
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Base-free, tunable, Au-catalyzed oxidative esterification of alcohols in continuous flow†
Felicity J.
Roberts
a,
Christian
Richard
a,
Fessehaye W.
Zemichael
a,
King Kuok (Mimi)
Hii
 *b,
Klaus
Hellgardt
*a,
Colin
Brennan
c and
David A.
Sale
c
*b,
Klaus
Hellgardt
*a,
Colin
Brennan
c and
David A.
Sale
c
aDepartment of Chemical Engineering, Imperial College London, Exhibition Road, South Kensington, London SW7 2AZ, UK. E-mail: k.hellgardt@imperial.ac.uk
bDepartment of Chemistry, Imperial College London, Exhibition Road, South Kensington, London SW7 2AZ, UK. E-mail: mimi.hii@imperial.ac.uk
cSyngenta, Process Studies Group, Jealotts Hill, Bracknell, Berkshire RG42 6EY, UK
First published on 22nd October 2018
Abstract
Under continuous flow conditions, hydrogen peroxide oxidizes primary alcohols (cinnamyl alcohol, decenol, decanol and benzyl alcohol) in methanol over Au/TiO2, without the need for added base. While the allylic alcohols afforded conjugated aldehydes, aliphatic and benzylic alcohols afforded acids or esters. Selectivity for either product can be achieved by adjusting the reaction parameters. Kinetic studies revealed that the formation of the easter is faster than that of the acid, due to a greater pre-organization (larger ln A) attributed to the more favourable formation of the hemiacetal intermediate.
Introduction
Esters are an important class of fine and speciality chemicals used in the production of fine chemicals.1 Given that alcohols can be easily obtained from petrochemicals as well as renewable feedstocks, oxidative transformations of these precursors are particularly attractive for delivering products that meet demand for sustainable, and ‘natural’ alternatives.The unique application of gold nanoparticles for selective liquid-phase oxidation reactions is a highly topical research area in catalysis.2 Under an O2 atmosphere (‘aerobic’), selective conversion of primary alcohols to aldehydes can be achieved without any additives. However, further oxidation to acids or esters often requires alkaline conditions (Scheme 1, eqn (1)).3 Stoichiometrically, two equivalents of O2 are required to produce acid derivatives from each alcohol. Given that O2 has low solubility in protic solvents (water and alcohols),4 aerobic reactions are often mass-transfer limited and have low turnover frequencies (TOF's); furthermore, implementing mixtures of O2 in flammable solvents also engenders additional safety concerns.5
 | ||
| Scheme 1 Oxidation of primary alcohols to carboxylic acid derivatives using O2 or H2O2 as oxidants. | ||
These issues may be circumvented by replacing O2 by H2O2: as the peroxide is readily available as a 30 wt% aqueous solution, a much higher effective concentration of the oxidant can be achieved in the reaction. Previously, it has been shown that primary alcohols can be converted into carboxylic acids under ‘base-free’ conditions using H2O2 as an oxidant.6 Prior to this work, the selective conversion of alcohols to methyl esters has not been demonstrated using H2O2 as an oxidant.
As part of our ‘Catalysis in Flow’ program, we have previously successfully implemented heterogeneous catalysis for the aerobic oxidation of alcohols to aldehydes and ketones under continuous flow.7 Herein, we will demonstrate that the approach can also be applied to the oxidation of alcohols to (conjugate) aldehydes, acids and esters, by using H2O2 as an oxidant. Under continuous flow conditions, the conversion of an aliphatic alcohol (1-decanol) to its acid or ester products can be achieved selectively, by judicious choice of the catalyst's particle size and reaction temperature. A study of the reaction kinetics, performed with benzyl alcohol as the model substrate, highlights the fundamental steps involved in the reaction, and factors that govern the reactivity and selectivity.
Results and discussion
Strong metal–support interactions (SMSI) are often implicated in aerobic oxidation reactions catalyzed by gold. In earlier work,8 smaller-sized (5.3 nm) Au nanoparticles (supported on ceria) have been reported to exhibit better catalytic performance than larger nanoparticles (7.4 nm) in the oxidation of benzyl alcohol to benzaldehyde, which was attributed to the greater oxygen vacancy defects on the smaller catalyst. In a more recent publication by Li and co-workers,9 the activity and selectivity of gold catalysts in oxidative esterification reactions are found to be enhanced by increasing the amount of surface acid–base sites.To test whether this is also applicable in the current system where H2O2 is used, two 1 wt% Au/TiO2 catalysts containing different nanoparticle sizes were compared: S1 was procured from commercial sources, while S2 was prepared by the deposition–precipitation (DP) method. The average sizes of the Au particles were analysed by transmission electron microscopy (TEM): while S1 had particle sizes of 3.6 nm, catalyst S2 contained bigger particles of approximately 4.7 nm (Fig. S1, ESI†), providing a useful comparison in these catalytic studies.
Catalytic reactions were initially performed in a commercial packed-bed reactor (XCube®). A diluted solution of cinnamyl alcohol (1) or trans-dec-2-en-1-ol (2) in a mixture of water–methanol, was delivered together with an aqueous solution of H2O2 (10 equiv.) through a preheated catalyst bed of S1 or S2 between 30–100 °C. The steady-state (single pass) conversion of these alcohols were subsequently recorded as a function of temperature (Fig. S4, ESI†). In both cases, the reactions afforded the conjugated aldehydes 3 and 4, respectively, as the major products (Scheme 2). As expected, the activity of the smaller catalyst S1 was higher than that of catalyst S2; single-pass conversions of ca. 80% for the oxidation of both substrates were obtained at 100 °C. At a residence time of 2.2 min, this corresponds to TOF values of between 22–29 s−1. These values are comparable to those previously reported for a similar reaction performed in a batch reactor, where a good conversion of cinnamyl alcohol (1) was achieved only with dropwise addition of a dilute solution (5%) of H2O2, to minimize the decomposition of the oxidant.6a
 | ||
| Scheme 2 Oxidation of allylic alcohols. | ||
No acid products were detected in the oxidation of allylic alcohols 1 and 2 – the formation of methyl esters was only detected in trace quantities at temperatures >85 °C. Attributing this to the inherent stability of these conjugated aldehydes towards 1,2-nucleophilic addition reactions, the oxidation of the aliphatic 1-decanol (5) was examined (Scheme 3). Compared to the oxidation of the allylic alcohols, the overall reaction of 5 was slower, furnishing a mixture of 1-decanoic acid (6) and methyl decanoate (7); the aldehyde intermediate was only observed in trace quantities, indicating that the second oxidation step is more facile than the first, in the absence of the conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond.
C bond.
 | ||
| Scheme 3 Oxidation of 1-decanol. | ||
Significantly, the distribution between the acid and ester products varies significantly with reaction temperature: using an excess of H2O2 (10 equiv.), acid formation is favoured at low temperatures, while increasing the reaction temperature promotes the formation of the methyl ester product. The selectivity can be further enhanced by using an equimolar amount of H2O2, where the exclusive formation of the acid can be achieved at temperatures ≤40 °C, while 100% selectivity for the ester can be attained at temperatures ≥70 °C (Fig. 1). Both 1-decanoic acid (‘capric acid’) and methyl decanoate are valuable ingredients used in formulations of cosmetics, flavours and fragrances, agrochemicals and even pharmaceuticals (as prodrugs). Hence, the ability to switch between the production of acid and ester simply by changing the reaction temperature of a continuous process can be attractive for industrial processes, as this will enable a manufacturer to react more quickly to fluctuating market demands.
 | ||
| Fig. 1 Selective oxidation of 1-decanol using catalyst S1 (1 equiv. of H2O2). | ||
To understand the fundamental processes governing the selectivity of the reaction, a series of experiments were performed in the next stage of this work, by recording changes in the single pass conversions and selectivity at different temperatures and residence times. To enable a wider range of flow rates, a custom-built Stokes flow reactor was used, where the reactants were delivered by syringe pumps (see Experimental section).
To facilitate direct comparisons with previous studies (where O2 was used as the terminal oxidant), the coupling between benzyl alcohol and methanol was studied.10 The oxidation of a mixture of benzyl alcohol in methanol (50 mM) and aq. H2O2 (100 mM) was initially passed through a catalyst bed containing 1 wt% Au/TiO2 at 70 °C. With a residence time of 13 s, 39% of the benzyl alcohol was converted into a mixture of three products: the major component was methyl benzoate (23%) accompanied by smaller amounts of benzaldehyde (8%) and benzoic acid (8%). Next, benzyl alcohol was replaced by benzaldehyde feed. Under the prescribed conditions, the formation of the methyl ester and acid was observed, but not benzyl alcohol, i.e. the oxidation of benzyl alcohol to benzaldehyde (via a transfer hydrogenation pathway) is irreversible. By the same token, methyl benzoate was not produced from a benzoic acid feed, indicating that uncatalyzed esterification of benzoic acid is not a significant process under these conditions. Similarly, uncatalyzed oxidation of benzaldehyde to benzoic acid was also comparatively slow (no significant reaction at 100 °C, τ = 5 min). Based on these observations, a reaction pathway consisting of sequential steps can be constructed (Scheme 4), whereby the benzyl alcohol is oxidized to the benzaldehyde intermediate, from which benzoic acid or methyl benzoate forms competitively, but not interchangeably.
 | ||
| Scheme 4 Sequential and competitive steps involved in the oxidative esterification of benzyl alcohol by H2O2 catalyzed by Au/TiO2. | ||
The scheme is reinforced by performing the experiment with benzyl alcohol and methanol at 70 °C with different residence times, affording the reaction profile shown in Fig. 2 – clearly showing the formation of a benzaldehyde intermediate, and its competitive conversion to the methyl ester or acid products.
 | ||
| Fig. 2 Reaction profile obtained at different residence times. Conditions: oxidation of benzyl alcohol (feed 1 = 50 mM in methanol) with H2O2 (feed 2 = 100 mM in H2O2) over Au/TiO2 at 70 °C, flow rates = 0.05–1 mL min−1. | ||
The decomposition of H2O2 is an important issue in any process that utilizes it as an oxidant. Au/TiO2 is also known to catalyse the process.11 Not only does it compromise efficiency, the evolution of the O2 by-product is also potentially hazardous. In this study, the consumption of H2O2 was monitored using TiOSO4 to provide a colorimetric test for the oxidant.12
At 0 °C, decomposition of a 250 mM solution of H2O2 in MeOH was complete upon exposure to the catalyst in 0.5 min, compared to control experiments conducted with other components of the catalyst (silica gel and TiO2), where the decomposition of H2O2 was insignificant below 70 °C (Fig. S6, ESI†).
Decomposition of H2O2 is a first order process, which obeys the rate law:
| ln[H2O2] = kt + ln[H2O2]0 |
By reducing the amount of catalyst (from 100 mg to 10 mg, to afford observable reaction rates within the range of flow rates), the decomposition of H2O2 was recorded over different flow rates and temperatures, and the data used to extrapolate rate constants, yielding an activation energy of 44 kJ mol−1 (Fig. S6 and S7, ESI†).
During the oxidative esterification reaction, a mass balance calculation shows that more H2O2 was consumed relative to the amounts of products obtained, indicating that competitive decomposition of H2O2 is significant. To rule out possible participation of O2 in the process, experiments were conducted at different pressures between 100 and 200 psi. No difference in conversion or selectivity was observed (Fig. 3); therefore, we surmised that the oxidation reactions are enacted by H2O2 alone.
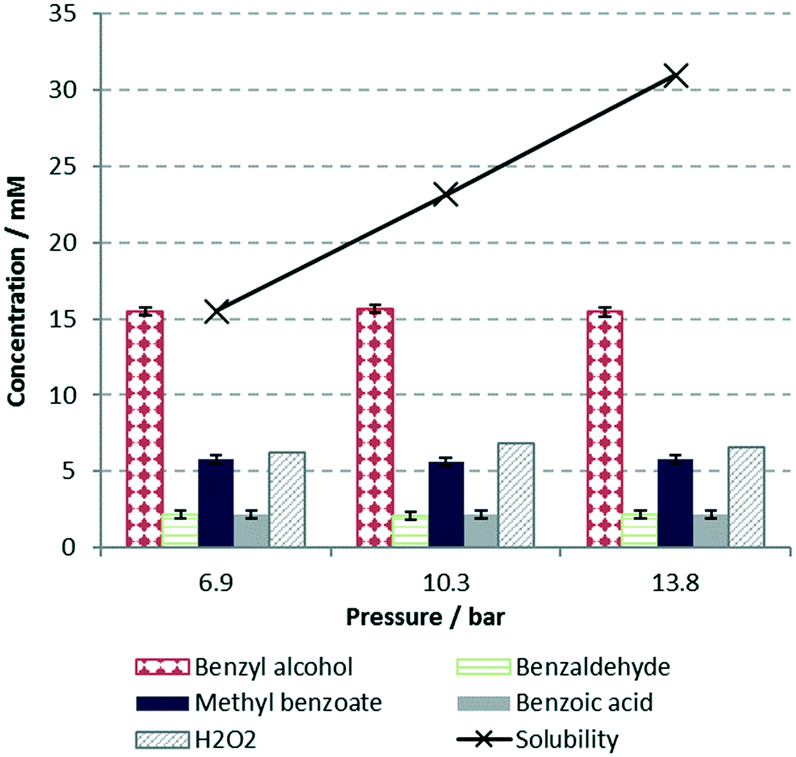 | ||
| Fig. 3 Effect of pressure on reaction outcome, plotted with the estimated solubility of O2, as predicted by Henry's law, using the Ostwald's coefficient reported by Tokunaga.4b Conditions: benzyl alcohol in MeOH/H2O (25 mM), 2 equiv. of H2O2, 70 °C, 200 mg Au/TiO2, τ = 13 seconds. | ||
A Hammett plot constructed by Fristrup et al.13 for the aerobic oxidation of alcohols to aldehydes over Au/TiO2 suggests that the rate-determining step involved the development of a partial positive charge on the benzylic carbon. In the present work, the rate of alcohol consumption at 70 °C was similarly measured for four para-substituted benzyl alcohols: 4-X-C6H4CH2OH; where X = Me, MeO, Cl and CF3. The resultant Hammett plot (Fig. 4) revealed a gradient ρ with a value of −0.94, which is similar to that reported for the aerobic process (where ρ = −1.01). Thus, it is reasonable to deduce that oxidations using hydrogen peroxide is also limited by the formation of the benzaldehyde intermediate.
 | ||
| Fig. 4 Hammett plot for the aldehyde formation step, where kX and kH are the k1 rate constants for the para-substituted (X = Me, MeO, Cl, CF3) and unsubstituted (X = H) benzyl alcohols, respectively. Reaction conditions: 25 mM alcohol in MeOH/H2O, 2 equivalents of H2O2, 100 mg Au/TiO2, 200 psi, 70 °C. | ||
A simplified kinetic model was constructed to deconvolute the catalytic system into four discreet processes (Scheme 5): the oxidation of benzyl alcohol with H2O2 to form the benzaldehyde intermediate (eqn (1)), which can form either the methyl ester or acid (eqn (2) and (3)). The decomposition of H2O2 was also included in the model (eqn (4)). All the reactions were assumed to be irreversible, and the number of catalyst sites, as well as methanol and water (both present in excess), are assumed to be constant and thus incorporated into the rate constants.
 | ||
| Scheme 5 Kinetic model and rate equations used in modelling. | ||
Experimental kinetic data were generated by recording the steady-state conversion of benzyl alcohol by varying residence times and reaction temperatures. Using an ordinary differential equation solver (Berkeley Madonna), fitting of the experimental data to the model above returned excellent fits (Fig. S9–S11, ESI†) and the extrapolated rate constants are given in Table 1.
| Rate constants | 50 °C | 70 °C | 100 °C | E a/kJ mol−1 | ln A |
|---|---|---|---|---|---|
| a Reaction conditions: 25 mM benzyl alcohol, 50 mM H2O2, 100 mg Au/TiO2 (45–53 μm), 200 psi, 0.8–2.5 mL min−1. | |||||
| k 1/M−1 L−1 | 1.2 | 3.0 | 5.7 | 30.3 | 11.6 |
| k 2/M−1 L−1 | 7.7 | 13.5 | 21.5 | 20.4 | 9.7 |
| k 3/M−1 L−1 | 4.5 | 7.9 | 9.4 | 14.4 | 7.0 |
| k 4/s−1 | 0.10 | 0.30 | 0.17 | — | — |
In the following we will discuss the rate constants and activation energies from the fitting exercise. These data need to be considered with caution as we show later that mass transfer effects contribute to the overall reactor/catalyst performance (Fig. 6). Nevertheless, since external mass transfer would be the first step in the sequence outlined in Scheme 4, it is completely valid to compare relative rates and activation energies. However, the actual values in Table 1 are likely to contain a mass transfer contribution, which will need to be addressed at a later stage.
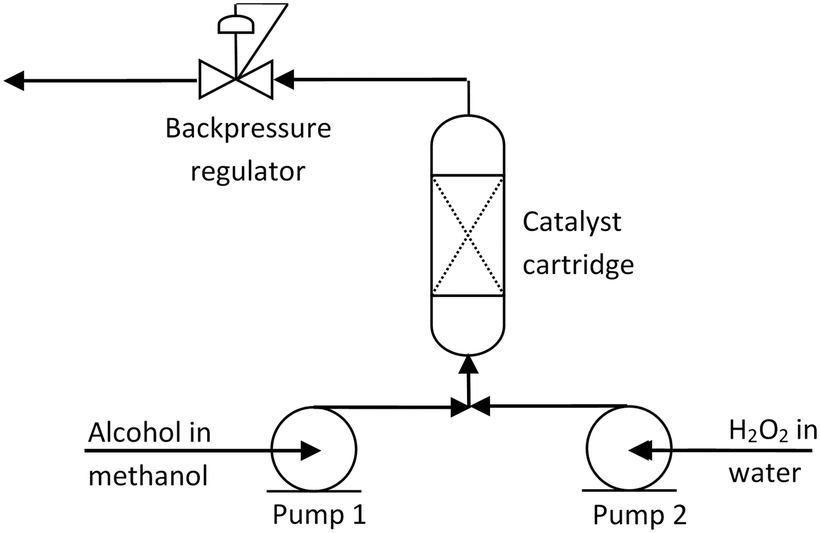 | ||
| Fig. 5 General schematic of the flow reactor used in the kinetic experiments. | ||
 | ||
| Fig. 6 Rate and (single-pass) conversion of alcohol vs. flow rate. | ||
In accordance with the Hammett study, the oxidation of alcohol to the aldehyde (k1) is rate-limiting. Interestingly, the decomposition of H2O2 is several orders of magnitude slower in the catalytic system, compared to that established in the absence of the reactants (Fig. S6, ESI†). This is attributed to a stronger affinity of the active sites towards organic substrates, compared to binding of H2O2 – implying that both oxidant and substrates are competing for the same catalyst sites. In a previous DFT study, the decomposition of H2O2 over Au/TiO2 was proposed to occur at the interface between TiO2(110) and Au(110).11 This supports the hypothesis that the oxidative transformation of primary alcohols occurs at the edge of the Au nanoparticle, where Au(110) surfaces dominate.
Notably, the activation energy calculated for the oxidation of benzyl alcohol to benzaldehyde is substantially lower (30.3 kJ mol−1) than that previously reported for the oxidation of 4-methyl benzyl alcohol using O2 and Au/CeO2 as the catalyst8 (59.5 kJ mol−1). We attribute this to the higher effective concentration of the oxidant, and better mass transfer of H2O2 to the active catalytic site (see Experimental section) compared to the diffusion of O2.
While the rate constants (k2) for ester formation are higher than those for acid formation (k3), it was somewhat surprising to find that their corresponding activation energies are also higher (20.4 vs. 14.4 kJ mol−1), which were verified independently by repeating the experiments using benzaldehyde as the precursor (Fig. S12–S15, ESI†); affording Ea values of 21.7 and 15.0 kJ mol−1 for the formation of ester and acid, respectively (Fig. S16, ESI†).
The higher Ea value for the ester formation may be due to the greater steric congestion in the tetrahedral intermediate I compared to II (Scheme 6), which affects the subsequent hydride abstraction step. The faster rate for ester formation is attributed to a higher pre-exponential factor (ln A), resulting from a more favourable formation of the hemiacetal intermediate I than the acetal II, as methanol is more nucleophilic than water. This is supported by reported values for the equilibrium constants corresponding to the formation of the hemiacetal (3.6 × 10−3 M−1), which is an order of magnitude greater than that for the formation of acetal (1.5 × 10−4 M−1).14
 | ||
| Scheme 6 Comparative condensation of benzaldehyde with O-electrophiles. | ||
Experimental
Catalyst preparation
Catalyst S1 was obtained from commercial sources.15 Catalyst S2 was prepared by following a procedure described by Haruta:16 An aqueous suspension of TiO2 P25 (Degussa 50 m2 g−1) was prepared and heated under continuous stirring to a temperature of 70 °C. An aqueous NH4OH solution (0.1 M) was added to adjust the pH to 9. This was followed by the addition of an aqueous solution of chloroauric acid (HAuCl4·3H2O, 2.53 mmol), at a rate of 50 mL h−1 with stirring. The pH of the solution was then readjusted to 9, and the reaction mixture was left to age for 2 h, while vigorous stirring was maintained at the reaction temperature. The solution was then cooled to room temperature. The solid was collected by suction filtration, and the catalyst was washed several times with deionized water to remove the chloride ions. After each wash, the filtrate was tested with AgNO3 to determine the presence of chloride ions (indicated by the formation of a white precipitate). The catalyst was then left to dry in a vacuum oven at 80 °C for 2 h, before it was calcined in air at 300 °C for 4 h. Catalysts were characterized for bulk elemental composition and for gold loading using X-ray florescence (XRF) spectroscopy. BET was employed for surface area and TEM for particle size measurements. Prior to use, S1 and S2 were ground, pelleted and sieved to give a particle size range between 38–53 μm.Oxidative esterification of alcohols and methanol
Experiments were performed in a commercially available packed-bed continuous flow reactor system (X-Cube®), described in our previous work.7 All reagents were used as received from commercial sources without purification, and all solvents were degassed. In a typical experiment, an equimolar or a 10-fold excess of aqueous H2O2 (50 or 500 mM) and the alcohol substrate (50 mM in methanol), were fed via two HPLC pumps into a tubular packed-bed reactor (70 mm × 4 mm) containing a packed bed of catalyst S1 or S2 (100 mg) sandwiched between two layers of quartz sand (ca. 3 cm each). Reactants were simultaneously fed into the reactor at 0.1, 0.2, 0.3 and 0.4 mL min−1. All four flow rates were operated at 30, 50, 70, 85 and 100 °C, at atmospheric pressure. Aliquots (2.0 mL) were collected at each flow rate, but only after the reactor had reached isothermal equilibrium and at least 4 mL of product had passed through the system and steady state had been achieved. An indicator paper was used to measure the approximate pH of the aqueous samples. The samples were then diluted with 2.0 mL of acetone to ensure homogeneity, and analysed by a GC-MS fitted with a Zebron Z8 wax plus column (30 m × 0.32 mm; temperature range 20–250/260 °C), using dimethyl benzamide as an internal standard. No catalytic activity was observed in repeated blank experiments with TiO2 (catalyst support), thus proving that the presence of gold is essential for catalysis.Benzyl alcohol oxidation (kinetic experiments)
Catalytic reactions with benzyl alcohol were carried out in a custom-built Stokes flow reactor described in our earlier work (Fig. 5),17 consisting of a PFA tube (100 × 4 × 6 mm) containing a packed catalyst bed inserted into an aluminium heater block. The reactor was fed by two syringe pumps, whose outputs were merged at a T-piece before entering the bed. Downstream from the packed bed was a thermocouple to measure the temperature of the fluid on exiting the bed, as well as a pressure gauge and backpressure regulator to pressurize the reactor up to 250 psi.The packed catalyst bed (S1) contained a layer of 1 wt% Au/TiO2 (100–200 mg, ground and sieved to 45–53 μm) sandwiched between silica (pore size: 60 Å, 40–63 μm technical grade). At the start of each experiment, one syringe pump was loaded with a solution of benzyl alcohol in methanol (50 mM), and the other with aq. H2O2 (100 mM, prepared from a 30 wt% solution). The pumps were started at 0.5 mL min−1 (providing a total flow rate of 1 mL min−1). Once the dead volume of the system had been displaced, the reactor was pressurised (typically to 200 psi) and then heated, after which the desired pump flow rates were set (between 0.8 and 2.5 mL min−1); and samples of effluent were collected. For kinetic experiments, three samples were collected at each flow rate.
Sample analysis: 0.1 mL of the effluent was diluted in 0.7 mL methanol containing an external standard (4.4 mM 4-chlorobenzyl alcohol), before analysis by HPLC using a C18 column (150 × 4.6 mm). Additionally, the peroxide concentration in the effluent was determined by adding 160 μL of effluent to 4 mL of a TiOSO4 solution (0.17 M in aq. 2 M H2SO4) and the absorbance of the resultant solution was measured at 410 nm, and compared to a calibration curve. The Peclet numbers (Pe) for the reactor was determined by tracer experiments, and the reactor was found to operate under plug flow conditions (ESI†).
Kinetic modelling
For the kinetic analysis, recovered catalysts (retained in the PFA tube) were re-used with no observable loss in catalyst activity; hence, catalyst deactivation was not observed within the experimental timescale. Kinetic modelling and data-fitting was performed using a differential equation solver.18 The reaction equations and initial concentrations, along with initial guesses of rate constants, were used as inputs. These data were inputted into the program to generate predicted concentration vs. time curves. In addition, experimental data were imported and used to float variables such as the rate constants, to produce a best fit. The solver employs a fixed step size Runge–Kutta integration method, with the errors measured as root-mean-squared differences between the experimental data and the model fits.Mass transfer resistance
Single-pass conversions of the alcohol were independent of catalyst particle size; however, the rate of alcohol consumption changed with flow rates (Fig. 6), indicating that the intrinsic rate of the oxidation was sufficiently fast for external mass transfer to be contributing. At 1 mL min−1 (the experimental flow rate), the reactions operated under a mixed regime. Thus, the reaction rate data contain mass transfer contributions, making the absolute rate constants and activation energies less accurate, but nevertheless still valid for comparisons between competing reactions. In theory, the flow rate could have been increased to ensure that measurements were made in the purely kinetic regime. However, attempts to do this led to unacceptable pressure drops and conversions of less than <20% which precluded accurate determination of product distributions.Conclusions
We have shown that H2O2 can be used as a more effective reagent than O2 for the oxidative conversion of alcohols to acid derivatives. No base is needed for the reactions, and faster turnovers can be achieved by increasing the effective concentration of the oxidant. Furthermore, selective oxidation of a primary alcohol to its acid or methyl ester (demonstrated using 1-decanol and benzyl alcohol) can be achieved by adjusting the catalyst particle size, reaction temperature and stoichiometry, thus allowing flexibility for industrial applications.Kinetic studies revealed that the formation of ester has a higher activation energy than the competing acid formation; however, this is offset by inherent predispositions in the system, attributed to a more favourable equilibrium concentration of the hemiacetal intermediate, expediting the formation of the ester at higher reaction temperatures.
Due to the fast nature of the oxidation reactions, typical laboratory reactors using high concentrations of oxidants will be operating in a mixed regime with some mass transfer contribution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Syngenta and the Pharmacat Consortium in supporting a EPSRC Doctoral Training Award to FJR.Notes and references
- J. H. Teles, I. Hermans, G. Franz and R. A. Sheldon, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000, DOI:10.1002/14356007.a18_261.pub2.
- (a) N. Dimitratos, J. A. Lopez-Sanchez and G. J. Hutchings, Chem. Sci., 2012, 3, 20–44 RSC; (b) T. Takei, T. Akita, I. Nakamura, T. Fujitani, M. Okumura, K. Okazaki, J. H. Huang, T. Ishida and M. Haruta, in Advances in Catalysis, ed. B. C. Gates and F. C. Jentoft, Elsevier, 2012, vol. 55, pp. 1–126 Search PubMed; (c) Y. Zhang, X. J. Cui, F. Shi and Y. Q. Deng, Chem. Rev., 2012, 112, 2467–2505 CrossRef CAS PubMed; (d) A. S. Sharma, H. Kaur and D. Shah, RSC Adv., 2016, 6, 28688–28727 RSC.
- C. P. Ferraz, M. A. S. Garcia, E. Teixeira-Neto and L. M. Rossi, RSC Adv., 2016, 6, 25279–25285 RSC.
- (a) T. Sato, Y. Hamada, M. Sumikawa, S. Araki and H. Yamamoto, Ind. Eng. Chem. Res., 2014, 53, 19331–19337 CrossRef CAS; (b) J. Tokunaga, J. Chem. Eng. Data, 1975, 20, 41–46 CrossRef CAS.
- A. Gavriilidis, A. Constantinou, K. Hellgardt, K. K. Hii, G. J. Hutchings, G. L. Brett, S. Kuhn and S. P. Marsden, React. Chem. Eng., 2016, 1, 595–612 RSC.
- (a) J. Ni, W.-J. Yu, L. He, H. Sun, Y. Cao, H.-Y. He and K.-N. Fan, Green Chem., 2009, 11, 756–759 RSC; (b) J. Ftouni, M. Penhoat, J.-S. Girardon, A. Addad, E. Payen and C. Rolando, Chem. Eng. J., 2013, 227, 103–110 CrossRef CAS; (c) L. A. Wang, W. B. Yi and C. Cai, ChemSusChem, 2010, 3, 1280–1284 CrossRef CAS PubMed; (d) T. Xie, M. Lu, W. W. Zhang and J. Li, J. Chem. Res., 2011, 397–399 CrossRef CAS.
- (a) J. B. Brazier, K. Hellgardt and K. K. Hii, React. Chem. Eng., 2017, 2, 60–67 RSC; (b) N. Zotova, K. Hellgardt, G. H. Kelsall, A. S. Jessiman and K. K. Hii, Green Chem., 2010, 12, 2157–2163 RSC.
- P. Sudarsanam, B. Mallesham, D. N. Durgasri and B. Reddy, J. Ind. Eng. Chem., 2014, 20, 3115–3121 CrossRef CAS.
- J. Gao, G. L. Fan, L. Yang, X. Z. Cao, P. Zhang and F. Li, ChemCatChem, 2017, 9, 1230–1241 CrossRef CAS.
- Methyl benzoate is used in flavourings and an insect pheromone, while benzoic acid is an anti-bacterial used in topical medicine.
- A. Thetford, G. J. Hutchings, S. H. Taylor and D. J. Willock, Proc. R. Soc. A, 2011, 467, 1885–1899 CrossRef CAS.
- (a) G. Eisenberg, Ind. Eng. Chem., 1943, 15, 327–328 CAS; (b) B. J. Deadman, K. Hellgardt and K. K. Hii, React. Chem. Eng., 2017, 2, 462–466 RSC.
- P. Fristrup, L. B. Johansen and C. H. Christensen, Catal. Lett., 2008, 120, 184–190 CrossRef CAS.
- J. P. Guthrie, Can. J. Chem., 1978, 56, 2342–2354 CrossRef CAS.
- At the beginning of the project, the catalyst S1 was purchased from the World Gold Council, who has since ceased to supply this catalyst, at which point we procured it from STREM (AuroLite™). We did not notice any difference between the performance of these catalysts in this work.
- S. Tsubota, M. Haruta, T. Kobayashi, A. Ueda and Y. Nakahara, in Studies in Surface Science and Catalysis, ed. G. Poncelet, P. A. Jacobs, P. Grange and B. Delmon, Elsevier, 1991, vol. 63, pp. 695–704 Search PubMed.
- J. B. Brazier, B. N. Nguyen, L. A. Adrio, E. M. Barreiro, W. P. Leong, M. A. Newton, S. J. A. Figueroa, K. Hellgardt and K. K. Hii, Catal. Today, 2014, 229, 95–103 CrossRef CAS.
- https://www.berkeleymadonna.com/ .
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental procedure, characterization of reactor, catalysts, and data. See DOI: 10.1039/c8re00085a |
| This journal is © The Royal Society of Chemistry 2018 |