
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA luminescent organic radical with two pyridyl groups: high photostability and dual stimuli-responsive properties, with theoretical analyses of photophysical processes†
Shun
Kimura
a,
Akira
Tanushi
a,
Tetsuro
Kusamoto
 *a,
Shuntaro
Kochi
b,
Tohru
Sato
bc and
Hiroshi
Nishihara
*a
*a,
Shuntaro
Kochi
b,
Tohru
Sato
bc and
Hiroshi
Nishihara
*a
aDepartment of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: kusamoto@chem.s.u-tokyo.ac.jp; nisihara@chem.s.u-tokyo.ac.jp
bDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto, 615-8510, Japan
cUnit of Elements Strategy Initiative for Catalysts & Batteries, Kyoto University, Nishikyo-ku, Kyoto, 615-8510, Japan
First published on 24th January 2018
Abstract
Luminescent monoradicals are expected to show unique properties based on their doublet state, where establishing a method to improve their photostability is an important issue for expanding their photofunctionality. We synthesized a highly photostable luminescent organic radical, the bis(3,5-dichloro-4-pyridyl)(2,4,6-trichlorophenyl)methyl radical (bisPyTM), containing two pyridyl groups on a tris(2,4,6-trichlorophenyl)methyl radical (TTM) skeleton. bisPyTM in dichloromethane exhibited fluorescence with an emission peak wavelength, λem, of 650 nm. We visually detected an emission (λem = 712 nm) from crystalline bisPyTM at 77 K, which is the first example of definite solid-state emission in a radical. Introducing the two nitrogen atoms into the TTM skeleton was shown to lower the energies of the frontier orbitals. The oscillator strength, f, of the electronic transition between the lowest excited state and the ground state, and the off-diagonal vibronic coupling constants (VCCs) were calculated theoretically for bisPyTM and the (3,5-dichloro-4-pyridyl)bis(2,4,6-trichlorophenyl)methyl radical (PyBTM). The calculated PyBTM to bisPyTM ratios for f or VCC agreed well with experimental radiative and non-radiative rate constants (kr and knr) ratios, respectively. This study shows that scaled kr and knr can be estimated and compared in this class of radicals using theoretical calculations, greatly advancing the prediction and design of their photofunctionality. The half-life of bisPyTM upon continuous UV light irradiation in dichloromethane was 47 or 3000 times longer those that of PyBTM (which contains one pyridyl group) and TTM (which has no pyridyl rings), respectively. The electrochemical and luminescent properties of bisPyTM were modulated in two stages using protons or B(C6F5)3.
Introduction
Luminescent molecules are attracting increasing attention because of their suitability for a wide variety of scientific and engineering applications, such as organic light-emitting diodes and chemosensors. Several emission mechanisms, such as fluorescence via the singlet excited state,1 phosphorescence from the triplet excited state,2 and thermally activated delayed fluorescence,3,4 have been used to obtain the desired device properties. In contrast with conventional luminescent molecules, which are closed-shell with singlet ground states, radicals with an unpaired electron have been the focus of research into next-generation luminophores.5–12 Radicals, in which fluorescence occurs from the lowest doublet excited state (D1) to the doublet ground state (D0), can exhibit unique characteristics, such as having long emission wavelengths without extended π-conjugation.13 Of particular importance in electroluminescent devices is the high efficiency of the internal electron (hole) → photon conversion ratio, up to 100%,14,15 which has been experimentally suggested recently.16 Although closed-shell fluorescent emitters suffer from quenching via the lowest triplet excited state (T1), which is statistically formed together with the lowest singlet excited state (S1) in a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, particular monoradicals do not suffer from this quenching problem because of the absence of excited states between D1 and D0 and the spin-allowed D1 → D0 transition.14
:1, particular monoradicals do not suffer from this quenching problem because of the absence of excited states between D1 and D0 and the spin-allowed D1 → D0 transition.14
The doublet luminescence of radicals has been investigated much less than that of closed-shell luminescent molecules, because of its rarity and low chemical stability under ambient conditions or in photoexcited states.17,18 Following studies on the emission properties of in situ-generated unstable diarylmethyl or triarylmethyl radicals,19–22 polychlorinated triarylmethyl (PTM) and tris(2,4,6-trichlorophenyl)methyl (TTM) radicals (Fig. 1) have been developed as luminescent radicals.23,24 They show excellent chemical stability under ambient conditions, although they decompose upon photoirradiation.17,18 Improving the photostability of radicals is an important step toward developing unique doublet-based photofunctionality or novel phenomena arising from the interplay between luminescent and magnetic (spin) properties.
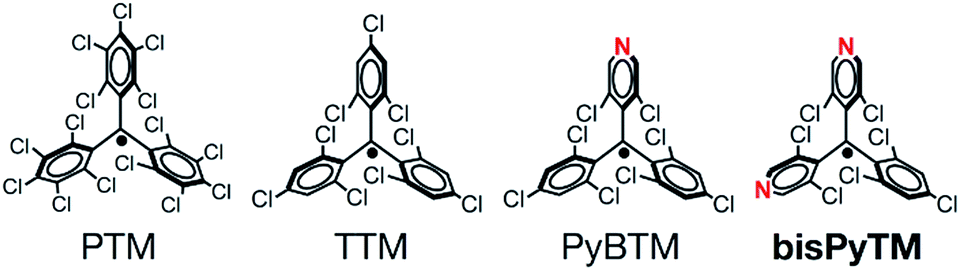 | ||
| Fig. 1 The molecular structures of PTM, TTM, PyBTM and bisPyTM. | ||
A drastic increase in the photostability of luminescent radicals has been achieved in the (3,5-dichloro-4-pyridyl)bis(2,4,6-trichlorophenyl)methyl radical (PyBTM; Fig. 1), prepared by our group.14 PyBTM has a pyridyl ring in the TTM skeleton and showed fluorescence with an absolute quantum yield (ϕem) of 0.01–0.03 in solution and a ϕem value of 0.26 in a rigid matrix at room temperature. Importantly, its stability upon UV irradiation was up to 115 times higher than that of TTM. The excellent photostability has expanded the scope of doublet-based photofunctionality in radicals. We have reported on stimuli responsiveness,25 the halogen atom substitution effect,26 and increased ϕem and photostability upon coordination to metal ions in PyBTM.27–29 Studies on PyBTM have suggested that introducing a pyridyl group may lead to radicals with improved photostability, owing to the lowering of energy levels around the frontier orbitals. However, the utility of this method is not well established.
In this study, we developed a new bis(3,5-dichloro-4-pyridyl)(2,4,6-trichlorophenyl)methyl radical (bisPyTM; Fig. 1), which has two pyridyl groups, and is stable and luminescent. We aimed to resolve two issues in understanding the fundamental properties of luminescent triarylmethyl radicals. The first is the effects of introducing an additional pyridyl ring into the PyBTM skeleton on the energies of the frontier orbitals, luminescent properties, and photostability. The increase in the number of pyridyl rings is expected to increase the photostability and to expand the variation of the coordination structure in relation to multistage chemical responses or high-dimensional assemblies.30,31
The second challenge is interpreting the luminescence characteristics and photophysical parameters, including the radiative and non-radiative rate constants (kr and knr), through theoretical calculations using density functional theory (DFT) and time-dependent (TD)-DFT. The technical challenges and limited number of compounds mean that joint theoretical and experimental studies on the relaxation processes of luminescent radicals in their excited state are still rare. However, conical intersection32 and back-electron transfer8 have been investigated theoretically as non-radiative relaxation processes of radicals in the excited state. We calculated the excited states and molecular and electronic structures of PyBTM and bisPyTM, and investigated non-radiative relaxation processes via vibronic coupling density (VCD) analysis33 to evaluate the rate of internal conversion. The differences in kr and knr between the two radicals can be explained through the difference in oscillator strengths and off-diagonal vibronic coupling constants (VCCs).
Results and discussion
Theoretical methods for estimating photophysical parameters
The relaxation process from the D1 state to the D0 state consists of radiative and non-radiative transitions (Scheme 1). Luminescence corresponds to the radiative D1 → D0 transition; thus, kr is proportional to the oscillator strength, f, of the transition for the D1 nuclear coordinates. The f value is calculated using TD-DFT in this study. In contrast, the non-radiative transition is expected to consist of internal conversion followed by vibrational relaxation in the radicals studied here (Scheme 1). Intersystem crossing from S1 to Tn states can also provide a non-radiative relaxation pathway in conventional closed-shell luminescent molecules. However, intersystem crossing from D1 to other excited states with different multiplicities, such as a quartet state (Q1), is negligible, because the Q1 state is energetically much higher than the D1 state.14 Thus, knr is determined mainly by the rate of internal conversion. Internal conversion has been suggested as a major pathway in the non-radiative decay of some radicals,21,28,34 however, no theoretical calculations have been performed to clarify the deactivation mechanism. In this study, VCD analysis of the luminescent radicals elucidated the mechanism of their non-radiative decay. This was suitable for our radicals, because it allowed us to evaluate the rate of internal conversion and to determine the vibrational modes that contribute to internal conversion. Internal conversion is caused by off-diagonal vibronic coupling. The rate of internal conversion is proportional to the square of the off-diagonal VCCs.33 The off-diagonal VCC of mode α between electronic states m and n is defined by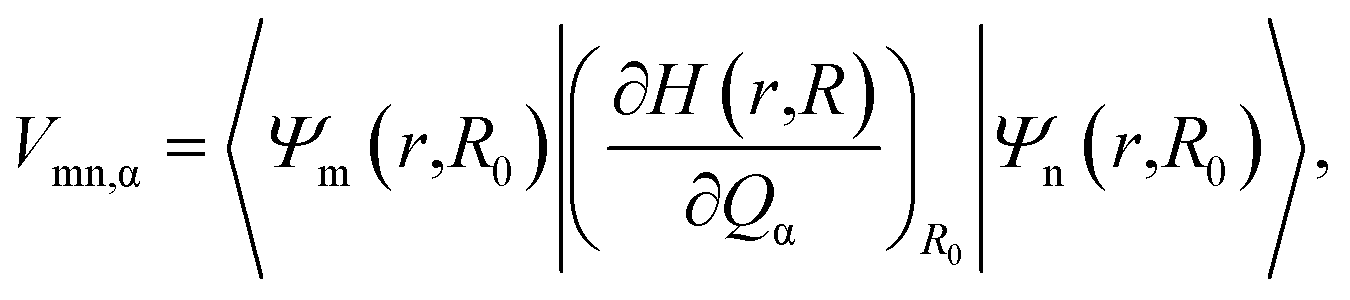 | (1) |
 | ||
| Scheme 1 Monoradical deactivation processes from the lowest excited (D1) state. | ||
The origin of a vibronic coupling in terms of the electronic and vibrational states can be visualized using VCD analysis,34 which provides insight into the vibronic coupling in a molecule. Furthermore, we can control vibronic couplings to suppress internal conversion. For instance, based on the VCD analysis of a non-emitting molecule, triphenylamine (TPA), Sato et al. reduced the off-diagonal VCCs by introducing non-emitting substituents to make the TPA moiety fluorescent.35,36 The VCD is defined by
| ηmn,α(ri) = ρmn(ri) × να(ri), | (2) |
 | (3) |
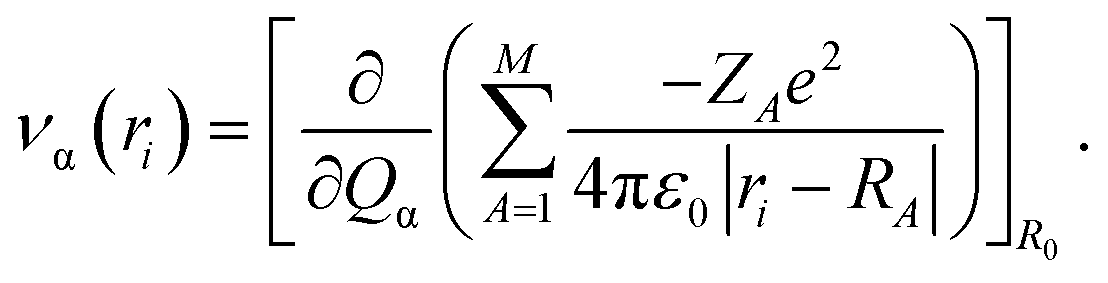 | (4) |
The spatial integral of the VCD is equal to the VCC:
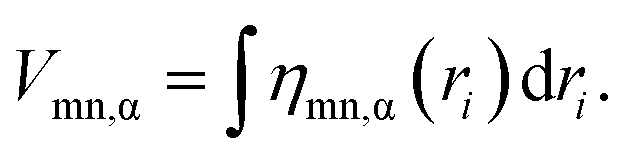 | (5) |
Using eqn (2) and (5), the origin of a VCC can be described using functions of η, ρ, and ν, which depend on position.
Synthesis and characterization
bisPyTM was prepared according to Scheme 2, and characterized via electron spin resonance (ESR) spectroscopy, high-resolution electrospray ionization mass spectrometry, and elemental analysis. The S = 1/2 spin per bisPyTM molecule was confirmed by comparing the value of the second integral of the ESR signal intensity with that of the reference compound, 4-hydroxy-TEMPO. The molecular structure of bisPyTM in the crystalline state was analyzed via single-crystal X-ray diffraction (Fig. 2). The central carbon atom, C1, is sp2 hybridized and sterically shielded by six halogen atoms. C1 and its three neighboring carbon atoms (C4, C9, and C15) lie in the same plane. The three aromatic rings are held in a propeller-like conformation due to steric hindrance from the chlorine atoms. The bond angles and dihedral angles (Table S1†) are similar to those of PyBTM.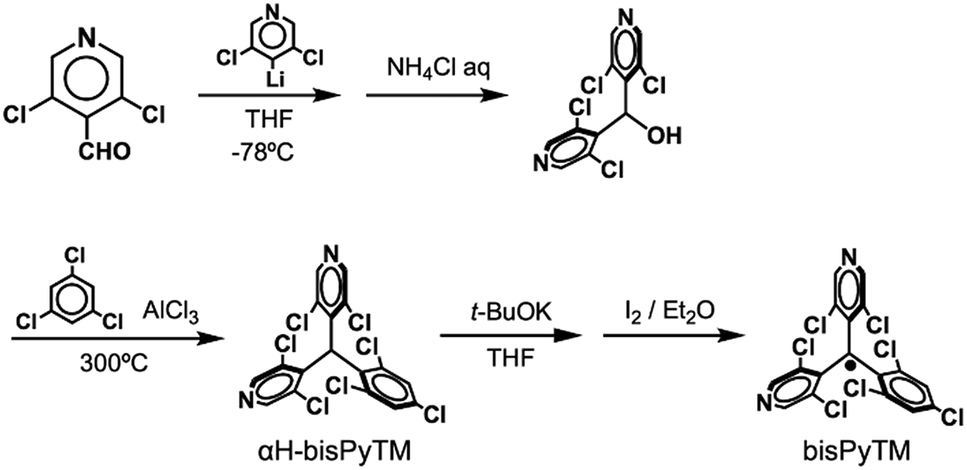 | ||
| Scheme 2 The synthetic route for bisPyTM. | ||
 | ||
| Fig. 2 (a) The molecular structure of bisPyTM in the crystalline state with thermal ellipsoids set at 50% probability, and (b) the view along the C1–C4 bond. | ||
ESR spectroscopy at 177 K
An ESR spectrum of bisPyTM in dichloromethane was measured at 177 K to estimate the spin density distribution over the bisPyTM skeleton (Fig. 3a). The spectrum showed a characteristic hyperfine structure with a g value of 2.004, similar to that of PyBTM. The spectrum was reproduced via computational simulation, considering hyperfine coupling with six 1H and two 14N atoms. The obtained hyperfine coupling constants (hccs) were qualitatively similar to those calculated via DFT (Table S2†). The results indicate that the spin density was delocalized over the molecule, including the central C1 atom and two pyridyl nitrogen atoms (Fig. 3b). The simulated hcc of 14N (0.108 mT) was comparable to that of PyBTM (0.115 mT). This result indicates that the spin density on the N atom was similar in bisPyTM and PyBTM;14 the densities estimated from the simulated hcc and DFT were 0.06 and 0.069, respectively.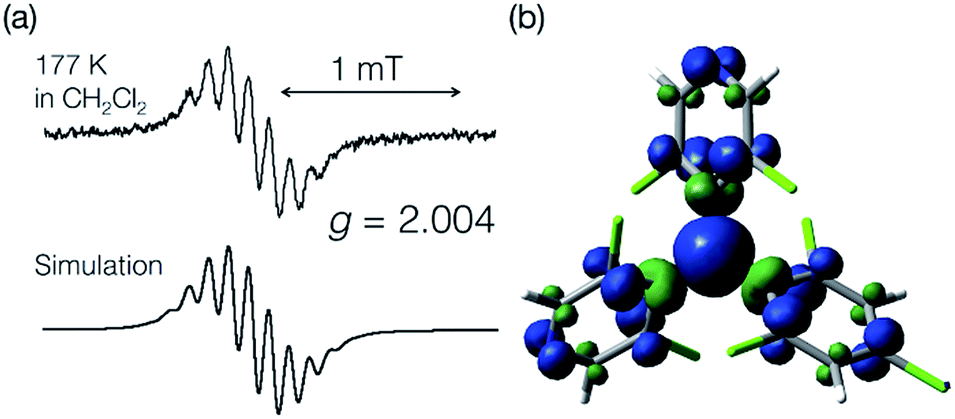 | ||
| Fig. 3 (a) An ESR spectrum of bisPyTM in CH2Cl2 at 177 K (top), and a computational simulation (bottom). (b) The spin density distribution on bisPyTM calculated via DFT (UB3LYP/6-31G(d,p)). | ||
Electronic structure in the ground state (D0 state)
The electronic properties of bisPyTM were calculated via DFT, to reveal the effects of introducing two pyridyl rings to the TTM framework on the distribution and energies around the frontier orbitals. The unrestricted B3LYP level of theory with 6-31G(d,p) basis sets was used, and the solvent effect (dichloromethane) was treated using the polarizable continuum model. The calculated molecular orbitals (MOs) of bisPyTM are shown in Fig. 4. The next highest occupied molecular orbital (NHOMO; 120α and β) and NHOMO−1 (119α and β) were assumed to be the bonding and antibonding combination of wavefunctions at the two pyridyl rings, which were distributed mainly on the two pyridyl rings. These two MOs were almost degenerate. The next lowest unoccupied molecular orbital (NLUMO; 122α and β) of bisPyTM was delocalized on the two pyridyl rings, whereas the singly occupied molecular orbital (SOMO; 121α and β) was located mainly on the central carbon atom with non-negligible delocalization on the three aromatic rings. The distribution of the MOs was similar to that in PyBTM, where the NHOMO and NLUMO were distributed mainly on the pyridyl ring, and the SOMO was located on the central carbon atom and extended to the aromatic rings. | ||
| Fig. 4 The molecular orbitals of bisPyTM and PyBTM in the D0 and D1 states, calculated via DFT (UB3LYP/6-31G(d,p)). The solvent (dichloromethane) effect is treated using the polarizable continuum model. | ||
The energy diagram shows that the energies of the frontier orbitals (119α–122α and 119β–122β) of the radicals all decreased as the number of pyridyl rings increased; bisPyTM had energies 0.10–0.20 eV lower than PyBTM. This decrease is expected to change the optical and redox properties and stabilities of the compounds, because the frontier orbital energies directly or indirectly determine the optical and redox properties.25 The lowering of the energy of the β-SOMO (121β for bisPyTM and 121β for PyBTM) was confirmed via cyclic voltammetry.
Cyclic voltammetry
The redox properties of the radicals were investigated via cyclic voltammetry. The cyclic voltammogram of bisPyTM in 0.1 M nBu4NClO4/CH2Cl2 (Fig. 5a and S1†) showed a reversible reduction wave at E0′ = −0.57 V vs. the ferrocenium/ferrocene (Fc+/Fc) redox couple, corresponding to the reduction of bisPyTM (Scheme 3a). This process was explained through the injection of an electron into the β-SOMO. The E0′ value of bisPyTM was more positive than those of PyBTM (−0.74 V) and TTM (−0.99 V) under identical conditions, suggesting that increasing the number of pyridyl rings increases the electron-accepting ability of the radical and decreases the energy level of the β-SOMO. This observation can be explained by the higher electronegativity of nitrogen compared to carbon.14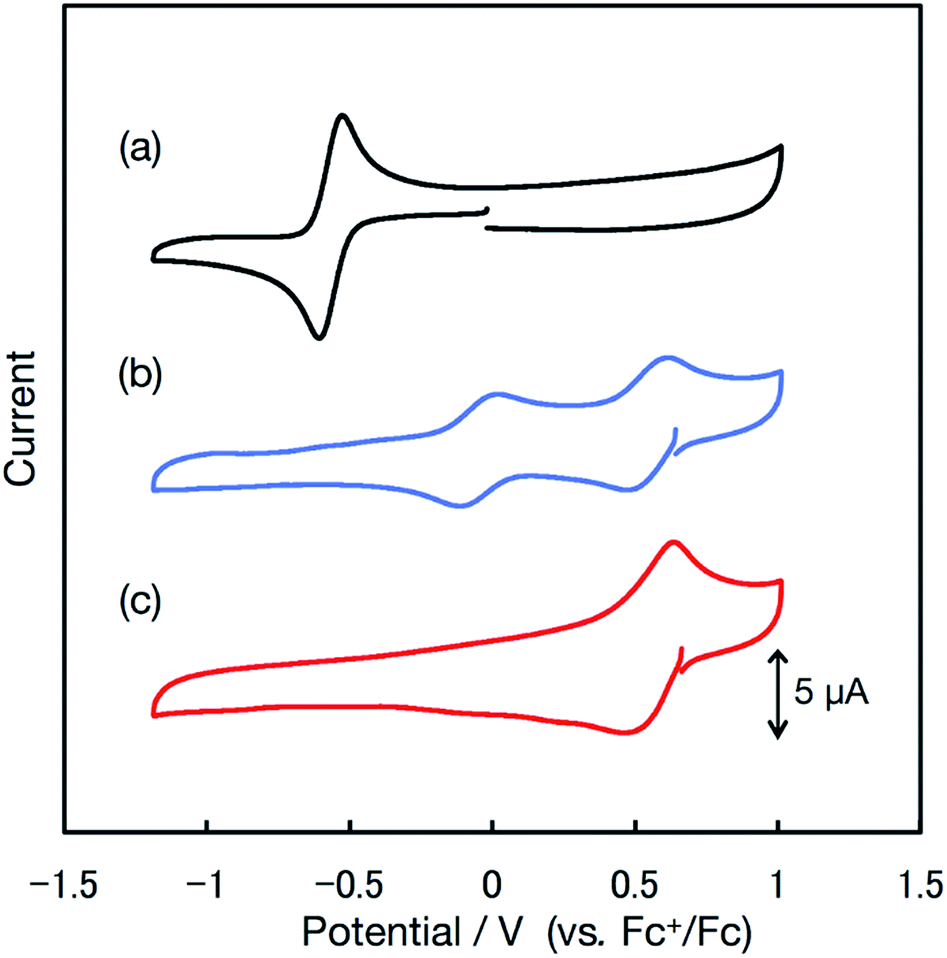 | ||
| Fig. 5 Cyclic voltammograms of bisPyTM (0.5 mM) in 0.1 M nBu4NClO4/CH2Cl2 at a scan rate of 0.1 V s−1 (a) with no additives and (b) with 2.0 eq. and (c) 5.0 eq. of TfOH. | ||
 | ||
| Scheme 3 Redox behaviors of (a)bisPyTM, (b)[HN-bisPyTM]+ and (c) [(HN)2-bisPyTM]2+. | ||
Optical properties
The UV/vis absorption spectrum of bisPyTM in dichloromethane displayed a strong near-UV band (λabs = 355 nm, transition from 121α to 122α and 123α) and a low-lying band (Fig. 6a). The lowest-energy transition band (transition from 120β to 121β) that formed the lowest excited state (D1 state) was observed at a peak wavelength of 536 nm. These transitions were assigned based on TD-DFT calculations (ESI†). The relative wavelengths and oscillator strengths for the calculated transition bands agreed with the absorption spectrum obtained experimentally, supporting the orbital assignments and the validity of the calculations. A β-electron-centered 120β → 121β electronic transition induced the D1 state, as in TTM, PyBTM, and PyBTM derivatives.14,25–29 | ||
| Fig. 6 (a) Absorption (blue line) and emission (red dotted line, λex = 355 nm) spectra of bisPyTM in dichloromethane (4.5 × 10−5 and 1.2 × 10−5 M, respectively) at room temperature, and an emission spectrum in the crystalline state at 77 K (pale red dashed line, λex = 355 nm). An enlarged portion of the absorption spectrum (10 fold) is shown for λ = 400 to 650 nm (pale blue line). (b) Emission spectra of αH-bisPyTM crystals doped with 3 (red) and 40 wt% (blue) bisPyTM. (c) Photograph of bisPyTM solution in dichloromethane under UV light at λ = 365 nm. (d) Photograph of bisPyTM crystals at 77 K under UV light at λ = 365 nm. (e) Photographs of αH-bisPyTM crystals doped with 3 (left) and 40 wt% (right) bisPyTM under UV light at λ = 365 nm. | ||
bisPyTM in dichloromethane displayed fluorescence, with an emission maximum wavelength, λem, of 650 nm upon excitation at λex = 355 nm (Fig. 6a and c). ϕem and the lifetime (τ) were measured as being 0.009 and 3.6 ns, respectively. This emission wavelength was strongly red-shifted compared with PyBTM and TTM, indicating the large structural change in bisPyTM in the excited state.
We estimated kr and knr of bisPyTM and PyBTM using eqn (6) and (7) (Table 1). kr of bisPyTM was smaller than that of PyBTM, whereas knr was considerably larger. The observed PyBTM to bisPyTM ratio of the kr values was consistent with results from the theoretical calculation described in the next section. The greater knr of bisPyTM was explained by accelerated internal conversion, which is also described in the next section.
| ϕem = kr/(kr + knr) | (6) |
| τ = 1/(kr + knr) | (7) |
| λ expabs1/nm | λ expabs2/nm | λ expem/nm | λ calcabs2/nm (f) | λ calcem/nm (f) | ϕ em | τ/ns | k r/×106 s−1 | k nr/×108 s−1 | |
|---|---|---|---|---|---|---|---|---|---|
| bisPyTM | 355 | 536 | 650 | 496 (0.0153) | 643 (0.0290) | 0.009(0) | 3.6 | 2.5 | 2.8 |
| PyBTM | 370 | 541 | 585 | 487 (0.0349) | 572 (0.0457) | 0.025(0) | 6.4 | 3.9 | 1.5 |
| TTM | 373 | 542 | 570 | — | — | 0.02 | 7.0 | 3 | 1.4 |
Surprisingly, bisPyTM exhibited luminescence both in solution and in the solid state. Deep red luminescence with λem = 712 nm was observed in the crystalline state (Fig. 6a and d) at 77 K. To the best of our knowledge, this is the first reported organic radical showing distinct luminescence in its crystalline state.37 We propose that quenching via intermolecular interactions in the crystalline state is weaker for bisPyTM than for the other radicals, explaining the luminescence in the crystalline state. Solid-state luminescence was also observed when we doped bisPyTM into the αH-bisPyTM crystal, the precursor for bisPyTM (Scheme 2). The emission wavelength of the 3 wt%-doped sample was shorter than that of bisPyTM in dichloromethane, which would be caused by reduced reorientation energy due to rigid packing. The emission wavelength was red-shifted, and the intensity decreased with increasing bisPyTM concentration (Fig. 6b and e), implying that an excimer was formed under highly doped conditions.38
Molecular and electronic structures in the lowest excited state (D1 state)
The molecular and electronic structures of bisPyTM in the D1 state were calculated via TD-DFT (UB3LYP/6-31G(d,p)) to clarify the characteristics of the radiative and non-radiative processes that occurred from the D1 state.In the optimized molecular structure of bisPyTM in its D1 state, the bond lengths between the central carbon atom and the aryl moieties (C1–C4, C1–C9, C1–C15) and between carbon atoms at the ortho and meta positions (C5–C6, C2–C3, C7–C8, C10–C11, C16–C17, C13–C14) were shorter than those in the D0 state, whereas the other C–C and C–N bonds were longer (Fig. 7a). The N1–C4–C1 and N2–C9–C1 bond angles in the two pyridyl rings decreased (Fig. 7b). These results indicate the increased quinoid character of the aryl groups as the aromaticity of the pyridyl rings decreased in the D1 state.
 | ||
| Fig. 7 A schematic diagram of the structural changes from the D0 to D1 states of bisPyTM and PyBTM: (a) changes in the bond lengths, in units of Å, and angles; and (b) changes in the N1–C4–C1 and N2–C9–C1 angles of bisPyTM. | ||
The MOs of bisPyTM in the D1 state in dichloromethane are shown in Fig. 4. The distribution of each MO is similar to that in the D0 state, except the α(β)-NHOMO (120α(β)). The distribution of the α(β)-NHOMO was similar to that of the α(β)-NHOMO−3 in the D0 state. The structural change between the D1 and the D0 states induced the energy level reordering of the energetically closed MOs.
The MOs of the reference compound, PyBTM, in the D1 state in dichloromethane were calculated (Fig. 4). The MO distributions were similar to those in the D0 state in the unoccupied orbitals, whereas modulation of the distribution was detected in the α(β)-NHOMO level. The α(β)-NHOMO was delocalized on the pyridyl group and two trichlorophenyl groups in the D0 state, whereas it was delocalized on the two trichlorophenyl groups in the D1 state. Introducing the solvent effect using the polarizable continuum model caused the reordering of energy levels and altered the distribution of MOs, including the α(β)-NHOMO. The α(β)-NHOMO was located at the pyridyl ring in the D1 state without the solvent effect, whereas it was distributed on the trichlorophenyl moieties with negligible extension to the pyridyl ring with the solvent effect (Fig. 4 and S2†). Because the β-NHOMO was directly involved in forming the D1 state, the change in the distribution on the β-NHOMO should affect the photophysical properties. These results demonstrate the importance of considering the solvent effect in these calculations.29 Therefore, we performed theoretical analysis considering the effects of dichloromethane.
Theoretical analyses of photophysical parameters
We estimated the rates of photophysical processes, kr and knr, for PyBTM and bisPyTM to explain the experimentally observed difference in ϕem. kr and knr can be evaluated by calculating the f values and off-diagonal VCCs between the D1 and the D0 states, as mentioned in the section ‘Theoretical methods for estimating photophysical parameters’. Off-diagonal VCDs indicate the vibrational modes that contribute to internal conversion.The f values for the D1 nuclear coordinates calculated using TD-DFT, considering the effects of the solvent (dichloromethane), were 0.0290 and 0.0457 for bisPyTM and PyBTM, respectively. The PyBTM to bisPyTM ratio of f calculated was 1.58. Neglecting the effects of Franck–Condon factors on the radiative rate constants,33 we compared this ratio with the observed ratio of the radiative rate constants. The observed ratio of PyBTM to bisPyTM was 1.6. The calculated f values reproduced the ratio of the observed kr values for the two radicals well.
Fig. 8 shows the off-diagonal VCCs and VCDs of bisPyTM and PyBTM. The VCCs of bisPyTM were smaller than those of common organic compounds. For instance, the maximum VCC in a triphenylamine derivative that is known as a non-fluorescent molecule is ca. 8 × 10−4 a.u.,35 while that in bisPyTM is less than 7 × 10−4 a.u. The square sum of the VCCs of bisPyTM was 1.9 × 10−6 a.u., which was larger than that of PyBTM (1.4 × 10−6 a.u.), reproducing the greater knr value of bisPyTM. The observed ratio of knr, knr (bisPyTM)/knr (PyBTM), was 1.9 (Table 1). Neglecting the effects of Franck–Condon factors on knr,33 we calculated the ratio of knr values from the square sums of the off-diagonal VCCs. The ratio of the square sum for bisPyTM to that for PyBTM is 1.36, which is reasonably consistent with the observed ratio. This difference in off-diagonal VCCs in bisPyTM and PyBTM came from the difference in overlap density, which arose from the distribution of the NHOMOs. The overlap density was distributed on the two trichlorophenyl moieties in PyBTM, whereas it was on the only trichlorophenyl moiety in bisPyTM; the greater localization in bisPyTM resulted in its larger VCD.
 | ||
| Fig. 8 Off-diagonal VCCs between the adiabatic D1 and D0 states for the normal mode of (a) bisPyTM and (d) PyBTM in dichloromethane. The overlap density (isovalue of ρ = 0.01) of (b) bisPyTM and (e) PyBTM, and the off-diagonal VCD between the adiabatic D1 and D0 states for selected vibrational modes of (c) bisPyTM and (f) PyBTM (isovalue of 4 × 10−6 a.u.). | ||
These results suggest that internal conversion was mainly attributed to a non-radiative decay process, and that the theoretical methods shown here enable us to estimate kr and knr for this class of radicals, greatly advancing the prediction and design of their photofunctionality.
Photostability
The low photostability of luminescent radicals is an important problem that must be overcome. The decay of fluorescence intensity was monitored and compared for bisPyTM and PyBTM upon continuous irradiation with light at each maximum absorption wavelength. Fig. 9 shows the decay in luminescence intensity for each compound, and the degree of decay represents the decomposition rate upon exposure to light. The estimated half-life (t1/2) in dichloromethane was 6.7 × 103 s; this value was 43 times larger than that of PyBTM (1.6 × 102 s). | ||
| Fig. 9 Plots showing the emission decay of bisPyTM, PyBTM, and TIPS pentacene in dichloromethane under continuous excitation, with light at λex = 355 nm (bisPyTM), 370 nm (PyBTM), and 310 nm (TIPS pentacene). The emission intensities are normalized. | ||
Considering our previous results,14 bisPyTM is expected to show a half-life 3000 times longer than TTM. When the photostability is defined as a product of the absorption coefficients (ε) at the irradiation wavelengths and t1/2, the photostability of bisPyTM was 30 times higher than PyBTM. In other aprotic solvents, bisPyTM was 4 to 44 times more stable (6 to 71 times longer half-lives) than PyBTM (Table 2). These results suggest that the photostability is greatly improved as the number of pyridyl groups in the TTM framework increases and the energy levels of the frontier orbitals are lowered.
| bisPyTMa | PyBTMb | TIPS pentacenec | bisPyTM/PyBTMd | bisPyTM/TIPS pentacened | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| t 1/2/s | ε /M−1 cm−1 | t 1/2 × ε | t 1/2/s | ε /M−1 cm−1 | t 1/2 × ε | t 1/2/s | ε /M−1 cm−1 | t 1/2 × ε | t 1/2/s | t 1/2 × ε | t 1/2/s | t 1/2 × ε | |
| a λ ex = 355 nm. b λ ex = 370 nm. c λ ex = 310 nm in dichloromethane, hexane, and chloroform; λex = 300 nm in acetonitrile; and λex = 330 nm in acetone. d A comparison of t1/2 and t1/2 × ε for the compounds. e Molar absorption coefficients at the irradiation wavelengths. | |||||||||||||
| Dichloromethane | 6.7(3) × 103 | 1.78 × 104 | 1.2 × 108 | 1.56(2) × 102 | 2.54 × 104 | 4.0 × 106 | 3.4(9) × 10 | 1.56 × 105 | 5.3 × 106 | 43 | 30 | 20 × 10 | 23 |
| Acetonitrile | 1.4(1) × 104 | 2.19 × 104 | 3.2 × 108 | 3.07(7) × 103 | 2.47 × 104 | 7.6 × 106 | 3.8(5) × 102 | — | — | 47 | 41 | 38 | — |
| Acetone | 4.0(4) × 103 | 1.36 × 104 | 5.5 × 107 | 6.4(1) × 103 | 2.26 × 104 | 1.4 × 107 | 1.1(7) × 102 | 2.30 × 104 | 2.6 × 106 | 6.3 | 3.8 | 35 | 21 |
| Hexane | 7.8(5) × 103 | 1.85 × 104 | 1.4 × 108 | 1.10(1) × 102 | 3.90 × 104 | 4.3 × 106 | 1.90(9) × 103 | 1.25 × 105 | 2.4 × 108 | 71 | 34 | 4.1 | 0.6 |
| Chloroform | 7.3(5) × 103 | 1.65 × 104 | 1.2 × 108 | 1.20(3) × 103 | 2.25 × 104 | 2.7 × 106 | 1.10(1) × 10 | 1.78 × 105 | 2.0 × 106 | 60 | 44 | 66 × 10 | 61 |
| Ethanol | 2.0(2) × 10 | 1.48 × 104 | 3.0 × 105 | 1.46(4) × 10 | 2.09 × 104 | 3.1 × 105 | — | — | — | 1.4 | 1.0 | — | — |
| Methanol | 1.8(2) × 10 | 1.23 × 104 | 2.2 × 105 | 2.0(3) × 10 | 2.25 × 104 | 4.4 × 105 | — | — | — | 0.90 | 0.49 | — | — |
t 1/2 values in the solvents were not simply scaled by solvent parameters such as the dielectric constant or dipole moment (Fig. S3†). This trend suggests that there is a specific interaction between the radical and solvent molecules that modulates the photostability. For example, exceptionally low t1/2 values in alcohols were detected for both PyBTM and bisPyTM. The low values may result from hydrogen bonds formed between nitrogen atoms on the radicals and hydrogen atoms in the EtOH or MeOH molecules, which accelerate decomposition in the photoexcited state.
For industrial applications, the photostabilities of bisPyTM and 6,13-bis(triisopropylsilylethynyl)pentacene (TIPS pentacene) were compared. TIPS pentacene is a closed-shell molecule commonly used in organic semiconductors as a benchmark material due to its high photostability and solubility.39–41 Its fluorescence peak maximum (λem = 650 nm upon excitation at λex = 360 nm) was similar to those of PyBTM and bisPyTM. Fig. 9 and Table 2 provide clear evidence for the superior photostability of bisPyTM; the photostability of the radical was 0.6 to 61 times higher (4 to 660 times larger t1/2 values) than TIPS pentacene. This result demonstrates the potential of the compound for practical applications.
Dual responsive properties
We aimed to control the optical and electrochemical properties of bisPyTM by using the two pyridyl nitrogen atoms in the molecular structure as chemical stimulus-responsive sites. First, we investigated the effects of adding a Brønsted–Lowry acid. The reversible response of the radical to protonation and deprotonation was confirmed via UV/vis absorption spectroscopy during an acid–base titration (Fig. 10a and c). A new transition band at λ = 409 nm appeared as the intensity of the absorption at λ = 355 nm decreased upon the addition of trifluoromethanesulfonic acid (TfOH). The initial spectral shape was almost recovered by adding triethylamine (NEt3). Protonation affected the luminescence properties; the fluorescence intensity decreased substantially upon addition of TfOH (Fig. 10b and d). PyBTM showed similar absorption and emission spectra upon protonation and deprotonation.14 | ||
| Fig. 10 (a) UV/vis absorption spectra of bisPyTM in CH2Cl2 following addition of TfOH: bisPyTM (red) + 0.5, 1.0, 1.5, 2.0, 2.5, and 3.0 (blue) eq. of TfOH. (b) Emission spectra of bisPyTM in CH2Cl2 following addition of TfOH (λex = 380 nm, isosbestic point wavelength): bisPyTM (red) + 0.5, 1.0, 2.0, and 3.0 (blue) eq. of TfOH. (c) UV/vis absorption spectra and (d) emission spectra of bisPyTM in CH2Cl2 following addition of NEt3 (λex = 380 nm): bisPyTM + 3.0 eq. of TfOH (blue) + 1.0, 2.0, and 3.0 (green) eq. of NEt3. (e) UV/vis absorption spectra and (f) emission spectra of bisPyTM in CH2Cl2 with addition of B(C6F5)3 (λex = 375 nm, average wavelength of two isosbestic points): bisPyTM (blue) + 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, and 3.5 (red) eq. of B(C6F5)3. Fresh solutions were prepared for each measurement. (g) UV/vis absorption spectra and (h) emission spectra of bisPyTM in CH2Cl2 with addition of NEt3 (λex = 375 nm): bisPyTM + 3.5 eq. of B(C6F5)3 (red) + 0, 0.5, 1, 2, 3 and 4 (green) eq. of NEt3. Fresh solutions were prepared for each measurement. | ||
The redox properties, examined via cyclic voltammetry, also changed drastically upon protonation. Upon the successive addition of TfOH, two new redox waves at E0′ = −0.05 and 0.55 V appeared, and only one redox wave at E0′ = 0.55 V was observed after the addition of excess TfOH (Fig. 5c). This result suggests a two-stage increase in the electron-accepting ability of the molecule resulting from single-to-double protonation of the two pyridyl rings. Accordingly, the redox waves at E0′ = −0.05 and 0.55 V were ascribed to the [HN-bisPyTM]+/[HN-bisPyTM] and [(HN)2-bisPyTM]2+/[(HN)2-bisPyTM]+ redox couples, respectively, where [HN-bisPyTM]+ and [(HN)2-bisPyTM]2+ are monoprotonated and diprotonated bisPyTM, respectively (Scheme 3). Monoprotonation or diprotonation shifted the reduction potential of bisPyTM by 0.52 or 1.12 V in the positive direction, respectively. Similar positive shifts were observed in the protonation or methylation of PyBTM,14,25 and were explained by a decrease in electron density upon protonation, which increased the electron-accepting ability.
The reduction potential of [(HN)2-bisPyTM]2+ was higher than that of F4TCNQ (2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane, E0′ = 0.16 V),42 which is commonly used as an electron acceptor or hole dopant in molecule-based devices and materials,43,44 showing that [(HN)2-bisPyTM]2+ functions as a superior electron acceptor (i.e., oxidizing agent).
Next, we used B(C6F5)3, which is a Lewis acid capable of binding to nitrogen atoms, as an external stimulus. Upon the addition of B(C6F5)3, two isosbestic points (λ = 371 and 378 nm for 0–1 and 1–3 eq., respectively) were obtained in the UV/vis spectra. These two points can be assigned as the equilibrium of bisPyTM and one B(C6F5)3 adduct, and that of one and two adducts, indicating the stepwise response of the two nitrogen atoms to B(C6F5)3 (Fig. 10e). The emission characteristics of bisPyTM also showed a two-stage response to B(C6F5)3. The emission maximum wavelengths were gradually red-shifted upon the addition of B(C6F5)3, and the resulting emission band at λ = 756 nm extended over 850 nm (Fig. 10f), suggesting the near-IR emission of [(C6F5)3BN]2-bisPyTM. The mechanism of the red shift is similar to that for (C6F5)3BN-PyBTM.25 Upon the addition of NEt3, the emission and UV/vis absorption spectra did not return to the initial states, indicating the incomplete recovery and partial decomposition of bisPyTM (Fig. 10g and h). These results show the partly reversible response of the radical to B(C6F5)3 and NEt3.
Conclusions
We prepared bisPyTM, a luminescent stable organic radical that has two pyridyl moieties. Upon continuous UV light irradiation, it exhibited a half-life in dichloromethane that was 43 and 200 times longer than those of PyBTM and TIPS pentacene, respectively. The photostabilities of the radicals were greatly improved as the number of pyridyl groups in the TTM skeleton increased. We confirmed the luminescence of crystalline bisPyTM at 77 K, which would be the first example of visible solid-state emission from a radical. The two nitrogen atoms of bisPyTM can act as chemical stimulus-responsive sites and react with TfOH and B(C6F5)3 to modulate the optical and electrochemical characteristics. The differences in the kr and knr values of bisPyTM and PyBTM were reproduced through theoretical calculations based on DFT, TD-DFT, and VCD analyses. The VCDs indicated that the difference in the off-diagonal VCCs in the two radicals comes from the difference in the overlap density, which originates from the distribution of the NHOMOs.Our results showed that introducing a pyridyl ring increases the photostability by lowering the energy levels of frontier orbitals. We also demonstrated that the scaled kr and knr values, important parameters that determine the photophysical properties, can be estimated and compared in this class of radicals via theoretical calculations. These findings contribute greatly to predicting the photophysical properties of luminescent radicals, thereby achieving high emission efficiency and controlled photofunctionality.
Experimental
Materials
All chemicals were reagent grade and purchased commercially. Water was purified using an AUTOPURE WD500 system (Yamato Scientific Co., Ltd). The acetonitrile (MeCN), dichloromethane (CH2Cl2), diethyl ether (Et2O), hexane, tetrahydrofuran (THF) and toluene used for syntheses were purified through an organic solvent purifier (Nikko Hansen Co., Ltd).Equipment
NMR spectra were recorded with a Bruker US500 spectrometer at room temperature. Chemical shifts in ppm were referenced using tetramethylsilane (0.00 ppm) as an internal standard. Microwave assisted synthesis was performed using a Biotage Initiator+. UV-vis-NIR absorption spectra were recorded with a JASCO V570 spectrophotometer. Steady-state emission spectra were recorded with HITACHI F-4500 and JASCO FP8600 spectrometers. Absolute photoluminescence quantum yields were measured using a Hamamatsu Photonics C9920-02G system. Fluorescence lifetime measurements were measured using a Hamamatsu Photonics Quantaurus-Tau C11367-02 system. ESR spectra were recorded with a JEOL JES-FA200 spectrometer. 4-Hydroxy-TEMPO was used as a standard for estimating the spin concentration. Observed g-values were calibrated with an Mn2+/MgO marker. Cyclic voltammetry was carried out under an argon atmosphere with a GC working electrode, a platinum wire counter electrode and an Ag+/Ag reference electrode (10 mM AgClO4 and 0.1 mM nBu4NClO4 in MeCN solution), with an ALS-650DT voltammetric analyzer. The platinum working electrode was polished with an alumina abrasive and washed with water and acetone with ultrasonication. The counter electrode was heated with an oxygen flame to remove organic compounds on the surface and washed with acetone. The reference electrode was washed with acetone. A Fc+/Fc redox couple was observed at 0.188 V under these experimental conditions. The potential of the voltammograms is shown relative to the Fc+/Fc redox couple.Single crystal X-ray crystallography
The data for single crystal X-ray diffraction analysis were collected at 113 K on a Rigaku AFC10 diffractometer, with a Rigaku Saturn CCD system equipped with a rotating-anode X-ray generator that emitted graphite-monochromated Mo Kα radiation (0.7107 Å). A suitable single crystal was mounted on a looped film (micromount) with liquid paraffin. An empirical absorption correction, using equivalent reflections and Lorentzian polarization correction, was performed with the program Crystal Clear 1.3.6. The structure was solved using SIR-9245 and the whole structure was refined against F2 with SHELXL-97.46 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were located in idealized positions and were refined using a riding model with fixed thermal parameters.
Computational details
DFT calculations were executed using the Gaussian 09 program package.47 The geometries of the compounds were optimized with the grid = ultrafine option without symmetry constraints, using the crystal structure coordinates as the starting structure. Calculations were performed using the unrestricted Becke three-parameter hybrid functional with the Lee–Yang–Parr correlation functional (B3LYP)48 with the 6-31G(d,p) basis set.49 Frequency calculations were carried out to ensure that the optimized geometries were minima on the potential energy surface, in which no imaginary frequencies were observed in any of the compounds. TD-DFT calculations were performed using UB3LYP to calculate the first 15 doublet transitions. The solvent effect was considered using the PCM model.50 VCCs and VCDs were calculated using our in-house codes.Evaluation of the stabilities of bisPyTM, PyBTM and TIPS pentacene under UV light
A solution (ca. 5 × 10−6 M, 1.0 mL) in a 1 cm optical path length quartz cell was bubbled with argon, sealed, and set in a JASCO FP8600 spectrometer. The intensity of luminescence at 650 and 585 nm was monitored, exciting with 355 and 370 nm light (the excitation slit was 20 nm, and the shutter control was off), for bisPyTM and PyBTM, respectively. For TIPS pentacene, the intensity of luminescence at 650 nm was monitored, exciting with 310 (in dichloromethane), 300 (in acetonitrile) and 330 (in acetone) nm light. The logarithm of fluorescence intensity versus time was plotted and the slope of an approximate line was estimated to be the rate of photolysis.Synthesis
Synthesis of bis(3,5-dichloro-4-pyridyl)(1,3,5-trichlorophenyl)methane (αH-bisPyTM): under an argon atmosphere, a mixture of bis(3,5-dichloro-4-pyridyl)methanol51 (7.78 g, 24.0 mmol), 1,3,5-trichlorobenzene (69.4 g, 382 mmol) and aluminum chloride (31.98 g, 240 mmol) was heated to 300 °C for 10 min via microwave irradiation. The resulting dark-brown mixture was cooled to room temperature, dissolved in CH2Cl2 and added to ice water. The mixture was neutralized using aqueous NaHCO3 and the organic layer was separated. The water layer was extracted with CH2Cl2 (3 times). The organic layers were combined, dried with Na2SO4, and evaporated. The crude product was purified via silica gel column chromatography (eluent: ethyl acetate/hexane = 1/4, v/v) and recrystallization from CH2Cl2/hexane. Preparative GPC yielded the pure product as a pale yellow solid (96 mg, 1%). The steric hindrance of methylene carbon atoms, instability of the reaction intermediate (methyl cation) and lowered electron densities of pyridine rings upon coordination to aluminum chloride can cause this low yield. The major product is bis(3,5-dichloro-4-pyridyl)methane, which would be produced by the decomposition of the starting material due to the severe reaction temperature, whereas the target compound was not formed at lower reaction temperatures. 1H NMR (CDCl3, 500 MHz): δ = 6.70 (s, 1H), 7.28 (d, 1H, J = 2.5 Hz), 7.41 (d, 1H, J = 2.5 Hz), 8.37 (s, 1H), 8.40 (s, 1H), 8.51 (s, 1H), 8.52 (s, 1H). GC-MS: m/z = 486 ([C17H7Cl7N2]+). Elem. anal. calcd for C17H6Cl7N2: C 41.89, H 1.45, N 5.75; found: C 41.67, H 1.80, N 5.67.Synthesis of the bis(3,5-dichloro-4-pyridyl)(1,3,5-trichlorophenyl)methyl radical (bisPyTM):14 under a nitrogen atmosphere, αH-bisPyTM (31.0 mg, 63.8 μmol) was dissolved in dry THF (3.0 mL). A 1 M solution of tBuOK in THF (130 μL) was added dropwise, and the color of the solution changed to red. The reaction mixture was stirred overnight in the dark. I2 (121 mg, 476 μmol) in dry diethyl ether (10.8 mL) was added dropwise and stirred for 2.25 h. The remaining I2 was reduced by washing with aq. 10% Na2S2O3 3 times. The water layer was extracted with diethyl ether once, and the combined organic layer was dried with Na2SO4. The red solution was filtered, evaporated, purified via Al2O3 column chromatography (eluent: diethyl ether/hexane = 1/4, v/v) and dried in vacuo to afford bisPyTM (28.1 mg, 91%) as a red solid. HRMS (negative ion mode ESI-TOF) m/z: [M]−. Calcd for C17H6Cl7N2: 484.8322; found 484.8303. Elem. anal. calcd for C17H6Cl7N2: C 41.98, H 1.24, N 5.76; found: C 41.73, H 1.83, N 5.31. ESR: the spin concentration of bisPyTM in toluene (1.2 × 10−5 M) was estimated by comparing the value of the twice-integration of the signal intensity with that of a reference sample (4-hydroxy-TEMPO in toluene; 1.5 × 10−5 M). The existence of S = 1/2 spin on one bisPyTM molecule was confirmed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present study was supported by JST CREST grant number JPMJCR15F2. This work was also supported by JSPS KAKENHI grant numbers JP26220801, JP16K13973, JP17H04870, and JP15K05607. T. K. is grateful to the Ogasawara Foundation for the Promotion of Science and Engineering and the Shorai Foundation for Science for financial support. S. K. acknowledges MERIT (the material education program for future leaders in Research, Industry, and Technology) in the MEXT Leading Graduate School Doctoral Program. This research was conducted using the HITACHI SR16000 system (yayoi) in the Information Technology Center, The University of Tokyo.Notes and references
- C. W. Tang and S. A. Vanslyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- M. A. Baldo, D. F. O'brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- V. Gamero, D. Velasco, S. Latorre, F. López-Calahorra, E. Brillas and L. Juliá, Tetrahedron Lett., 2006, 47, 2305–2309 CrossRef CAS.
- D. Velasco, S. Castellanos, M. López, F. López-Calahorra, E. Brillas and L. Juliá, J. Org. Chem., 2007, 72, 7523–7532 CrossRef CAS PubMed.
- L. Fajarí, R. Papoular, M. Reig, E. Brillas, J. L. Jorda, O. Vallcorba, J. Rius, D. Velasco and L. Juliá, J. Org. Chem., 2014, 79, 1771–1777 CrossRef PubMed.
- A. Heckmann, S. Dümmler, J. Pauli, M. Margraf, J. Köhler, D. Stich, C. Lambert, I. Fischer and U. Resch-genger, J. Phys. Chem. C, 2009, 113, 20958–20966 CAS.
- E. V. Tretyakov, V. F. Plyusnin, A. O. Suvorova, S. V. Larionov, S. A. Popov, O. V. Antonova, E. M. Zueva, D. V. Stass, A. S. Bogomyakov, G. V. Romanenko and V. I. Ovcharenko, J. Lumin., 2014, 148, 33–38 CrossRef CAS.
- Y. Gao, A. Obolda, M. Zhang and F. Li, Dyes Pigm., 2017, 139, 644–650 CrossRef CAS.
- E. Neier, R. Arias, N. Rady, S. Venkatesan, T. W. Hudnall and A. Zakhidov, Org. Electron., 2017, 44, 126–131 CrossRef CAS.
- Y. Beldjoudi, I. Osorio-Román, M. A. Nascimento and J. M. Rawson, J. Mater. Chem. C, 2017, 5, 2794–2799 RSC.
- H. Namai, H. Ikeda, Y. Hoshi, N. Kato, Y. Morishita and K. Mizuno, J. Am. Chem. Soc., 2007, 129, 9032–9036 CrossRef CAS PubMed.
- Y. Hattori, T. Kusamoto and H. Nishihara, Angew. Chem., Int. Ed., 2014, 53, 11845–11848 CrossRef CAS PubMed.
- Q. Peng, A. Obolda, M. Zhang and F. Li, Angew. Chem., Int. Ed., 2015, 54, 7091–7095 CrossRef CAS PubMed.
- A. Obolda, X. Ai, M. Zhang and F. Li, ACS Appl. Mater. Interfaces, 2016, 8, 35472–35478 CAS.
- M. A. Fox, E. Gaillard and C. Chen, J. Am. Chem. Soc., 1987, 109, 7088–7094 CrossRef CAS.
- S. R. Ruberu and M. A. Fox, J. Phys. Chem., 1993, 97, 143–149 CrossRef CAS.
- M. Sakamoto, X. Cai, M. Hara, S. Tojo, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2005, 127, 3702–3703 CrossRef CAS PubMed.
- J. C. Scaiano, L. J. Johnston, W. G. McGimpsey and D. Weir, Acc. Chem. Res., 1988, 21, 22–29 CrossRef CAS.
- D. Meisel, P. K. Das, G. L. Hug, K. Bhattacharyya and R. W. Fessenden, J. Am. Chem. Soc., 1986, 108, 4706–4710 CrossRef CAS.
- P. Brodard, A. Sarbach, J. Gumy, T. Bally and E. Vauthey, J. Phys. Chem. A, 2001, 105, 6594–6601 CrossRef CAS.
- M. Ballester, J. Riera, J. Castatier, C. Badfa and J. M. Monsó, J. Am. Chem. Soc., 1971, 93, 2215–2225 CrossRef CAS.
- O. Armet, J. Veciana, C. Rovira, J. Riera, J. Castaner, E. Molins, J. Rius, C. Miravitlles, S. Olivella and J. Brichfeus, J. Phys. Chem., 1987, 91, 5608–5616 CrossRef CAS.
- T. Kusamoto, S. Kimura, Y. Ogino, C. Ohde and H. Nishihara, Chem.–Eur. J., 2016, 22, 17725–17733 CrossRef CAS PubMed.
- Y. Hattori, T. Kusamoto and H. Nishihara, RSC Adv., 2015, 5, 64802–64805 RSC.
- Y. Hattori, T. Kusamoto and H. Nishihara, Angew. Chem., Int. Ed., 2015, 54, 3731–3734 CrossRef CAS PubMed.
- Y. Hattori, T. Kusamoto, T. Sato and H. Nishihara, Chem. Commun., 2016, 440, 908–912 Search PubMed.
- Y. Ogino, T. Kusamoto, Y. Hattori, M. Shimada, M. Tsuchiya, Y. Yamanoi, E. Nishibori, K. Sugimoto and H. Nishihara, Inorg. Chem., 2017, 56, 3909–3915 CrossRef CAS PubMed.
- H. Iwamura and N. Koga, Pure Appl. Chem., 1999, 71, 231–238 CrossRef CAS.
- T. K. Maji, R. Matsuda and S. Kitagawa, Nat. Mater., 2007, 6, 142–148 CrossRef CAS PubMed.
- J. Grilj, E. N. Laricheva, M. Olivucci and E. Vauthey, Angew. Chem., Int. Ed., 2011, 50, 4496–4498 CrossRef CAS PubMed.
- M. Uejima, T. Sato, D. Yokoyama, K. Tanaka and J.-W. Park, Phys. Chem. Chem. Phys., 2014, 16, 14244–14256 RSC.
- D. T. Breslin and M. A. Fox, J. Phys. Chem., 1994, 98, 408–411 CrossRef CAS.
- Y. Kameoka, M. Uebe, A. Ito, T. Sato and K. Tanaka, Chem. Phys. Lett., 2014, 615, 44–49 CrossRef CAS.
- M. Uebe, A. Ito, Y. Kameoka, T. Sato and K. Tanaka, Chem. Phys. Lett., 2015, 633, 190–194 CrossRef CAS.
- Emission spectra of radicals in the solid state at 5 K, very low temperature, were reported. (a) R. Beaulac, D. Luneau and C. Reber, Chem. Phys. Lett., 2005, 405, 153–158 CrossRef CAS; (b) R. Beaulac, G. Bussière, C. Reber, C. Lescop and D. Luneau, New J. Chem., 2003, 27, 1200–1206 RSC.
- D. Blasi, D. M. Nikolaidou, F. Terenziani, I. Ratera and J. Veciana, Phys. Chem. Chem. Phys., 2017, 19, 9313–9319 RSC.
- A. Shimizu, A. Ito and Y. Teki, Chem. Commun., 2016, 52, 2889–2892 RSC.
- A. Maliakal, K. Raghavachari, H. Katz, E. Chandross and T. Siegrist, Chem. Mater., 2004, 16, 4980–4986 CrossRef CAS.
- I. Kaur, W. Jia, R. P. Kopreski, S. Selvarasah, M. R. Dokmeci, C. Pramanik, N. E. McGruer and G. P. Miller, J. Am. Chem. Soc., 2008, 130, 16274–16286 CrossRef CAS PubMed.
- M. Kivala, C. Boudon, J. P. Gisselbreeht, B. Enko, P. Seiler, B. M. Imke, N. Langer, P. D. Jarowski, G. Geseheidt and F. Diederich, Chem.–Eur. J., 2009, 15, 4111–4123 CrossRef CAS PubMed.
- M. Pfeiffer, K. Leo, X. Zhou, J. S. Huang, M. Hofmann, A. Werner and J. Blochwitz-Nimoth, Org. Electron., 2003, 4, 89–103 CrossRef CAS.
- K. Walzer, B. Männig, M. Pfeiffer and K. Leo, Chem. Rev., 2007, 107, 1233–1271 CrossRef CAS PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- T. Itoh, A. Takada, K. Hirai and H. Tomioka, Org. Lett., 2005, 7, 811–814 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal structure data, ESR spectral data, DFT and TD-DFT calculations data, spectroscopic data, quantum yield and photostability in various solvents, electrochemical data (PDF). CCDC 1559962. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04034b |
| This journal is © The Royal Society of Chemistry 2018 |