
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
NMR-based investigations of acyl-functionalized piperazines concerning their conformational behavior in solution†
Robert Wodtke‡
a,
Janine Steinberg‡ab,
Martin Köckerling c,
Reik Löser*ab and
Constantin Mamat*ab
c,
Reik Löser*ab and
Constantin Mamat*ab
aInstitut für Radiopharmazeutische Krebsforschung, Helmholtz-Zentrum Dresden-Rossendorf, Bautzner Landstraße 400, D-01328 Dresden, Germany. E-mail: r.loeser@hzdr.de; c.mamat@hzdr.de
bFakultät Chemie und Lebensmittelchemie, Technische Universität Dresden, D-01062 Dresden, Germany
cInstitut für Chemie – Anorganische Festkörperchemie, Universität Rostock, Albert-Einstein-Straße 4a, D-18059 Rostock, Germany
First published on 6th December 2018
Abstract
Selected N-benzoylated piperazine compounds were synthesized to study their conformational behavior using temperature-dependent 1H NMR spectroscopy. All investigated piperazines occur as conformers at room temperature resulting from the restricted rotation of the partial amide double bond. In the case of selected mono-N-benzoylated and unsymmetrically N,N′-substituted derivatives, the appearance of the 1H NMR spectrum was further shaped by the limited interconversion of the piperazine chair conformations. Therefore, two different coalescence points TC were determined and their resulting activation energy barriers ΔG‡ were calculated to be between 56 and 80 kJ mol−1. In most of the cases, TC and ΔG‡ for the amide site appeared to be higher than the corresponding values for the ring inversion. The influences of substituents on rotational and inversion barriers were analyzed by correlation to Hammett constants. The obtained results are discussed and interpreted in the context of literature data. An additional aryl substituent connected at the amine site led to reduced rotational and inversion barriers compared to the free secondary amine. To support and evidence the findings from the NMR analyses, single crystals of selected piperazines were obtained and XRD analyses were performed. To underline the results, two potential TGase 2 inhibitors were investigated showing energy barriers with similar values.
Introduction
Piperazines belong to the important class of saturated heterocyclic compounds in organic chemistry containing two nitrogen atoms.1 The piperazine scaffold has been integrated as a privileged structure and is frequently found in bioactive and pharmacologically relevant compounds across a number of different therapeutic areas. A wide variety of N-functionalized piperazines serve as skeletons in pesticides, pharmaceuticals2–4 such as analgesics5 and other drugs,6,7 scaffolds in medicinal chemistry,8–10 intermediates in organic reactions,11,12 ligands for complexation purposes,13–15 for peptide syntheses and modifications16,17 as well as building blocks in material sciences e.g. for polymerizations.18 Furthermore, piperazines are also in use for the synthesis of labeling compounds and building blocks, e.g., for inserting fluorescent dyes19,20 or for the introduction of radionuclides21–23 into biologically active molecules. The understanding of the conformational behavior of these functionalized piperazines is important not only to explain biological and/or pharmacological effects, but also for applications in material sciences.Among the different N-functionalized piperazines, N-acylated piperazines exhibit a complex conformational behavior due to the hindered rotation of the amide bond in addition to the conformational phenomena of the piperazine ring itself. Regarding the first phenomenon, the hindered rotation of the C–N bond in secondary and tertiary amides is well known and has far-reaching implications towards peptide and protein folding.24 However, the NMR properties of tertiary benzamides were only rarely studied in the past with N,N-dimethyl benzamides25–27 and N,N-diethyl benzamides28 representing the best characterized compounds. Furthermore, the structural properties and conformational dynamics of tertiary amides including 1-acylpiperazines have been investigated by other spectroscopic methods within several studies.29,30 Recently, the NMR behavior of selected benzoylpiperazines, which were used as prosthetic groups for 18F-labeling, was investigated.21
To shed further light on the conformations of benzoylated piperazines including substituent effects at the benzoyl residue and different moieties at the piperazine ring, novel symmetrically and unsymmetrically N,N′-functionalized piperazine derivatives were synthesized and characterized in terms of their particular NMR behavior.
Results and discussion
To get a deeper understanding on the NMR behavior of acylated piperazines in terms of their conformation at room temperature, three groups of N-acylated piperazine derivatives were synthesized. Different substituted benzoyl moieties were used for functionalization to show their influence on the formation of rotamers and the resulting rotation barrier. The first group consists of mono N-acylated compounds 3a–i, the second group contains symmetrically N,N′-diacylated compounds 4a–f and the third group consists of N-acylated-N′-arylated derivatives 6a–k. The first and second group of benzoylated piperazines was obtained by the reaction of piperazine (1) in excess to the respective benzoyl chlorides 2a–i in a one-pot reaction. Both, the mono-N-acylated compounds 3a–i and the N,N′-diacylated compounds 4a–f were yielded from the reaction mixture.31 Most of the compounds are known from the literature and were proven by 1H and 13C NMR analyses. An overview of the synthesis route to mono-N-substituted and symmetrically N,N-disubstituted derivatives is given in Scheme 1. To determine the influence of an aryl moiety containing the electron withdrawing nitro group at the N′-position of the piperazine, a third set of compounds 6a–d, f, h–k was synthesized by using 1.1 equivalents of 1-(4-nitrophenyl)piperazine (5) and 1 equivalent of the respective benzoyl chloride 2a–d, f, h–k in anhydrous THF using triethylamine as base. Compounds 6a–d, f, h–k were obtained in yields ranging from 37 to 98%. Their structural identities were verified by 1H and 13C NMR analyses. An overview of the synthesis route to these derivatives is given in Scheme 2. | ||
| Scheme 1 Synthesis of functionalized piperazines 3a–i and 4a–f used for the NMR studies. | ||
 | ||
| Scheme 2 Synthesis of functionalized piperazines 6a–d, f, h–k used in the NMR studies. | ||
NMR experiments
To determine the conformational behavior of these piperazine compounds, three different phenomena have to be considered (Fig. 1). The first results from the reduced rotation of the N–C-bond located in the amide moiety formed by the benzoyl residue and one or both of the tertiary piperazine nitrogen atoms. This is due to the partial double bond character, which results in the occurrence of two different rotamers. This effect is known from simple dialkylated carboxylic acid amides and also tertiary benzamides.25,28,32,33 The second phenomenon regards the conformation of the piperazine ring itself. Like cyclohexanes, piperazine rings can change their conformations (e.g. chair, half-chair, boat, twist-boat). Even though systematic experimental studies are lacking, the energetic barriers for the transformation of the chair conformations seem to be higher for piperazines than for cyclohexanes. This can be concluded from comparing the ring inversion barriers of cis-1,4-dimethylcyclohexane and N,N′-dimethylpiperazine, which are 40.0 kJ mol−1 in dichlorodifluoromethane34 and 55.7 kJ mol−1 in dichloromethane,35 respectively. The third phenomenon describes the pyramidal inversion of the nitrogen atom of the amine moiety; however, this should not play a major role.36 Subsequently, symmetrically disubstituted piperazines are discussed at first, followed by the mono-substituted and unsymmetrically substituted piperazines, due to the increasing complexity of the conformational behavior. | ||
| Fig. 1 Conformational dynamics of benzoylated piperazines. | ||
Symmetrically disubstituted piperazines
To investigate the conformational behavior of symmetrically N,N′-disubstituted derivatives 4a–f, 1H-NMR spectra were recorded in CDCl3 and DMSO-d6. Normally, one summarized signal for the protons (NCH2) of the piperazine ring would be expected according to the symmetry of the molecule and the flexibility of the piperazine ring. This is only the case at elevated temperatures (>25–30 °C) when the molecules are flexible enough to rotate. At deeper temperatures (below 25 °C), four independent signals were found (e.g. for 4a: δ = 3.40, 3.55, 3.74, 3.90 ppm at −10 °C). H,H-COSY spectra measured in CDCl3 revealed that the inner two NCH2 signals couple in most of the cases (Fig. 2).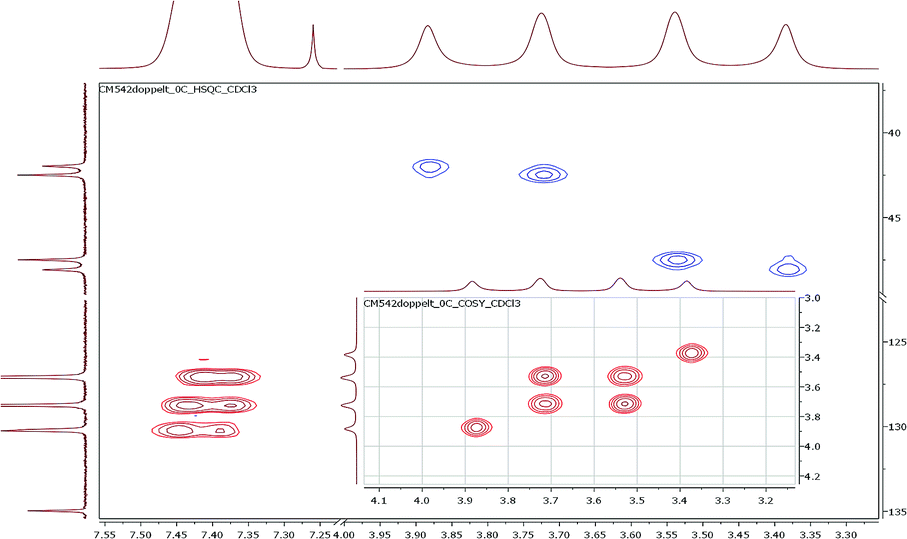 | ||
| Fig. 2 Part of the 1H,13C-HSQC and H,H-COSY (inset) spectra using the symmetrically disubstituted compound 4a at 0 °C. | ||
These signals belong to the anti (trans) isomer whereas the two outer NCH2 signals belong to the syn (cis) isomer according to the results obtained by Sahoo and Chand (Fig. 3).14 The 1H–13C-HSQC spectrum of compound 4a measured at 0 °C verifies this behavior. Additionally, all 13C NMR spectra show also four independent signals (4a syn: δ = 42.0 and 48.1 ppm and anti: δ = 42.5 and 47.5 ppm at −10 °C) at low temperatures. This further evidenced the formation of two different rotational isomers. The next indication is given by the study of the methyl group and the signals in the aromatic region of 4-methylbenzoyl compound 4b. In this case, both doublets of the aromatic moiety and the methyl group (δ = 2.35 and 2.40 ppm) occur twice at temperatures below −10 °C (Fig. 4).
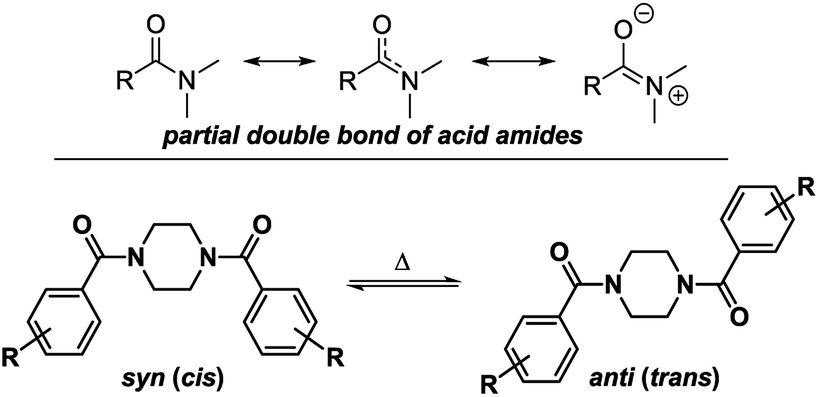 | ||
| Fig. 3 Possible rotational conformers as a result of the partial double bond character of the formed amide for symmetrically disubstituted piperazines 4a–f. | ||
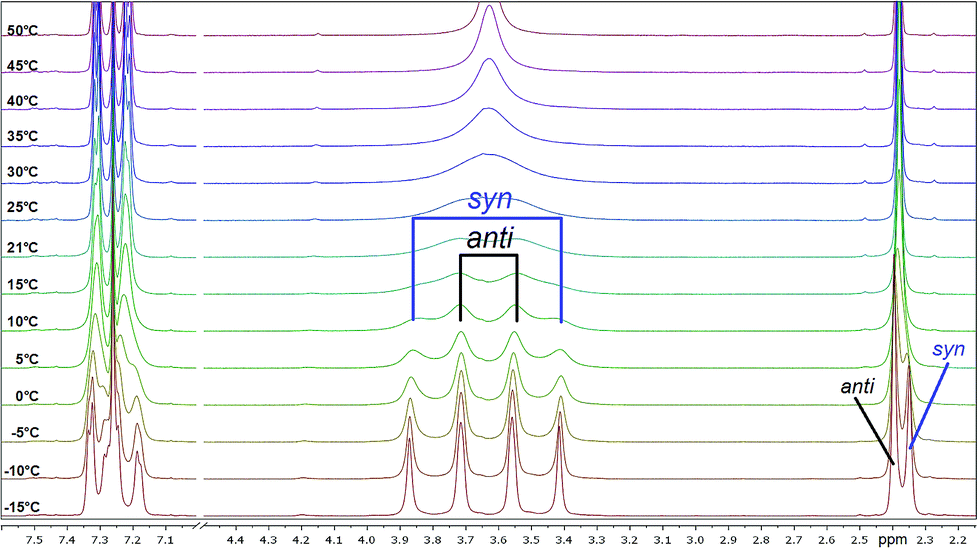 | ||
| Fig. 4 Temperature-dependent 1H NMR spectra of 4b measured in CDCl3 (region of interest from 2.00 to 7.6 ppm is shown). | ||
All investigated symmetrically disubstituted piperazines 4a–f show the interconversion between these two isomers: syn (cis) and anti (trans). In this case, one coalescence temperature TC was observed, which is dependent on the solvent (Fig. 4). By warming the NMR sample, the four signals with an integration value of two hydrogen atoms each combine to one signal with a value of 8 at the coalescence point TC resulting from a free rotation of the amide bonds present in the molecules. This allows the calculation of the Gibbs free activation energy ΔG‡. In general, the exchange rate at the coalescence temperature TC is given by the equation kexc = πΔν/21/2.37,38 As a result, the Gibbs free activation energy for the rotation of the amide bond can be calculated using the Eyring eqn (II).39 The results are presented in Table 1.
| No. | R | Ratio anti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :syn :syn |
Δν [Hz] | kexc [Hz] | TC [K] | ΔG‡ [kJ mol−1] |
|---|---|---|---|---|---|---|
| a Not detectable; (a) anti; (s) syn. | ||||||
| 4a | H | 1.4:1 |
111.1 (a) | 246.8 (a) | 306.15 | 61.0 (a) |
| 299.4 (s) | 665.1 (s) | 58.5 (s) | ||||
| 4b | 4-Me | 1.5:1 |
94.2 (a) | 209.3 (a) | 299.15 | 60.0 (a) |
| 274.1(s) | 608.9 (s) | 57.3 (s) | ||||
| 4c | 4-OMe | — | n.d.a | — | 280.15 | — |
| 4d | 4-F | 1.5:1 |
103.2 (a) | 229.3 (a) | 296.15 | 59.1 (a) |
| 272.0 (s) | 604.2 (s) | 56.8 (s) | ||||
| 4e | 4-Cl | 1.6:1 |
108.6 (a) | 241.2 (a) | 300.15 | 59.8 (a) |
| 284.5 (s) | 632.0 (s) | 57.4 (s) | ||||
| 4f | 4-Br | 1.5:1 |
108.8 (a) | 241.7 (a) | 304.15 | 60.7 (a) |
| 285.9 (s) | 635.1 (s) | 58.2 (s) | ||||
| 4g | 4-I | 109.9 (a) | 244.1 (a) | 305.15 | 60.9 (a) | |
| 287.2 (s) | 638 (s) | 58.4 (s) | ||||
Different Δν values each for the syn and for the anti derivatives were determined whereas only one TC was found leading to different ΔG‡ values for the syn and the anti isomer. Notably, it was not possible to determine Δν values for the methoxy compound 4c in CDCl3 and for all compounds 4a–f in DMSO-d6 due to the limited cooling capacity of the NMR cooling unit and too low coalescence temperatures in relation to the melting point of the solvent, respectively.
As shown in Fig. 4, a clear distinction between the syn (cis) and anti (trans) rotamers was observed at low temperatures (<0 °C). Four independent signals occurred in the 1H NMR spectra of all compounds measured in CDCl3 with the anti isomer representing the energetically favored rotamer as a similar ratio of approx. 3:2 was determined for all compounds. In accordance to these results, all anti isomers have also a higher rotation barrier (59.1–61.5 kJ mol−1) than the corresponding syn derivatives (56.7–59.1 kJ mol−1) (Table 1).
Mono-substituted piperazines
Mono-N-benzoylated piperazines 3a–i contain only one amide bond. The other nitrogen site of the piperazine ring is unsubstituted and can therefore be considered as a typical secondary amine. Interestingly, also four signals occur at 25 °C for most of these compounds, two for the CH2 groups next to the amide moiety and two for the CH2 groups adjacent to the amine residue. As an example, the spectrum of 3b shows four broad signals (δ = 2.81, 2.91, 3.41, 3.70 ppm in CDCl3 at 25 °C) for the NCH2 groups of the piperazine moiety (Fig. 5). An evaluation of a H,H-COSY spectrum measured in CDCl3 showed again an independent coupling of two NCH2 groups in 3a. In this case, the equatorial proton of the amide site couples with the equatorial proton of the amine site of the piperazine ring and both axial protons couple.40 Furthermore, the 1H,13C-HSQC spectra showed the independent coupling of the protons to the appropriate carbon signals (ESI†). Additionally, broad signals for the carbons of the NCH2 groups (e.g. 3b: δ = 43.3, 46.2, 49.0 ppm) are found when analyzing the 13C NMR spectra.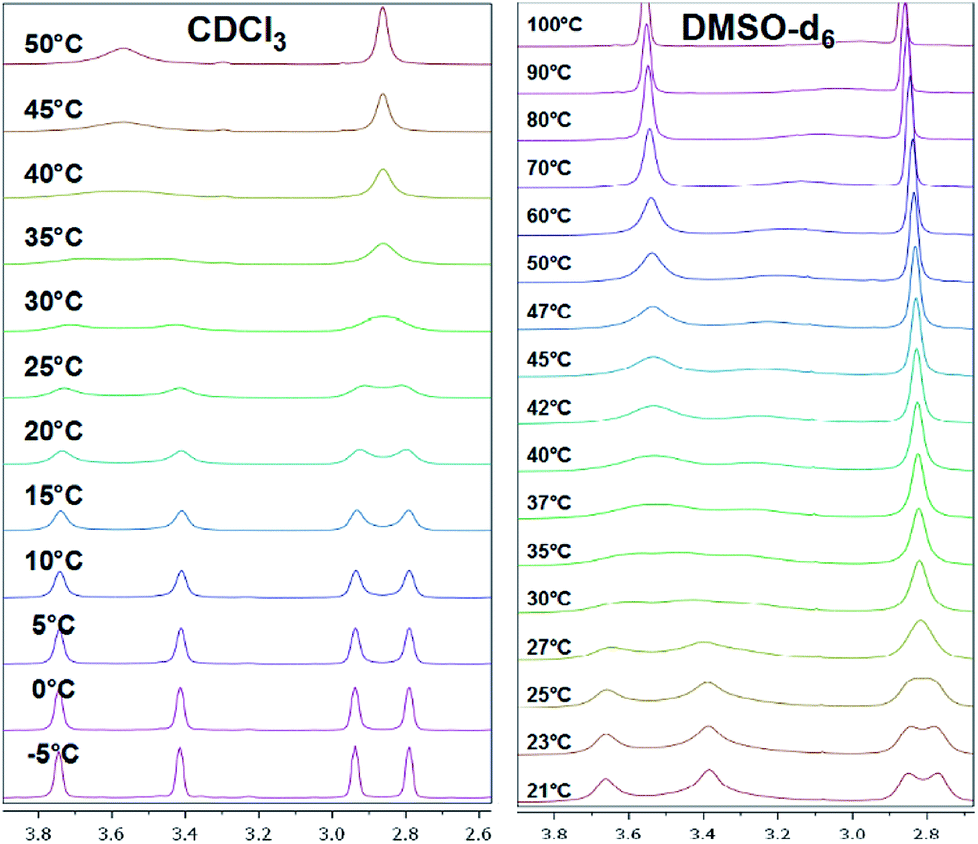 | ||
| Fig. 5 Temperature-dependent 1H NMR spectra of 3b measured in CDCl3 (left) and DMSO-d6 (right, only piperazine region from 2.6 to 4.0 ppm is shown). | ||
Two effects are responsible for this behavior of the mono-N-benzoylated piperazines 3a–i. Two different conformers (rotamers) occur due to the limited rotation of the C–N amide bond resulting from its partial double bond character (splitting of the NCH2 signals at the amide nitrogen atom). Thus, two distinct sets of signals are observed in the 1H NMR spectrum resulting from the different chemical environment that is faced by the substituents attached to the amide nitrogen atom as already shown for the symmetrically disubstituted compounds 4a–f. In general, this behavior of symmetric N,N-dialkylamide spin systems is describable as first-order process on the NMR time scale.41
The second effect is related to the hindered interconversion of the two piperazine ring chair conformations (Fig. 1, bottom). In the present case, this process is also decreased at room temperature (splitting of the NCH2 signals at the amine nitrogen atom). For simple non-acylated piperazines42–44 and morpholines45 the observation of distinct conformers by NMR is restricted to low temperatures (<−10 °C). The different energy barriers of both effects resulted in two different coalescence points. To further investigate the conformational behavior of the piperazines and to determine these energies, temperature-dependent NMR experiments39 were performed for all derivatives in CDCl3 and in DMSO-d6. Results are presented in Tables 2 and 3.
| No. | R | Δν [Hz] | kexc [Hz] | TC [K] | ΔG‡ [kJ mol−1] |
|---|---|---|---|---|---|
| 3a | H | 90.9 (amine) | 246.8 (amine) | 308.15 (amine) | 62.0 (amine) |
| 217.8 (amide) | 665.1 (amide) | 318.15 (amide) | 61.7 (amide) | ||
| 3b | 4-Me | 88.5 (amine) | 196.6 (amine) | 303.15 (amine) | 61.0 (amine) |
| 198.3 (amide) | 440.5 (amide) | 313.15 (amide) | 61.0 (amide) | ||
| 3c | 4-OMe | 80.0 (amine) | 177.7 (amine) | 288.15 (amine) | 58.1 (amine) |
| 165.1 (amide) | 366.8 (amide) | 295.15 (amide) | 57.8 (amide) | ||
| 3d | 4-F | 83.3 (amine) | 185 (amine) | 298.15 (amine) | 60.1 (amine) |
| 204 (amide) | 453.2 (amide) | 309.15 (amide) | 60.1 (amide) | ||
| 3e | 4-Cl | 86.6 (amine) | 192.4 (amine) | 305.15 (amine) | 61.5 (amine) |
| 217.1 (amide) | 482.3 (amide) | 319.15 (amide) | 62.0 (amide) | ||
| 3f | 4-Br | 87.5 (amine) | 194.4 (amine) | 304.15 (amine) | 61.2 (amine) |
| 214.1 (amide) | 475.6 (amide) | 316.15 (amide) | 61.4 (amide) | ||
| 3g | 4-I | 87.3 (amine) | 193.9 (amine) | 310.15 (amine) | 62.5 (amine) |
| 215.0 (amide) | 477.6 (amide) | 315.15 (amide) | 61.2 (amide) | ||
| 3h | 4-NO2 | 92.14 (amine) | 204.7 (amine) | 320.15 (amine) | 64.4 (amine) |
| 267.2 (amide) | 596.6 (amide) | n.d. (amide) | n.d. (amide) | ||
| 3i | 3-Br | 81.4 (amine) | 180.8 (amine) | 309.15 (amine) | 62.5 (amine) |
| 215.13 (amide) | 477.9 (amide) | 319.15 (amide) | 62.0 (amide) |
| No. | R | Δν [Hz] | kexc [Hz] | TC [K] | ΔG‡ [kJ mol−1] |
|---|---|---|---|---|---|
| 3b | 4-Me | 48.28 (amine) | 107.3 (amine) | 299.15 (amine) | 61.6 (amine) |
| 166.58 (amide) | 370.0 (amide) | 309.15 (amide) | 60.6 (amide) | ||
| 3d | 4-F | n.d. (amine) | n.d. (amine) | 296.15 (amine) | n.d. (amine) |
| 168.9 (amide) | 375.2 (amide) | 308.15 (amide) | 60.4 (amide) | ||
| 3e | 4-Cl | 59.36 (amine) | 131.9 (amine) | 307.15 (amine) | 62.8 (amine) |
| 190.68 (amide) | 423.6 (amide) | 321.15 (amide) | 62.7 (amide) | ||
| 3f | 4-Br | 59.6 (amine) | 132.4 (amine) | 305.15 (amine) | 62.4 (amine) |
| 189.1 (amide) | 420.1 (amide) | 330.15 (amide) | 64.6 (amide) | ||
| 3g | 4-I | 58.2 (amine) | 129.3 (amine) | 304.15 (amine) | 62.2 (amine) |
| 183.2 (amide) | 407 (amide) | 318.15 (amide) | 62.2 (amide) | ||
| 3h | 4-NO2 | 75.5 (amine) | 167.7 (amine) | 325.15 (amine) | 66.0 (amine) |
| 236.1 (amide) | 524.5 (amide) | 338.15 (amide) | 65.6 (amide) | ||
| 3i | 3-Br | 62.60 (amine) | 139.1 (amine) | 311.65 (amine) | 63.7 (amine) |
| 196.23 (amide) | 435.9 (amide) | 328.15 (amide) | 64.1 (amide) |
When monitoring the piperazine derivatives 3a–i over a range of up to 80 K, the four signals of the NCH2 groups gradually disappear and coalesce to the two expected signals (one signal for the amide site and one for the amine site of the piperazine ring) at increased temperatures (TC,amine = 303 K, TC,amide = 313 K for 3b in CDCl3). The determined ΔG‡ values for both barriers lie between 57 and 66 kJ mol−1 with higher values obtained in DMSO-d6 (Tables 2 and 3). The determined values for the amide bond rotation barriers are in agreement to those reported for other benzamides, which are in the range of 57–73 kJ mol−1.46–48 The observed solvent effect on the rotational barriers is also in accord to literature reports and can be explained by the fact that more polar solvent molecules can better stabilize the zwitterionic canonical form of the amide bond (Fig. 3 top right).25–27,49
As obvious from Tables 2 and 3, the ΔG‡ values for the amide bond and for the amine site strongly depend on the substituent at the benzoyl residue. Generally, substituents in meta and para position to the amide bond influence its rotational barrier by inductive and mesomeric effects via the π-system of the phenyl ring. To quantify this interaction, correlation analyses were performed using Hammett σ constants (σm/p, σp−, σp+). For DMSO-d6, the correlations of the ΔG‡ values versus σm/p are shown in Fig. 6 (for further correlations see ESI†). All correlations resulted in fair to good linear relationships with a positive slope. Thus, the rotational barriers increase with increasing electron withdrawing character of the substituents defined by their σm/p values. Consequently, substituents with a higher electron withdrawing effect promote the formation of the zwitterionic form of the amide bond and/or destabilize the transition state of the isomerization, in which the carbonyl bond is oriented perpendicular to the nitrogen lone electron pair. Both effects will lead to a higher activation enthalpy for the C–N bond rotation. Worth of note, for the activation energies determined in DMSO-d6, similar correlations were obtained for the different Hammett σ constants, whereas for the energies determined in CDCl3, the σp+ values, which describe the capacity of substituents to delocalize a positive charge,50 give significant better correlations than using the other Hammett σ constants for both amide and amine sites (see ESI†). This result can be interpreted in the context of the fact that the zwitterionic ground state is less stabilized in this solvent compared to DMSO-d6 and therefore the donating effect of the para-substituent to the positively charged carbonyl carbon in the transition state is more predominant in CDCl3.
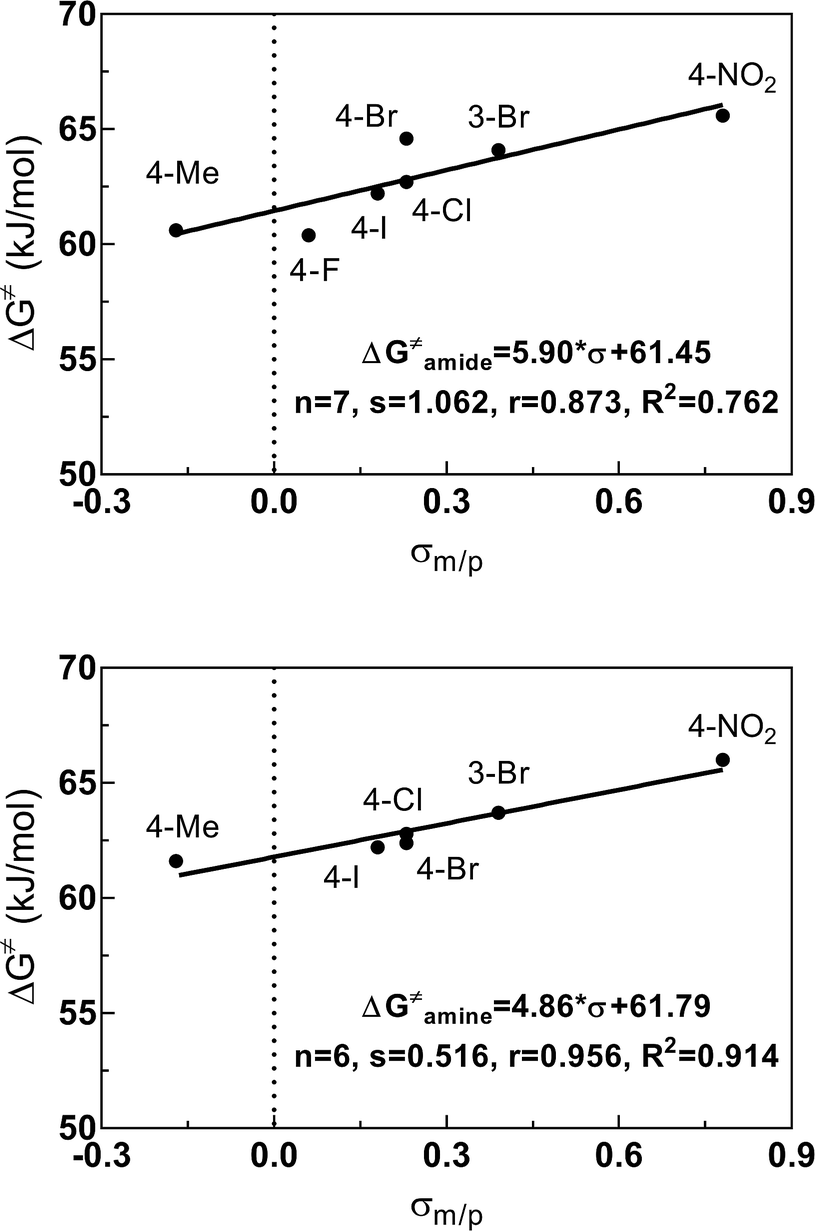 | ||
| Fig. 6 Relationship between the electronic properties (σm/p) of the substituents at the benzoyl residue and the activation energies ΔG‡ for the amide (top) and amine (bottom) sites of compounds 3 determined in DMSO-d6. Plot of ΔG‡ = f(σm/p) using the ΔG‡ values listed in Table 3 and the following values for σm: 0.39 (3-Br) and for σp: −0.17 (Me), 0.06 (F), 0.23 (Cl), 0.23 (Br), 0.18 (I) and 0.78 (NO2).51 Regression analysis was performed by linear regression. | ||
The derived relationships are in accordance to studies on the rotational barriers for the tertiary amide bond in N,N-dimethyl benzamides25–27 and N,N-diethylbenzamides.28 The strengthened zwitterionic character of the amide bond in turn results in a reduced conformational flexibility of the piperazine ring and requires higher energies for the ring conversion. Worth of note, irrespective of the kind of electronic substituent parameter, the free activation enthalpies for the ring inversion barriers (ΔG‡ of amine) gave significantly better correlations than the barriers for amide bond rotation. To the best of our knowledge, systematic investigations of the substituent effects on the ring inversion barriers in piperazines have not been reported so far. The observed trend that the free activation enthalpies increase with higher electron pull of the substituent at the benzoyl moiety is in contrast to results obtained for N-substituted morpholines, for which a higher degree of conjugation at the ring nitrogen results in lower ring inversions barriers.52–54 This effect has been mainly attributed to larger bond angles around the ring nitrogen upon introduction of acyl substituents, which diminishes the angle strain in the transition state. Furthermore, we hypothesize that the stereoelectronic gauche effect, which should be in operation in both piperazine and morpholine rings, acts towards stabilization of the chair conformations. In morpholines, the gauche effect should act mainly through charge transfer from the equatorial σC–H orbitals at C-3 and C-5 into the anti-bonding 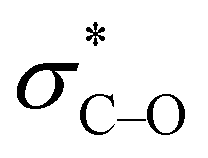 orbitals (Fig. 7). Consequently, substituents at nitrogen with increasing electron pull will lower this hyperconjugation as the electron density in the respective σC–H orbitals is reduced, and therefore the ring inversion barriers are decreased. Thus, ring flattening and decreased hyperconjugation act additively.
orbitals (Fig. 7). Consequently, substituents at nitrogen with increasing electron pull will lower this hyperconjugation as the electron density in the respective σC–H orbitals is reduced, and therefore the ring inversion barriers are decreased. Thus, ring flattening and decreased hyperconjugation act additively.
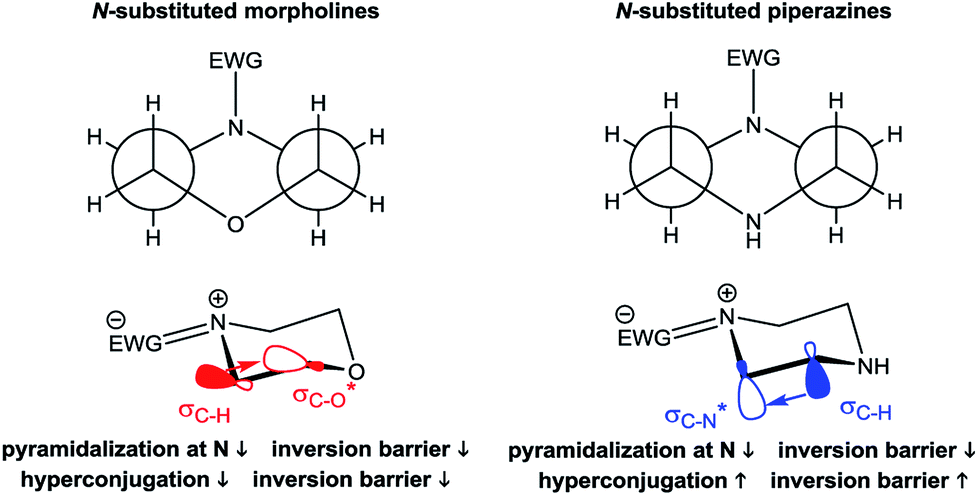 | ||
| Fig. 7 Proposed relationship between ring flattening and hyperconjugation (gauche effect) in N-substituted morpholines and piperazines. The Newman projections shown on top are indicating the gauche arrangement of both heteroatoms in the ring. Below are shown the chair models and the main direction of charge transfer by the gauche effect and the influence of electron-withdrawing groups (EWG) at the ring nitrogen. See text for further explanation. | ||
In contrast, in the piperazine derivatives of compound series 3, in which the oxygen atom is replaced by less electronegative nitrogen, the charge transfer by the gauche effect is acting mainly in the counter direction, i.e. from the σC–H orbitals at C-3 and C-5 into the anti-bonding 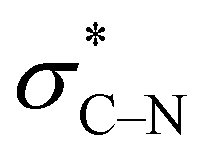 orbitals involving the N-1 nitrogen. Higher positive partial charge at N-1 should increase this hyperconjugation, as it has been found for 2-fluoroethylamine derivatives,55 and therefore the chair form should be more stabilized by this effect. Consequently, the influence of electron-withdrawing group at the nitrogen in N-monosubstituted piperazines on ring flattening and hyperconjugation by the gauche effect are acting in opposite directions. In this context, it is worth of note that the activation energy for N-benzoylmorpholine is with 31.0 kJ mol−1 (in dichlorofluoromethane) considerably lower than that found for compound 3a (61.02 kJ mol−1 in CDCl3).52–54 The influence of the substituents on the gauche effect seems to be stronger than that on ring flattening with regards to the inversion barriers in piperazines, which results in a correlation with positive slope between the Hammett substituent constants and the free activation enthalpies for ring inversion (Fig. 6 and ESI†). This effect can be further illustrated by comparing the ring inversion barriers of methoxy- and nitro-substituted compounds 3c and 3h in CDCl3, which exhibit the lowest and highest values for ΔG‡, respectively (see Table 2; 58.08 vs. 64.43 kJ mol−1).
orbitals involving the N-1 nitrogen. Higher positive partial charge at N-1 should increase this hyperconjugation, as it has been found for 2-fluoroethylamine derivatives,55 and therefore the chair form should be more stabilized by this effect. Consequently, the influence of electron-withdrawing group at the nitrogen in N-monosubstituted piperazines on ring flattening and hyperconjugation by the gauche effect are acting in opposite directions. In this context, it is worth of note that the activation energy for N-benzoylmorpholine is with 31.0 kJ mol−1 (in dichlorofluoromethane) considerably lower than that found for compound 3a (61.02 kJ mol−1 in CDCl3).52–54 The influence of the substituents on the gauche effect seems to be stronger than that on ring flattening with regards to the inversion barriers in piperazines, which results in a correlation with positive slope between the Hammett substituent constants and the free activation enthalpies for ring inversion (Fig. 6 and ESI†). This effect can be further illustrated by comparing the ring inversion barriers of methoxy- and nitro-substituted compounds 3c and 3h in CDCl3, which exhibit the lowest and highest values for ΔG‡, respectively (see Table 2; 58.08 vs. 64.43 kJ mol−1).
Unsymmetrically substituted piperazines
Based on the results for the mono-substituted piperazines, we were interested in the NMR behavior of mono-benzoyl-substituted piperazines where the secondary amine group is replaced by a tertiary amine group. For this purpose, 4-nitrophenylpiperazine was reacted with different benzoyl chlorides to yield the series of analogues 6a–k. Dynamic NMR experiments were performed in CDCl3. Again, four signals occur at 25 °C for most of these compounds.For the meta and para substituted derivatives, the ΔG‡ values for the rotation of the amide bond and ring inversion were derived (Table 4). Generally, the activation energies for both phenomena are up to 2.5 kJ mol−1 lower compared to those energies for the respective mono-substituted piperazines 3. The presence of the phenyl group at the former secondary amine results in a partial double bond character of the N–C1,Phenyl bond due to delocalization of the nitrogen lone pair towards the substituent. The nitro group in para position to the piperazine nitrogen (push–pull system) further favors this delocalization. This increases the bond angle around the nitrogen atom and lowers the ring inversion barriers (ring flattening). Simultaneously, the electron density within the σC–H orbital is reduced, which might decline the charge transfer to the 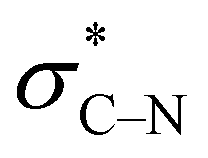 orbital and thus, lowers the barrier for ring inversion (Fig. 7). Similar to compounds 3, correlation analyses of the ΔG‡ values with the Hammett σ constants resulted in linear relationships with positive slopes. This indicates, that the gauche effect is still predominant compared to the ring flattening.
orbital and thus, lowers the barrier for ring inversion (Fig. 7). Similar to compounds 3, correlation analyses of the ΔG‡ values with the Hammett σ constants resulted in linear relationships with positive slopes. This indicates, that the gauche effect is still predominant compared to the ring flattening.
| No. | R | Solvent | Δν [Hz] | kexc [Hz] | TC [K] | ΔG‡ [kJ mol−1] |
|---|---|---|---|---|---|---|
| a Coalescence of two protons from one of the NCH2 groups of the amide site, complete coalescence of the amide site was not detectable. | ||||||
| 6a | H | CDCl3 | 99.0 (amine) | 219.9 (amine) | 299.15 (amine) | 59.9 (amine) |
| 183.9 (amide) | 408.5 (amide) | 307.15 (amide) | 60.0 (amide) | |||
| 6b | 4-Me | CDCl3 | 96.3 (amine) | 213.9 (amine) | 294.15 (amine) | 58.9 (amine) |
| 160.2 (amide) | 355.9 (amide) | 302.15 (amide) | 59.3 (amide) | |||
| 6c | 4-OMe | CDCl3 | 85.9 (amine) | 190.8 (amine) | 281.15 (amine) | 56.4 (amine) |
| 126.6 (amide) | 281.2 (amide) | 286.15 (amide) | 56.6 (amide) | |||
| 6d | 4-F | CDCl3 | 90.2 (amine) | 200.4 (amine) | 291.15 (amine) | 58.4 (amine) |
| 166.5 (amide) | 369.9 (amide) | 297.15 (amide) | 58.2 (amide) | |||
| 6f | 4-Br | CDCl3 | 92.5 (amine) | 205.5 (amine) | 297.15 (amine) | 59.6 (amine) |
| 177.0 (amide) | 393.2 (amide) | 305.15 (amide) | 59.6 (amide) | |||
| 6h | 4-NO2 | CDCl3 | 99.9 (amine) | 221.9 (amine) | 309.15 (amine) | 61.9 (amine) |
| 232.0 (amide) | 515.4 (amide) | 323.15 (amide) | 62.6 (amide) | |||
| 6i | 3-Br | CDCl3 | 88.3 (amine) | 196.2 (amine) | 300.15 (amine) | 60.4 (amine) |
| 183.0 (amide) | 406.5 (amide) | 311.15 (amide) | 60.8 (amide) | |||
| 6j | 2-Br | DMSO-d6 | 74.5 (amine) | 165.5 (amine) | >393.15 (amine) | — (amine) |
| 6k | 2-NO2 | CDCl3 | 232.4a | 516.3 | 312.15a | 60.4 |
| 6k | 2-NO2 | DMSO-d6 | 105.3 (amine) | 233.9 (amine) | 391.15 (amine) | 78.9 (amine) |
| 262.7 (amide) | 583.6 (amide) | >393.15 (amide) | — (amide) | |||
Furthermore, the substituent parameter σp+ gives again significantly better correlations than parameters σp and σp− for both amide and amine sites (ESI†). The three bromobenzoyl derivatives 6f, 6i, and 6j were used to compare the influence of the position (ortho, meta and para). Changing the position of the bromine atom from para (6f) to meta (6i) did not result in significantly different rotational barriers of the amide bond as the value of ΔG‡ is approx. 60 kJ mol−1 for both compounds. In contrast, a different chemical shift pattern of the NCH2 groups in CDCl3 and DMSO-d6 was found for 6j due to the steric hindrance of the bromine in ortho position and therefore a reduced rotation about the Cipso–C(O) bond.56 Thus, additional NMR spectra of 6j were recorded in acetonitrile-d3, acetone-d3, benzene-d6, and methanol-d4 (Fig. 8, ESI†), which revealed a different splitting of the NCH2 protons depending on the solvent. In CDCl3 the protons are further split (ESI†) whereas in DMSO-d6 three broadened signals and a triplet with two protons each was found. H,H-COSY revealed, that two of them coupled indicating that the two downfield-shifted signals are belonging to the amide site and the two other belonging to the amine site of the piperazine ring.
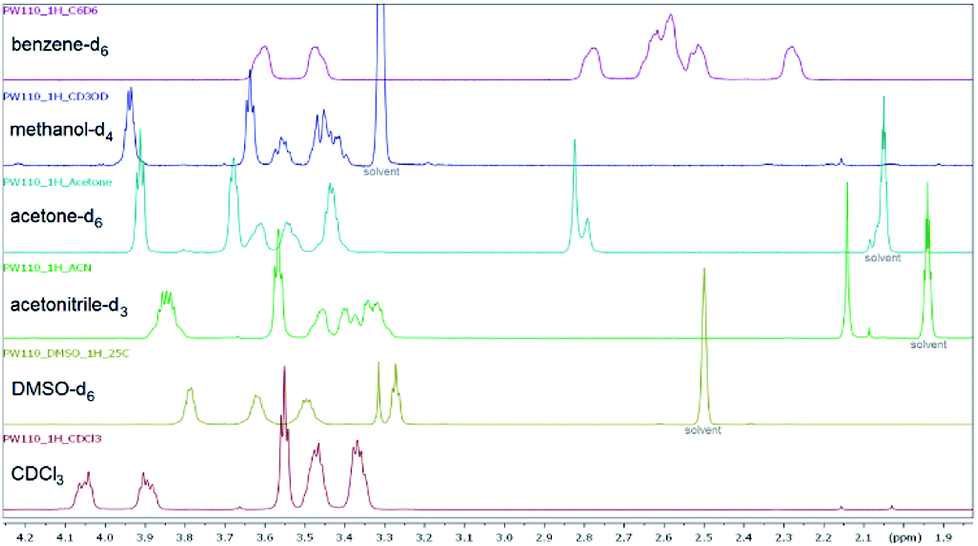 | ||
| Fig. 8 1H NMR spectra (aliphatic region) of ortho-bromo derivative 6j measured in six different deuterated solvents at 25 °C to show the pattern of the piperazine protons in dependence of the solvent. | ||
A similar situation was found for the nitro derivatives 6k and 6h. Derivative 6k, with the nitro group in ortho position, shows five independent signals in the 1H NMR spectrum at temperatures below 38 °C in contrast to para-nitro derivative 6h which shows the expected four signals when measured in CDCl3. Interestingly, both compounds 6k and 6h show four signals when measured in DMSO-d6. This can be explicated by similar mesomeric and electronic effects of both compounds making ortho and para position comparable. Thus, the behavior of 6k can be again ascribed to the additional steric hindrance of the nitro group in ortho position of the benzoyl residue resulting in additional rotamers due to the hindered rotation of the Cipso–C(O) bond. As also found for ortho-bromo compound 6j, two protons from one of the NCH2 groups of the amide site are split and a coalescence temperature of TC = 41 °C and a ΔG‡ = 60.4 kJ mol−1 was found (ESI†). The complete coalescence of amide site and amine site, respectively, of 6j and 6k was not reached due to the limitations imposed by the experimental setup.
To obtain further insight into the causes for the limited rotation of the amide bond and ring inversion as studied by NMR, single crystals of 4d, 6d, 6f, and 6i were used to determine the solid-state structures of these compounds. They all consist of neutral molecules, packed in monoclinic lattices with space groups P21/n (4d and 6d), P21/c (6i), and C2/c (6f). The molecular structures with atom numbering schemes are shown in Fig. 9. Whereas the molecules of 6d, 6f, and 6i, which comprise the asymmetric unit, are located on general positions and not located on any symmetry element (besides identity), the center of the piperazine ring of 4d is located on an inversion center, such that the molecule has inversion symmetry. For compounds 6d, 6f, and 6i, two conformers exist in a 1:1 ratio, which are not superimposable due to the inversion centers in the crystal which are located outside of the molecules. Fig. 10 shows the superposition of the two ringconformers of 6d, showing the different orientations, especially of the piperazine moieties, when the 4-nitro-phenyl rings are fitted on top of each other. For compound 6i the structure is affected by some disorder, which could best be described using a split model. Relevant crystallographic details of all structures are collected in the ESI.†
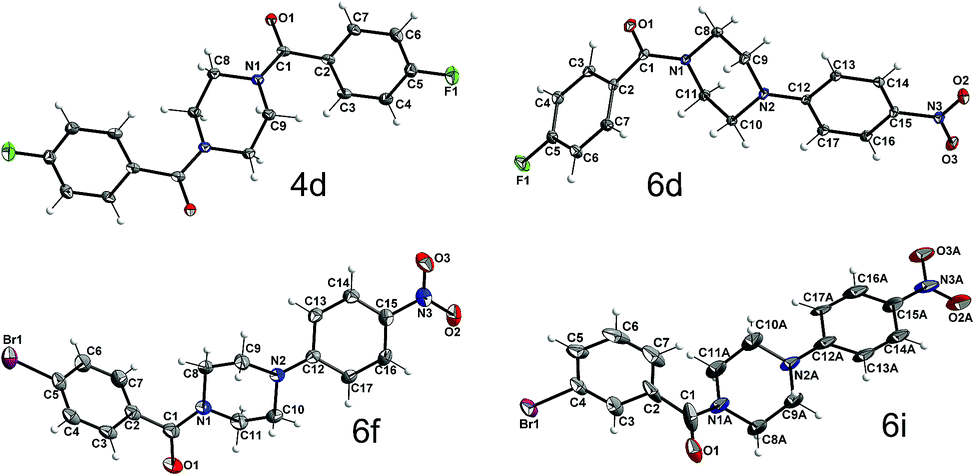 | ||
| Fig. 9 Molecular structures of 4d, 6d, 6f, and 6i with atom labeling scheme (ORTEP plots, thermal displacement ellipsoids are shown with 50% probability at −150 °C). | ||
 | ||
| Fig. 10 Superimposition fit of the two conformers, which exist in the ratio of 1:1 in the solid-state structure of compound 6d (label A marks the atoms of the second conformer; not all labels shown). | ||
An interesting question concerning these structures is if the rotational barrier as observed in the NMR spectra (see above) goes along with structural indication of at least double bond character of the carbonyl C–N bond of compounds 6d, 6f, and 6i. An inspection of the bond lengths and angles, exemplarily for 6d, shows that the N1 as well as the N2 atoms are surrounded by carbon atoms in a close to planar manner. N1 deviates out of the plane formed by C1, C8 and C11 carbon atoms by 0.07 Å. A similar value of 0.02 Å was determined for the distance of N2 from the plane defined by C9, C10, and C12. The average C–N–C bond angles at the piperazine nitrogen atoms are found to be close to 120° (119.8° for N1 and 120.0° for N2). In this context, it should be noted that for the tertiary amine nitrogen in 4-(3-butyn-1-yl)-1-(4-fluorobenzoyl)piperazine, an average C–N–C bond angle of 109.8° was recently determined from a single-crystal structure of this compound,21 which clearly highlights the broadening of the bond angle by the 4-nitrophenyl group and therefore the considerable depyramidalization of the corresponding nitrogen atom. Moreover, the increased flattening of the piperazine ring by the 4-nitrophenyl substituent is illustrated by comparing the bond angle at the piperazine nitrogen atom inside the ring (C9(A)–N2(A)–C10(A)); Table 5. Whereas for 4-(3-butyn-1-yl)-1-(4-fluorobenzoyl)-piperazine a bond angle of 109.0° was determined, those values for compounds 6d, f, i were significantly higher (111.4–113.8°). The distinct involvement of the amide and amine nitrogen in partial double bonds is further expressed by the respective bond lengths, as the bond length of N1 to the carbonyl C1 atom is significantly shorter than to the other two C atoms, N1–C1: 1.355(1) Å vs. N1–C8: 1.460(1) Å, N1–C11: 1.462(1) Å. Similarly, the N2 contact to the aromatic carbon atom C12 is shorter than the other two contacts: N2–C12: 1.367(1) Å vs. N2–C9: 1.463(1), N2–C10: 1.524(1) Å. In contrast, a length of 1.464 Å was determined for the corresponding C–N bond in 4-(3-butyn-1-yl)-1-(4-fluorobenzoyl)piperazine.21 These structural features imply the overlap of the free electron pair of N2 with the π-system of the electron-deficient nitrophenyl residue and explain the lower ring inversion barriers of compounds 6 compared to their monosubstituted analogues 3 because of the flattened C–N–C bond angle. Moreover, the increased positive charge at the amine nitrogen might attenuate the hyperconjugation shown in Fig. 7 and therefore contribute to the destabilization of the chair conformation. Selected geometrical data of all four XRD structurally characterized compounds are summarized in Table 5.
| 4d | 6d | 6f | 6i | |
|---|---|---|---|---|
| a Δ(plane) N1: deviation of the position of N1 out of the plane of surrounding three C atoms.b For 6i: the labels for N1, N2, and C8–C16 need to be replaced by N1A, N2A, C8A–C16A, because of the atom naming in the disorder split model. | ||||
| Δ(plane) N1/Åa | 0.08 | 0.07 | 0.01 | 0.00 |
| Δ(plane) N2/Å | — | 0.02 | 0.18 | 0.237 |
| Av. bond angle N1/° | 119.7 | 119.8 | 120.0 | 120.0 |
| Av. bond angle N2/° | — | 120.0 | 118.5 | 117.3 |
| Bond angle C9(A)–N2(A)–C10(A) | — | 112.2 | 111.4 | 113.8 |
| Bond angle C9(A)–N2(A)–C12(A) | — | 123.7 | 122.0 | 118.4 |
| Bond angle C10(A)–N2(A)–C12(A) | — | 124.1 | 122.0 | 119.7 |
| N1–C1/Åb | 1.3569(8) | 1.355(1) | 1.356(2) | 1.373(6) |
| N1–C8/Å | 1.4588(8) | 1.460(1) | 1.456(2) | 1.458(7) |
| N1–C11/Å | 1.4593(8) | 1.462(1) | 1.452(2) | 1.474(6) |
| N2–C12/Å | — | 1.367(1) | 1.372(2) | 1.394(9) |
| N2–C9/Å | — | 1.463(1) | 1.463(2) | 1.465(6) |
| N2–C10/Å | — | 1.524(1) | 1.456(2) | 1.46(1) |
Conformations of inhibitors for transglutaminase 2 (TGase 2)
Amongst others, the structural element of N,N′-unsymmetrically substituted piperazines is found in TGase 2-inhibitors57–59 as shown in Scheme 3 (the structural element is highlighted in blue). The beneficial influence of the piperazine ring as linker between the benzoyl moiety and the remaining parts of the molecule on the inhibitory activity towards TGase 2 has been demonstrated.58 Fluorine derivative 8 is a potential 18F-radiotracer for PET (positron emission tomography) imaging of TGase 2 when fluorine-18 (β+-emitter, half life 110 min) is introduced and compound 7 could be potentially used as precursor (starting compound) for radiolabeling via nucleophilic introduction of [18F]fluoride. The preparation and analyses of both compounds 7 and 8 is published elsewhere together with their inhibitory activities.57 | ||
| Scheme 3 Structures of TGase 2 inhibitors 7 and 8. | ||
As expected from the previous investigations, two rotamers were observed for compounds 7 and 8 due to the partial double bond character of tertiary amides that involve the piperazine ring. A good indication for this phenomenon is the proton of the α-carbon atom in the lysine residue. In case of compound 7, two broad singlets were observed for this proton at 25 °C in CDCl3 as well as in DMSO-d6. The determination of the rotation barriers for both compounds was accomplished in CDCl3 on the basis of the signals for the CαH proton. The temperature- dependent spectra of fluorine compound 8 are presented in Fig. 11. The coalescence temperature TC was found at 7 °C for fluorine compound 8 and at 28 °C for nitro compound 7 in CDCl3. The rotation barrier ΔG‡ was calculated to be 57.4 kJ mol−1 for 8 and 61.7 kJ mol−1 for 7. The higher rotation barrier for compound 7 is in accordance to the previous results for compounds 3 and 6, as the nitro group increases the partial double bond character of the benzoyl amide bond compared to fluorine.
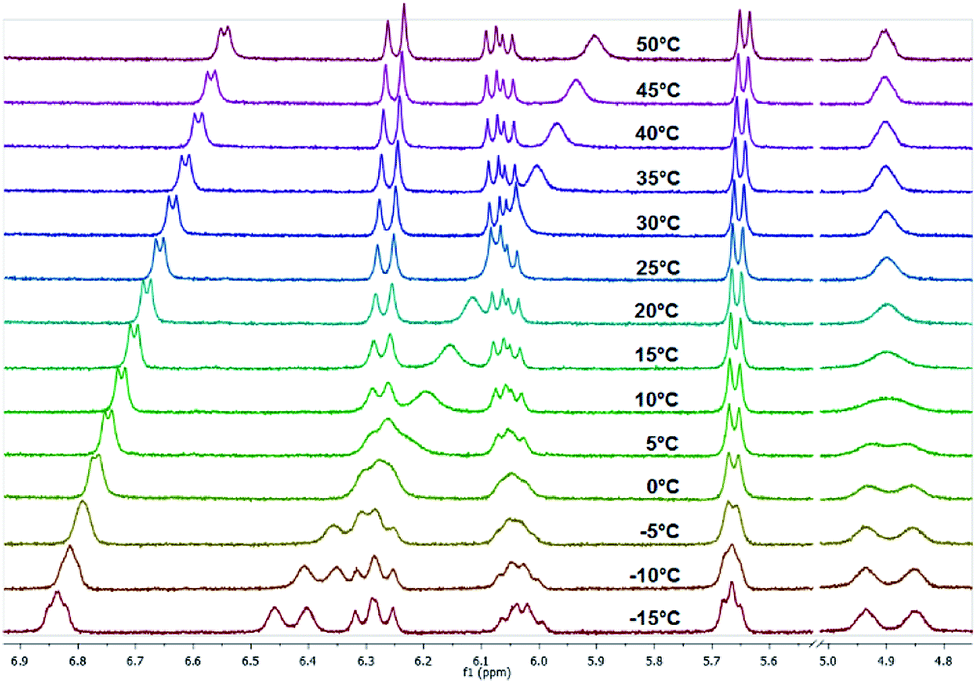 | ||
| Fig. 11 Temperature-dependent 1H NMR spectra of fluorine compound 8 measured in CDCl3 (region of interest from 4.70 to 7.00 ppm is shown). | ||
Experimental
General
All chemicals were purchased from commercial suppliers and used without further purification unless otherwise specified. Anhydrous THF was purchased from Acros. NMR spectra of all compounds were recorded on an Agilent DD2-400 MHz or DD2-600 MHz NMR spectrometer with ProbeOne. Chemical shifts of the 1H, 19F, and 13C spectra were reported in parts per million (ppm) using TMS as internal standard for 1H/13C and CFCl3 for 19F spectra at 25 °C unless otherwise stated. Spectra were calibrated to their solvent signals. Dynamic NMR measurements were carried out in a temperature range of 253–393 K using the Agilent VT accessory with digital temperature controller. TC was estimated from the NMR spectrum showing the collapse of the methylene signals from the piperazine moiety and Δν values were extracted from the maximum split signals of the piperazine methylene protons, which was not varied with decreasing temperature. Values of the exchange rate, kexc, were calculated using equation kexc = πΔν/21/2.37,38 Subsequently, values of the free activation energy, ΔG‡, were obtained by solving the Eyring eqn (I) for ΔG‡ (II).37,39 In accordance to other studies on amide cis/trans isomerization, the transmission coefficient, κ, was assumed to be 1.60
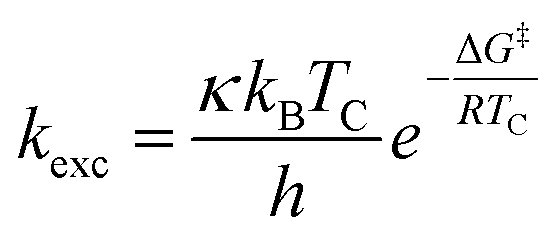 | (I) |
 | (II) |
Mass spectrometric (MS) data were obtained on a Xevo TQ-S mass spectrometer (Waters) by electron spray ionization (ESI). The melting points were determined on a Galen III melting point apparatus (Cambridge Instruments & Leica) and are uncorrected. Microanalyses were carried out with a LECO CHNS 932 elemental analyzer. Chromatographic separations and TLC detections were performed using Merck Silica Gel 60 (63–200 μm) and Merck Silica Gel 60 F254 sheets, respectively. TLCs were developed by visualization under UV light (λ = 254 nm). Single crystal X-ray diffraction data of 4d, 6d, 6f, and 6i were collected with a Bruker-Nonius Apex-II-diffractometer using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). Diffraction measurements were done at −150 °C (4d, 6d, 6i) or 23 °C (6f). The structures was solved by direct methods and refined against F2 by full-matrix least-squares using the program suites from G. M. Sheldrick.61–63 All non-hydrogen atoms were refined anisotropically; all hydrogen atoms were placed on geometrically calculated positions and refined using riding models. CCDC-1816843 (4d), CCDC-1588729 (6d), CCDC-1825578 (6f), and CCDC-1838271 (6i) contain the supplementary crystallographic data.
Chemistry
:ethyl acetate 1:1) to yield 6a as yellow solid (150 mg, 73%). Mp 153–156 °C; Rf = 0.30 (petroleum ether:ethyl acetate 1:2); 1H NMR (400 MHz, CDCl3): δ = 3.33–4.04 (m, 8H, NCH2), 6.83 (d, 3J = 9.3 Hz, 2H, Ar–H), 7.40–7.49 (m, 5H, Ar–H), 8.13 (d, 3J = 9.3 Hz, 2H, Ar–H) ppm; 13C NMR (101 MHz, CDCl3): δ = 42.1 (br. s, NCH2), 47.3 (br. s, NCH2), 113.3, 126.1, 127.3, 128.8, (4× CHAr), 130.3, 135.2, 139.3, 154.6 (4× CAr), 170.7 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). MS (ESI+) m/z: 312 (M+ + H, 100%).:ethyl acetate 1:1) to yield 6a as yellow solid (120 mg, 56%). Mp 193 °C; Rf = 0.20 (petroleum ether:ethyl acetate 1:2); 1H NMR (600 MHz, CDCl3): δ = 2.39 (s, 3H, CH3), 3.33–4.00 (m, 8H, NCH2), 6.82 (d, 3J = 9.1 Hz, 2H, Ar–H), 7.24 (d, 3J = 7.8 Hz, 2H, Ar–H), 7.34 (d, 3J = 9.1 Hz, 2H, Ar–H), 8.12 (d, 3J = 9.1 Hz, 2H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 21.6 (CH3), 41.9 (br. s, NCH2), 47.2 (br. s, NCH2), 113.2, 126.0, 127.4, 129.4 (4× CHAr), 132.2, 139.2, 140.6, 154.6 (4× CAr), 170.9 (CO). MS (ESI+) m/z: 326 (M+ + H, 100%).:ethyl acetate 1:1) to yield 6c as yellow solid (220 mg, 98%). Mp 133 °C; Rf = 0.24 (petroleum ether:ethyl acetate 1:2); 1H NMR (600 MHz, CDCl3): δ = 3.46 (br. s, 4H, NCH2), 3.70–3.90 (m, 7H, NCH2 + CH3), 6.82 (d, 3J = 9.3 Hz, 2H, Ar–H), 6.93 (d, 3J = 8.6 Hz, 2H, Ar–H), 7.42 (d, 3J = 8.6 Hz, 2H, Ar–H), 8.12 (d, 3J = 9.3 Hz, 2H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 47.3 (br. s, NCH2), 55.5 (CH3), 113.2, 114.0, 126.1, 129.4 (4× CHAr), 127.1, 139.2, 154.6, 161.3 (4× CAr), 170.7 (CO). MS (ESI+) m/z: 342 (M+ + H, 100%).:ethyl acetate 1:1) to yield 6c as yellow solid (200 mg, 93%). Mp 173 °C; Rf = 0.52 (chloroform:ethanol 20:1); 1H NMR (600 MHz, CDCl3): δ = 3.31–4.02 (m, 8H, NCH2), 6.83 (d, 3J = 8.8 Hz, 2H, Ar–H), 7.12 (t, 3J = 8.4 Hz, 3JH,F = 8.4 Hz, 2H, Ar–H), 7.42 (d, 3J = 8.4 Hz, 4JH,F = 5.3 Hz, 2H, Ar–H), 8.12 (d, 3J = 8.8 Hz, 2H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 44.7, 50.0 (2× br. s, NCH2), 115.8 (CHAr), 118.4 (d, 2JC,F = 21.9 Hz, CHAr), 128.6 (CHAr), 132.2 (d, 3JC,F = 8.3 Hz, CHAr), 131.7 (d, 4JC,F = 3.4 Hz, CAr), 157.1 (CAr), 166.3 (d, 1JC,F = 246.4 Hz, CAr), 172.3 (CO); 19F NMR (565 MHz, CDCl3): δ = −109.4. MS (ESI+) m/z: 330 (M+ + H, 100%).:ethyl acetate 1:1) to yield 6i as yellow solid (145 mg, 37%). Mp 187–189 °C; Rf = 0.24 (petroleum ether:ethyl acetate 1:1); 1H NMR (400 MHz, CDCl3): δ = 3.25–4.01 (m, 8H, NCH2), 6.82 (d, 3J = 9.4 Hz, 2H, Ar–H), 7.32 (d, 3J = 7.6 Hz, 3J = 8.4 Hz, 1H, Ar–H), 7.57 (d, 3J = 8.4 Hz, 1H, Ar–H), 8.11 (d, 3J = 9.4 Hz, 2H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 41.9 (br. s, NCH2), 47.3 (br. s, NCH2), 113.3, 126.0, 129.0, 132.0 (4× CHAr), 124.7, 134.0, 139.3, 154.5 (CAr), 169.7 (CO). MS (ESI+) m/z: 390 (M+ + H, 35Br 100%), 392 (M+ + H, 37Br, 100%).:ethyl acetate 1:1) to yield 6x as yellow solid (190 mg, 81%). Mp 145 °C; Rf = 0.14 (petroleum ether:ethyl acetate 1:1); 1H NMR (400 MHz, CDCl3): δ = 3.27–3.73 (m, 6H, NCH2), 3.97 (br. s, 2H, NCH2), 6.86 (d, 3J = 9.3 Hz, 2H, Ar–H), 7.63 (d, 3J = 8.8 Hz, 2H, Ar–H), 8.16 (d, 3J = 9.3 Hz, 2H, Ar–H), 8.32 (d, 3J = 8.8 Hz, 2H, Ar–H), ppm; 13C NMR (150 MHz, CDCl3): δ = 47.2 (br. s, NCH2), 47.8 (br. s, NCH2), 113.7, 124.2, 126.1, 128.4, (4× CHAr), 126.1, 141.3, 148.8, 154.4 (4× CAr), 168.3 (CO). MS (ESI+) m/z: 357 (M+ + H, 100%).:ethyl acetate 1:1) to yield 6i as yellow solid (240 mg, 93%). Mp 133 °C; Rf = 0.40 (petroleum ether:ethyl acetate 1:2); 1H NMR (400 MHz, CDCl3): δ = 3.25–4.07 (m, 8H, NCH2), 6.84 (d, 3J = 9.3 Hz, 2H, Ar–H), 7.32 (dd, 3J = 7.6 Hz, 3J = 8.4 Hz, 1H, Ar–H), 7.37 (d, 3J = 7.6 Hz, 1H, Ar–H), 7.57–7.62 (m, 2H, Ar–H), 8.13 (d, 3J = 9.3 Hz, 2H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 41.9 (br. s, NCH2), 47.3 (br. s, NCH2), 113.4, 122.9, 125.8, 126.1, 130.3, 130.4, 133.4, 137.2, 139.4, 168.9 (CO). MS (ESI+) m/z: 390 (M+ + H, 35Br 100%), 392 (M+ + H, 37Br, 100%).:ethyl acetate 1:1) to yield 6j as yellow solid (210 mg, 54%). Mp 179 °C; Rf = 0.20 (petroleum ether:ethyl acetate 1:1); 1H NMR (600 MHz, CDCl3): δ = 3.32–3.41 (m, 2H, NCH2), 3.42–3.51 (m, 2H, NCH2), 3.55 (t, 3J = 5.2 Hz, 2H, NCH2), 3.85–3.94 (m, 1H, NCH2), 4.01–4.09 (m, 1H, NCH2), 6.82 (d, 3J = 9.4 Hz, 2H, Ar–H), 7.26–7.32 (m, 2H, Ar–H), 7.39 (t, 3J = 7.4 Hz, 1H, Ar–H), 7.60 (d, 3J = 8.3 Hz, 2H, Ar–H), 8.11 (d, 3J = 9.4 Hz, 2H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 43.7, 48.6, 49.5, 50.1 (4× NCH2), 115.9 (CHAr), 121.8 (CAr), 128.6, 130.4, 130.6, 133.3, 135.6 (5× CHAr), 140.0, 141.8, 157.1 (3× CAr), 170.4 (CO). MS (ESI+) m/z: 390 (M+ + H, 35Br 100%), 392 (M+ + H, 37Br, 100%).:ethyl acetate 1:1) to yield 6k as yellow solid (200 mg, 85%). Mp 182–186 °C; Rf = 0.09 (petroleum ether:ethyl acetate 1:1); 1H NMR (600 MHz, CDCl3): δ = 3.37–3.47 (m, 4H, NCH2), 3.57–3.64 (m, 2H, NCH2), 3.80–3.96 (br. m, 1H, NCH2), 3.85–3.94 (m, 1H, NCH2), 4.08–4.25 (br. m, 1H, NCH2), 6.85 (d, 3J = 9.3 Hz, 2H, Ar–H), 7.44 (d, 3J = 7.6 Hz, 1H, Ar–H), 7.63 (t, 3J = 8.3 Hz, 1H, Ar–H), 7.76 (t, 3J = 7.4 Hz, 1H, Ar–H), 8.15 (d, 3J = 9.3 Hz, 2H, Ar–H), 8.23 (d, 3J = 8.3 Hz, 1H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 29.9, 41.5, 46.2, 47.1 (4× NCH2), 115.9 (CHAr), 113.8, 123.6 (2× CAr), 125.1, 126.1, 128.2, 130.4, 134.8 (4× CHAr), 142.8, 145.6 (2× CAr), 166.8 (CO). MS (ESI+) m/z: 357 (M+ + H, 100%).
O). MS (ESI+) m/z: 312 (M+ + H, 100%).:ethyl acetate 1:1) to yield 6a as yellow solid (120 mg, 56%). Mp 193 °C; Rf = 0.20 (petroleum ether:ethyl acetate 1:2); 1H NMR (600 MHz, CDCl3): δ = 2.39 (s, 3H, CH3), 3.33–4.00 (m, 8H, NCH2), 6.82 (d, 3J = 9.1 Hz, 2H, Ar–H), 7.24 (d, 3J = 7.8 Hz, 2H, Ar–H), 7.34 (d, 3J = 9.1 Hz, 2H, Ar–H), 8.12 (d, 3J = 9.1 Hz, 2H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 21.6 (CH3), 41.9 (br. s, NCH2), 47.2 (br. s, NCH2), 113.2, 126.0, 127.4, 129.4 (4× CHAr), 132.2, 139.2, 140.6, 154.6 (4× CAr), 170.9 (CO). MS (ESI+) m/z: 326 (M+ + H, 100%).:ethyl acetate 1:1) to yield 6c as yellow solid (220 mg, 98%). Mp 133 °C; Rf = 0.24 (petroleum ether:ethyl acetate 1:2); 1H NMR (600 MHz, CDCl3): δ = 3.46 (br. s, 4H, NCH2), 3.70–3.90 (m, 7H, NCH2 + CH3), 6.82 (d, 3J = 9.3 Hz, 2H, Ar–H), 6.93 (d, 3J = 8.6 Hz, 2H, Ar–H), 7.42 (d, 3J = 8.6 Hz, 2H, Ar–H), 8.12 (d, 3J = 9.3 Hz, 2H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 47.3 (br. s, NCH2), 55.5 (CH3), 113.2, 114.0, 126.1, 129.4 (4× CHAr), 127.1, 139.2, 154.6, 161.3 (4× CAr), 170.7 (CO). MS (ESI+) m/z: 342 (M+ + H, 100%).:ethyl acetate 1:1) to yield 6c as yellow solid (200 mg, 93%). Mp 173 °C; Rf = 0.52 (chloroform:ethanol 20:1); 1H NMR (600 MHz, CDCl3): δ = 3.31–4.02 (m, 8H, NCH2), 6.83 (d, 3J = 8.8 Hz, 2H, Ar–H), 7.12 (t, 3J = 8.4 Hz, 3JH,F = 8.4 Hz, 2H, Ar–H), 7.42 (d, 3J = 8.4 Hz, 4JH,F = 5.3 Hz, 2H, Ar–H), 8.12 (d, 3J = 8.8 Hz, 2H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 44.7, 50.0 (2× br. s, NCH2), 115.8 (CHAr), 118.4 (d, 2JC,F = 21.9 Hz, CHAr), 128.6 (CHAr), 132.2 (d, 3JC,F = 8.3 Hz, CHAr), 131.7 (d, 4JC,F = 3.4 Hz, CAr), 157.1 (CAr), 166.3 (d, 1JC,F = 246.4 Hz, CAr), 172.3 (CO); 19F NMR (565 MHz, CDCl3): δ = −109.4. MS (ESI+) m/z: 330 (M+ + H, 100%).:ethyl acetate 1:1) to yield 6i as yellow solid (145 mg, 37%). Mp 187–189 °C; Rf = 0.24 (petroleum ether:ethyl acetate 1:1); 1H NMR (400 MHz, CDCl3): δ = 3.25–4.01 (m, 8H, NCH2), 6.82 (d, 3J = 9.4 Hz, 2H, Ar–H), 7.32 (d, 3J = 7.6 Hz, 3J = 8.4 Hz, 1H, Ar–H), 7.57 (d, 3J = 8.4 Hz, 1H, Ar–H), 8.11 (d, 3J = 9.4 Hz, 2H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 41.9 (br. s, NCH2), 47.3 (br. s, NCH2), 113.3, 126.0, 129.0, 132.0 (4× CHAr), 124.7, 134.0, 139.3, 154.5 (CAr), 169.7 (CO). MS (ESI+) m/z: 390 (M+ + H, 35Br 100%), 392 (M+ + H, 37Br, 100%).:ethyl acetate 1:1) to yield 6x as yellow solid (190 mg, 81%). Mp 145 °C; Rf = 0.14 (petroleum ether:ethyl acetate 1:1); 1H NMR (400 MHz, CDCl3): δ = 3.27–3.73 (m, 6H, NCH2), 3.97 (br. s, 2H, NCH2), 6.86 (d, 3J = 9.3 Hz, 2H, Ar–H), 7.63 (d, 3J = 8.8 Hz, 2H, Ar–H), 8.16 (d, 3J = 9.3 Hz, 2H, Ar–H), 8.32 (d, 3J = 8.8 Hz, 2H, Ar–H), ppm; 13C NMR (150 MHz, CDCl3): δ = 47.2 (br. s, NCH2), 47.8 (br. s, NCH2), 113.7, 124.2, 126.1, 128.4, (4× CHAr), 126.1, 141.3, 148.8, 154.4 (4× CAr), 168.3 (CO). MS (ESI+) m/z: 357 (M+ + H, 100%).:ethyl acetate 1:1) to yield 6i as yellow solid (240 mg, 93%). Mp 133 °C; Rf = 0.40 (petroleum ether:ethyl acetate 1:2); 1H NMR (400 MHz, CDCl3): δ = 3.25–4.07 (m, 8H, NCH2), 6.84 (d, 3J = 9.3 Hz, 2H, Ar–H), 7.32 (dd, 3J = 7.6 Hz, 3J = 8.4 Hz, 1H, Ar–H), 7.37 (d, 3J = 7.6 Hz, 1H, Ar–H), 7.57–7.62 (m, 2H, Ar–H), 8.13 (d, 3J = 9.3 Hz, 2H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 41.9 (br. s, NCH2), 47.3 (br. s, NCH2), 113.4, 122.9, 125.8, 126.1, 130.3, 130.4, 133.4, 137.2, 139.4, 168.9 (CO). MS (ESI+) m/z: 390 (M+ + H, 35Br 100%), 392 (M+ + H, 37Br, 100%).:ethyl acetate 1:1) to yield 6j as yellow solid (210 mg, 54%). Mp 179 °C; Rf = 0.20 (petroleum ether:ethyl acetate 1:1); 1H NMR (600 MHz, CDCl3): δ = 3.32–3.41 (m, 2H, NCH2), 3.42–3.51 (m, 2H, NCH2), 3.55 (t, 3J = 5.2 Hz, 2H, NCH2), 3.85–3.94 (m, 1H, NCH2), 4.01–4.09 (m, 1H, NCH2), 6.82 (d, 3J = 9.4 Hz, 2H, Ar–H), 7.26–7.32 (m, 2H, Ar–H), 7.39 (t, 3J = 7.4 Hz, 1H, Ar–H), 7.60 (d, 3J = 8.3 Hz, 2H, Ar–H), 8.11 (d, 3J = 9.4 Hz, 2H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 43.7, 48.6, 49.5, 50.1 (4× NCH2), 115.9 (CHAr), 121.8 (CAr), 128.6, 130.4, 130.6, 133.3, 135.6 (5× CHAr), 140.0, 141.8, 157.1 (3× CAr), 170.4 (CO). MS (ESI+) m/z: 390 (M+ + H, 35Br 100%), 392 (M+ + H, 37Br, 100%).:ethyl acetate 1:1) to yield 6k as yellow solid (200 mg, 85%). Mp 182–186 °C; Rf = 0.09 (petroleum ether:ethyl acetate 1:1); 1H NMR (600 MHz, CDCl3): δ = 3.37–3.47 (m, 4H, NCH2), 3.57–3.64 (m, 2H, NCH2), 3.80–3.96 (br. m, 1H, NCH2), 3.85–3.94 (m, 1H, NCH2), 4.08–4.25 (br. m, 1H, NCH2), 6.85 (d, 3J = 9.3 Hz, 2H, Ar–H), 7.44 (d, 3J = 7.6 Hz, 1H, Ar–H), 7.63 (t, 3J = 8.3 Hz, 1H, Ar–H), 7.76 (t, 3J = 7.4 Hz, 1H, Ar–H), 8.15 (d, 3J = 9.3 Hz, 2H, Ar–H), 8.23 (d, 3J = 8.3 Hz, 1H, Ar–H) ppm; 13C NMR (150 MHz, CDCl3): δ = 29.9, 41.5, 46.2, 47.1 (4× NCH2), 115.9 (CHAr), 113.8, 123.6 (2× CAr), 125.1, 126.1, 128.2, 130.4, 134.8 (4× CHAr), 142.8, 145.6 (2× CAr), 166.8 (CO). MS (ESI+) m/z: 357 (M+ + H, 100%).Conclusions
We systematically investigated the influence of various functional groups and their position at the benzoyl residue on the 1H NMR behavior and conformational transitions in benzamide systems containing piperazines. For mono-benzoyl substituted piperazines, positive correlations of the activation energies for amide bond rotation and ring inversion with the Hammett σ constants of the substituents at the benzoyl group have been derived. The remarkable correlation between these parameters and the inversion barrier in piperazines has been discussed considering reported findings for N-substituted morpholines. In this context, a model for the relationship of ring flattening and gauche effect and their impact on the ring inversion barrier is proposed. This sheds light on the complex influences on energetic barriers for conformational transitions in acylated heterocycloaliphates, which are commonly encountered structural elements in active pharmaceutical ingredients and potential drug molecules. Furthermore, activation energies were determined for benzoyl substituted 4-nitrophenyl piperazines, which were slightly lower compared to the monosubstituted series with substituent effects that are in accordance to the results obtained for mono-benzoyl substituted piperazines. Furthermore, the influence of the substituent position at the benzoyl moiety on the 1H NMR behavior was demonstrated for this compound series.Additionally, two TGase 2 inhibitors as pharmacologically relevant compounds exhibiting benzoylpiperazine partial structures were investigated. ΔG‡ values of 61.7 kJ mol−1 and 57.4 kJ mol−1 were found for the compounds containing the 4-nitrobenzoyl and 4-fluorobenzoyl residues, respectively.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is part of the diploma thesis of Janine Steinberg (TU Dresden, Faculty of Chemistry and Food Chemistry).References
- K. E. Gettys, Z. Ye and M. Dai, Synthesis, 2017, 49, 2589 CrossRef CAS.
- D. P. Katz, J. Deruiter, D. Bhattacharya, M. Ahuja, S. Bhattacharya, C. R. Clark, V. Suppiramaniam and M. Dhanasekaran, Drug Alcohol Depend., 2016, 164, 1 CrossRef CAS.
- M. Shaquiquzzaman, G. Verma, A. Marella, M. Akhter, W. Akhtar, M. F. Khan, S. Tasneem and M. M. Alam, Eur. J. Med. Chem., 2015, 102, 487 CrossRef CAS PubMed.
- L. Shen, Y. H. Ye, X. T. Wang, H. L. Zhu, C. Xu, Y. C. Song, H. Li and R. X. Tan, Chem.–Eur. J., 2006, 12, 4393 CrossRef CAS PubMed.
- T. Irikura, K. Masuzawa, K. Nishino, M. Kitagawa, H. Uchida, N. Ichinoseki and M. Ito, J. Med. Chem., 1968, 11, 801 CrossRef CAS.
- A. K. Rathi, R. Syed, H. S. Shin and R. V. Patel, Expert Opin. Ther. Pat., 2016, 26, 777 CrossRef CAS.
- D. A. Abou El-Ella, M. M. Hussein, R. A. Serya, R. M. Abdel Naby, A. M. Al-Abd, D. O. Saleh, W. I. El-Eraky and K. A. Abouzid, Bioorg. Chem., 2014, 54, 21 CrossRef CAS PubMed.
- R. Patel and S. Park, Mini-Rev. Med. Chem., 2013, 13, 1579 CrossRef CAS.
- P. Miniyar, P. Murumkar, P. Patil, M. Barmade and K. Bothara, Mini-Rev. Med. Chem., 2013, 13, 1607 CrossRef CAS.
- M. Font, E. Ardaiz, L. Cordeu, E. Cubedo, J. Garcia-Foncillas, C. Sanmartin and J. A. Palop, Bioorg. Med. Chem., 2006, 14, 1942 CrossRef CAS.
- A. Dömling and Y. Huang, Synthesis, 2010, 2859 CrossRef.
- B. Kundu, Curr. Opin. Drug Discovery Dev., 2003, 6, 815 CAS.
- B. Zhao, Y. Xiao, D. Yuan, C. Lu and Y. Yao, Dalton Trans., 2016, 45, 3880 RSC.
- H. S. Sahoo and D. K. Chand, Dalton Trans., 2010, 39, 7223 RSC.
- D. J. Barnes, R. L. Chapman, R. S. Vagg and E. C. Watton, J. Chem. Eng. Data, 1978, 23, 349 CrossRef CAS.
- S. A. Andronati, T. L. Karaseva and A. A. Krysko, Curr. Med. Chem., 2004, 11, 1183 CrossRef CAS.
- D. A. Horton, G. T. Bourne and M. L. Smythe, Mol. Diversity, 2000, 5, 289 CrossRef CAS.
- Z. Huang and W. Shi, Prog. Org. Coat., 2007, 59, 312 CrossRef CAS.
- R. Wodtke, J. König, A. Pigorsch, M. Köckerling and C. Mamat, Dyes Pigm., 2015, 113, 263 CrossRef CAS.
- J.-M. Heldt, O. Kerzendörfer, C. Mamat, F. Starke, H.-J. Pietzsch and J. Steinbach, Synlett, 2013, 24, 432 CrossRef CAS.
- C. Mamat, M. Pretze, M. Gott and M. Köckerling, Beilstein J. Org. Chem., 2016, 12, 2478 CrossRef CAS.
- M. Pretze and C. Mamat, J. Fluorine Chem., 2013, 150, 25 CrossRef CAS.
- L. Lemoucheux, J. Rouden, M. Ibazizene, F. Sobrio and M. C. Lasne, J. Org. Chem., 2003, 68, 7289 CrossRef CAS.
- C. Dugave and L. Demange, Chem. Rev., 2003, 103, 2475 CrossRef CAS PubMed.
- L. M. Jackman, T. E. Kavanagh and R. C. Haddon, Org. Magn. Reson., 1969, 1, 109 CrossRef CAS.
- G. Alberghina, F. A. Bottino, S. Fisichella and C. Arnone, J. Chem. Res. (M), 1985, 1201 Search PubMed.
- G. Alberghina, F. A. Bottino, S. Fisichella and C. Arnone, J. Chem. Res. (S), 1985, 108 CAS.
- M. M. Turnbull, D. J. Nelson, W. Lekouses, M. L. Sarnov, K. A. Tartarini and T.-k. Huang, Tetrahedron, 1990, 46, 6613 CrossRef CAS.
- N. Emir, M. Bilge, M. Tursun, G. Kesan and C. Parlak, Spectrochim. Acta, Part A, 2014, 127, 388 CrossRef CAS.
- F. D. Lewis and E. L. Burch, J. Phys. Chem., 1996, 100, 4055 CrossRef CAS.
- S. K. Verma, B. N. Acharya and M. P. Kaushik, Org. Lett., 2010, 12, 4232 CrossRef CAS.
- M. Feigel, J. Phys. Chem., 1983, 87, 3054 CrossRef CAS.
- R. M. Hammaker and B. A. Gugler, J. Mol. Spectrosc., 1965, 17, 356 CrossRef CAS.
- H.-J. Schneider, R. Price and T. Keller, Angew. Chem., Int. Ed., 1971, 10, 730 CrossRef CAS.
- L. W. Reeves and K. O. Strømme, J. Chem. Phys., 1961, 34, 1711 CrossRef CAS.
- A. Rauk, L. C. Allen and K. Mislow, Angew. Chem., Int. Ed., 1970, 9, 400 CrossRef CAS.
- H. Friebolin, Basic One- and Two-Dimensional NMR Spectroscopy, Wiley-VCH, Weinheim, 2010, p. 319 Search PubMed.
- F. P. Gasparro and N. H. Kolodny, J. Chem. Educ., 1977, 54, 258 CrossRef CAS.
- L. M. Jackman, in Dynamic Nuclear Magnetic Resonance Spectroscopy, ed. L. M. Jackman and F. A. Cotton, Academic Press, New York, 1975, p. 204 Search PubMed.
- M. Karplus, J. Am. Chem. Soc., 1963, 85, 2870 CrossRef CAS.
- E. A. Skorupska, R. B. Nazarski, M. Ciechańska, A. Jóźwiak and A. Kłys, Tetrahedron, 2013, 69, 8147 CrossRef CAS.
- V. Hietapelto, R. S. Laitinen, J. Pursiainen and E. Rahkamaa, Acta Chem. Scand., 1999, 53, 7 CrossRef CAS.
- R. K. Harris and R. A. Spragg, Chem. Commun., 1966, 314 RSC.
- R. G. Lett, L. Petrakis, A. F. Ellis and R. K. Jensen, J. Phys. Chem., 1970, 74, 2816 CrossRef CAS.
- R. Baskar, C. Baby, M. S. Moni and K. Subramanian, J. Mol. Struct., 2013, 1040, 90 CrossRef CAS.
- E. A. Noe and M. Raban, J. Am. Chem. Soc., 1975, 97, 5811 CrossRef CAS.
- W. E. Stewart and T. H. Siddall, Chem. Rev., 1970, 70, 517 CrossRef CAS.
- H. Kessler, Angew. Chem., Int. Ed., 1970, 9, 219 CrossRef CAS.
- K. B. Wiberg, Acc. Chem. Res., 1999, 32, 922 CrossRef CAS.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, USA, 2006, pp. 445–453 Search PubMed.
- C. Hansch and A. Leo, Exploring QSAR Fundamentals and Applications in Chemistry and Biology, American Chemical Society, Washington, 1995, pp. 1–24 Search PubMed.
- A. R. Modarresi-Alam, H. A. Amirazizi, H. Bagheri, H. R. Bijanzadeh and E. Kleinpeter, J. Org. Chem., 2009, 74, 4740 CrossRef CAS.
- P. Le Cam and J. Sandström, Chem. Scr., 1971, 1, 65 CAS.
- L. Lunazzi, D. Casarini, M. A. Cremonini and J. E. Anderson, Tetrahedron, 1991, 47, 7465 CrossRef CAS.
- C. Thiehoff, Y. P. Rey and R. Gilmour, Isr. J. Chem., 2017, 57, 92 CrossRef CAS.
- E. Bisz, A. Piontek, B. Dziuk, R. Szostak and M. Szostak, J. Org. Chem., 2018, 83, 3159 CrossRef CAS.
- R. Wodtke, C. Hauser, G. Ruiz-Gomez, E. Jäckel, D. Bauer, M. Lohse, A. Wong, J. Pufe, F. A. Ludwig, S. Fischer, S. Hauser, D. Greif, M. T. Pisabarro, J. Pietzsch, M. Pietsch and R. Löser, J. Med. Chem., 2018, 61, 4528 CrossRef CAS PubMed.
- A. Akbar, N. M. R. McNeil, M. R. Albert, V. Ta, G. Adhikary, K. Bourgeois, R. L. Eckert and J. W. Keillor, J. Med. Chem., 2017, 60, 7910 CrossRef CAS.
- J. Wityak, M. E. Prime, F. A. Brookfield, S. M. Courtney, S. Erfan, S. Johnsen, P. D. Johnson, M. Li, R. W. Marston, L. Reed, D. Vaidya, S. Schaertl, A. Pedret-Dunn, M. Beconi, D. Macdonald, I. Muñoz-Sanjuan and C. Dominguez, ACS Med. Chem. Lett., 2012, 3, 1024 CrossRef CAS.
- A. Skoglöf, I. Nilsson, S. Gustafsson, J. Deinum and P. O. Göthe, Biochim. Biophys. Acta, 1990, 1041, 22 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- G. M. Sheldrick, SHELXL 2014, University of Göttingen, Germany, 2014 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS.
- S. Kumari, C. B. Mishra and M. Tiwari, Bioorg. Med. Chem. Lett., 2015, 25, 1092 CrossRef CAS.
- P. Nordeman, L. R. Odell and M. Larhed, J. Org. Chem., 2012, 77, 11393 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Temperature-dependent NMR spectra. CCDC 1816843, 1588729, 1825578 and 1838271. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra09152h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |