
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStimuli-responsive thiol-epoxy networks with photo-switchable bulk and surface properties†
A. Romanoa,
I. Roppolo a,
M. Gieblerb,
K. Dietlikerc,
Š. Možinad,
P. Šketde,
I. Mühlbacherb,
S. Schlögl*b and
M. Sangermanoa
a,
M. Gieblerb,
K. Dietlikerc,
Š. Možinad,
P. Šketde,
I. Mühlbacherb,
S. Schlögl*b and
M. Sangermanoa
aDepartment of Applied Science and Technology, Politecnico di Torino, Corso Duca degli Abruzzi 24, 10129 Torino, Italy
bPolymer Competence Center Leoben GmbH, Roseggerstraße 12, A-8700 Leoben, Austria. E-mail: sandra.schloegl@pccl.at
cDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog Weg 1-5/10, CH-8093 Zürich, Switzerland
dSlovenian NMR Center, National Institute of Chemistry, Hajdrihova 19, 1000 Ljubljana, Slovenia
eEN-FIST Center of Excellence, Trg OF 13, 1000 Ljubljana, Slovenia
First published on 14th December 2018
Abstract
In the present work, the versatile nature of o-nitrobenzyl chemistry is used to alter bulk and surface properties of thiol-epoxy networks. By introducing an irreversibly photocleavable chromophore into the click network, material properties such as wettability, solubility and crosslink density are switched locally by light of a defined wavelength. The synthesis of photo-responsive thiol-epoxy networks follows a photobase-catalyzed nucleophilic ring opening of epoxy monomers with photolabile o-nitrobenzyl ester (o-NBE) groups across multi-functional thiols. To ensure temporal control of the curing reaction, a photolatent base is employed releasing a strong amidine-type base upon light exposure, which acts as an efficient catalyst for the thiol epoxy addition reaction. The spectral sensitivity of the photolatent base is extended to the visible light region by adding a selected photosensitizer to the resin formulation. Thus, in the case of photoactivation of the crosslinking reaction the photorelease of the base does not interfere with the absorbance of the o-NBE groups. Once the network has been formed, the susceptibility of the o-NBE groups towards photocleavage reactions is used for a well-defined network degradation upon UV exposure. Sol–gel analysis evidences the formation of soluble species, which is exploited to inscribe positive tone micropatterns by photolithography. Along with the localized tuning of network structure, the irreversible photoreaction is exploited to change the surface wettability of thiol-epoxy networks. The contact angle of water significantly decreases upon UV exposure due to the photo-induced formation of hydrophilic cleavage products enabling the inscription of domains with different surface wettability by photolithography.
Introduction
In 2001 Sharpless and co-workers introduced the concept of click chemistry, referring to a group of reactions in organic chemistry that are characterized by fast reaction rates, regioselectivity, high yields and mild reaction conditions.1 This pioneering work has become the starting point for extensive use of click reactions in the design of structural polymers with passive function.2 Recently, there has also been a growing interest in the application of click reactions in the design of stimuli-responsive polymers, which change their material properties as a function of a selected stimulus (e.g. light or temperature).3One prominent example of a click reaction is the nucleophilic ring-opening reaction of strained heterocyclic electrophiles such as the ring opening of epoxides with thiols yielding β-hydroxythio-ethers.4,5 In the presence of a sufficiently strong base catalyst the reaction occurs at low temperature and within short reaction time (see Fig. 1). In the initial step of the catalyzed thiol-epoxy click reaction, the thiol group is deprotonated by a base catalyst. A thiolate anion is formed, which undergoes a nucleophilic addition (SN2) reaction across the less hindered site of the epoxy moiety. The generated alkoxide anion is then quenched by the thiols or the presence of protic compounds in the system leading to the formation of a β-hydroxythio-ether link. The protonation step is thermodynamically driven and crucial for the click reaction as it prevents the homopolymerization of the epoxy groups.
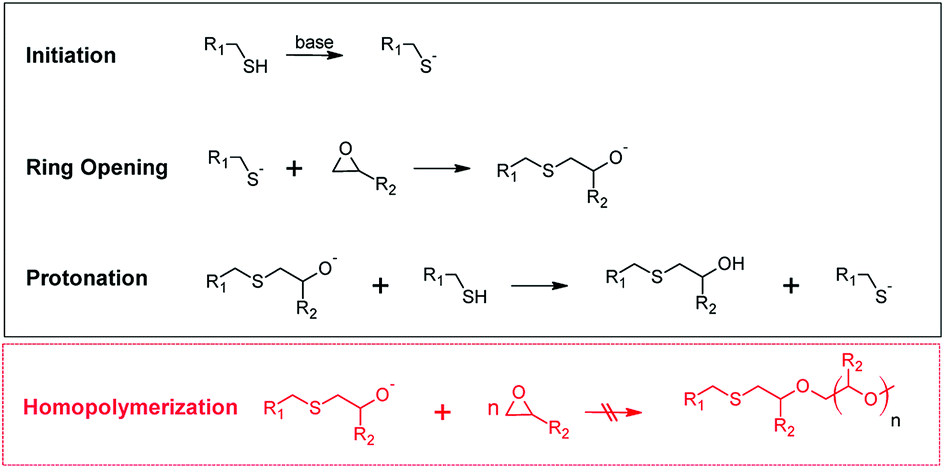 | ||
| Fig. 1 Reaction mechanism of the base-catalyzed nucleophilic addition reaction between thiol and epoxy groups. Homopolymerization of the epoxy groups does not occur due to the thermodynamically favored protonation step.5 | ||
The thiol-epoxy addition reaction is autocatalytic, since the formed secondary hydroxyl groups accelerate the ring-opening of the epoxy moieties.6
Thiol-epoxy reactions have gained increased attention in the preparation of functional polymers and polymer networks as a large number of low molecular weight epoxides and thiol compounds are commercially available.7,8 The variety of polymeric architectures ranges from the synthesis of linear polymers9 and dendrimers10 to the growth of polymer brushes onto various substrates.11 The formed secondary hydroxyl groups are often exploited as reactive anchor groups for post-modification or post-polymerization reactions, which typically involve esterification reactions with activated carboxylic acids or the formation of carbamates with isocyanate derivatives.12
Along with polymer synthesis and surface modification, thiol-epoxy chemistry is also used for the preparation of polymer networks and hydrogels.13 In particular, the interest of thiol-epoxy networks for application in adhesives, coatings or composites is steadily growing, since the materials are characterized by a high optical transparency and flexibility.14 However, thiol-epoxy formulations suffer from poor storage stability as the nucleophilic attack of commonly applied base catalysts (e.g. tertiary amines) cannot be controlled temporally.15 One approach to overcome this drawback is the use of nucleophilic tertiary amine catalysts that comprise a poor basicity. Due to a slow activation, the resin formulations can be prepared and processed within a reasonable time whilst the strong auto-accelerating effect ensures a complete curing at low temperature after application.7,16
Another promising strategy towards an increased shelf life of thiol-epoxy resins involves the employment of latent catalysts that are activated by an external stimulus such as temperature and/or UV-light.17 With respect to photolatent bases, catalysts such as tertiary amines, 1,5-diazabicyclo-[4.3.0]non-5-ene (DBN), tetramethylguanidine or 1,8-diazabicyclo-[5.4.0]undec-5-ene (DBU) are released upon UV exposure.18,19 Sangermano and co-workers demonstrated the optically triggered curing of thiol-epoxy formulations and the formation of hybrid coatings due to the photo-release of DBN.20 Other promising classes of photolatent bases involve quaternary ammonium salts of phenylglycoxylic acid or thioxanthone acetic acid.21
Advancing from thiol-epoxy networks that provide a passive function, the present work aims at the design of stimuli-responsive thiol-epoxy systems that change both their bulk and surface properties upon UV exposure.
Following the idea of a photolatent thiol-epoxy system with photo-responsive properties, a resin formulation (see Fig. 2) was developed containing a multi-functional thiol monomer (TMPMP), a bi-functional epoxy crosslinker (epoxy-NBE) with a photosensitive o-nitrobenzyl ester (o-NBE) group and a photolatent base PLB. Since the UV absorption profile of the photolatent base and the photosensitive o-NBE group are overlapping, a photosensitizer (ITX) was added to the formulation to shift the absorption window of the photolatent base to the visible light region. In the presence of ITX, the base-catalyzed curing reaction was triggered upon visible light exposure (λ > 400 nm) without inducing a premature cleavage of the photolabile o-NBE groups. Once formed, the thiol-epoxy networks were cleaved across the o-NBE links upon irradiation with light in the UV-A spectral region. The photo-induced cleavage reaction led to a distinctive increase in the solubility and wettability of the polymer networks as polar cleavage products are formed.22 Previous work on photocleavable networks showed that the UV-induced increase in solubility is more pronounced in lower crosslinked networks since less crosslinks have to be cleaved to form soluble species.23 Thus, in the current study a mono-functional epoxy monomer (epoxy-GME), which can act as chain extender, since it increases the molecular weight between two crosslink points.
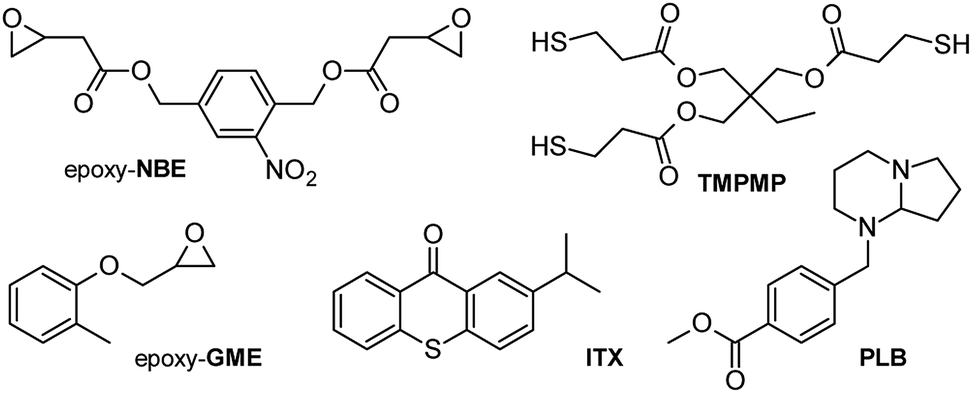 | ||
| Fig. 2 Components used in photo-responsive thiol-epoxy resin formulations. | ||
In previous work, we demonstrated the fabrication of positive tone photoresists by controlled degradation of thermally and photochemically cured epoxy based networks containing o-NBE functionalities upon UV exposure.23–25 In terms of the cationic ring opening, it should be noted, that epoxy monomers bearing o-NBE groups show a low reactivity and thus, an additional post-curing step at elevated temperature is required for efficient chain propagation and full conversion of the monomers.25,26
In the current work, we extended the concept of photocleavable networks to thiol-epoxy click systems. Whilst epoxy monomers with o-NBE groups suffer from low reactivity in cationic ring opening reactions, the base-catalyzed nucleophilic ring opening with thiols is reasonably efficient. The optically triggered change in the bulk properties involving the formation of soluble products due to network degradation was studied in detail and exploited for inscribing positive tone relief structures into the cured thiol-epoxy films by photolithography. Advancing from bulk properties, the photocleavage of the o-NBE group was further applied to facilitate irreversible changes in the surface wettability of thiol-epoxy networks.
Several studies report the controlled switching of surface properties by the photocleavage of o-NBE chromophores.27 In the majority of the concepts o-NBE groups are cleaved off from polymer chains leading to an increase of the surface's hydrophilicity due to the formation of polar cleavage products. Huck et al. described the synthesis of polymer brushes from 4,5-dimethoxy-2-nitrobenzyl methacrylate that comprised an advancing water contact angle of 75°.28 UV exposure of the polymer brushes resulted in a significant decrease of the contact angle (67°) due to the conversion of the ester to acid groups.
Encouraged by these studies we exploited the photosensitivity of the o-NBE links to spatially control surface wettability in thiol-epoxy networks. A distinctive increase of the surface polarity was observed due to the photo-induced formation of hydrophilic cleavage products, which enables a switching of the wettability over a broad range.
Results and discussion
Curing of photo-responsive thiol-epoxy networks
For the preparation of photo-responsive thiol-epoxy networks, epoxy-NBE was mixed with a stoichiometric concentration of the tri-functional thiol. A photolatent DBN derivative (PLB, see Fig. 2) was added as base catalyst, since amidine structures are well-known for their high nucleophilicity and basicity. Compared to tertiary amines, their basicity is three to four orders of magnitudes higher, which makes them interesting candidates for base-catalyzed crosslinking reactions. In the present study, a latent DBN structure was obtained by protecting the nitrogen with a photoremovable unit, yielding an N-benzylated photolatent amine. By possessing isolated secondary and tertiary amine groups, the photolatent amine comprises a low basicity with a pKa of 8.96.18 Upon UV exposure the protecting group is removed by a photoinitiated oxidation reaction and DBN is released. Owed to the conjugative interaction of the two nitrogen atoms across the carbon–nitrogen double bond, DBN is a much stronger base (pKa = 13.41) than the non-illuminated photolatent amidine precursor, and efficiently catalyses the nucleophilic ring opening of the epoxy groups.18It should be noted that the spectral sensitivity of PLB is in the UV-B region and thus, interferes with the absorbance of the o-NBE groups.18 To avoid a premature cleavage of the photosensitive o-NBE links during the photocuring, the photosensitivity of the photolatent base has to be extended towards longer wavelengths. Previous work has shown that the absorption characteristics of photolatent DBN derivatives can be adjusted over a wide range, either by introducing selected substituents onto the aromatic ring, or by adding various sensitizers (e.g. aromatic ketones or thioxanthone derivatives).20 To enable the curing of the photo-responsive thiol-epoxy resin formulation upon visible light exposure, ITX was added as a photosensitizer in the present study. Thus, curing and cleavage of the networks are orthogonal processes that proceeded at different wavelengths, which is crucial for the successful light-triggered modulation of network properties (see Fig. 3).
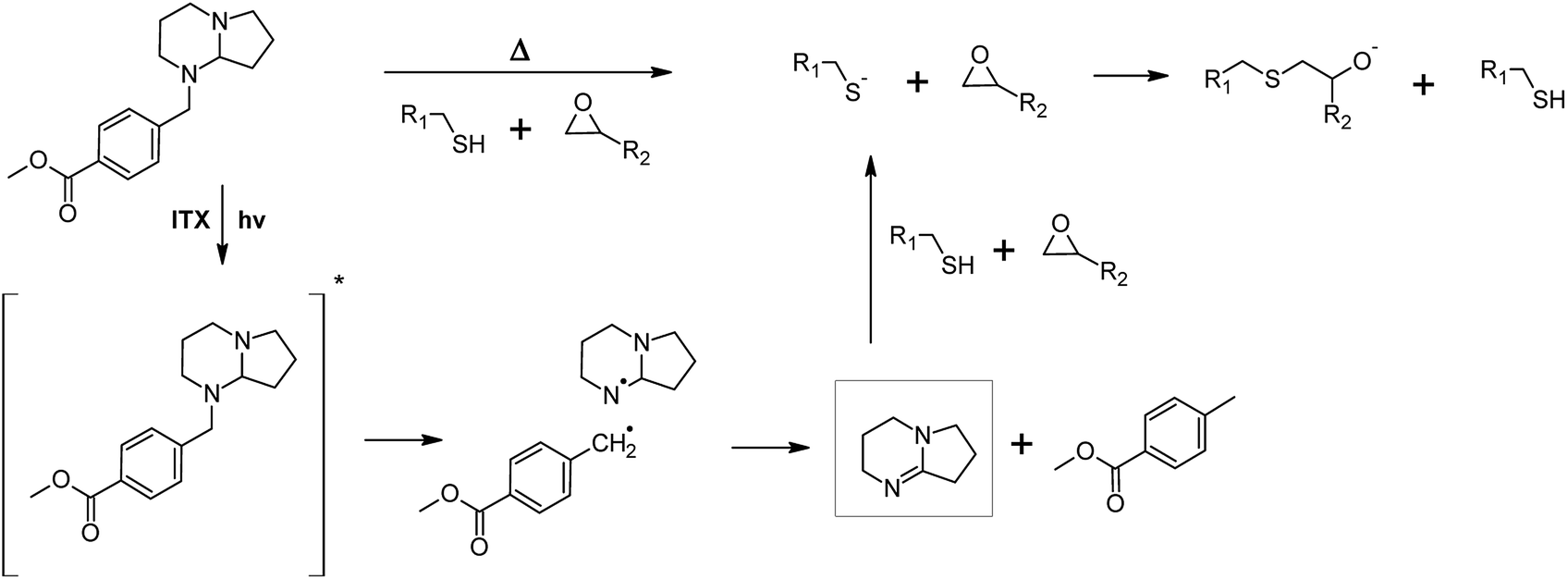 | ||
| Fig. 3 Thermally and photochemically triggered base-catalyzed curing of photo-responsive thiol-epoxy networks in the presence of a photolatent base and ITX as photosensitizer, which shifts the spectral sensitivity of the photolatent base towards the visible light region.20 | ||
Fig. 4a provides the FT-IR spectra of formulation PI4 (for the exact composition of the formulations used, see Table 1 in the experimental part) comprising a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio of thiol and epoxy functionalities at a photolatent base and ITX concentration of 4 wt%, prior to and after visible light exposure. The successful network formation by the photo-induced nucleophilic ring-opening reaction is confirmed by the depletion of the characteristic thiol absorption band at 2580 cm−1 (see insert I in Fig. 4a) and the epoxy signal at 906 cm−1 (see insert III in Fig. 4a). The decrease of the functional groups is accompanied by the appearance of the OH absorption band between 3200 and 3600 cm−1 (see insert II in Fig. 4a) giving rise to the formation of β-hydroxythio-ether links.
:1 stoichiometric ratio of thiol and epoxy functionalities at a photolatent base and ITX concentration of 4 wt%, prior to and after visible light exposure. The successful network formation by the photo-induced nucleophilic ring-opening reaction is confirmed by the depletion of the characteristic thiol absorption band at 2580 cm−1 (see insert I in Fig. 4a) and the epoxy signal at 906 cm−1 (see insert III in Fig. 4a). The decrease of the functional groups is accompanied by the appearance of the OH absorption band between 3200 and 3600 cm−1 (see insert II in Fig. 4a) giving rise to the formation of β-hydroxythio-ether links.
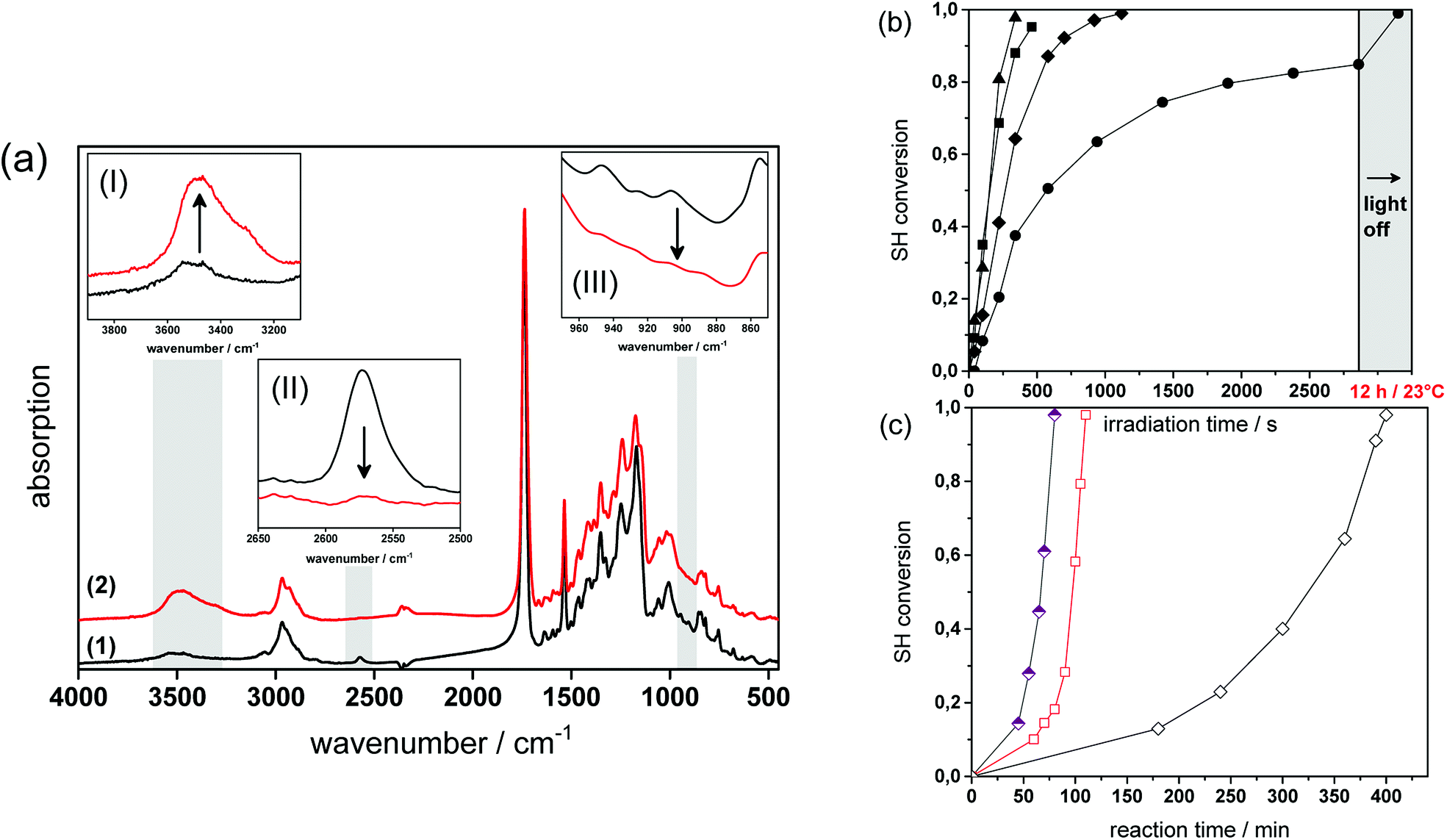 | ||
| Fig. 4 (a) FT-IR spectra of PI4 (1) prior to and (2) after photo-curing (1.8 J cm−2, λ > 400 nm, air). Inserts show the IR bands of the characteristic (I) OH, (II) SH and (III) epoxy groups magnified out of the FT-IR spectra. Normalized depletion of (b) the thiol absorption band (2580 cm−1) versus illumination time (1.8 J cm−2, λ > 400 nm, air). Resin formulations contained varying amounts of photolatent base and ITX: Pl2 (solid circles); Pl3 (solid diamonds); Pl4 (solid squares) and Pl5 (solid triangles). (c) Normalized depletion of the thiol absorption band (2580 cm−1) of non-illuminated resin formulation PI4 versus reaction time. The reaction temperature amounted to 23 (open diamonds), 50 (open squares) and 70 °C (half open diamonds). | ||
The curing kinetics of the ring opening reaction was studied by following the depletion of the functional thiol and epoxy groups upon prolonged visible light exposure. However, the conversion of the epoxy groups could not be calculated accurately due to peak overlapping (see insert III in Fig. 4a). Consequently, the curing reaction was monitored by the depletion of the thiol peak at 2580 cm−1.17,29 The influence of the photolatent base content (2–5 wt%) on the curing kinetics was studied by keeping the weight ratio between photolatent base and ITX constant at 1:1 (see Fig. 4b). As expected, the results revealed that both reaction rate as well as final monomer conversion increase with rising base concentration, since a higher content of DBN is released deprotonating a larger number of thiol groups. In particular, at a photolatent base content of 2 wt% a final thiol conversion of 83% is obtained upon 50 min visible light exposure. In contrast, nearly full thiol conversion (98%) is accomplished within 8 min at a base concentration of 5 wt%. It should be noted that dark reactions proceed within the illuminated resins and full monomer conversion is observed in each resin formulation independent of the base concentration after storing the films at room temperature overnight (see Fig. 4b).
Since the pKa of the N-benzylated photolatent base is in the range of some tertiary amines it might be also capable to catalyse the thiol-epoxy reaction under dark storage conditions.18 Fig. 4c shows the thiol conversion of the non-illuminated resin formulation PI4 as a function of the reaction temperature (23, 50 and 70 °C) and the reaction time. At room temperature the thiol conversion of the non-illuminated resin formulation remains below 15% within 3 hours.
However, upon prolonged storage time the curing reaction is also catalyzed by the less reactive N-benzylated photolatent base and once initiated, the reaction proceeds rather quickly due to the autocatalytic nature of the nucleophilic ring opening reaction. This autoacceleration effect is nicely demonstrated by the fact that after 240 min in the dark the thiol conversion is <15%, while it reaches nearly full conversion (97%) after 400 min (at 23 °C). At 70 °C, the base-catalyzed reaction of the N-benzylated photolatent base is significantly accelerated and 96% conversion of the thiol moieties is obtained after 80 min only.
From the results of the FT-IR studies it can be concluded that the pKa difference between the N-benzylated photolatent base and the photo-released DBN is large enough to ensure fast reaction rates upon visible light illumination whilst ensuring a sufficient storage stability of the resin formulation at room temperature of over three hours.
Regarding the light induced modification of the cured material, previous studies demonstrated that the efficiency of the cleavage is higher in networks with lower cross-linking density since less crosslinks must be cleaved for the formation of soluble cleavage products.25,26 To decrease the network density of the thiol-epoxy systems, the photocuring was also carried out in the presence of a mono-functional epoxy monomer (epoxy-GME). In particular, 5 and 7 mol% of epoxy-GME (see Fig. 2) was added to the resin formulation while keeping the stoichiometric ratio between thiol and epoxy groups at 1:1.
Due to the addition of epoxy-GME the glass transition temperature (Tg) of the cured thiol-epoxy networks slightly decreased from 15 to 11 °C (see Table 1).
However, FT-IR results revealed that the low amount of mono-functional epoxy monomers within the thiol-epoxy resins does not influence reaction rate or final monomer conversion of the nucleophilic ring opening reaction (see Fig. S1 in ESI†).
Cleavage of photo-responsive thiol-epoxy networks
The photocleavage of photo-responsive thiol-epoxy networks was followed in thin films, which were drop-cast on Si wafers. The cured samples were illuminated with UV-light at wavelengths below 400 nm to induce the cleavage reaction of the o-NBE links, which proceeds via biradical intermediates formed by β-hydrogen abstraction by the excited nitro group analogous to the Norrish-type II reaction (see Fig. 5a).30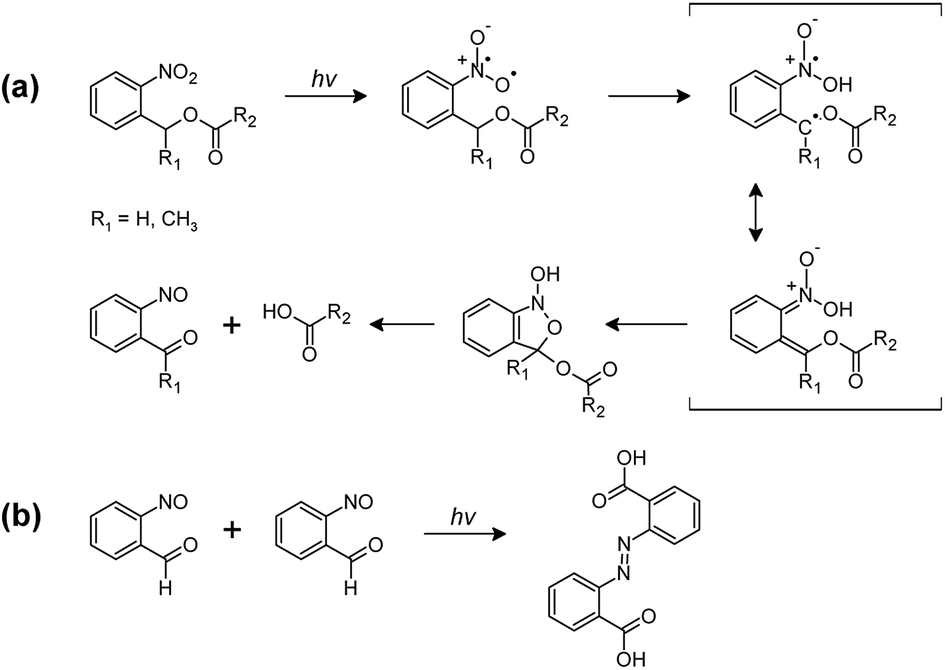 | ||
| Fig. 5 (a) Primary and (b) secondary photoreactions of o-NBE derivatives.31–33 | ||
In the triplet state, a proton abstraction from the methylene or methine carbon in γ-position takes place by one oxygen atom from the nitro group, which is followed by the generation of an aci-nitro tautomer in the ground state. The latter can undergo cyclization to form a benzoisoxaline derivative. The resonance stabilized five-membered ring is subsequently cleaved yielding a carboxylic acid and an o-nitrosobenzaldehyde as primary photoproducts. In secondary photoreactions the formed o-nitrosobenzaldehyde dimerizes and is able to form azobenzene groups (see Fig. 5b).31–33
In Fig. 6a, FT-IR spectra of formulation PI4 are compared prior to and after UV exposure. The UV induced conversion of nitro moieties to nitroso groups is indicated by the decrease of the two characteristic NO2 signals at 1537 cm−1 (asym. stretching) and 1348 cm−1 (sym. stretching). Moreover, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O absorption band (1635–1802 cm−1) is broadening, which is related to the formation of the characteristic cleavage products such as carboxylic acids.
O absorption band (1635–1802 cm−1) is broadening, which is related to the formation of the characteristic cleavage products such as carboxylic acids.
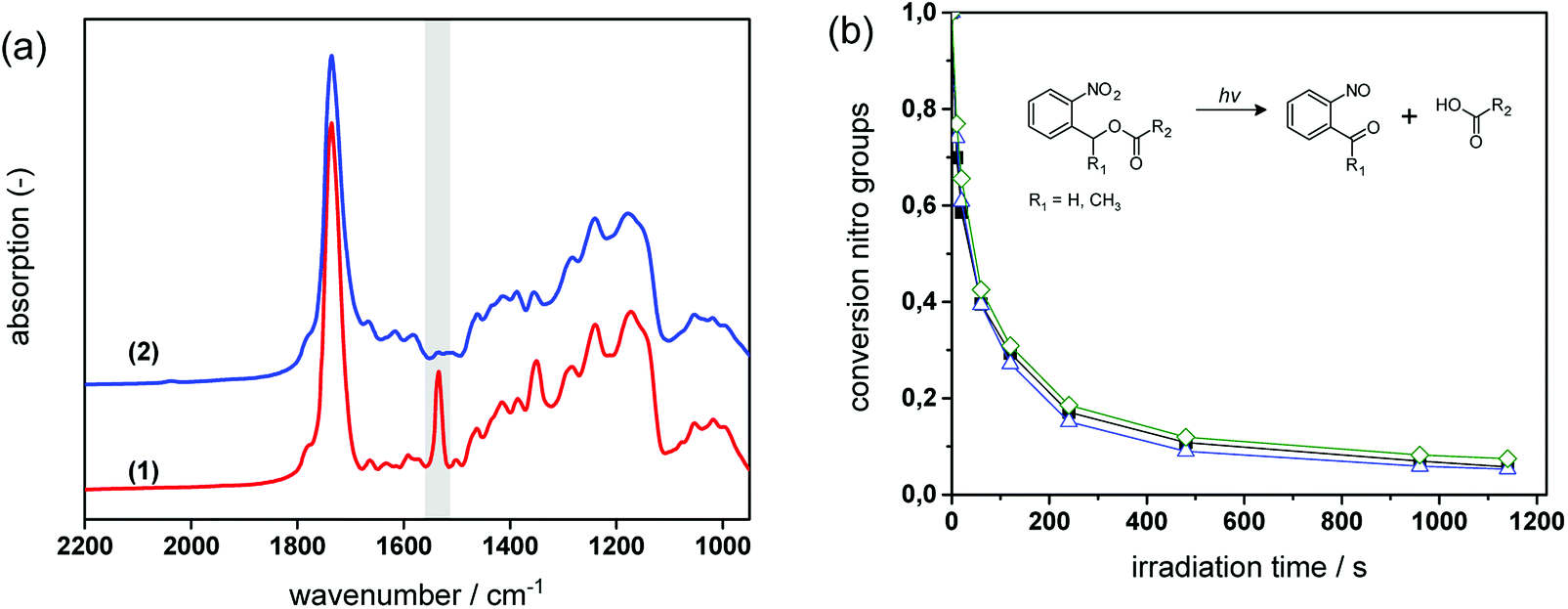 | ||
| Fig. 6 (a) FT-IR spectra of cured PI4 (1) prior to and (2) after photo-induced cleavage of the o-NBE links (85 J cm−2, λ < 400 nm, N2). (b) Following the photocleavage kinetics of the o-NBE links by FT-IR spectroscopy upon prolonged UV exposure: decrease of the normalized intensity of the nitro band at 1537 cm−1 in PI4 (solid squares), PI4-G5 (open diamonds) and PI4-G7 (open triangles). | ||
For cured thiol-epoxy resin formulations with varying amount of epoxy-GME, the cleavage kinetics was determined by following the depletion of the NO2 signal at 1537 cm−1 upon prolonged UV exposure (see Fig. 6b). From the FT-IR studies it can be obtained that the photocleavage proceeds rapidly in thiol-epoxy click networks and nearly full conversion of the nitro groups is observed upon 16 min UV exposure. As demonstrated in previous work the photocleavage kinetics of o-NBE links is strongly governed by film thickness (i.e. penetration depth of the incident light) and mobility of the chromophore.26 Owed to the thioether link, thiol-epoxy networks are typically characterized by a high flexibility and low glass transition temperature.34 This ensures a high mobility of the o-NBE groups leading to a fast conversion of the functional groups.
In highly mobile polymer networks bearing o-NBE groups, the UV-induced conversion of nitro to nitroso moieties is less influenced by the concentration of chain extenders (and thus, the crosslink density of the network).25,26 This behavior is also observed in photo-responsive thiol-epoxy networks which comprise a glass transition temperature that does not exceed 15 °C (see Table 1). From the FT-IR kinetics (see Fig. 6b) it can be obtained that both reaction rate and final conversion of the nitro groups are not significantly affected by the epoxy-GME content (0, 5 and 7 mol%).
To study the change in solubility properties sol–gel analysis of thiol-epoxy formulations with varying amount of epoxy-GME was carried out (see Fig. 7a). As mono-functional epoxy monomer, epoxy-GME can act as chain extender, since it increases the molecular weight between two crosslink points.23 In the absence of the mono-functional epoxy monomer (PI4), the gel fraction of the cured thiol-epoxy networks amounted to 98%. By adding 5 or 7 mol% epoxy-GME (PI4-G5, PI4-G7), the gel content decreased to 95 and 89%, respectively. The results indicate that epoxy-GME does not only affect the network structure but also the gel fraction, as a higher content of soluble species (e.g. oligomers) is formed in the presence of the mono-functional epoxy monomer.
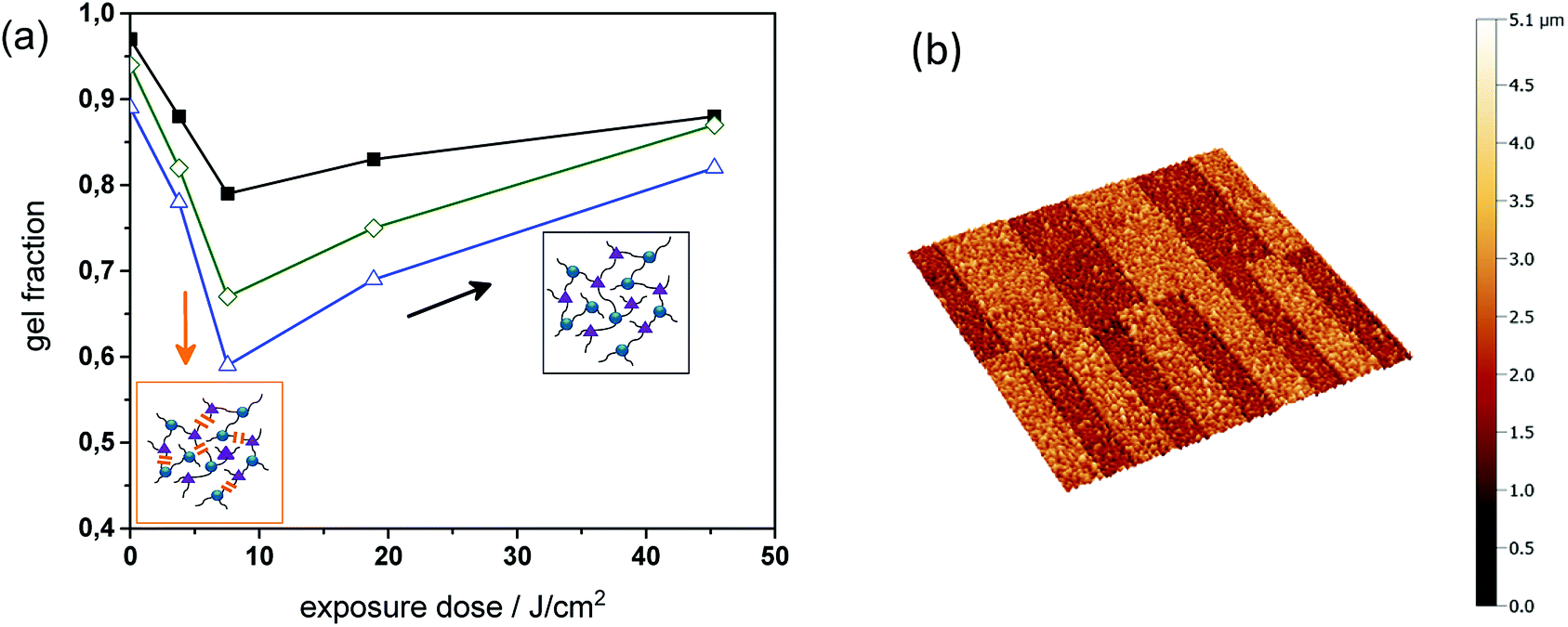 | ||
| Fig. 7 (a) Gel fraction of PI4, PI4-G5 and PI4-G7 versus exposure dose (75 mW cm−2, λ < 400 nm, N2) as obtained from FT-IR measurements. (b) Confocal micrograph of positive-tone relief structures (100 and 50 μm lines and spaces) inscribed into PI4-G5 by photolithography after the development in tetrahydrofuran. | ||
In a subsequent step, the cured films were UV irradiated and the gel fraction was monitored upon prolonged UV exposure. Independent on the epoxy-GME concentration the gel fraction of the photo-responsive thiol-epoxy networks followed the same trend. Upon a UV exposure of 7.5 J cm−2, the gel content steadily decreased corresponding to the photo-induced formation of soluble species.
However, if the exposure dose exceeded 7.5 J cm−2, the gel content increased and ranged between 82 and 88% after a UV exposure with 45 J cm−2 indicating a reformation of covalent links, which may be attributed to the photoinduced crosslinking via azobenzene group formation as shown in reaction (b) in Fig. 5. This phenomenon was also observed in photocleavable polyether and thiol-ene networks.25,26
To get an insight into the structure of the cleavage products formed in photo-responsive thiol-epoxy click networks, solid state NMR experiments were performed. In particular, the influence of the exposure dose and the atmosphere, in which the UV irradiation had been carried out (air versus nitrogen), was studied. 13C solid state NMR analysis of cured but not illuminated networks did not reveal any cleavage products (see Fig. S2 in ESI†). By UV illuminating the network with an exposure dose of 100 J cm−2 (compared to sol–gel analysis higher exposure doses were chosen since the film thickness of the samples was one order of magnitude larger) signals at δ 166 and 157 ppm, belonging to carbon atoms of a carbonyl ester and to carbon atoms linked with a nitro group, respectively, decreased (cf. Fig. S3a and b†). Simultaneously, new signals emerged at δ 172 and 147 ppm, which are related to carboxylic acids and carbon atoms linked with an azo group. Moreover, the formation of the azobenzene side products is independent on the atmosphere, since the signal was found in spectra of thiol-epoxy networks, which were either illuminated under air (see Fig. S3a†) or nitrogen (see Fig. S3b†). However, the intensity of the signal at δ 172 ppm (carboxylic acid) was slightly higher if the irradiation was performed under air. This increase is attributed to additional photo-oxidation reactions, since the kinetics of the cleavage reaction is not significantly affected by the presence of oxygen. At a higher exposure dose (300 J cm−2) this behaviour is even more pronounced (see Fig. S4a and b†). However, in none of the spectra a signal related to an aldehyde group was observed around δ 190 ppm. This result confirms that the aldehyde as primary photocleavage product rapidly undergoes secondary reactions involving the formation of azobenzene (dimerization) and carboxylic acid (oxidation).
The results of the sol–gel analysis further revealed that the relative decrease of the gel content is more pronounced in thiol-epoxy networks containing a higher amount of epoxy-GME. In particular, in the formulation containing no epoxy-GME (PI4) the relative decrease of the gel content was 24%, whilst it is up to 29 and 34% in the presence of 5 and 7 mol% epoxy-GME, respectively. This behavior is expected since an increasing epoxy-GME concentration corresponds to a decreasing crosslink density and in lower crosslinked networks fewer crosslinks have be cleaved before soluble products are formed.
Since the photodegradation was reasonably efficient in PI4-G5, the spatial control of solubility properties was exploited to inscribe micro-sized patterns within cured thiol-epoxy networks by using photolithography. For sample preparation, thin films of resin PI4-G5 were spin cast onto Si wafers. Once the resin formulation was photo-cured, positive-tone relief structures were obtained by UV exposure through a quartz-chromium mask, followed by a development step, washing off the photodecomposition products. The non-exposed areas remained insoluble during the development in tetrahydrofuran, whilst the illuminated areas became partly soluble due to the photolysis of the o-NBE links. Relief patterns with a structure size of 50 and 100 μm were obtained, which were visualized by confocal microscopy (see Fig. 7b).
The photocleavage of covalent crosslinks and the re-crosslinking of the thiol-epoxy network at higher exposure doses was further demonstrated by nanoindentation measurements. The data were analyzed according to the “Oliver & Pharr Method”, in which mechanical properties are derived from the unloading-part of the load–displacement curve.35 In the present work, the indentation hardness (HIT) was determined by taking an average of 15 individual indents over a 300 × 600 μm area. For sample preparation, the formulation PI4-G5 was cast on glass slides and the change in the hardness was measured in the cured and photocleaved state (UV exposure with 7.5 and 100 J cm−2). The results reveal that HIT distinctively decreases from 33.02 ± 1.05 MPa to 19.21 ± 2.42 MPa due to the photo-induced network degradation at a low exposure dose (7.5 J cm−2). However, upon prolonged UV exposure (100 J cm−2) a substantial increase in surface hardness is observed. HIT amounts to 40.78 ± 4.67 MPa and even surpasses the indentation hardness of the PI4-G5 network in the cured state. The indentation experiments correlate well with the solid state NMR and sol–gel measurements and confirm that the photo-responsive thiol-epoxy networks are prone to re-crosslinking reactions at higher exposure dose.
Light-triggered modulation of surface wettability
In addition to an irreversible transformation of bulk properties, the photo-responsive nature of the o-NBE links in thiol-epoxy click networks was used for altering the surface wettability. Since polar species such as carboxylic acids are formed upon photocleavage, the surface polarity of thin polymer films is expected to be switched conveniently.For sample preparation, thin films from resin PI4-G5 on silicon wafer were obtained by a spin-cast process. After curing, the films comprised a water contact angle of 77 ± 3°, which is in the range of typical amine-cured epoxy resins.36 In a subsequent step, the cured thiol-epoxy films were UV illuminated to induce the isomerization reaction of the o-NBE links. The illumination was carried out either under nitrogen or air to study the influence of the atmosphere on the photo-switchable wettability. The change of the contact angle as a function of the exposure time and atmosphere is displayed in Fig. 8a. In nitrogen atmosphere the water contact angle steadily decreases with rising exposure dose and reaches a plateau (62 ± 4°) if the UV exposure dose exceeds 41 J cm−2. This behavior is explained by the photo-induced cleavage of the o-NBE links yielding polar groups, as shown by solid state NMR experiments. At a certain exposure dose the o-NBE groups on the surface of the thiol-epoxy network are fully cleaved and thus, no further change of the contact angle is observed. Since the irradiation is carried out under inert conditions, the surface is also not prone to photo-oxidation reactions, which would affect the surface polarity.
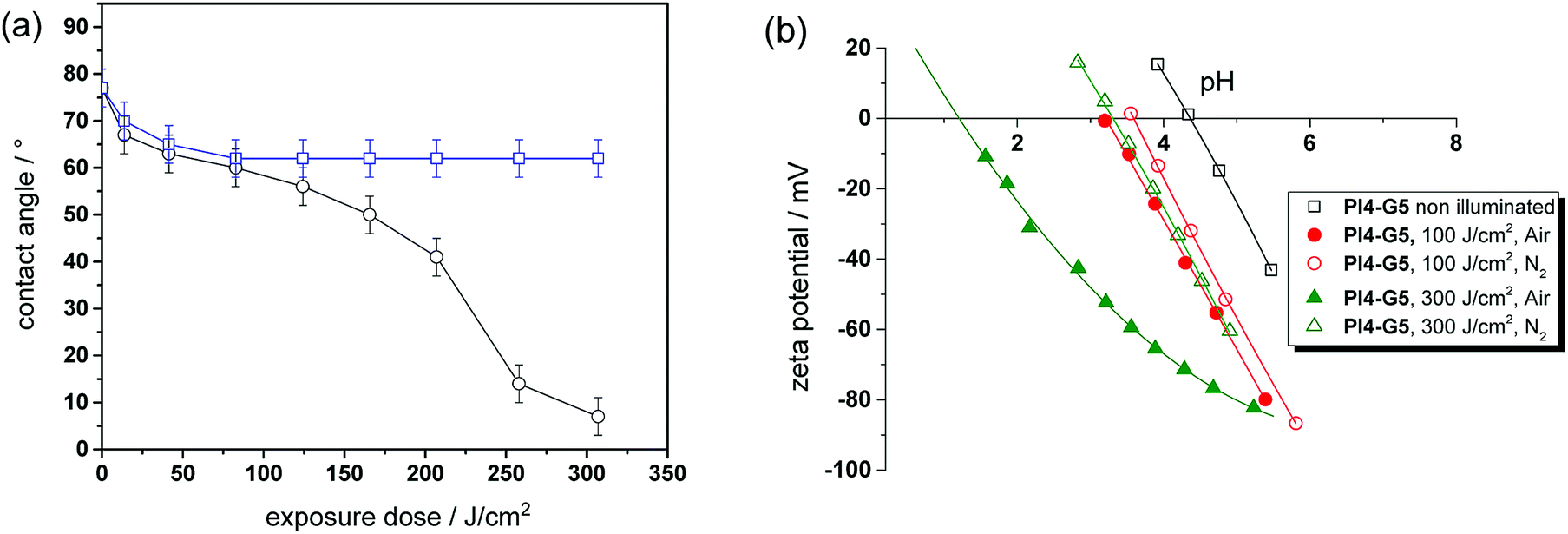 | ||
| Fig. 8 (a) Water contact angle of cured PI4-G5 versus exposure dose (138 mW cm−2, λ < 400 nm). UV exposure was carried out either in air (open circles) or nitrogen (open squares). (b) Zeta potential as a function of the pH value of cured PI4-G5 versus exposure dose (138 mW cm−2, λ < 400 nm) and atmosphere (nitrogen versus air). | ||
In contrast, the UV irradiation under air enables a distinctive shift of surface wettability due to the combined processes of photocleavage of the o-NBE groups and photo-oxidation. From the kinetics of the contact angle over exposure time three different regimes can be distinguished. In the first stage, a sharp decrease of the contact angle from 77 to 60 ± 4° is observed at low exposure doses. This behavior was also detected in the experiments carried out in inert atmosphere, but the decrease of the water contact angle is more pronounced under air due to the additional photo-oxidation of the surface. Between 41 and 207 J cm−2, the decrease of the contact angle slows down but does not reach a stable plateau due to the ongoing photo-oxidation. If the exposure dose exceeds 260 J cm−2, the decrease of the water contact angle is accelerated until the surface becomes fully wettable and it is no longer possible to record the static contact angle anymore. Comparing the kinetics of both experiments (air versus nitrogen atmosphere) the results suggest that the third regime is mainly controlled by photo-oxidation.
In addition to contact angle measurements, zeta potential experiments were performed to study the changes in surface polarity in dependence on the applied UV irradiation step in more detail (see Fig. 8b). The isoelectric point (IEP) of the cured thiol-epoxy network amounts to 4.35, which is in the range of uncharged polymer surfaces such as polyolefins (IEP ∼ 4). The results further indicate the absence of unreacted thiol moieties on the surface of the cured thiol-epoxy formulation since the IEP of surfaces bearing –SH groups is in the range of 3.37 Upon UV exposure in inert atmosphere, the IEP shifts to lower values, which confirms the formation of acidic groups. It is interesting to note, that a gradual decrease of the IEP is detectable with zeta-potential measurements at prolonged UV irradiation, whilst a plateau of the contact angle data is observed. This can be mainly related to the high sensitivity of the zeta-potential to small changes of the chemical surface composition in comparison to the less sensitive static contact angle.
However, the shift of the IEP is far more pronounced if the UV irradiation is carried out under air, which correlates well with the contact angle data. In particular at high exposure doses (under air), the IEP drops to 1.25. This gives rise to an acidic surface due to photo-oxidation reactions, which are expected to become the dominating reaction pathway at higher exposure doses. From the results it can be concluded that the versatility of o-NBE chemistry in combination with classic photo-oxidation enables a convenient switching of surface wettability as a function of the exposure dose and atmosphere.
Experimental
Materials and chemicals
(2-Nitro-1,4-phenylene) bis(methylene) bis(2-(oxiran-2-yl)acetate) (epoxy-NBE) as photosensitive monomer was synthesized as previously reported in ref. 24. Glycidyl 2-methylphenyl ether (epoxy-GME), 2-isopropyl thioxanthone (ITX) as photo-sensitizer and all other chemicals were purchased from Sigma-Aldrich (St. Louis, US) and were used without further purification. The photolatent base 4-(hexahydro-pyrrolo[1,2-a]pyrimidin-1-ylmethyl)-benzoic acid methyl ester (PLB) used in equal (1:1) weight percentage (2, 3, 4 and 5 wt%) with the ITX was a gift from BASF (Switzerland). The structures of the monomers, crosslinker and photoinitiating system are displayed in Fig. 2.
Preparation of thiol-epoxy formulations
Resin formulations were prepared by mixing the trifunctional thiol (TMPMP) with an equal molar epoxy bond concentration of epoxy-NBE and epoxy-GME (0, 5%, 7%). PLB and ITX were added in equal (1:1) weight percentage (2, 3, 4 and 5 wt%) to the formulation. The resin mixture was stirred at 50 °C for 45 min to dissolve the PLB and the ITX. The composition of the different thiol-epoxy resin formulations is summarized in Table 1.
Characterization of the curing and cleavage kinetics
The photocuring of the monomers and the photocleavage of the corresponding thiol-epoxy networks were monitored by FTIR spectroscopy employing a Nicolet iS50 FT-IR spectrometer (Thermo Scientific, Milano, Italy) in transmission mode, with a resolution of 4 cm−1 and 16 scans. The spectra were recorded in a wavenumber range between 450 and 4000 cm−1 and the absorption peak areas were calculated with OMNIC software. For sample preparation few drops of each resin formulation were placed on a silicon wafer and spread with a film applicator bar of 4 μm. The thin films were illuminated with a Hamamatsu LC8 lamp with visible bulb and a cut-off filter below 400 nm in air for 460 s. The light intensity amounted to 4 mW cm−2 (λ > 400). For the subsequent cleavage studies the cured samples were irradiated with a medium pressure mercury lamp: Hamamatsu LC30, equipped with an optical waveguide; and with a light intensity on the surface of the sample of about 75 mW cm−2 (λ = 250–470 nm) under nitrogen atmosphere and the cleavage reaction was monitored by FT-IR spectra taken after selected exposure times.For determining the formed cleavage products, solid-state NMR experiments were performed on an Agilent Technologies NMR System 600 MHz NMR spectrometer equipped with 3.2 mm NB Double Resonance HX MAS Solids Probe. Larmor frequencies of carbon and proton nuclei were 150.75 MHz and 599.46 MHz. Samples were packed into zirconia rotors. The cross polarisation (CP) and magic-angle spinning (MAS) solid-state NMR were used in 13C experiments. Acquisition time and relaxation delay for 13C experiments were 0.025 and 3 s, respectively. Spectra were recorded at spinning rates of 10 and 16 kHz. 13C chemical shifts were externally referenced to the adamantane peak (δ 38.3 ppm relative to tetramethylsilane). For the sample preparation, PI4-G5 was cast on glass slides with a coating knife (120 μm film thickness) and photo-cured as described in the previous paragraph. Subsequently, UV cleavage of the samples was carried out as described in the previous paragraph in either air or nitrogen atmosphere. For the solid state NMR measurements, the cured material was peeled off from the glass slide.
Preparation and characterization of sol–gel analysis and patterned films
For sol–gel analysis, thin films of formulation PI4-G5 were coated on Si wafer and photocured as described in the previous section. The cured sample were then UV irradiated for inducing the photocleavage using the Hamamatsu LC30 and with a light intensity on the surface of the sample of about 75 mW cm−2 (λ = 250–470 nm).The soluble species were then extracted by immersing the sample in tetrahydrofuran for 10 min. The gel fraction was quantitatively evaluated by FT-IR spectroscopy (Thermo Scientific in transmission mode) comparing the area of the ester peak (1752 cm−1), using the equation: gel fraction gf = A0/Ag × 100 where A0 and Ag equals to the area of ester prior to and after the developing of soluble species.
The photolithographic patterning was conducted with a quartz-chromium mask in contact mode. For the sample preparation of positive tone patterns, the resin of the PI4-G5 formulation (see Table 1) was spin-cast on silicon wafer and cured as described in the previous chapter. The subsequent patterning of the polymer networks was performed with the quartz chromium mask in contact mode using UV light with the medium pressure mercury lamp (Hamamatsu LC30, 75 mW cm−2; λ = 250–470 nm; N2).
The exposure dose amounted to 7.5 J cm−2 and after UV illumination the polymer films were developed by dipping the samples for a few seconds in tetrahydrofuran.
The patterned films were examined by confocal microscopy (MicroProf®, Fries Research and Technology, Germany). The measuring frequency was 2000 Hz and the measuring speed amounted to 607 μm s−1. The resolution in height was 10 nm and a lateral resolution <2.5 μm was accomplished. In addition, the resolution of the patterned film was evaluated by scanning electron microscopy (Auriga 60, Zeiss, Germany) operating at 20 kV after gold sputtering using an Agar Sputter Coater (Agar Scientific, United Kingdom).
Differential scanning calorimetry measurements were performed with a Mettler-Toledo DSC 821e (United States) applying a nitrogen flow of 20 mL min−1. The cured thiol-epoxy networks were heated from −20 to 100 °C with a heating rate of 20 °C min−1. The glass transition temperature (Tg) was taken from the second heating run and was read as the midpoint in heat capacity.
Surface characterization
The switching of wettability was analyzed with contact angle measurements performed by using a drop shape analysis system, DSA 100, from Krüss GmbH (Hamburg, Germany). Thin films of resin formulation PI4-G5 were spin-cast on Si wafer in order to obtain a flat and homogeneous resin layer and thus, avoiding that the surface energy is influenced by surface roughness. The cured sample was UV irradiated with a medium pressure mercury lamp: Hamamatsu LC30 (138 mW cm−2; λ = 250–470 nm; N2 and air) in order to facilitate the formation of hydrophilic cleavage species. The wettability of the irradiated sample was calculated with contact angle measurements (water was used as test liquid) few minutes (cooling down the surface temperature of the illuminated surface) after the irradiation.Zeta potential measurements were performed with an Electrokinetic Analyser (EKA, Anton Paar, Graz, Austria) to determine the isoelectric point. The streaming potential method was applied to obtain the zeta potential of the thiol-epoxy surfaces prior to and after UV exposure in 1 mM KCl. The zeta potential was measured starting from natural pH to lower acidic values by adding 50 mM HCl using an autotitrating unit (RTU, Anton Paar, Graz, Austria).
The nanoindentation experiments were performed with the Ultra Nanoindentation Tester from Anton Paar (Graz, Austria) using a pyramidal-shaped Berkovich tip. The contact force amounted to 50 μN whilst the maximum indentation force was set to 1000 μN, with a loading rate of 6000 μN min−1 and an unloading rate of 6000 μN min−1. A 30 s hold segment was applied at the maximum load to obtain reliable stress–strain curves. The distance between two measurements was 150 μm and 15 indents were performed over an area of 300 × 600 μm to record the mechanical properties over a larger length scale and to get reliable results. For the determination of the indentation hardness (HIT) the Oliver–Pharr method was used.35
Conclusions
Photocleavable thiol-epoxy networks were prepared by base catalyzed ring opening reaction of multi-functional thiols across bi-functional epoxy monomers bearing photo-sensitive o-NBE links. Photo-triggered curing of the UV sensitive epoxy monomer was accomplished with a photolatent base and ITX as sensitizer, which shifted the initiating window to the visible light region. Photocuring proceeded rapidly due to the highly nucleophilic and basic nature of the photo-released base (DBN) and nearly full conversion of the thiol groups was observed after 8 min of visible light exposure. At room temperature the non-illuminated resin formulation is stable over three hours but at elevated temperature (70 °C), the base-catalyzed reaction of the N-benzylated photolatent base is highly efficient and it turned out that 96% of the thiol moieties are consumed after 80 min.The subsequent photo-induced network degradation upon UV exposure was confirmed by sol–gel analysis and nanoindentation experiments. The formation of soluble species is compromised by side reactions leading to a regeneration of covalent crosslinks upon prolonged UV exposure. In particular, the formation of azobenzene side products was confirmed by solid state NMR experiments. The addition of 5 and 7 mol% mono-functional epoxides improves the yield of soluble species from 24 to 29 and 34%, respectively. The photocleavage is reasonable efficient to inscribe positive-tone relief patterns within the cured thiol-epoxy networks by photolithography.
Besides the wavelength dependent change in network properties, the photosensitive nature of the thiol-epoxy networks was used to alter their surface wettability. In particular, contact angle and zeta-potential measurements revealed that the photo-induced formation of polar cleavage products such as carboxylic acids leads to a significant enhancement of the surface polarity and wettability. The results further showed that the decrease of the water contact angle is influenced by both exposure dose as well as the atmosphere in which the UV illumination is carried out. Whilst under nitrogen the change in the contact angel does not exceed 15°, the surface of the thiol-epoxy networks becomes fully wettable if the UV irradiation is performed under air due to additional photo-oxidation of the surface.
In an upcoming publication, the combined tuning of surface and bulk properties will be exploited to create micro-sized polymer structures with tailored and localized changes of the surface polarity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was performed at the Polymer Competence Center Leoben GmbH (PCCL, Austria) within the framework of the COMET-program of the Federal Ministry for Transport, Innovation and Technology and Federal Ministry for Economy, Family and Youth with contributions by the Chair of Chemistry of Polymeric Materials (Montanuniversitaet Leoben, Austria). The PCCL is funded by the Austrian Government and the State Governments of Styria, Upper and Lower Austria. Part of the research was also performed within the K-Project 'PolyTherm' at the Polymer Competence Center Leoben GmbH (PCCL, Austria) within the framework of the COMET-program of the Federal Ministry for Transport, Innovation and Technology and Federal Ministry for Digital and Economic Affairs. Funding is provided by the Austrian Government and the State Government of Styria. The research was further supported by the project RETINA which is being implemented and co-financed from the European Union – European Regional Development Fund in the frame of the Cooperation Programme Interreg V-A Slovenia-Austria in the programme period 2014–2020. In addition, the authors thank Simon Kaiser (PCCL) for characterizing the patterned films with confocal microscopy.References
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., 2001, 113, 2056–2075 CrossRef.
- (a) H. Nandivada, X. Jiang and J. Lahann, Adv. Mater., 2007, 19, 2197–2208 CrossRef; (b) J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- (a) J. Zhuang, M. R. Gordon, J. Ventura, L. Li and S. Thayumanavan, Chem. Soc. Rev., 2013, 42, 7421–7435 RSC; (b) W. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572–2590 CrossRef.
- A. O. Konuray, X. Fernández-Francos and X. Ramis, Polym. Chem., 2017, 8, 5934–5947 RSC.
- M. C. Stuparu and A. Khan, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3057–3070 CrossRef.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- X. Fernández-Francos, A.-O. Konuray, A. Belmonte, S. De la Flor, À. Serra and X. Ramis, Polym. Chem., 2016, 7, 2280–2290 RSC.
- (a) M. Sangermano, M. Cerrone, G. Colucci, I. Roppolo and R. Acosta Ortiz, Polym. Int., 2010, 59(8), 1046–1051 Search PubMed; (b) I. Isarn, X. Ramis, F. Ferrando and A. Serra, Polymers, 2018, 10, 277 CrossRef.
- S. Binder, I. Gadwal, A. Bielmann and A. Khan, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2040–2046 CrossRef.
- I. Gadwal and A. Khan, RSC Adv., 2015, 5, 43961–43964 RSC.
- (a) W. J. Yang, K.-G. Neoh, E.-T. Kang, S. Lay-Ming Teo and D. Rittschof, Polym. Chem., 2013, 4, 3105 RSC; (b) S. B. Rahane, R. M. Hensarling, B. J. Sparks, C. M. Stafford and D. L. Patton, J. Mater. Chem., 2012, 22, 932–943 RSC; (c) R. Iwata, R. Satoh, Y. Iwasaki and K. Akiyoshi, Colloids Surf., B, 2008, 62, 288–298 CrossRef.
- (a) K. A. Günay, P. Theato and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1–28 CrossRef; (b) M. Benaglia, A. Alberti, L. Giorgini, F. Magnoni and S. Tozzi, Polym. Chem., 2013, 4, 124–132 RSC; (c) E. M. Muzammil, A. Khan and M. C. Stuparu, RSC Adv., 2017, 7, 55874–55884 RSC.
- (a) N. Cengiz, J. Rao, A. Sanyal and A. Khan, Chem. Commun., 2013, 49, 11191–11193 RSC; (b) J. A. Carioscia, J. W. Stansbury and C. N. Bowman, Polymer, 2007, 48, 1526–1532 CrossRef CAS.
- (a) C. F. Carlborg, A. Vastesson, Y. Liu, W. van der Wijngaart, M. Johansson and T. Haraldsson, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2604–2615 CrossRef CAS; (b) O. Korychenska, C. Acebo, M. Bezuglyi, A. Serra and J. V. Grazulevicius, React. Funct. Polym., 2016, 106, 86–92 CrossRef CAS; (c) M. C. Stuparu and A. Khan, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3057–3070 CrossRef CAS.
- D. Guzmán, X. Ramis, X. Fernández-Francos and A. Serra, Eur. Polym. J., 2014, 59, 377–386 CrossRef.
- R. M. Loureiro, T. C. Amarelo, S. P. Abuin, E. R. Soulé and R. J. Williams, Thermochim. Acta, 2015, 616, 79–86 CrossRef CAS.
- A. O. Konuray, X. Fernández-Francos and X. Ramis, Polymer, 2017, 116, 191–203 CrossRef CAS.
- K. Dietliker, T. Jung, K. Studer and J. Benkhoff, Chimia, 2007, 61, 655–660 CrossRef.
- K. Arimitsu and R. Endo, Chem. Mater., 2013, 25, 4461–4463 CrossRef CAS.
- M. Sangermano, A. Vitale and K. Dietliker, Polymer, 2014, 55, 1628–1635 CrossRef CAS.
- (a) H. Salmi, X. Allonas, C. Ley, A. Defoin and A. Ak, Polym. Chem., 2014, 5, 6577–6583 RSC; (b) X. Dong, P. Hu, G. Zhu, Z. Li, R. Liu and X. Liu, RSC Adv., 2015, 5, 53342–53348 RSC.
- A. P. Pelliccioli and J. Wirz, Photochem. Photobiol. Sci., 2002, 1, 441–458 RSC.
- M. Giebler, S. V. Radl, M. Ast, S. Kaiser, T. Griesser, W. Kern and S. Schlögl, J. Polym. Sci., Part A: Polym. Chem., 2018, 8, 52 Search PubMed.
- S. Radl, M. Kreimer, J. Manhart, T. Griesser, A. Moser, G. Pinter, G. Kalinka, W. Kern and S. Schlögl, Polymer, 2015, 69, 159–168 CrossRef CAS.
- S. Radl, I. Roppolo, K. Pölzl, M. Ast, J. Spreitz, T. Griesser, W. Kern, S. Schlögl and M. Sangermano, Polymer, 2017, 109, 349–357 CrossRef CAS.
- S. V. Radl, C. Schipfer, S. Kaiser, A. Moser, B. Kaynak, W. Kern and S. Schlögl, Polym. Chem., 2017, 8, 1562–1572 RSC.
- N. Wagner and P. Theato, Polymer, 2014, 55, 3436–3453 CrossRef CAS.
- A. A. Brown, O. Azzaroni, L. M. Fidalgo and W. T. S. Huck, Soft Matter, 2009, 5, 2738 RSC.
- Y. H. Zhao, D. Vuluga, L. Lecamp and F. Burel, RSC Adv., 2016, 6, 32098–32105 RSC.
- A. P. Pelliccioli and J. Wirz, Photochem. Photobiol. Sci., 2002, 1, 441–458 RSC.
- E. Reichmanis, B. C. Smith and R. Gooden, J. Polym. Sci., Polym. Chem. Ed., 1985, 23, 1–8 CrossRef CAS.
- V. N. Rajasekharan Pillai, Synthesis, 1980, 1980, 1–26 CrossRef.
- H. Barzynski and D. Sänger, Angew. Makromol. Chem., 1981, 93, 131–141 CrossRef CAS.
- (a) A. Belmonte, C. Russo, V. Ambrogi, X. Fernández-Francos and S. de La Flor, Polymers, 2017, 9, 113 CrossRef; (b) D. Guzmán, X. Ramis, X. Fernández-Francos and A. Serra, Polymers, 2015, 7, 680–694 CrossRef.
- W. C. Oliver and G. M. Pharr, J. Mater. Res., 1992, 7, 1564–1583 CrossRef CAS.
- S.-J. Park, F.-L. Jin and C. Lee, Mater. Sci. Eng., A, 2005, 402, 335–340 CrossRef.
- (a) C.-H. Kuo, H.-Y. Chang, C.-P. Liu, S.-H. Lee, Y.-W. You and J.-J. Shyue, Phys. Chem. Chem. Phys., 2011, 13, 3649 RSC; (b) J. Manhart, D. Lenko, I. Mühlbacher, A. Hausberger, R. Schaller, A. Holzner, W. Kern and S. Schlögl, Eur. Polym. J., 2015, 66, 236–246 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Results on solid state NMR spectroscopy and FT-IR spectra. See DOI: 10.1039/c8ra08937j |
| This journal is © The Royal Society of Chemistry 2018 |