
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrowave irradiation: a green approach for the synthesis of functionalized N-methyl-1,4-dihydropyridines†
M. Musawwer Khan *a,
Saigala,
Sarfaraz Khana,
Sumbulunnisan Shareefa and
Subash C. Sahoob
*a,
Saigala,
Sarfaraz Khana,
Sumbulunnisan Shareefa and
Subash C. Sahoob
aDepartment of Chemistry, Aligarh Muslim University, Aligarh 202002, India. E-mail: musawwer@gmail.com
bDepartment of Chemistry, Center of Advanced Studies in Chemistry, Panjab University, Chandigarh-160014, India
First published on 14th December 2018
Abstract
An eco-friendly and cost-effective, microwave-assisted green approach has been developed for the synthesis of diverse functionalized N-methyl-1,4-dihydropyridines (1,4-DHPs). This pseudo three-component reaction was carried out between two equivalents of (E)-N-methyl-1-(methylthio)-2-nitroethenamine (NMSM) and one equivalent of aromatic aldehydes under microwave irradiation at 100 °C without catalyst and solvent. Short reaction times, avoidance of toxic solvents or expensive, metallic and corrosive catalysts and no need for column chromatographic purification are among the valuable features of the presented method. Moreover, the “greenness” of the method was evaluated within the ambits of the defined green metrics such as atom economy, carbon efficiency, E-factor, reaction mass efficiency, overall efficiency, process mass intensity and solvent intensity and the method exhibited a good to excellent score.
Introduction
The development of novel, green and sustainable synthetic procedures has received substantial attention because of the growing environmental pollution and its overwhelming effect on human life. Therefore, the design of synthetic protocols with efficient atom economy under catalyst- and solvent-free conditions based on the microwave utilizing multicomponent reaction (MCR) strategy has become a central theme in the evolution of green chemistry.1 MCRs have the ability to generate conveniently only one product from three or more components in a single synthetic operation with multiple bond formation and high atom economy.2 Over the past decade, several classes of heterocycles including natural and unnatural compounds have been successfully synthesized through MCRs.3 Owing to the simple experimental processes and one-pot procedure, the application of catalytic methods involving MCR has gained remarkable attention as it allows simple and straightforward access to novel heterocyclic libraries of small molecules.4 MCRs are now renowned and considered as an efficient tool for generating complex and diverse molecules of medicinal importance.5 They also have some inherent advantages such as high bond efficiency, convergence and step economy, short synthetic period, reduction in waste products, high selectivity as well as operational simplicity.6 On the other hand, the microwave irradiation assisted synthetic protocol has also gained importance, mainly in developing sustainable solvent- and catalyst-free alternatives to laborious reactions. Additionally, these approaches give better results in contrast to traditional synthesis in terms of waste reduction, extreme drop in reaction times, and relatively isolated pure products, thereby avoiding tedious column chromatographic purification. Hence they embody a prevailing green alternative to conventional synthesis.7Among the nitrogen containing heterocyclic compounds, functionalized 1,4-dihydropyridines are considered as invaluable scaffolds because of their abundance in natural products8 and exhibit a variety of biological activities such as antihypertension,9 anticancer,10 antioxidant,11 antiviral,12 antitumor,13 anti-inflammatory,14 analgesic15 and potential calcium channel antagonist activities.16 1,4-Dihydropyridines (1,4-DHPs), particularly 4-aryl-substituted-1,4-dihydropyridines, also possess various pharmacological activities like vasodilatory, anti-atherosclerotic, bronchodilatory, antiradical, antitumor and anti-diabetic effects.17 Extensive studies have revealed that these compounds display other therapeutic properties including HIV protease inhibition, radioprotection, neuroprotection, platelet and antiaggregatory activities etc.18 For example, compound I was found to be an anticonvulsant while compound II exhibits antileishmanial properties.19
Recently, sugar derived 1,4-dihydropyridine III was revealed to show antitumor activity against two types of tumor cell lines, HepG2 and HeLa (Fig. 1).20 Therefore, the preparation of 1,4-dihydropyridines is one of the most attractive areas of research for synthetic and medicinal chemists due to their diverse applications. For the synthesis of 1,4-DHPs, starting from the classical Hantzsch reaction,21 a number of protocols have been reported.22
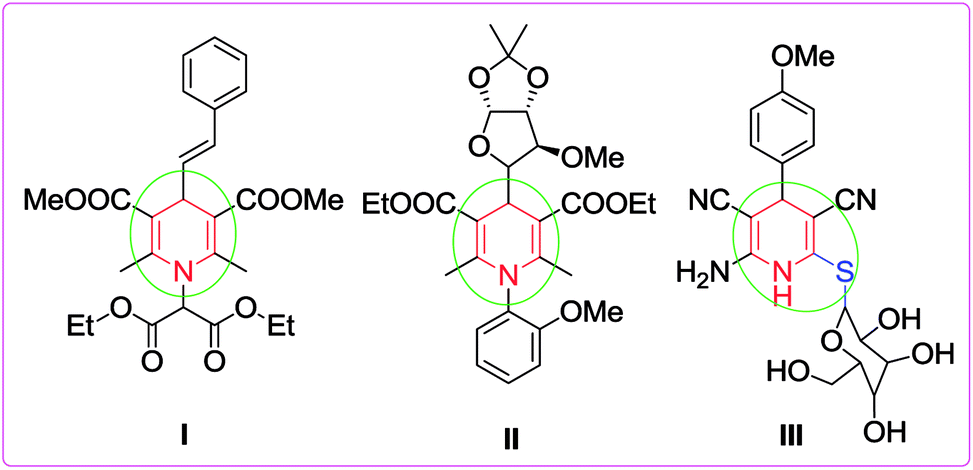 | ||
| Fig. 1 Some biologically active highly substituted 1,4-DHPs. | ||
In recent times, (E)-N-methyl-1-(methylthio)-2-nitroethenamine (NMSM) has been found to be a very attractive building block for the synthesis of a variety of nitrogen and oxygen containing heterocyclic compounds.23 To the best of our knowledge, only one protocol has been reported in the literature so far for the synthesis of N-methyl-1,4-dihydropyridines by using NMSM and aromatic aldehydes in presence of a 2-aminopyridine catalytic system.24 However, the above method suffered from some limitations, like very long reaction time, used of toxic catalyst, used of solvent, and lower yields for some of the products. Hence, the need to develop a rapid, efficient, and environmentally benign synthetic protocol for the preparation of such 1,4-DHP derivatives is paramount and justified.
Very recently, we have reported a green approach for the synthesis of 4H-chromen-5-ones by using NMSM, dimedone and aromatic aldehydes under catalyst- and solvent-free conditions25 and our group also developed green protocols for the synthesis of biologically active heterocycles such as N-substituted-1,4-dihydropyridines, coumarin-fused 1,4-dihydropyridines, tetrahydropyridines, functionalized furan-2-one and pyrrol-2-one.26 In continuation of our research interest to develop newer synthetic methodologies under greener conditions, this paper describes an environmentally benign and sustainable method for the synthesis of highly functionalized N-methyl-1,4-DHPs under microwave irradiation without catalyst and solvent as depicted in Scheme 1.
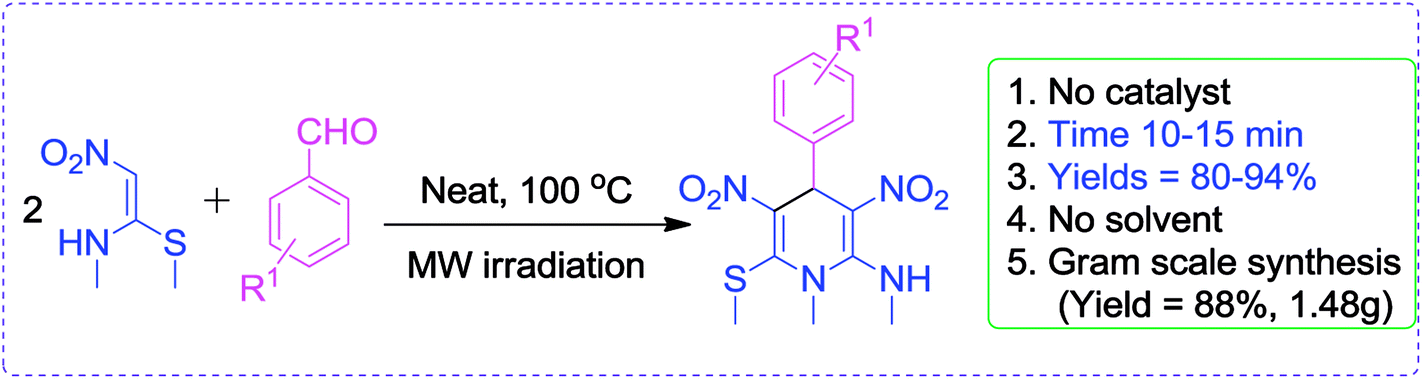 | ||
| Scheme 1 Synthesis of highly functionalized N-methyl-1,4-dihydropyridines. | ||
Results and discussion
In our initial studies, a trial reaction was performed with benzaldehyde 1a (1.0 mmol) and NMSM 2a (2.0 mmol) in the presence of 10 mol% ZnCl2 under refluxing ethanol. The reaction progress and consumption of starting materials was checked by TLC. After completion of the reaction as monitored by TLC, the highly substituted N-methyl-1,4-DHP derivative 3a was isolated in 62% yield and the product was characterized by physical properties24 and spectroscopic techniques including IR, 1H and 13C NMR (Table 1, entry 1). Next, we focused on an optimization study to obtain the best results in terms of yield and time. To this end, different catalysts including FeCl3, SnCl2·2H2O, p-toluenesulfonic acid (p-TSA), piperidine and DBU (10 mol%) were screened in the model reaction under refluxing ethanol. Unfortunately, we did not obtain any improvement in results (Table 1, entries 2–6). Next, we attempted the same reaction in the presence of 10 mol% ZnCl2 under solvent-free conditions at 80 °C and isolated 70% yield of the product (Table 1, entry 7). Similarly, we have also executed the same reaction under microwave irradiation at 80 °C without solvent in the presence of 10 mol% ZnCl2, and isolated 79% yield of the product within 20 min. Then the same reaction was also attempted in catalyst- and solvent-free conditions under microwave irradiation at 80 °C. Unexpectedly, this gave a better result in comparison with the above experiments (Table 1, entry 9). Next, to optimize the reaction temperature of this transformation, the above model reaction was executed at different temperatures including 90 °C, 100 °C, 105 °C, 110 °C and 120 °C under microwave irradiation excluding catalyst and solvent (Table 1, entries 10–14). From the above observations, the reaction performed without catalyst and solvent under microwave irradiation at 100 °C emerged as the optimal reaction conditions in terms of the yields and time (Table 1, entry 11). The optimization of reaction yields vs. the reaction temperature is depicted in Fig. 2.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | Temp. (°C) | Time | Yieldb (%) |
| a Reaction conditions: NMSM (1a 2.0 mmol), benzaldehyde (2a 1.0 mmol).b Isolated yields. μw = microwave irradiation. | |||||
| 1 | ZnCl2 (10) | EtOH | 80 | 5 h | 62 |
| 2 | FeCl3 (10) | EtOH | 80 | 7 h | 52 |
| 3 | SnCl2·2H2O (10) | EtOH | 80 | 8 h | 57 |
| 4 | p-TSA (10) | EtOH | 80 | 12 h | 32 |
| 5 | Piperidine (10) | EtOH | 80 | 15 h | 22 |
| 6 | DBU (10) | EtOH | 80 | 10 h | 10 |
| 7 | ZnCl2 (10) | Neat | 80 | 50 min | 70 |
| 8 | ZnCl2 (10) | Neat | 80 (μw) | 20 min | 79 |
| 9 | None | Neat | 80 (μw) | 20 min | 82 |
| 10 | None | Neat | 90 (μw) | 15 min | 89 |
| 11 | None | Neat | 100 (μw) | 10 min | 94 |
| 12 | None | Neat | 105 (μw) | 10 min | 93 |
| 13 | None | Neat | 110 (μw) | 10 min | 91 |
| 14 | None | Neat | 120 (μw) | 10 min | 89 |
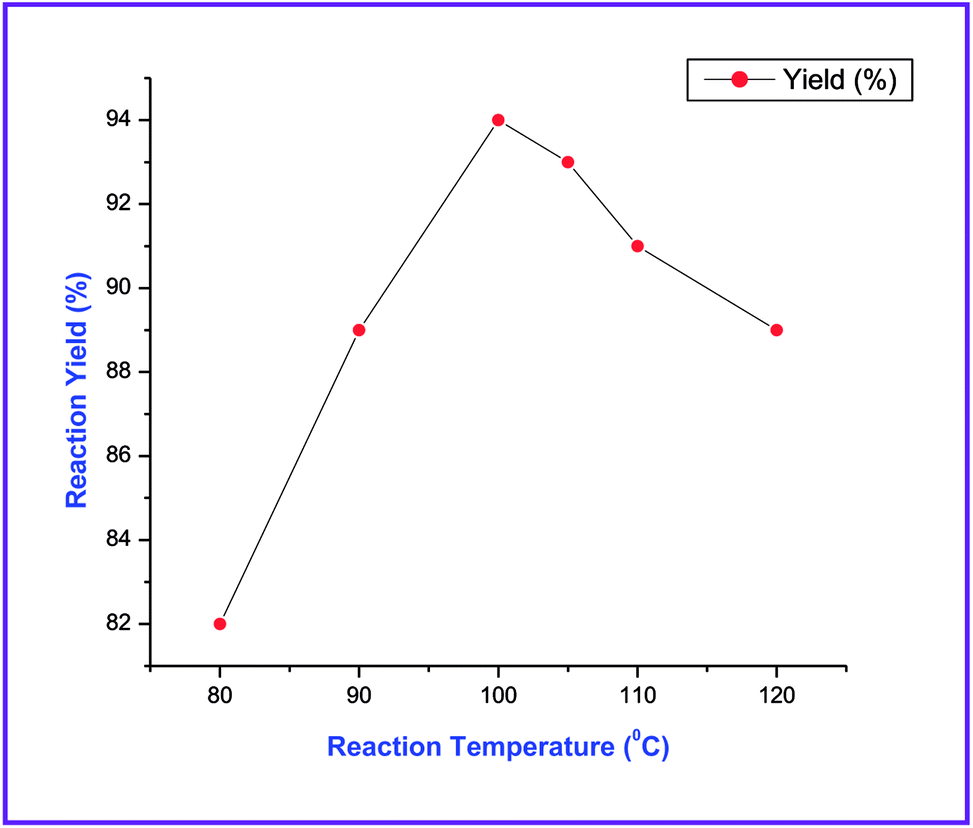 | ||
| Fig. 2 % yield of reaction vs. reaction temperature for the synthesis of 3a. | ||
After establishing the optimized conditions, we examined the reaction of NMSM with 4-nitrobenzaldehyde and obtained the desired N-methyl-1,4-dihydropyridine derivative 3b in 85% yield within 10 min. Next, we proceeded to investigate the substrate scope for the presented method with various aromatic aldehydes having substituents including NO2, Cl, Br, F, OMe, Me and Et under the optimal reaction conditions, and the reaction time and yield of the isolated products 3c–3o are summarized in Table 2.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ar | Product | Time | % yieldb | Mp (°C) | Reported mp (°C)24 |
a NMSM and aromatic aldehydes in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, microwave irradiation (200 W) at 100 °C.b Isolated yields. :1 ratio, microwave irradiation (200 W) at 100 °C.b Isolated yields. |
||||||
| 1 |  |
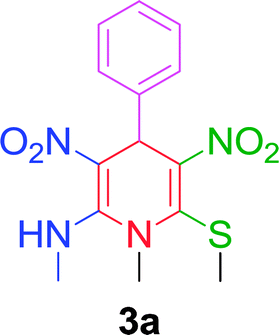 |
10 min | 94 | 198–200 | 201 |
| 2 |  |
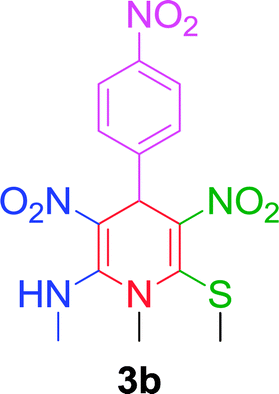 |
10 min | 85 | 218–220 | — |
| 3 | 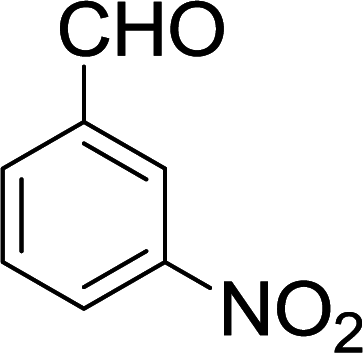 |
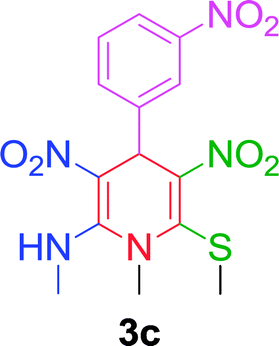 |
15 min | 86 | 216–218 | — |
| 4 | 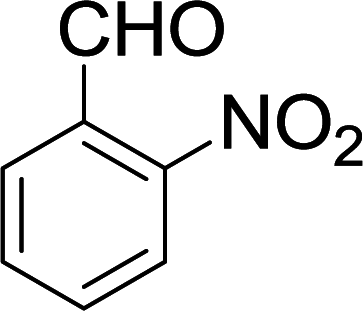 |
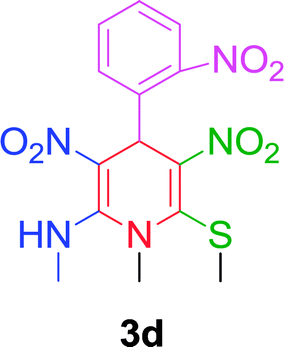 |
15 min | 85 | 226–228 | 226 |
| 5 |  |
 |
10 min | 87 | 210–212 | 213 |
| 6 |  |
 |
10 min | 85 | 208–210 | — |
| 7 | 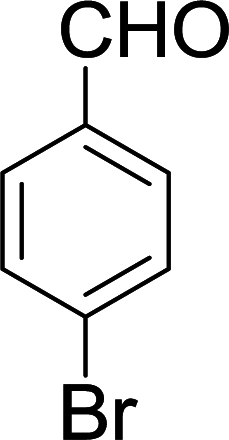 |
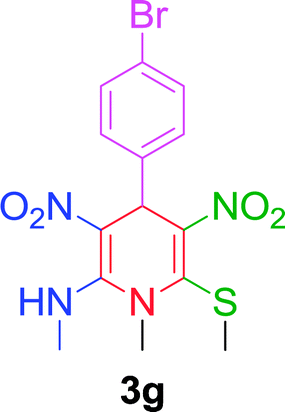 |
10 min | 86 | 218–220 | 219 |
| 8 | 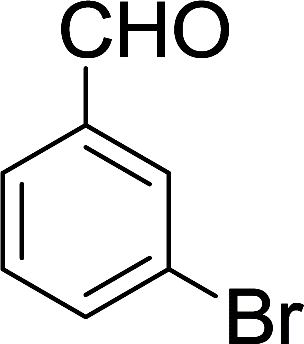 |
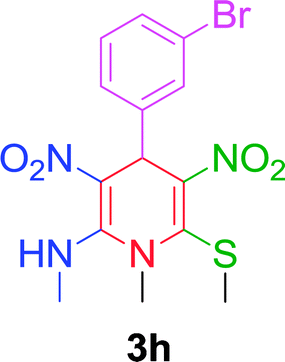 |
15 min | 88 | 206–208 | — |
| 9 |  |
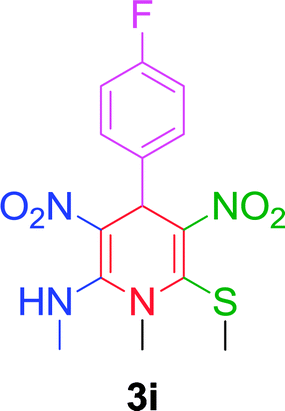 |
10 min | 90 | 230–232 | 230 |
| 10 |  |
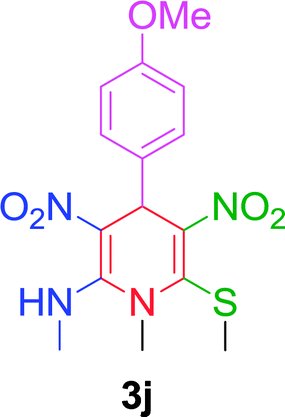 |
10 min | 92 | 200–202 | 203 |
| 11 | 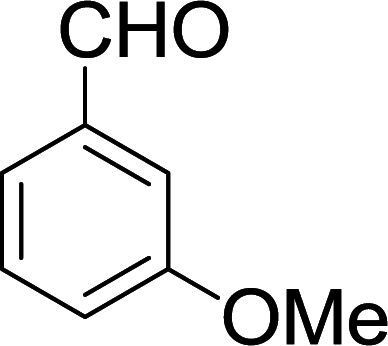 |
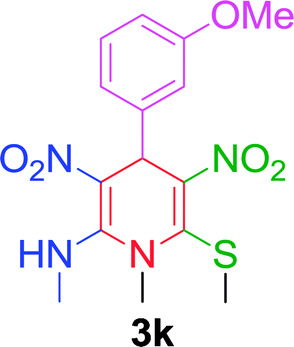 |
15 min | 88 | 236–238 | — |
| 12 | 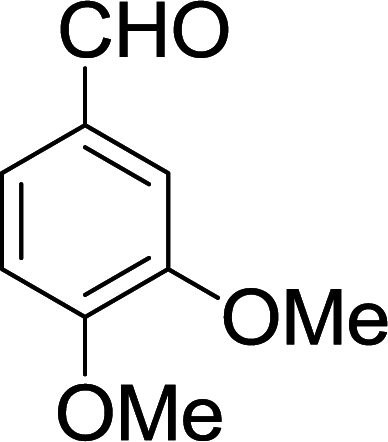 |
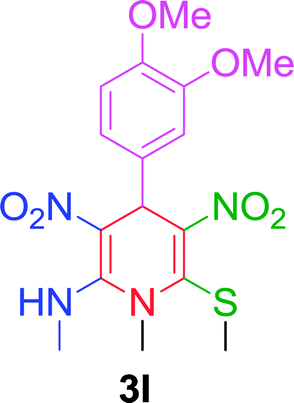 |
10 min | 90 | 242–244 | — |
| 13 | 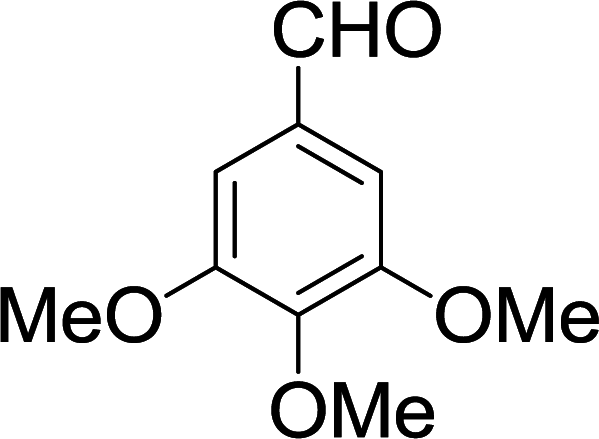 |
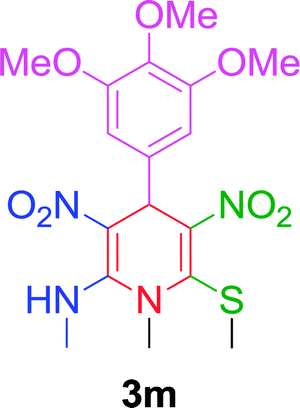 |
10 min | 92 | 260–262 | — |
| 14 | 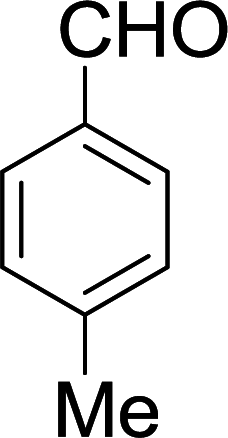 |
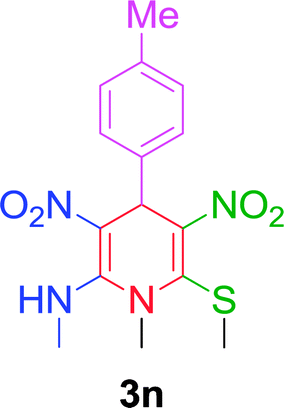 |
10 min | 94 | 212–214 | 214 |
| 15 | 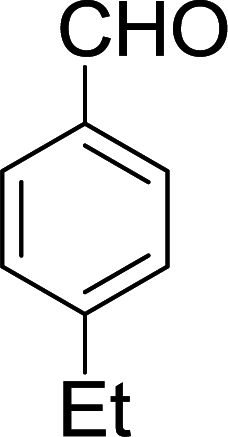 |
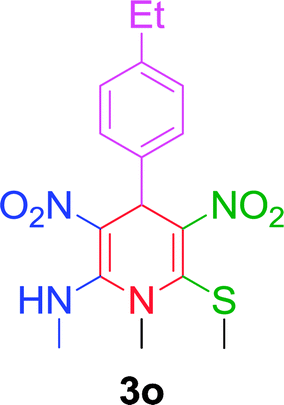 |
10 min | 92 | 236–238 | — |
| 16 | 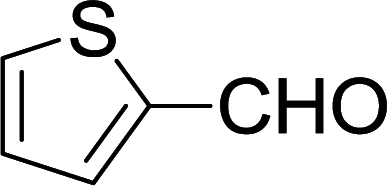 |
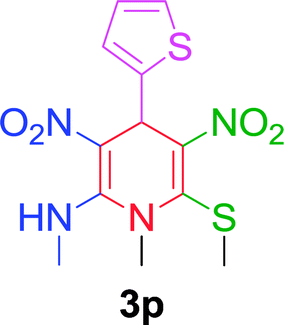 |
15 min | 86 | 208–210 | 209 |
| 17 |  |
 |
15 min | 80 | 158–160 | 149 |
| 18 |  |
 |
15 min | 86 | 228–230 | 229 |
| 19 |  |
 |
10 min | 88 | 222–224 | 217 |
In general, the reaction proceeded smoothly under the optimized conditions to afford the desired products in good to excellent yields. It was observed that several functionalities, including both electron donating and electron withdrawing groups, in the aromatic ring were compatible with the reaction conditions.
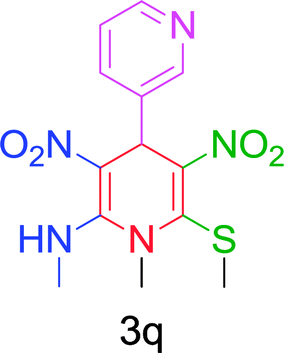
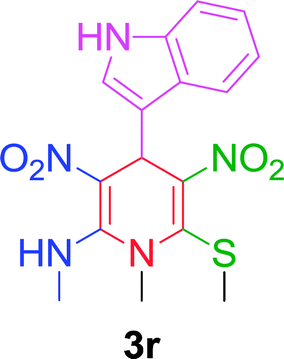
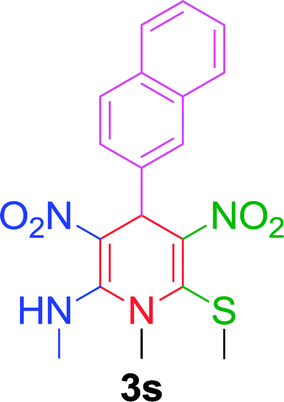
Furthermore, the reactions of NMSM with heteroaryl aldehydes including thiophene-2-carbaldehyde, pyridine-3-carbaldehyde and indole-3-carbaldehyde were executed under this protocol and yielded the corresponding products 3p–3r in 80–86% yield. In addition, 2-naphthaldehyde also gave the desired product 3s in 88% yield on heating with NMSM under microwave irradiation at 100 °C as shown in Table 2. After achieving excellent results in terms of yields and time, we moved to practical testing and explored our protocol on the gram-scale level under the optimized conditions.
The gram scale preparation of 3a was carried out, wherein benzaldehyde (5.0 mmol) successfully reacted with NMSM (10.0 mmol) under the optimized conditions to yield the desired product in 88% yield (1.48 g) after 15 min as depicted in Scheme 1. The newly synthesized compounds were confirmed by their melting points, spectroscopic techniques (FTIR, 1H NMR, 13C NMR, HRMS) and elemental analysis, and matched well with the reported data.24 The spectroscopic data are in good agreement with the proposed structure and some characteristic NMR peaks assigned for compound 3n are shown in Fig. S2 (see ESI†). Finally, the structure as well as the relative stereochemistry of 1,4-dihydropyridine 3n was confirmed by X-ray crystallographic analysis (CCDC 1841358). From the crystal structure, it was found that the 1,4-dihydropyridine ring adopted a boat conformation in which the flag pole positions 1 and 4 are occupied by the –N–CH3 and –Ph–CH3 group respectively. The ORTEP representation of compound 3n is presented in Fig. 3.
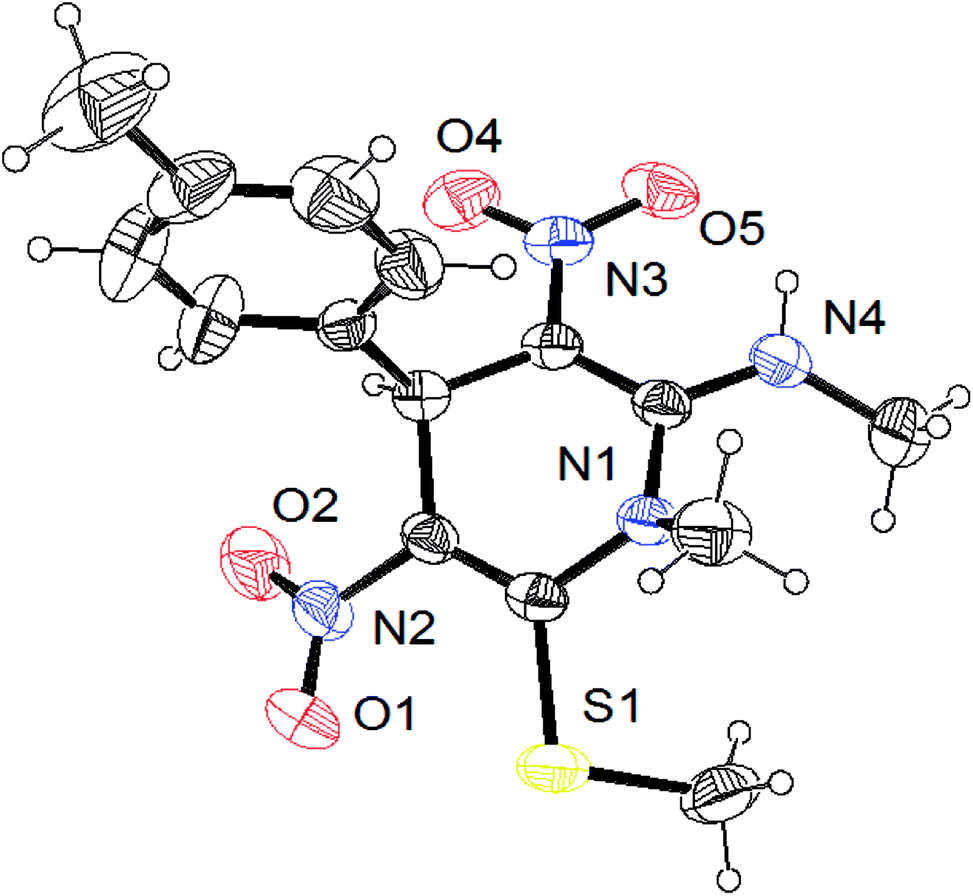 | ||
| Fig. 3 ORTEP diagram of compound 3n (CCDC 1841358). | ||
A single crystal was mounted on a glass fibre and used for data collection. A metallic light yellow coloured crystal of compound 3n was grown by slow evaporation of acetonitrile solvent for X-ray analysis. The single crystal X-ray diffraction data were collected from a SuperNova diffractometer, single source at offset/far, with a HyPix3000 detector using graphite-monochromated MoKα radiation (λ = 0.71073 Å) at 293 K. The crystallized compound has a triclinic crystal system, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , and unit cell dimensions a = 7.3596(3) Å, b = 9.2464(6) Å, c = 14.1726(5) Å. The unit cell packing of the compound 3n is shown in Fig. 4.
, and unit cell dimensions a = 7.3596(3) Å, b = 9.2464(6) Å, c = 14.1726(5) Å. The unit cell packing of the compound 3n is shown in Fig. 4.
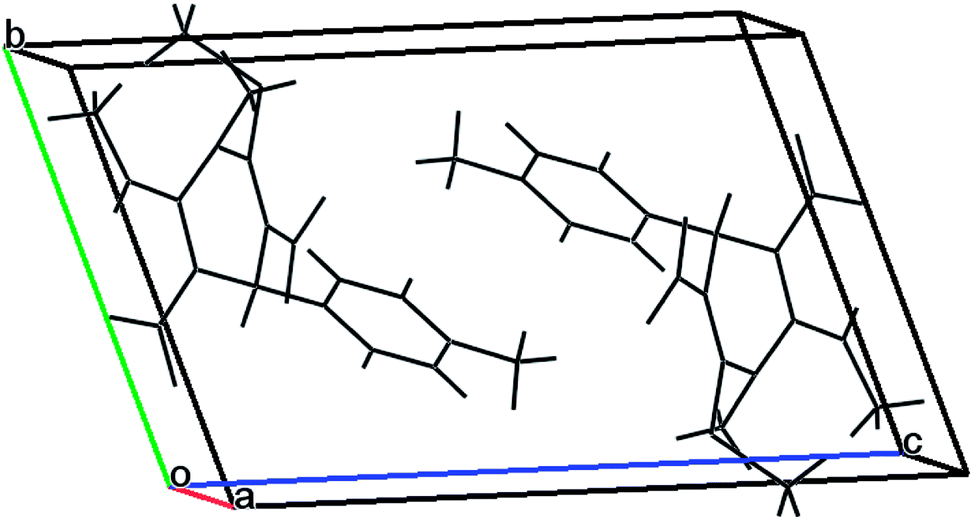 | ||
| Fig. 4 Unit cell packing of compound 3n. | ||
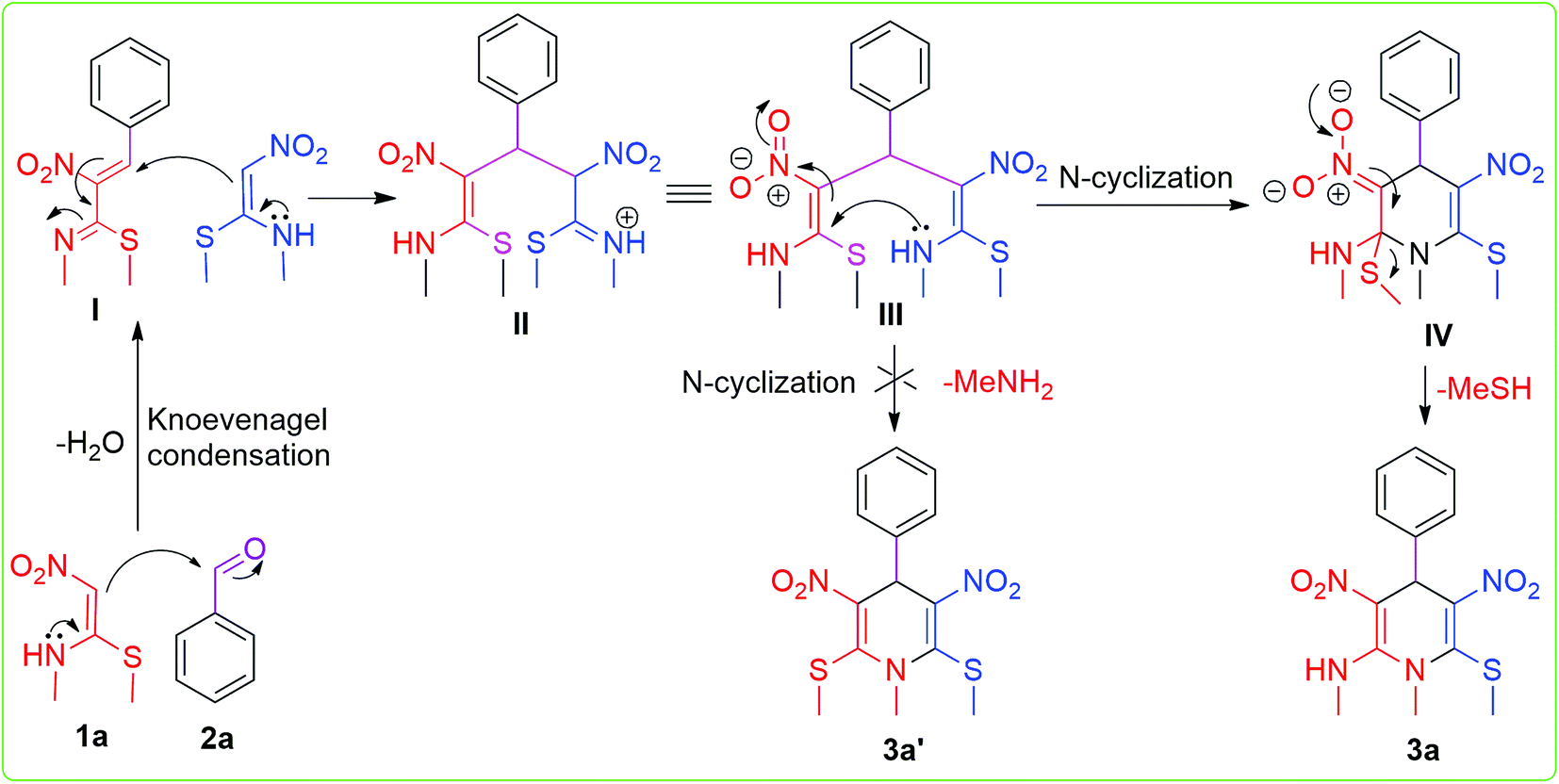
Finally, a plausible reaction mechanism for the synthesis of N-methyl-1,4-dihydropyridine was proposed, as shown in Scheme 2. We consider that the first step involves the Knoevenagel condensation of one molecule of NMSM and aromatic aldehyde to form intermediate I. To study the formation of proposed intermediate I and to isolate it, we have also conducted a reaction with NMSM (1.0 mmol) and benzaldehyde (1.0 mmol) under the optimised reaction conditions, but unfortunately we unable to isolate the intermediate I. In this control case we obtained the proposed product in lower yield. Nonetheless, we proceed on the assumption that the intermediate I was formed, which acted as a Michael acceptor and reacted with the Michael donor compound NMSM.23b,24
 | ||
| Scheme 2 Plausible reaction mechanism for the synthesis of N-methyl-1,4-DHPs. | ||
Another molecule of NMSM reacted with Knoevenagel product I and formed Michael adduct II, which underwent rotation to give conformer III. Then, the key intermediate III underwent intra-molecular N-cyclization to form species IV. Finally the species IV afforded the desired product by the elimination of –MeSH. In contrast the elimination of –MeNH2 and formation of 3a′ (2,6-bismethylthio derivative) were not observed. Comparing the leaving capacity of –MeNH2 and –MeSH, the latter is more labile and has a greater leaving tendency.
In the context of green chemistry, it was required to validate our synthetic method as environmentally benign, which was performed through the evaluation of green chemistry metrics for this one-pot synthesis of N-methyl-1,4-dihydropyridines.27–29 Several green metrics, including atom economy (AE), carbon efficiency (CE), reaction mass efficiency (RME), overall efficiency (OE), process mass intensity (PMI), E-factor and solvent intensity (SI), have been evaluated for all the synthesized compounds, which empowers us to assess the present synthetic method in terms of waste, carbon efficiency and energy usage. The calculated values of the green metrics of all the synthesized compounds are summarized in Table S2 (see ESI†).
The calculated values, including high atom economy (83–86%), carbon efficiency (93–94%), overall efficiency (85–94%), and small E-factor (4–3), solvent intensity (4–3) and PMI (5–4), endorse this presented method as green and sustainable.
Conclusion
In summary, the present study demonstrated the development of a rapid, sustainable, environmentally benign and highly efficient synthetic protocol for the preparation of N-methyl-1,4-dihydropyridine derivatives under microwave irradiation without catalyst and solvent. This protocol offers noteworthy advantages such as good to excellent yields, short reaction time, avoidance of toxic catalyst and solvent, wide range of applicability, purification by crystallization, easily available starting materials, low cost, clean reaction profiles and good amenability with the principles of green chemistry. In addition, the presented methodology produced smaller amounts of waste, with good to excellent values of green chemistry metrics (AE, CE, RME, OE, PMI, SI and E-factor), and our method stands as an exemplary choice in this framework.Experimental section
General information
Commercially available solvents and chemicals were obtained from suppliers and used to execute this work as received unless otherwise mentioned. All the reactions were performed on a Monowave 300 microwave synthesizer (Anton Paar) by employing a glass vial G10. TLC analysis was performed using silica gel GF-254 from SRL. Stuart digital melting point apparatus (SMP10) was used for the measurement of melting points, which are uncorrected. FTIR spectra were recorded in potassium bromide pellets on a Perkin-Elmer 10.4.00 IR spectrophotometer. 1H-NMR and 13C-NMR spectral analysis were carried out on Bruker (Avance-II 400 MHz, Varian-AS400 NMR) spectrometers using TMS as an internal standard and DMSO-d6 as a solvent. Crystal data were collected with a SuperNova diffractometer, single source at offset/far, with a HyPix3000 detector (CCD) at 293 K using graphite monochromated MoKα radiation (λ = 0.71073 Å). Elemental analysis was performed on a Perkin-Elmer-2400 CHN/S analyser and Thermo Scientific (FLASH 2000) analyser while high resolution mass spectra were recorded on an ORBITRAP mass analyser (Thermo Scientific, Q Exactive) under electrospray ionisation (ESI) conditions.General procedure for the preparation of N-methyl-1,4-dihydropyridines (3a–s)
A mixture of aromatic aldehydes (1.0 mmol) and (E)-N-methyl-1-(methylthio)-2-nitroethenamine (NMSM) (2.0 mmol) was taken in a glass vial G10 and irradiated with microwave radiation (200 W) at 100 °C on a Monowave 300 synthesizer for 10–15 min. The reaction progress and consumption of starting materials were checked by TLC. After completion of the reaction as indicated by TLC, the resulting precipitate-containing glass vial was withdrawn from the microwave oven and allowed to cool. Then the precipitate was washed with 1.5 mL of cold ethanol and dried. Recrystallization was performed from hot ethanol to provide the pure products.Spectral data of synthesized compounds
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Saigal and SK acknowledge the CSIR and UGC, New Delhi respectively, for their Fellowship. S. C. Sahoo acknowledge to DST-FIST for single crystal XRD facility. The authors are also thankful to IIT Patna, NCL Pune for providing NMR and HRMS facility. The authors are also grateful to the Chairman, Department of Chemistry, AMU, Aligarh, for providing necessary research facilities to complete this work. We are also thankful to the referees for their valuable suggestions.Notes and references
- (a) R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC; (b) P. Gupta and S. Paul, Catal. Today, 2014, 236, 153–170 CrossRef CAS; (c) R. C. Cioc, E. Ruijter and R. V. A. Orru, Green Chem., 2014, 16, 2958–2975 RSC; (d) C. de Graaff, E. Ruijter and R. V. A. Orru, Chem. Soc. Rev., 2012, 41, 3969–4009 RSC.
- (a) H. G. O. Alvim, D. L. J. Pinheiro, V. H. Carvalho-Silva, M. Fioramonte, F. C. Gozzo, W. A. da Silva, G. W. Amarante and B. A. D. Neto, J. Org. Chem., 2018, 83, 12143–12153 CrossRef CAS PubMed; (b) L. Zeng, B. Huang, Y. Shen and S. Cui, Org. Lett., 2018, 20, 3460–3464 CrossRef CAS PubMed; (c) B. H. Rotstein, S. Zaretsky, V. Rai and A. K. Yudin, Chem. Rev., 2014, 114, 8323–8359 CrossRef CAS PubMed; (d) J. Zhu and H. Bienayme, Multicomponent reactions, Wiley-VCH, Weinheim, Germany, 1st edn, 2005 CrossRef.
- (a) H. G. O. Alvim, J. R. Correa, J. A. F. Assumpcao, W. A. da Silva, M. O. Rodrigues, J. L. de Macedo, M. Fioramonte, F. C. Gozzo, C. C. Gatto and B. A. D. Neto, J. Org. Chem., 2018, 83, 4044–4053 CrossRef CAS PubMed; (b) B. B. Toure and D. G. Hall, Chem. Rev., 2009, 109, 4439–4486 CrossRef CAS PubMed; (c) D. J. Ramon and M. Yus, Angew. Chem., Int. Ed., 2005, 44, 1602–1634 CrossRef CAS PubMed; (d) J.-P. Wan and Y. Liu, Curr. Org. Chem., 2011, 15, 2758–2773 CrossRef CAS.
- (a) A. K. Bagdi, S. Santra, K. Monir and A. Hajra, Chem. Commun., 2015, 51, 1555–1575 RSC; (b) A. C. Boukis, K. Reiter, M. Frolich, D. Hofheinz and M. A. R. Meier, Nat. Commun., 2018, 9, 1439–1449 CrossRef PubMed; (c) N. Parikh, S. R. Roy, K. Seth, A. Kumar and A. K. Chakraborti, Synthesis, 2016, 48, 547–556 CAS.
- (a) M. M. Khan, R. Yousuf, S. Khan and Shafiullah, RSC Adv., 2015, 5, 57883–57905 RSC; (b) L. F. Titze and A. Modi, Med. Res. Rev., 2000, 20, 304–322 CrossRef; (c) I. Ugi, A. Dömling and B. Werner, J. Heterocycl. Chem., 2000, 37, 647–658 CrossRef CAS; (d) P. A. Tempest, Curr. Opin. Drug Discovery Dev., 2005, 8, 776–788 CAS; (e) H. Bienayme, C. Hulme, G. Oddon and P. Schmitt, Chem.–Eur. J., 2000, 6, 3321–3329 CrossRef CAS.
- (a) M. M. Khan, S. Khan, Saigal and S. Iqbal, RSC Adv., 2016, 6, 42045–42061 RSC; (b) J. P. Wan, L. Gan and Y. T. Liu, Org. Biomol. Chem., 2017, 15, 9031–9043 RSC; (c) Y. L. Gu, Green Chem., 2012, 14, 2091–2128 RSC.
- (a) C. Villa, M. T. Genta, A. Bargagna, E. Mariani and A. Loupy, Green Chem., 2001, 3, 196–200 RSC; (b) S. Paul, M. Gupta, R. Gupta and A. Loupy, Tetrahedron Lett., 2001, 42, 3827–3829 CrossRef CAS; (c) M. B. Gawande, S. N. Shelke, R. Zboril and R. S. Varma, Acc. Chem. Res., 2014, 47, 1338–1348 CrossRef CAS PubMed; (d) A. Sarkar, S. Santra, S. K. Kundu, A. Hajra, G. V. Zyryanov, O. N. Chupakhin, V. N. Charushin and A. Majee, Green Chem., 2016, 18, 4475–4525 RSC; (e) A. Vass, J. Dudas, J. Toth and R. S. Varma, Tetrahedron Lett., 2001, 42, 5347–5349 CrossRef CAS; (f) S. Roshandel, S. C. Suri, J. C. Marcischak, G. Rasul and G. K. S. Prakash, Green Chem., 2018, 20, 3700–3704 RSC.
- (a) A. Pröbstle, A. Neszmelyi, G. Jerkovich, H. Wagner and R. Bauer, Nat. Prod. Lett., 2006, 4, 235–240 CrossRef; (b) A. Hantzsch, Chem. Ber., 1881, 14, 1637–1638 CrossRef.
- D. J. Triggle, D. D. Langs and R. A. Janis, Med. Res. Rev., 1989, 9, 123–180 CrossRef CAS.
- M. Kawase, A. Shah, H. Gaveriya, N. Motohashi, H. Sakagami and A. J. Varga, Bioorg. Med. Chem., 2002, 10, 1051–1055 CrossRef CAS.
- (a) R. P. Mason, I. T. Mak, M. W. Trumbore and P. E. Mason, Am. J. Cardiol., 1999, 84, 16–22 CrossRef; (b) O. Aruoma, C. Smith, R. Cecchini, P. Evans and B. Halliwell, Biochem. Pharmacol., 1991, 42, 735–743 CrossRef CAS.
- A. Hilgeroth, Mini-Rev. Med. Chem., 2002, 2, 235–245 CrossRef CAS PubMed.
- C. Safak and R. Simsek, Mini-Rev. Med. Chem., 2006, 6, 747–755 CrossRef CAS.
- S. Bahekar and D. Shinde, Acta Pharm., 2002, 52, 281–287 CAS.
- S. Gullapalli and P. Ramarao, Neuropharmacology, 2002, 42, 467–475 CrossRef CAS PubMed.
- (a) R. Shan, C. Velaskez and E. E. Knaus, J. Med. Chem., 2004, 47, 254–261 CrossRef CAS PubMed; (b) D. J. Triggle and D. Rampe, Trends Pharmacol. Sci., 1989, 10, 507–511 CrossRef CAS PubMed; (c) H. Bocker and F. P. Guengerich, J. Med. Chem., 1986, 29, 1596–1603 CrossRef PubMed; (d) A. Sausins and G. Duburs, Heterocycles, 1988, 27, 269–289 CrossRef CAS.
- (a) X. Zhou, R. Coburn and M. Morris, J. Pharm. Sci., 2005, 94, 2256–2265 CrossRef CAS PubMed; (b) X. F. Zhou, L. Zhang, E. Tseng, E. S. Ramsay, J. J. Schentag, R. A. Coburn and M. E. Morris, Drug Metab. Dispos., 2005, 33, 32132–321328 CrossRef PubMed; (c) K. Pajuste, Z. Hyvonen, O. Petrichenko, D. Kaldre, M. Rucins, B. Cekavicus, V. Ose, B. Skrivele, M. Gosteva, E. M. Picardat, M. Plotniece, A. Sobolev, G. Duburs, M. Ruponen and A. Plotniece, New J. Chem., 2013, 37, 3062–3075 RSC.
- (a) E. Fasani, M. Fagnoni, D. Dondi and A. Albini, J. Org. Chem., 2006, 71, 2037–2045 CrossRef CAS PubMed; (b) I. O. Donkor, X. Zhou, J. Schmidt, K. C. Agrawal and V. Kishore, Bioorg. Med. Chem., 1998, 6, 563–568 CrossRef CAS PubMed; (c) T. Straub, C. Boesenberg, V. Gekeler and F. Boege, Biochemistry, 1997, 36, 10777–10783 CrossRef CAS PubMed; (d) A. Hilgeroth and H. Lilie, Eur. J. Med. Chem., 2003, 38, 495–499 CrossRef CAS.
- (a) G. Prasanthi, K. V. S. R. G. Prasad and K. Bharathi, Eur. J. Med. Chem., 2014, 73, 97–104 CrossRef CAS PubMed; (b) V. P. Pandey, S. S. Bisht, M. Mishra, A. Kumar, M. I. Siddiqi, A. Verma, M. Mittal, S. A. Sane, S. Gupta and R. P. Tripathi, Eur. J. Med. Chem., 2010, 45, 2381–2388 CrossRef CAS PubMed.
- H. A. S. Abbas, W. A. El-Sayed and N. M. Fathi, Eur. J. Med. Chem., 2010, 45, 973–982 CrossRef CAS.
- (a) A. Hantzsch, Justus Liebigs Ann. Chem., 1882, 215, 1–82 CrossRef; (b) C. Simon, T. Constantieux and J. Rodriguez, Eur. J. Org. Chem., 2004, 24, 4957–4980 CrossRef; (c) P. Langer, Chem.–Eur. J., 2001, 7, 3858–3866 CrossRef CAS.
- (a) V. K. Sharma and S. K. Singh, RSC Adv., 2017, 7, 2682–2732 RSC; (b) J.-P. Wan and Y. Liu, RSC Adv., 2012, 2, 9763–9777 RSC; (c) J. Sun, Q. Wu, E.-Y. Xia and C.-G. Yan, Eur. J. Org. Chem., 2012, 35, 1976–1983 CrossRef; (d) L. Ohberg and J. Westman, Synlett, 2001, 8, 1296–1298 CrossRef; (e) D. Bandyopadhyay, S. Maldonado and B. K. Banik, Molecules, 2012, 17, 2643–2662 CrossRef CAS.
- (a) F. Rahimi, H. Hosseini, M. Bayat and Y. T. Jeong, Tetrahedron, 2018, 59, 818–822 CrossRef CAS; (b) A. M. Jadhav, Y. I. Kim, K. T. Lim and Y. T. Jeong, Tetrahedron, 2018, 59, 554–557 CrossRef CAS; (c) A. M. Jadhav, S. K. Krishnammagari, J. T. Kim and Y. T. Jeong, Tetrahedron, 2017, 73, 5163–5169 CrossRef CAS; (d) M. V. Reddy, G. D. Reddy, J. T. Kim and Y. T. Jeong, Tetrahedron, 2016, 72, 6484–6491 CrossRef CAS; (e) J. Mao, J. Wang, W. Zhang, Z. Li, J. Zhu and C. Guo, ARKIVOC, 2016,(iii), 171–186 Search PubMed; (f) B. Balachandra, S. Shanmugam, T. Muneewarahand and M. Ramakritinan, RSC Adv., 2015, 5, 64781–64789 RSC; (g) P. Gunasekaran, P. Prasanna and S. Perumal, Tetrahedron Lett., 2014, 55, 329–332 CrossRef CAS; (h) N. Poomathi, J. Kamalraja, S. Mayakrishnan, D. Muralidharan and P. T. Perumal, Synlett, 2014, 25, 708–712 CrossRef CAS; (i) S. Sivakumar, R. R. Kumar, P. Elumalai, Q. N. Ahmed and A. K. Padala, ACS Comb. Sci., 2013, 15, 631–638 CrossRef PubMed; (j) K. Hajiyeva, A. Ismiev, M. Franz, M. Schmidtmann, J. Martens and A. Maharramov, Synth. Commun., 2017, 47, 1–5 CrossRef.
- H. S. P. Rao and A. Parthiban, Org. Biomol. Chem., 2014, 12, 6223–6238 RSC.
- M. M. Khan, Saigal, S. Shareef, S. Khan and S. C. Sahoo, Synth. Commun., 2018, 48, 2683–2694 CrossRef CAS.
- (a) M. M. Khan, Saigal, S. Khan, S. Shareef and M. Danish, ChemistrySelect, 2018, 3, 6830–6835 CrossRef CAS; (b) M. M. Khan, Saigal, S. Khan, S. Shareef and S. Hussain, ChemistrySelect, 2018, 3, 2261–2266 CrossRef CAS; (c) M. M. Khan, S. Khan, Saigal and S. C. Sahoo, ChemistrySelect, 2018, 3, 1371–1380 CrossRef CAS; (d) M. M. Khan, S. Khan, S. Iqbal, Saigal and R. Yousuf, New J. Chem., 2016, 40, 7504–7512 RSC.
- Green metrics reviews, see: (a) F. Roschangar, R. A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752–768 RSC; (b) J. Augé, Green Chem., 2008, 10, 225–231 RSC; (c) C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef CAS.
- (a) X. Zhang, G. Dhawan, A. Muthengi, S. Liu, W. Wang, M. Legris and W. Zhang, Green Chem., 2017, 19, 3851–3855 RSC; (b) P. Wadhwa, T. Kaur and A. Sharma, RSC Adv., 2015, 5, 44353–44360 RSC.
- C. Jiménez-González, D. J. C. Constable and C. S. Ponder, Chem. Soc. Rev., 2012, 41, 1485–1498 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1841358. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra09155b |
| This journal is © The Royal Society of Chemistry 2018 |