, S.-Q. He and Y.-C. Chen, Org. Lett., 2007, 9, 3671–3674 CrossRef CAS PubMed; (c) Y. Hayashi, S. Aratake, Y. Imai, K. Hibino, Q.-Y. Chen, J. Yamaguchi and T. Uchimaru, Chem.–Asian J., 2008, 3, 225–232 CrossRef CAS PubMed; (d) Y. J. Lim and D. Y. Kim, Bull. Korean Chem. Soc., 2013, 34, 1955–1956 CrossRef CAS.
- Only three methods of electrophilic amination of unbranched aromatic ketones are reported: (a) T.-Y. Liu, H.-L. Cui, Y. Zhang, K. Jiang, W. Du, S.-Q. He and Y.-C. Chen, Org. Lett., 2007, 9, 3671–3674 CrossRef CAS PubMed; (b) W. Li, X. Liu, X. Hao, X. Hu, Y. Chu, W. Cao, S. Qin, C. Hu, L. Lin and X. Feng, J. Am. Chem. Soc., 2011, 133, 15268–15271 CrossRef CAS PubMed; (c) M. Shang, X. Wang, S. M. Koo, J. Youn, J. Z. Chan, W. Yao, B. T. Hastings and M. Wasa, J. Am. Chem. Soc., 2017, 139, 95–98 CrossRef CAS PubMed; (d) For a related nucleophilic amination strategy that can yield tertiary alpha-amino ketones see: D. H. Miles, J. Guasch and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 7632–7635 CrossRef CAS PubMed; (e) For a recent non-direct decarboxylative method see: Y. Wei, H.-X. Zhang, J.-L. Zeng, J. Nie and J.-A. Ma, Org. Lett., 2017, 19, 2162–2165 CrossRef CAS PubMed.
- T. Takeda and M. Terada, J. Am. Chem. Soc., 2013, 135, 15306–15309 CrossRef CAS PubMed.
- (a) X. Yang and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 3205–3208 CrossRef CAS PubMed; (b) For a subsequent report under very similar phosphoric acid conditions see: G. A. Shevchenko, G. Pupo and B. List, Synlett, 2015, 26, 1413–1416 CrossRef CAS.
- B. M. Trost and H. Ito, J. Am. Chem. Soc., 2000, 122, 12003–12004 CrossRef CAS.
- For a review, see: B. M. Trost and M. J. Barlett, Acc. Chem. Res., 2015, 48, 688–701 CrossRef CAS PubMed.
- (a) P. Magnus, N. Garizi, K. A. Seibert and A. Ornholt, Org. Lett., 2009, 11, 5646–5648 CrossRef CAS PubMed; (b) For a recent use of this method involving the products of ketone alpha-amination, see: B. M. Trost and J. S. Tracy, Org. Lett., 2017, 19, 2630–2633 CrossRef CAS PubMed.
- P. Gmeiner and B. Bollinger, Liebigs Ann. Chem., 1992, 273–278 CrossRef CAS.
- E. A. Abourashed, A. T. El-Alfy, I. A. Khan and L. Walker, Phytother. Res., 2003, 17, 703–712 CrossRef CAS PubMed.
- V. A. Cullum, J. B. Farmer, D. Jack and G. P. Levy, Br. J. Pharmacol., 1969, 35, 141–151 CrossRef CAS PubMed.
- J. H. Jones, P. S. Anderson, J. J. Baldwin, B. V. Clineschmidt, D. E. McClure, G. F. Lundell, W. C. Randall, G. E. Martin, M. Williams, J. M. Hirschfield, G. Smith and P. K. Lumma, J. Med. Chem., 1984, 27, 1607–1613 CrossRef CAS PubMed.
- (a) B. M. Trost, T. Saget, A. Lerchen and C. Hung, Angew. Chem., Int. Ed., 2016, 55, 781–784 CrossRef CAS PubMed; (b) B. M. Trost, T. Saget and C. Hung, J. Am. Chem. Soc., 2016, 138, 3659–3662 CrossRef CAS PubMed; (c) B. M. Trost, T. Saget and C. Hung, Angew. Chem., Int. Ed., 2017, 56, 2440–2444 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, and characterization data. See DOI: 10.1039/c8sc00147b |
| This journal is © The Royal Society of Chemistry 2018 |