DOI:
10.1039/C7SC04732K
(Edge Article)
Chem. Sci., 2018,
9, 1989-1995
A water-soluble supramolecular complex that mimics the heme/copper hetero-binuclear site of cytochrome c oxidase†‡
Received
2nd November 2017
, Accepted 12th January 2018
First published on 15th January 2018
Abstract
In mitochondria, cytochrome c oxidase (CcO) catalyses the reduction of oxygen (O2) to water by using a heme/copper hetero-binuclear active site. Here we report a highly efficient supramolecular approach for the construction of a water-soluble biomimetic model for the active site of CcO. A tridentate copper(II) complex was fixed onto 5,10,15,20-tetrakis(4-sulfonatophenyl)porphinatoiron(III) (FeIIITPPS) through supramolecular complexation between FeIIITPPS and a per-O-methylated β-cyclodextrin dimer linked by a (2,2′:6′,2′′-terpyridyl)copper(II) complex (CuIITerpyCD2). The reduced FeIITPPS/CuITerpyCD2 complex reacted with O2 in an aqueous solution at pH 7 and 25 °C to form a superoxo-type FeIII–O2−/CuI complex in a manner similar to CcO. The pH-dependent autoxidation of the O2 complex suggests that water molecules gathered at the distal Cu site are possibly involved in the FeIII–O2−/CuI superoxo complex in an aqueous solution. Electrochemical analysis using a rotating disk electrode demonstrated the role of the FeTPPS/CuTerpyCD2 hetero-binuclear structure in the catalytic O2 reduction reaction.
Introduction
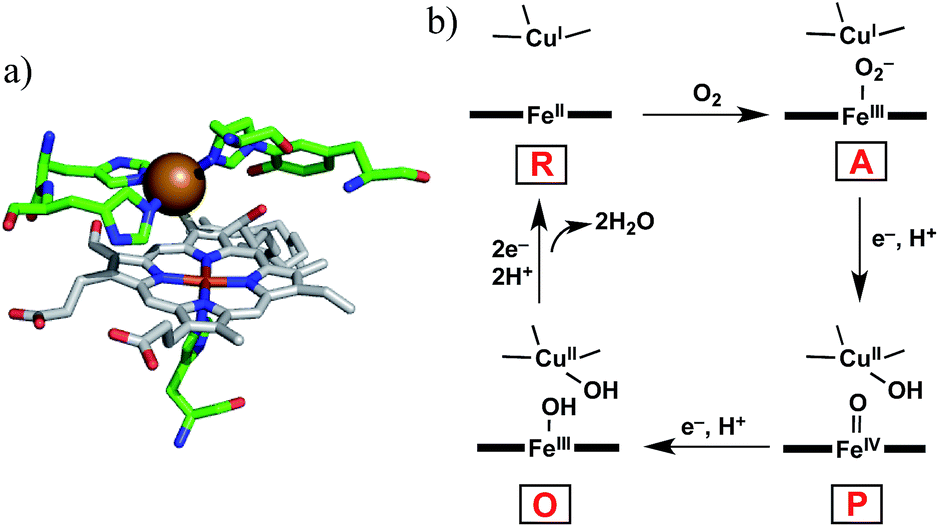
Cytochrome c oxidase (CcO) is the terminal enzyme in the mitochondrial respiratory chain. CcO consumes most of the molecular oxygen (O2) processed by living organisms by reducing it to water (H2O).1 The four-electron/four-proton reduction process (O2 + 4e− + 4H+ → 2H2O) takes place at the heme a3/CuB hetero-binuclear active centre of CcO (Fig. 1a).1–5 For the catalytic O2 reduction reaction, the reaction mechanism schematically depicted in Fig. 1b has been proposed.1–3 In the catalytic cycle, the fully reduced heme a3/CuB site (FeII/CuI, compound R) reacts with O2 to form an oxymyoglobin-like superoxo complex of heme a3 (FeIII–O2−/CuI, compound A).3,6 Compound A is rapidly (∼0.5 ms) converted to an oxoferryl intermediate (FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/CuII–OH, compound P) via O–O bond cleavage assisted by H atom injection from a vicinal tyrosine residue.3–6 Mechanistic investigations have suggested that one or more water molecules near the bound O2 can facilitate the conversion of compound A to compound P.7,8
O/CuII–OH, compound P) via O–O bond cleavage assisted by H atom injection from a vicinal tyrosine residue.3–6 Mechanistic investigations have suggested that one or more water molecules near the bound O2 can facilitate the conversion of compound A to compound P.7,8
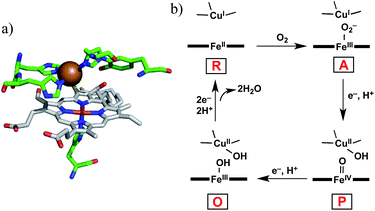 |
| | Fig. 1 (a) Heme a3/CuB hetero-binuclear active site of CcO (PDB ID: 1OCO) and (b) the simplified mechanism for the O2 reduction reaction catalysed by CcO. | |
To understand the reaction mechanism, synthetic heme/copper models have been constructed using tetraarylporphinatoiron(II) (PFeII) combined with CuI complexes (CuILn, where L is a nitrogen donor ligand; n (coordination number) = 3 or 4).4,5 However, upon oxygenation of the PFeII/CuILn model systems in anhydrous organic solvents, μ-peroxo-type bridged structures, i.e., PFeIII–O2–CuIILn complexes, tend to form instead of compound A-like superoxo species.9–12 In native CcO, the μ-peroxo-type bridged structure has not been experimentally identified, although it has been proposed as a transitional precursor of compound P.3,12,13 The structural differences between the native and model systems (superoxo vs. μ-peroxo)14 might be attributed to the influence of water.7,8,13 A model study by Naruta and co-workers demonstrated that the μ-peroxo complex (PFeIII–O2–CuIIL3) formed at −70 °C was converted to the superoxo complex (PFeIII–O2−/CuIL3) at −30 °C by the action of water molecules.15 In native CcO, highly ordered water molecules have been detected in the vicinity of heme a3/CuB.7,16 A quantum chemical calculation suggested that a water molecule in the vicinity of CuB decreases the energy barrier of the transformation of compound A to compound P.8 In this context, a water-soluble PFeII/CuILn model compound would be useful to investigate the role of water on the reactivity of the Fe/Cu hetero-binuclear complex with O2. However, very few heme/copper mimics functioning under aqueous conditions have been prepared so far, except for the system constructed in the engineered heme pocket of myoglobin.17,18
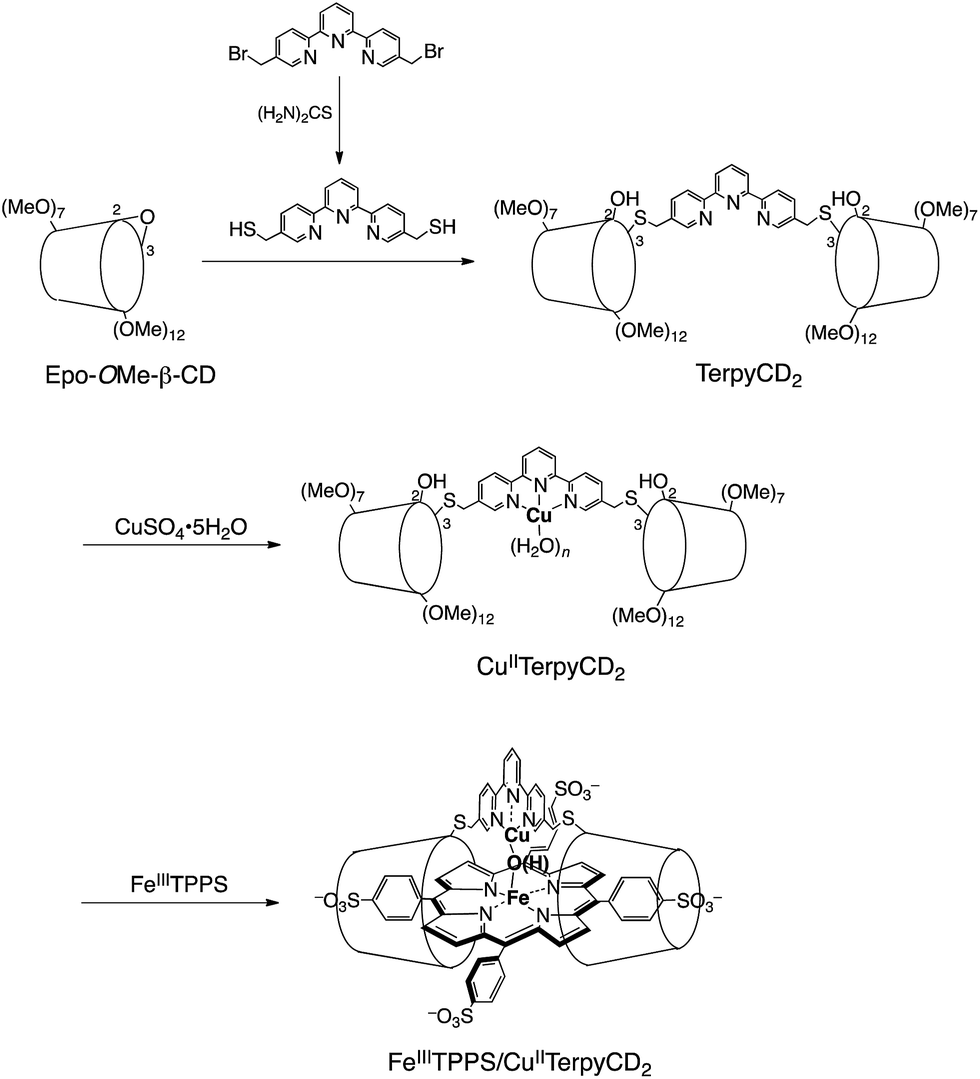
In this study, we describe an aqueous synthetic PFe/CuL3 hetero-binuclear model system built on a porphyrin/cyclodextrin supramolecular complex (Scheme 1). This system takes advantage of the very stable formation of a self-assembling 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 complex of 5,10,15,20-tetrakis(4-sulfonatophenyl)porphinatoiron (FeTPPS) with per-O-methylated β-cyclodextrins (CDs).19 We have previously studied the porphyrin/cyclodextrin complexes as simple biomimetic models of heme proteins that function under aqueous conditions,20–23 where the molecular cage of per-O-methylated β-CDs provided a microscopic hydrophobic environment for FeTPPS similar to the heme pocket of heme proteins.24 Here, we have synthesised a per-O-methylated β-CD dimer linked by a CuII–terpyridine complex (CuIITerpyCD2, Scheme 1) to replicate the distal tridentate CuB site of CcO. The structural characterisation of the supramolecular FeTPPS/CuTerpyCD2 complex and its reactivity towards O2 are described.
:2 complex of 5,10,15,20-tetrakis(4-sulfonatophenyl)porphinatoiron (FeTPPS) with per-O-methylated β-cyclodextrins (CDs).19 We have previously studied the porphyrin/cyclodextrin complexes as simple biomimetic models of heme proteins that function under aqueous conditions,20–23 where the molecular cage of per-O-methylated β-CDs provided a microscopic hydrophobic environment for FeTPPS similar to the heme pocket of heme proteins.24 Here, we have synthesised a per-O-methylated β-CD dimer linked by a CuII–terpyridine complex (CuIITerpyCD2, Scheme 1) to replicate the distal tridentate CuB site of CcO. The structural characterisation of the supramolecular FeTPPS/CuTerpyCD2 complex and its reactivity towards O2 are described.
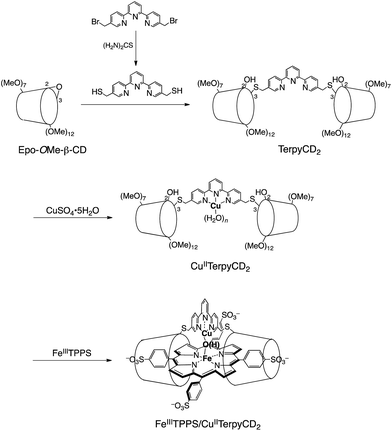 |
| | Scheme 1 Preparation of the supramolecular FeIIITPPS/CuIITerpyCD2 complex. | |
Results and discussion
Synthesis of a water-soluble FeIII/CuII hetero-binuclear complex
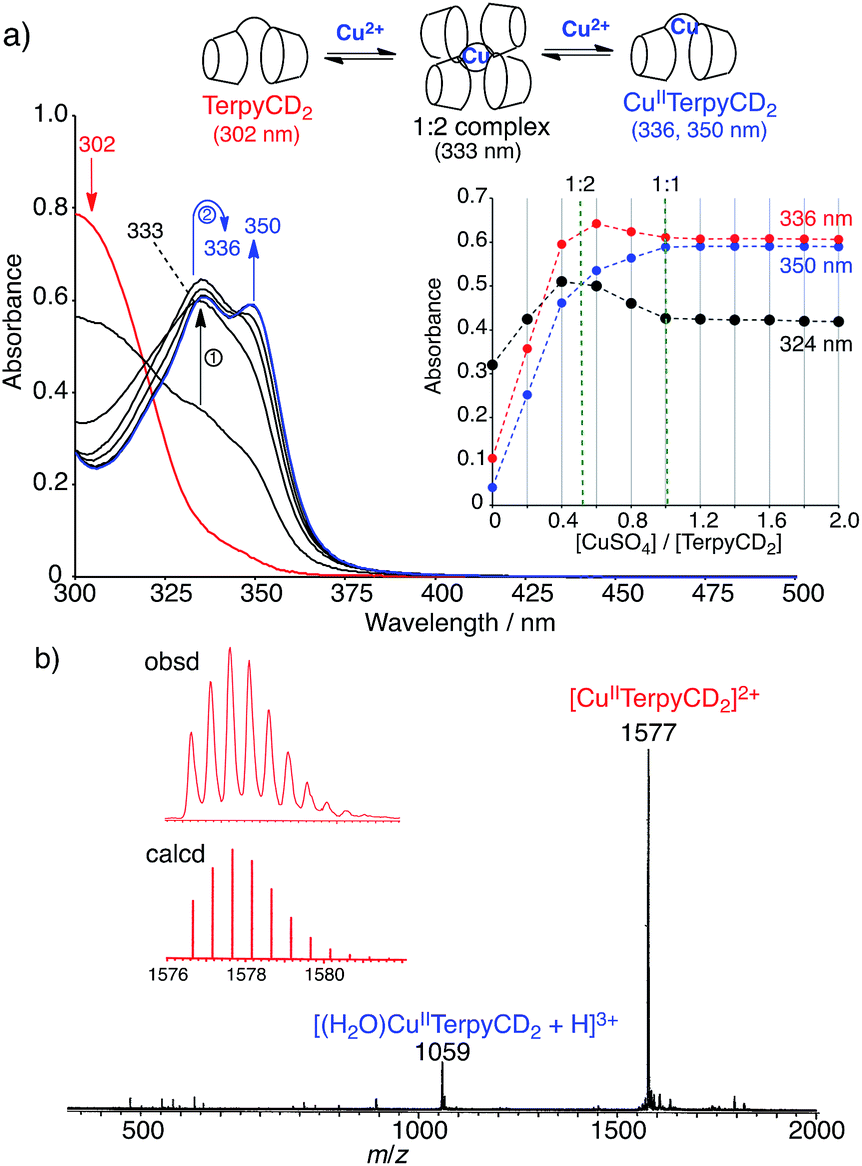
The synthetic route of a supramolecular FeIIITPPS/CuIITerpyCD2 complex is shown in Scheme 1 and experimental details are described in (ESI‡). Briefly, the terpyridyl ligand was inserted as a linker of the CD dimer (TerpyCD2) by the reaction of 5,5′′-bis(mercaptomethyl)-2,2′:6′,2′′-terpyridine with 2,3-monoepoxy-per-O-methylated β-CD (Epo-OMe-β-CD).20 The addition of CuSO4·5H2O to TerpyCD2 in an aqueous solution generated two absorption bands at 336 and 350 nm (Fig. 2a), which corresponded to the ligand to metal charge transfer bands of the terpyridyl–CuII 1:1 complex.25 In the UV-vis titration, a biphasic spectral change was observed (Fig. 2a inset), indicating that the 1:2 complex of Cu2+ with TerpyCD2 (λmax = 333 nm) was first formed and then it was converted to the thermodynamically stable 1:1 complex upon further addition of Cu2+. The spectral changes were completed at one equivalent of Cu2+. The complexation between TerpyCD2 and Cu2+ was also monitored by electrospray mass spectroscopy. In the 1:1 mixture of CuSO4 and TerpyCD2 in H2O, the 1:1 complex (CuIITerpyCD2) was observed at m/z 1577 and 1059 (Fig. 2b), which corresponds to [CuIITerpyCD2]2+ and [(H2O)CuIITerpyCD2 + H]3+, respectively. The 1:2 complex was also detected as a small ion peak when the 1:2 mixture of CuSO4 and TerpyCD2 in H2O was analysed by electrospray mass spectroscopy (data not shown).
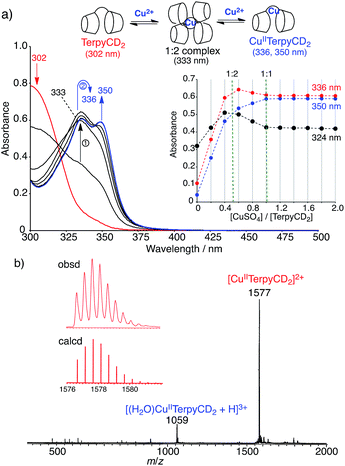 |
| | Fig. 2 Complexation of TerpyCD2 with Cu2+ in aqueous solution. (a) UV-vis spectral change of TerpyCD2 (33 μM) upon stepwise addition of CuSO4 in water at 25 °C. The inset shows changes in absorbances as a function of [CuSO4]/[TerpyCD2]. The biphasic titration curve indicates transient formation of the 1:2 complex before forming the thermodynamically stable 1:1 complex (CuIITerpyCD2) during the titration. (b) Electrospray mass spectrum (positive mode) of the 1:1 mixture of TerpyCD2 and CuSO4 in H2O. The inset shows the simulated isotope distribution patterns for the [CuIITerpyCD2]2+ complex. | |
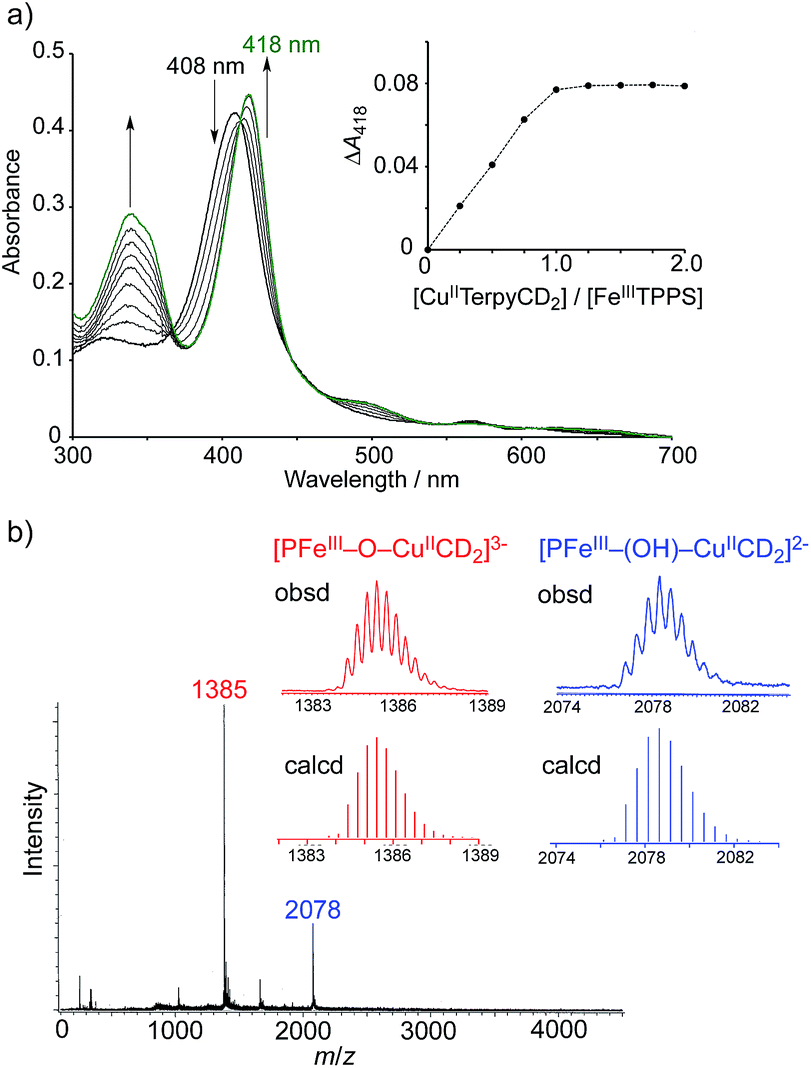
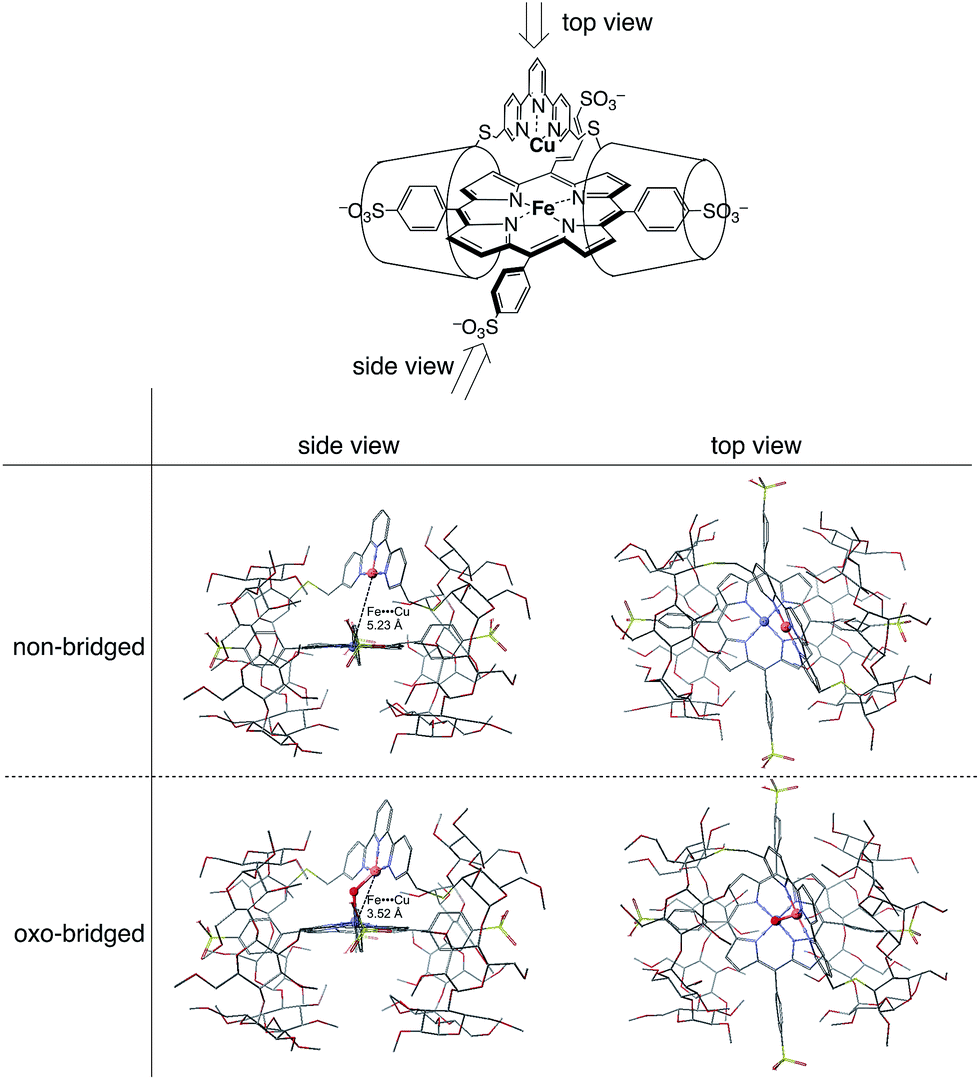
The CuIITerpyCD2 complex was then titrated with FeIIITPPS (Fig. 3a). The Soret band of FeIIITPPS shifted from 408 nm to 418 nm, indicating that a μ-oxo-dimer of FeIIITPPS dissociated to the monomeric monohydroxo complex (FeIII(OH−)TPPS)19 through interaction with CuIITerpyCD2. The spectral changes were completed upon addition of one equivalent of CuIITerpyCD2 to FeIIITPPS, indicating a quantitative 1:1 complexation. The obtained complex was then analysed by electrospray mass spectroscopy. The two main ion peaks were detected at m/z 1385 and 2078 as tri- and di-anionic species, respectively (Fig. 3b). Considering total charges of the complexes, the peaks at m/z 1385 and 2078 were assigned to the μ-oxo and μ-hydroxo FeIIITPPS/CuIITerpyCD2 complexes, i.e., [PFeIII–O–CuIICD2]3− and [PFeIII–(OH)–CuIICD2]2−, respectively. The assignments were confirmed by isotope pattern simulations (Fig. 3b inset). Evidence of the μ-oxo (FeIII–O–CuII) structure was also provided by its characteristic absorption bands at 453 and 567 nm, which appeared when the pH of the solution was increased (Fig. S3‡). The red-shifted Soret band at alkaline conditions indicates formation of the PFeIII–O–CuII μ-oxo complex.26–28 The pH titration revealed the acid–base equilibrium of [PFeIII–O–CuIICD2]3− and [PFeIII–(OH)–CuIICD2]2− with pKa = 8.8. This pKa value is consistent with that previously predicted by Karlin and Blackburn (pKa = 8 ± 2.5).28 The electron paramagnetic resonance (EPR) spectra showed significantly weak signals at g = 6.09 and 2.08 in the FeIIITPPS/CuIITerpyCD2 complex (Fig. S4‡) because of the antiferromagnetic coupling between the two metal ions as a result of their close proximity. The optimized molecular structure (Fig. 4) also illustrates the proximity of Fe and Cu ions in the FeIIITPPS/CuIITerpyCD2 complex; the Fe/Cu distances for the non-bridged and oxo-bridged forms are 5.23 and 3.52 Å, respectively. The distances are similar to those in native CcO, in which the oxidised heme a3/CuB distance were found in the range of 4.4–4.9 Å.4
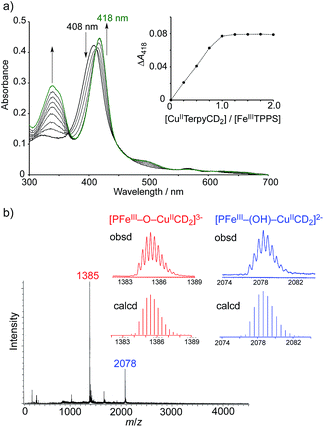 |
| | Fig. 3 Characterisation of the supramolecular FeIIITPPS/CuIITerpyCD2 complex. (a) UV-vis spectral changes of FeIIITPPS (3 μM) upon stepwise addition of CuIITerpyCD2 in 0.05 M phosphate buffer at pH 7.0 and 25 °C. The inset shows the changes in absorbance at 418 nm as a function of the molar ratio ([CuIITerpyCD2]/[FeIIITPPS]). (b) Electrospray mass spectrum (negative mode) of the 1:1 mixture of FeIIITPPS and CuIITerPyCD2 in H2O. The inset shows the simulated isotope distribution patterns for the μ-oxo- and μ-hydroxo-bridged FeIIITPPS/CuIITerpyCD2 complexes. | |
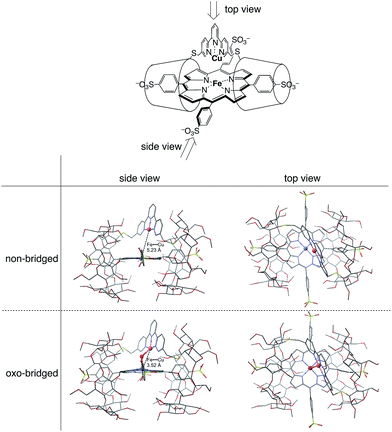 |
| | Fig. 4 Optimized molecular structures of the FeTPPS/CuTerpyCD2 inclusion complexes in the Fe/Cu non-bridged and Fe/Cu oxo-bridged forms. The models are shown from both side and top views. Hydrogen atoms are omitted for clarity. Molecular mechanics calculations were carried out using CONFLEX/MM3 (extensive search) parameters in Scigress version 2.2.1 software program (Fujitsu). | |
Characterisation of an O2 adduct of the FeII/CuI complex
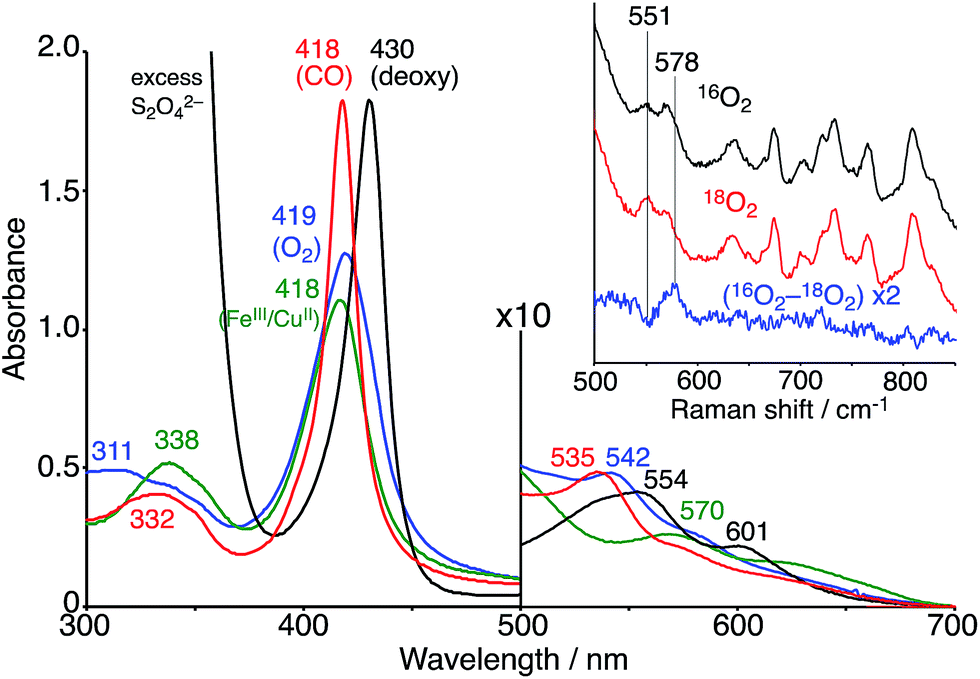
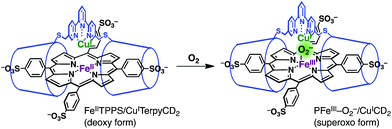
The FeIIITPPS/CuIITerpyCD2 complex was reduced with excess sodium dithionite (Na2S2O4) to obtain the fully reduced [PFeII/CuICD2]3− complex in the deoxy state in an O2-free solution (λmax at 430, 554, and 601 nm, Fig. 5, black line). The dissolved O2 in the solution was completely consumed by excess dithionite, and the redox potential of dithionite is negative enough to reduce both FeIII and CuII to FeII and CuI.29,30 After the reduction, the solution was passed through a short gel-filtration column (Sephadex G-25) under aerobic conditions to remove excess S2O42− and its oxidised products. The UV-vis spectrum of the resulting solution showed absorption maxima at 419 nm and 542 nm (Fig. 5, blue line); the Q-band was very different from that of the oxidised state (FeIIITPPS/CuIITerpyCD2, λmax (Q-band) = 570 nm, green line) and similar to that of the O2 complex of the previously reported FeIITPPS/CD dimer system.20 Introduction of CO gas into the solution caused further spectral changes with absorption maxima at 418 nm and 535 nm (Fig. 5, red line). The sharp Soret band is characteristic of the CO–FeIITPPS complex,20 indicating that a ligand exchange from O2 to CO occurs in this system.
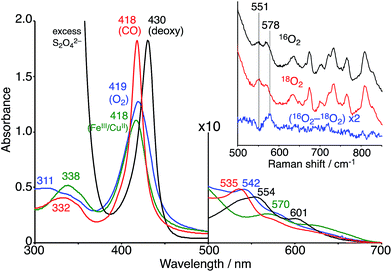 |
| | Fig. 5 UV-vis spectra of the FeIIITPPS/CuIITerpyCD2 (green) and its reduced FeIITPPS/CuITerpyCD2 complexes in the deoxy (black), oxy (blue) and CO (red) forms in 0.05 M phosphate buffer at pH 7.0 and 25 °C. The inset shows the resonance Raman spectra of the FeIITPPS/CuITerpyCD2 complexes obtained by excitation at 405 nm under 16O2 atmosphere (black), 18O2 atmosphere (red), and the difference 16O2–18O2 (blue). Conditions: 0.05 M phosphate buffer at pH 7.0, 77 K (frozen solution). | |
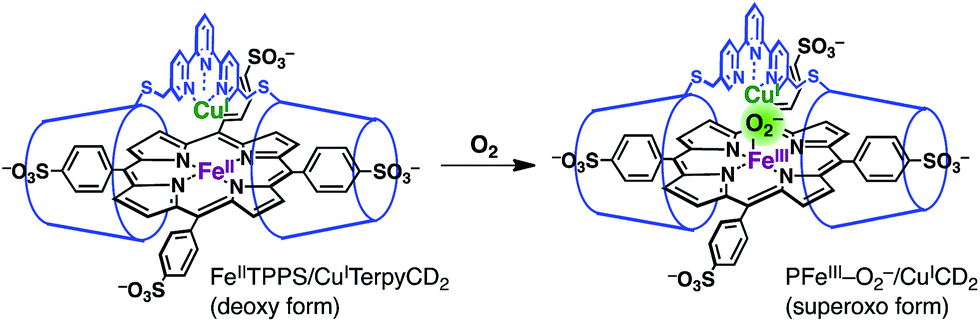
The O2 complex was further characterized by EPR and resonance Raman (rR) spectroscopic analyses. The EPR spectrum of the O2 adduct of FeIITPPS/CuITerpyCD2 measured at 77 K was completely silent (Fig. S4‡), which was consistent with the spectra of other O2 complexes of the PFeII/CuILn hetero-binuclear systems.31–33 The rR analysis at 77 K (frozen solution of the O2 adduct) using 405 nm excitation revealed a characteristic band at 578 cm−1, which shifted to 551 cm−1 under an 18O2 atmosphere (Fig. 5 inset). The isotope shift (Δν = 27 cm−1) corresponds to the expected value for the νFe–O stretching mode.15 The wavenumber is quite similar to those of the PFeIII–O2−/CuILn superoxo complexes in the previously reported native34 and synthetic model systems as listed in Table 1.14,15,35 Furthermore, the O–O bond stretching mode (νO–O) was not enhanced in this system. This is a relevant observation as the νO–O band is often observed in the range of 750–900 cm−1 in the PFeIII–O2–CuIILn μ-peroxo complexes, but not in the case of the FeIII–O2−/CuILn superoxo complexes (Table 1).14,15,35–37 Based on the rR data, the configuration of the present O2-adduct of FeIITPPS/CuITerpyCD2 is assigned as the superoxo-type PFeIII–O2−/CuIL3 complex (Fig. 6), which is the same coordination mode as in compound A of native CcO.3,14,38
Table 1 The Fe–O and O–O stretching frequencies (νFe–O/cm−1, νO–O/cm−1) in the O2 complexes of native CcO and synthetic PFe/CuLn compounds
|
|
ν
Fe–O/cm−1 16O2 (18O2) |
ν
O–O/cm−1 16O2 (18O2) |
Medium |
|
Ref. 34.
Ref. 6.
Ref. 14.
Ref. 35.
Ref. 15.
This work.
Ref. 37.
Ref. 36.
|
|
Superoxo group
|
| CcO (beef heart)a |
572 (548) |
— |
H2O, pH 7.4 |
| CcO (bovine heart)b |
571 (545) |
— |
H2O, pH 7.2 |
| Fe/Cu[NMePr]c |
570 (544) |
— |
CH2Cl2 |
| FeCuArOHd |
575 (549) |
— |
DMF |
| [(LN4-OH)Cu/Fe(TMPIm)]e |
574 (548) |
— |
CH3CN/THF |
| FeTPPS/CuTerpyCD2f |
578 (551) |
— |
H2O, pH 7.0 |
|
|
μ-Peroxo group
|
| LS-4DCHImg |
585, 591 (564) |
876, 863 (820) |
MeTHF |
| [LOHFe/Cu]h |
— |
799 (752) |
CH3CN/toluene |
| [(LN4-OH)Cu/Fe(TMPIm)]e |
611 (584) |
787, 803 (751) |
CH3CN/THF |
 |
| | Fig. 6 Oxygenation of the FeIITPPS/CuITerpyCD2 complex to form a superoxo PFeIII–O2−/CuICD2 complex. | |
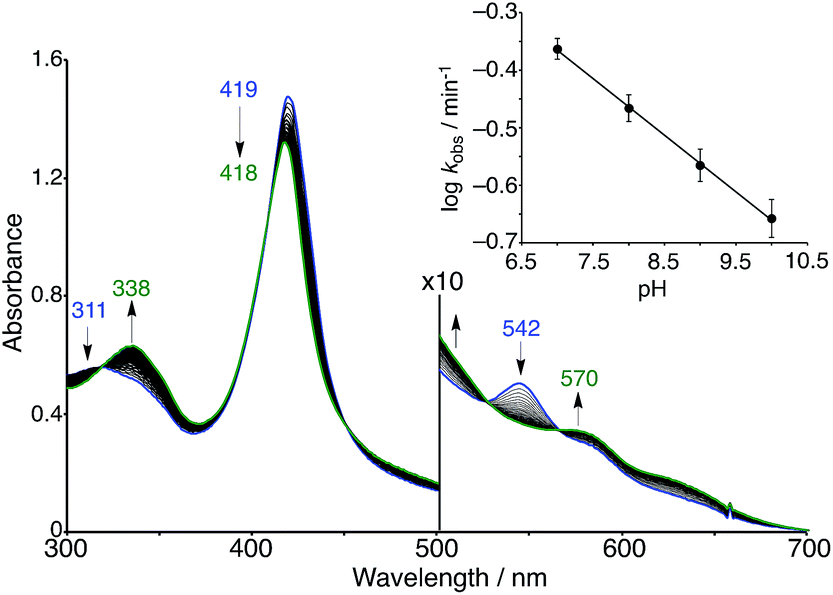
The superoxo PFeIII–O2−/CuICD2 complex was gradually converted to another state when the solution was allowed to stand at pH 7 and 25 °C under aerobic conditions (Fig. 7). The absorption spectra showed several isosbestic points and the final spectrum (shown as a green line in Fig. 7) was coincident to that of the oxidised FeIIITPPS/CuIITerpyCD2 complex (Fig. 5). EPR spectral changes also support oxidation of the superoxo PFeIII–O2−/CuICD2 species to the FeIIITPPS/CuIITerpyCD2 complex (Fig. S4‡). The first-order rate constants (kobs) for the conversion were determined from the absorbance change at various pH conditions. Interestingly, the superoxo complex was more rapidly converted at lower pH (Fig. 7 inset). The linear pH/logkobs dependency at pH 7–10 (slope = −0.11) suggests that the conversion is partially accelerated by a proton-coupled process.39 Collman et al. have reported that the rate of the O2 reduction catalysed by their PFe/CuLn model complex is pH-dependent and increases at lower pH.40 We have previously reported that the autoxidation rate of the O2 complex in the PFeII/CD dimer system without any distal functions is independent of pH in the neutral pH region (7–10), whereas it is accelerated at pH below 6 and above 10.24 Therefore, the pH-rate dependency at the neutral pH region suggests that the water molecules gathered at the distal Cu site promote the conversion of the PFeIII–O2−/CuICD2 complex to the oxidised PFeIII–(OH)–CuIICD2 complex.
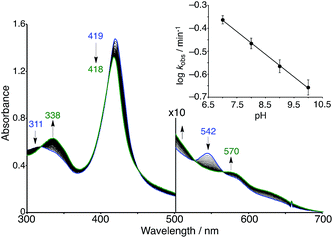 |
| | Fig. 7 Spontaneous conversion of the superoxo (PFeIII–O2−/CuICD2) complex in 0.05 M phosphate buffer at pH 7.0 and 25 °C. The spectrum was recorded at 15 s intervals. The inset shows the logarithmic first-order rate constants (kobs) for the conversion as a function of the pH of the solution. | |
The quantum chemical study on native CcO8 proposes that a water molecule coordinating to the distal copper ion facilitates the conversion of compound A to compound P through the formation of the hydroperoxo FeIII–OOH intermediate that has not been experimentally detected. Thus, the involvement of a water molecule in the present PFeIII–O2−/CuICD2 complex is likely to occur. In addition, molecular modelling suggests that a water molecule bound to the distal copper ion can induce protonation of the superoxo complex (Fig. 8a), where the methoxy groups of the CD dimer are suitable to provide two hydrogen bonding sites to the water. The pH-dependent decomposition of the superoxo complex, as shown in Fig. 7, might be explained by the acid–base equilibrium of the water molecule (Fig. 8b), where the proton-donation to the superoxo complex is likely to induce the O–O bond cleavage as proposed in CcO8 and/or the proton-assisted autoxidation reaction similar to myoglobin.41,42
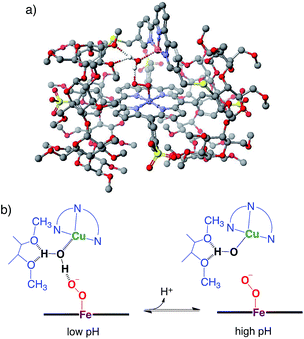 |
| | Fig. 8 The superoxo PFeIII–O2−/CuICD2 complex with a water molecule. (a) The molecular model constructed using CONFLEX/MM3 (extensive search) parameters in Scigress version 2.2.1 software program (Fujitsu). Hydrogen atoms, except for water, are omitted for clarity. (b) The possible acid–base equilibrium of the water, where the proton-donation to the superoxo PFeIII–O2− moiety is likely to occur at low pH. | |
The O2 binding in the present complex was practically irreversible; the O2 complex of FeIITPPS/CuITerpyCD2 was never converted to its FeII/CuI deoxy complex, even when the O2 complex once formed was dissolved in a deoxygenated buffer (Fig. S5‡). In contrast, the deoxy complex was observed in the FeIITPPS/TerpyCD2 complex without copper under the same experimental conditions.43 This result indicates that the O2 bound to PFeII is tightly held by the distal CuIL3 complex, as previously demonstrated by the Fe/Cu superoxo complex.14 The tight O2 binding was also confirmed by observing ligand exchange with CO. The ligand exchange occurred slowly over ∼30 min when the Fe/Cu superoxo complex was dissolved in a CO saturated buffer (Fig. S5‡), whereas it occurred instantaneously in the absence of distal Cu complex or in the absence of O2 (Fig. S5‡). The ligand exchange of O2 with CO also rapidly occurs in the previous FeIITPPS/CD dimer systems.20,24 The significantly slow ligand exchange of PFeII–O2−/CuIL3 with CO caused by distal Cu complex might be related to the lower CO/O2 affinity ratio of native CcO (0.1) in comparison to that of myoglobin (20–50) or haemoglobin (200–250).44
Electrochemical analysis for the O2 reduction
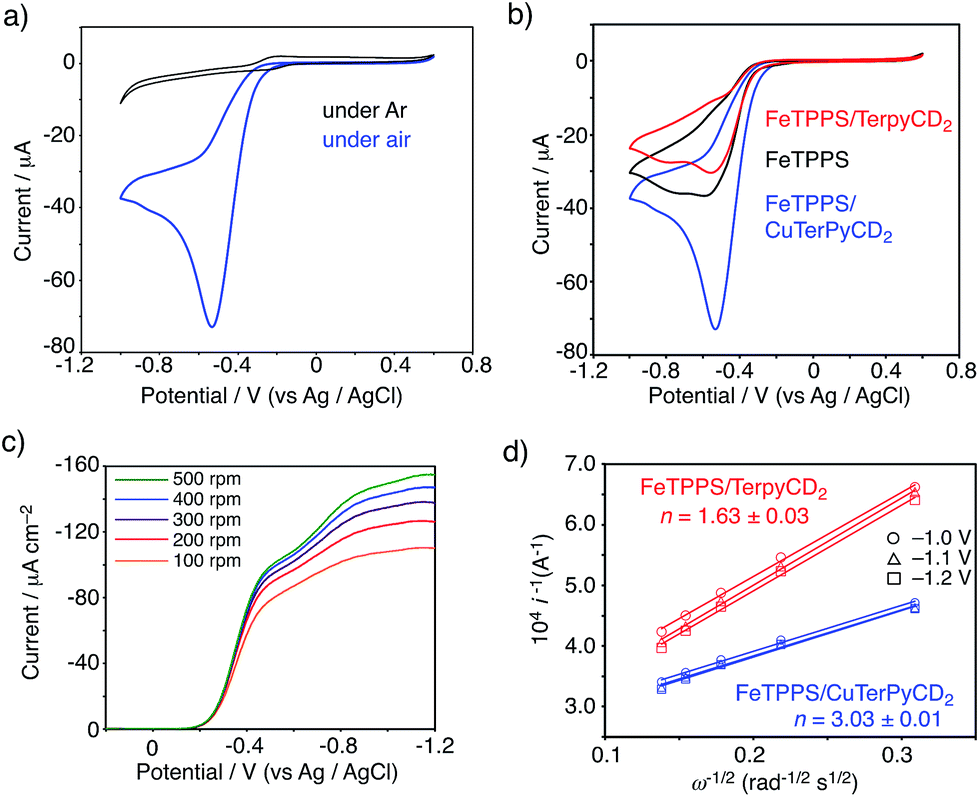
To evaluate the CcO-like function of this system, we monitored the electrocatalytic O2 reduction reaction.45–47 The cyclic voltammogram (CV) of the FeIIITPPS/CuIITerpyCD2 complex immobilized on a glassy carbon electrode showed a reversible redox couple at E1/2 = −0.21 V (vs. Ag/AgCl) in a deoxygenated buffer solution (under Ar, Fig. 9a, black line). The result is similar to those of the previously reported PFe/CuLn hetero-binuclear systems; the FeIII/FeII and CuII/CuI redox waves appear at the same potentials.31,46 In an air-saturated buffer, the CV of the FeIIITPPS/CuIITerpyCD2 complex showed a large catalytic current below −0.25 V because of O2 reduction (Fig. 9a, blue line). A comparison of the CVs of the FeIIITPPS/CuIITerpyCD2 complex with those of the reference samples, i.e., FeIIITPPS and FeIIITPPS/TerpyCD2 (Fig. 9b), clearly indicates the effect of the Fe/Cu hetero-binuclear structure in the O2 reduction; the FeIIITPPS/CuIITerpyCD2 complex showed a very large catalytic current starting from a lower onset potential (ΔEonset = −40 mV). The O2 reduction process was then studied by linear sweep voltammetry (LSV) using a rotating disk electrode (RDE, Fig. 9c). The LSVs of the FeIIITPPS/CuIITerpyCD2 and FeIIITPPS/TerpyCD2 complexes showed diffusion limited catalytic O2-reduction currents below −1.0 V vs. Ag/AgCl. In the case of FeTPPS without the CD dimer, the current was never saturated in LSV due to a slow reaction rate of the iron porphyrin with O2 on the disk electrode (Fig. S6‡). The saturated currents observed in the FeIIITPPS/CuIITerpyCD2 and FeIIITPPS/TerpyCD2 complexes at various rotation rates were analysed using the Koutecky–Levich equation to determine the average number of electrons (n) used in the O2 reduction (Fig. 9d).48 A significant increase in the n value was observed for the Fe/Cu hetero-binuclear complex (n = 3.03 ± 0.01) compared to the control sample without copper (n = 1.63 ± 0.03).49 Therefore, we conclude that the terpyridyl Cu complex associated with FeTPPS in our model system facilitates the catalytic O2 reduction as an electron source, as proposed in the mechanism of native CcO3 and as proven using the synthetic model systems.5,45,48
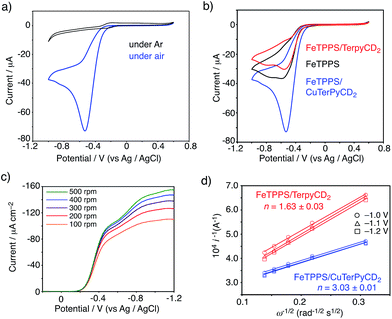 |
| | Fig. 9 (a, b) CV of the FeTPPS/CuTerpyCD2 complex and its reference samples absorbed on the glassy carbon electrode with Nafion (5 wt% dispersion, 10 μL) in pH 7 phosphate buffer at a scan rate of 0.1 V s−1 using Ag/AgCl and Pt wire as the reference counter electrodes, respectively. (c) LSV data for the FeTPPS/CuTerpyCD2 complex (10 nmol) coated with Nafion (5 wt% dispersion, 10 μL) on a glassy carbon electrode in air saturated pH 7.0 phosphate buffer at a scan rate of 10 mV s−1 at multiple rotations using Ag/AgCl and a Pt wire as the reference and counter electrodes, respectively. (d) Koutecky–Levich plots for the FeTPPS/CuTerpyCD2 and FeTPPS/TerpyCD2 complexes at the potentials of −1.0, −1.1 and −1.2 V to determine the average number of electrons (n) used for the O2 reduction reaction. | |
Conclusions
In conclusion, we have synthesized a water-soluble biomimetic model complex for the heme a3/CuB hetero-binuclear active centre of CcO by utilizing a supramolecular complexation, and characterised its reactivity with O2. To the best of our knowledge, this is the first example of a totally synthetic CcO model that works in a completely aqueous solution. In common with compound A of native CcO, we have identified the PFeIII–O2−/CuICD2 superoxo complex as the O2 adduct in our model system in aqueous solution, whereas the PFeIII–O2–CuIILn μ-peroxo complexes tend to form in the other synthetic model systems in anhydrous organic solvents. The pH-dependent conversion of the PFeIII–O2−/CuICD2 superoxo complex to its oxidised μ-hydroxo PFeIII–(OH)–CuIICD2 complex suggested the involvement of water molecules in the formation of the superoxo complex in aqueous solution. We believe that our aqueous model system will help to clarify the long-standing arguments with regard to the native and synthetic model systems in CcO chemistry.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We thank (late) Prof. Takashi Ogura (University of Hyogo) for the use of rR instruments and helpful discussion. This work was financially supported by MEXT/JSPS KAKENHI (Grant No. 15H02569, 16K13092, 17H02208), the MEXT-Supported Program for the Strategic Research Foundation at Private Universities (2015–2019), the Naito Foundation, Iketani Science and Technology Foundation, and Suntory Foundation for Life Sciences. JW and TH thank financial support from the bilateral France–Japan ANR-JST program TMOL“Molecular Technology” project “MECANO” AI\R.1 4.JTIC.OOO2.O 1.
Notes and references
- G. T. Babcock, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12971–12973 CrossRef CAS.
- S. Ferguson-Miller and G. T. Babcock, Chem. Rev., 1996, 96, 2889–2907 CrossRef CAS PubMed.
- S. Yoshizawa and A. Shimada, Chem. Rev., 2015, 115, 1936–1989 CrossRef PubMed.
- E. Kim, E. E. Chufán, K. Kamaraj and K. D. Karlin, Chem. Rev., 2004, 104, 1077–1133 CrossRef CAS PubMed.
- J. P. Collman, R. Boulatov, C. J. Sunderland and L. Fu, Chem. Rev., 2004, 104, 561–588 CrossRef CAS PubMed.
- T. Ogura, S. Hirota, D. A. Proshlyakov, K. Shinzawa-Itoh, S. Yoshikawa and T. Kitagawa, J. Am. Chem. Soc., 1996, 118, 5443–5449 CrossRef CAS.
- K. Muramoto, K. Ohta, K. Shinzawa-Itoh, K. Kanda, M. Taniguchi, H. Nabekura, E. Yamashita, T. Tsukihara and S. Yoshikawa, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7740–7745 CrossRef CAS PubMed.
- M. R. A. Blomberg, P. E. M. Siegbahn, G. T. Babcock and M. Wilkström, J. Am. Chem. Soc., 2000, 122, 12848–12858 CrossRef CAS.
- E. Kim, J. Shearer, S. Lu, P. Moënne-Loccoz, M. E. Helton, S. Kaderli, A. D. Zuberbühler and K. D. Karlin, J. Am. Chem. Soc., 2004, 126, 12716–12717 CrossRef CAS PubMed.
- T. Chishiro, Y. Shimazaki, F. Tani, Y. Tachi, Y. Naruta, S. Karasawa, S. Hayami and Y. Maeda, Angew. Chem., Int. Ed., 2003, 42, 2788–2791 CrossRef CAS PubMed.
- E. E. Chufán, S. C. Puiu and K. D. Karlin, Acc. Chem. Res., 2007, 40, 563–572 CrossRef PubMed.
- S. Hematian, I. Garcia-Bosch and K. D. Karlin, Acc. Chem. Res., 2015, 48, 2462–2474 CrossRef CAS PubMed.
- A. W. Schaefer, M. T. Kieber-Emmons, S. M. Adam, K. D. Karlin and E. I. Solomon, J. Am. Chem. Soc., 2017, 139, 7958–7973 CrossRef CAS PubMed.
- J. P. Collman, C. J. Sunderland, K. E. Berg, M. A. Vance and E. I. Solomon, J. Am. Chem. Soc., 2003, 125, 6648–6649 CrossRef CAS PubMed.
- J.-G. Liu, Y. Naruta and F. Tani, Angew. Chem., Int. Ed., 2005, 44, 1836–1840 CrossRef CAS PubMed.
- M. Ralle, M. L. Verkhovskaya, J. E. Morgan, M. I. Verkhovsky, M. Wilkström and N. J. Blackburn, Biochemisty, 1999, 38, 7185–7194 CrossRef CAS PubMed.
- J. A. Sigman, B. C. Kwok and Y. Lu, J. Am. Chem. Soc., 2000, 122, 8192–8196 CrossRef CAS.
- A. Bhagi-Damodaran, M. A. Michael, Q. Zhu, J. Reed, B. A. Sandoval, E. N. Mirts, S. Chakraborty, P. Moënne-Loccoz, Y. Zhang and Y. Lu, Nat. Chem., 2017, 9, 257–263 CrossRef CAS PubMed.
- K. Kano, H. Kitagishi, S. Tamura and A. Yamada, J. Am. Chem. Soc., 2004, 126, 15202–15210 CrossRef CAS PubMed.
- K. Kano, H. Kitagishi, M. Kodera and S. Hirota, Angew. Chem., Int. Ed., 2005, 44, 435–438 CrossRef CAS PubMed.
- H. Kitagishi, M. Tamaki, T. Ueda, S. Hirota, T. Ohta, Y. Naruta and K. Kano, J. Am. Chem. Soc., 2010, 132, 16730–16732 CrossRef CAS PubMed.
- K. Watanabe, H. Kitagishi and K. Kano, Angew. Chem., Int. Ed., 2013, 52, 6894–6897 CrossRef CAS PubMed.
- H. Kitagishi, S. Kurosawa and K. Kano, Chem.–Asian J., 2016, 11, 3213–3219 CrossRef CAS PubMed.
- K. Kano, H. Kitagishi, C. Dagallier, M. Kodera, T. Matsuo, T. Hayashi, Y. Hisaeda and S. Hirota, Inorg. Chem., 2006, 45, 4448–4460 CrossRef CAS PubMed.
- V. M. Manikandamathavan, V. Rajapandian, A. J. Freddy, T. Weyhermüller, V. Subramanian and B. U. Nair, Eur. J. Med. Chem., 2012, 57, 449–458 CrossRef CAS PubMed.
- M.-A. Kopf, Y.-M. Neuhold, A. D. Zuberbühler and K. D. Karlin, Inorg. Chem., 1999, 38, 3093–3102 CrossRef CAS.
- E. Kim, M. E. Helton, I. M. Wasser, K. D. Karlin, S. Lu, H.-w. Huang, P. Moënne-Loccoz, C. D. Incarvito, A. L. Rheingold, M. Honecker, S. Kaderli and A. D. Züberbuhler, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3623–3628 CrossRef CAS PubMed.
- S. Fox, A. Nanthakumar, M. Wilkström, K. D. Karlin and N. J. Blackburn, J. Am. Chem. Soc., 1996, 118, 24–34 CrossRef CAS.
- S. G. Mayhew, Eur. J. Biochem., 1978, 85, 535–547 CrossRef CAS PubMed.
- B. Liu, Y. Chen, T. Doukov, S. M. Soltis, C. D. Stout and J. A. Fee, Biochemistry, 2009, 48, 820–826 CrossRef CAS PubMed.
- Z. Halime, H. Kotani, Y. Li, S. Fukuzumi and K. D. Karlin, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13990–13994 CrossRef CAS PubMed.
- P. Vorburger, M. Lo, S. Choua, M. Bernard, F. Melin, N. Oueslati, C. Boudon, M. Elhabiri, J. A. Wytko, P. Hellwig and J. Weiss, Inorg. Chim. Acta, 2017, 468, 232–238 CrossRef CAS.
- C. Kahlfuss, J. A. Wytko and J. Weiss, ChemPlusChem, 2017, 4, 584–594 CrossRef.
- C. Varotsis, W. H. Woodruff and G. T. Babcock, J. Biol. Chem., 1990, 265, 11131–11136 CAS.
- J. P. Collman, R. A. Decréau, Y. Yan, J. Yoon and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 5794–5795 CrossRef CAS PubMed.
- J.-G. Liu, Y. Naruta, F. Tani, T. Chishiro and Y. Tachi, Chem. Commun., 2004, 120–121 RSC.
- S. M. Adam, I. Garcia-Bosch, A. W. Schaefer, S. K. Sharma, M. A. Siegler, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2017, 139, 472–481 CrossRef CAS PubMed.
- The νFe–O stretching frequency of 578 cm−1 is higher than the reported values for the penta-coordinated PFeIII–O2− complexes, indicating that the Lewis acidity of the distal Cu complex significantly alters the Fe–O bonding. For the νFe–O data of PFeIII–O2− complexes, see: K. M. Vogel, P. M. Kozlowski, M. Z. Zgierski and T. G. Spiro, J. Am. Chem. Soc., 1999, 121, 9915–9921 CrossRef CAS.
- If this conversion would have been completely conjugated with the proton transfer, the slope of the pH/logkobs plot would be −1.0.
- J. P. Collman, S. Ghosh, A. Dey, R. A. Decréau and Y. Yang, J. Am. Chem. Soc., 2009, 131, 5034–5035 CrossRef CAS PubMed.
- K. Shikama, Chem. Rev., 1998, 98, 1357–1373 CrossRef CAS PubMed.
- In the present system, any intermediate species ascribed to ferryloxo (FeIVO) complex could not be observed during conversion from the superoxo to the oxidised complexes in aqueous solution at room temperature.
- The O2 complex was also formed in the FeIITPPS/TerpyCD2 complex without copper. The UV-vis spectrum of this complex (λmax = 419 and 542 nm) was found to be consistent with that of the PFeIII–O2−/CuICD2 superoxo complex, which further supports the absence of a direct interaction between the distal CuI and the bound O2 in the PFeIII–O2−/CuICD2 complex.
- R. Motterlini and R. Foresti, Am. J. Physiol.: Cell Physiol., 2017, 312, C302–C313 CrossRef PubMed.
- J. P. Collman, N. K. Devaraj, R. A. Decréau, Y. Yang, Y.-L. Yan, W. Ebina, T. A. Eberspacher and C. E. D. Chidsey, Science, 2007, 315, 1565–1568 CrossRef CAS PubMed.
- R. Boulatov, J. P. Collman, I. M. Shiryaeva and C. J. Sunderland, J. Am. Chem. Soc., 2002, 124, 11923–11935 CrossRef CAS PubMed.
- F. Melin, A. Trivella, M. Lo, C. Ruzié, I. Hijazi, N. Oueslati, J. A. Wytko, B. Boitrel, C. Boudon, P. Hellwig and J. Weiss, J. Inorg. Biochem., 2012, 108, 196–202 CrossRef CAS PubMed.
- S. Chatterjee, K. Sengupta, S. Hematian, K. D. Karlin and A. Dey, J. Am. Chem. Soc., 2015, 137, 12897–12905 CrossRef CAS PubMed.
- The selectivity of the electrocatalytic O2 reduction by the FeTPPS/CuTerpyCD2 and its copper free complexes could not be determined using a rotating ring disk electrode system because of very low currents detected at the ring electrode.
Footnotes |
| † This paper is dedicated to (late) Professor Takashi Ogura. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04732k |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Daiki
Shimoji
a,
Takehiro
Ohta
*a,
Daiki
Shimoji
a,
Takehiro
Ohta