
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen synthesis of PbCrO4 nanostructures using gum of ferula assa-foetida for enhancement of visible-light photocatalytic activity
Rahele Zhianiab,
Ali Es-haghic,
Seyed Mohsen Sadeghzadeh *ab and
Farzaneh Shamsaab
*ab and
Farzaneh Shamsaab
aNew Materials Technology and Processing Research Center, Department of Chemistry, Neyshabur Branch, Islamic Azad University, Neyshabur, Iran
bYoung Researchers and Elite Club, Neyshabur Branch, Islamic Azad University, Neyshabur, Iran. E-mail: seyedmohsen_sadeghzadeh@yahoo.com
cDepartment of Biology, Mashhad Branch, Islamic Azad University, Mashhad, Iran
First published on 6th December 2018
Abstract
Photocatalytic selective oxidation has attracted considerable attention as an environmentally friendly strategy for organic transformations. Some methods have been reported for the photocatalytic oxidation of sulfides into sulfoxides in recent years. However, the practical application of these processes is undermined by several challenges, such as low selectivity, sluggish reaction rates, requirement of UV-light irradiation, use of additives, and instability of the photocatalyst. Pure monoclinic lead chromate nanoparticles were prepared via a new simple way as Pb and Cr sources. PbCrO4 NPs were synthesized via a green method in the presence of gum of ferula assa-foetida from Pb(NO3)2 and CrCl3 as lead and chromium resources, respectively. The structural analysis of the samples confirmed the formation of PbCrO4 nanostructures in the range of 30 ± 5 nm. The PbCrO4 nanocatalyst was thoroughly characterized by powder X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), and energy dispersive X-ray spectroscopy (EDX) study. Considering the large ionic internal character and high mechanical and thermal stability as well as long-term colloidal stability, this system can be considered as a perfect nanocatalyst by using the host–guest approach. A green and ecofriendly method for oxidation of sulfides to sulfones in the presence of O2 as an oxidant was examined for the synthesised PbCrO4 NPs. The easy and applied reusability of the catalyst was observed after the completion of the reaction under visible-light irradiation.
Introduction
The oxidation reaction is one of the most vital protocols for changing raw materials into products.1–4 Oxidation reactions have some desired features of a ‘green’ procedure in terms of reduced waste production and play key roles in the industry.5–9 There are many methods for conventional oxidants for oxidation of sulfide.10–19 O2 is clean, inexpensive, easily accessible and eco-friendly as its side product is only water. The over-oxidation of sulfides resulting in sulfone formation is a common problem. In this regard, intense efforts have been assigned to the expansion of oxidation reactions using green methods and recyclable catalysts that co-produce safe waste.20,21 Photocatalytic developments, which use visible light as a great and easily accessible source of energy for catalytic reactions, have been considered to be significant for a sustainable future.22,23There are some reports on the use of molecular oxygen for the photocatalytic oxidation of sulfides into sulfoxides. Chen et al. reported photocatalytic systems that exhibited aerobic oxidation of sulfides into sulfoxides by using titanium(IV) oxide as the catalyst.24–26 These methods used methanol as the solvent, and it was also oxidized to formaldehyde. Also, Li et al. conducted photocatalytic oxidation of sulfides on Pt/BiVO4 in water under visible-light illumination.27
In the past decades, inorganic nanomaterials have attracted significant interest because of their exclusive properties such as optical, electronic and magnetic properties and potential applications in nanodevices.28–32 There are four types of reported oxides in Pb–Cr–O ternary system: PbCrO4, Pb2CrO5, Pb5CrO8 and Pb11CrO16. Three compounds with Pb(PbO)nCrO4 (n = 0, 1, 4) composition are known to have optoelectronic properties. PbCrO4 crystallizes in two structures such as stable monoclinic and unstable orthorhombic.33 PbCrO4 is an important solid material that is applied as a photosensitizer; it has a stable monoclinic structure and is yellow pigment because of its thermal stability and electrical properties.34 Lead chromate is a significant photocatalyst material. It has been widely applied in decorative and protective systems and mass coloration of fibers, plastics and elastomers.35,36 Also, lead chromate has been applied as a host material for humidity-sensing resistors, photosensitizers and other devices.37
Given our continued interest in nanocatalysis and catalyst development for organic reactions,38–40 we offer a new, low-temperature and facile route to synthesize nanostructured PbCrO4 utilizing gum of ferula assa-foetida. To the best of our knowledge, it is our first effort to exploit gum of ferula assa-foetida as a novel and renewable catalyst to produce nanostructured PbCrO4. The fuel exploited for the fabrication of the nanostructured PbCrO4 in this research is environmentally friendly and non-toxic. Gum of ferula assa-foetida is rich in ferulic acid as well as disulfite that can play the role of reductant; also, it may be responsible for control over shape and size due to the formation of favorable steric hindrance effect in the synthesis process. The reported method can be easily applied for large-scale production of nanostructured PbCrO4. Therefore, herein, we have used the advantages of the properties of PbCrO4 NPs for the photocatalytic oxidation of sulfide under visible-light irradiation.
Experimental
General procedure for the preparation of PbCrO4 NPs
Here, 0.5 g of lead nitrate and 1.21 g of chromium chloride were dissolved in distilled water. Then, 2 mmol of citric acid was added to the mixed solution and the pH of the above solution was adjusted to 9 by adding tetraethylenepentamine (TEPA) drop-wise; in the next step, 2 mmol of ethylene glycol as the esterification agent was added to the resultant solution. Increasing temperature and evaporation of the gel-like solution led to the preparation of a porous solid mass. One g of gum of ferula assa-foetida was mixed with prepared powders and calcined at 700 °C for 3 h.General procedure for the oxidation of sulfide
Here, 0.1 mg of PbCrO4 NPs, 3 mmol of thioanisole and 0.1 mmol of triethylamine were added to 5 mL of CH3OH. After the reaction mixture was stirred for 30 min in dark to reach the adsorption equilibrium, O2 was purged into the vessel to raise the initial pressure to 0.1 MPa. The reaction mixture was magnetically stirred and illuminated with λ > 400 nm visible light irradiation at room temperature. At the end of reaction, the PbCrO4 photocatalyst particles were separated from the reaction mixture by filtration and the products were quantitatively analyzed by gas chromatography (GC) equipped with a flame ionization detector (FID) using chlorobenzene as the internal standard.Results and discussion
Crystallite size and respective phase composition of the samples were evaluated by XRD. The crystalline structures of the as-synthesized PbCrO4 NPs were detected by XRD analysis. Fig. 1 shows the XRD pattern of PbCrO4 NPs synthesized with Pb(NO3)2 and CrCl3. The presence of peaks in the XRD pattern of PbCrO4 is consistent with the monoclinic structure, which is in agreement with JCPDS standard cards no. 08-0210. PbCrO4 belongs to the space group P21/n and could be identified in some cases by the (011), (120), and (200) peaks, of which the (011) is most distinct from those of other phases. Also, (120) is the most intense line for PbCrO4. To prove the correct preparation of PbCrO4, EDX pattern was obtained for the sample (Fig. 2). As seen in the spectrum, the sample is composed of Cr, Pb, and O. SEM was used to study the size and morphology of PbCrO4. The SEM image of PbCrO4 showed that the prepared catalyst is nanometer quasi-spherical with an average diameter of 30 ± 5 nm (Fig. 3a). Also, the TEM image of PbCrO4 is presented in Fig. 3b. It clearly shows well dispersion of the prepared catalyst and several larger structures, which can be ascribed to the aggregation/coalescence of each nanoparticle. Table 1 shows the values of BET surface area, pore size, and pore volume for PbCrO4. As for PbCrO4, the BET surface area, total pore volume, and pore diameter (nm) are obtained as 719 m2 g−1, 2.73 cm3 g−1, and 24.82 nm, respectively.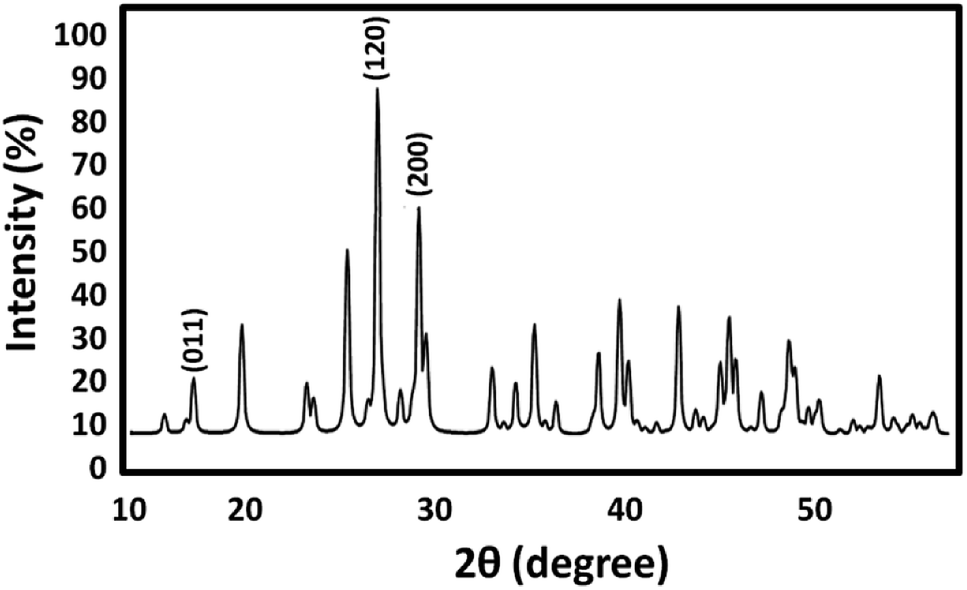 | ||
| Fig. 1 XRD analysis of PbCrO4 NPs. | ||
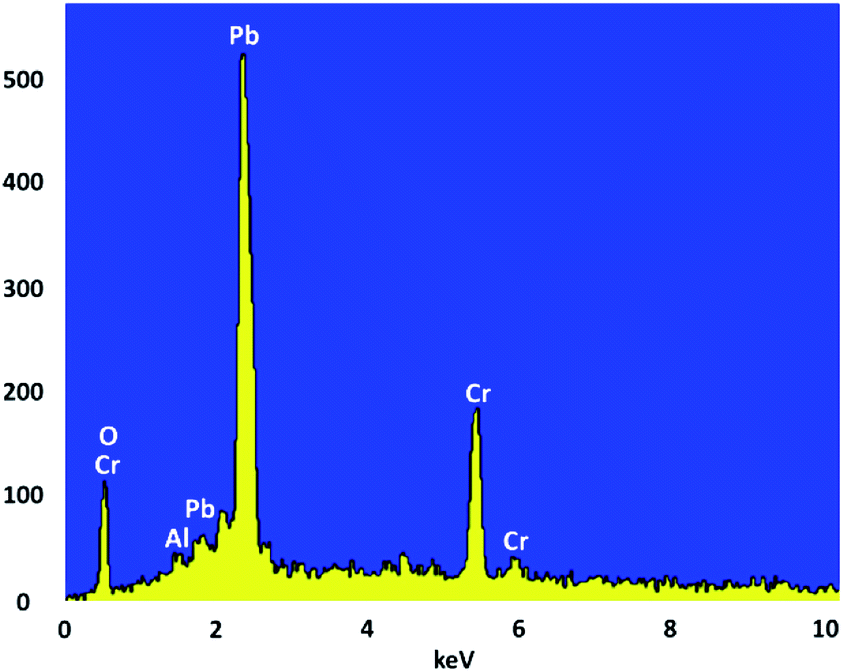 | ||
| Fig. 2 The EDX spectra of PbCrO4 NPs. | ||
 | ||
| Fig. 3 (a) SEM images of PbCrO4 NPs; (b) TEM images of PbCrO4 NPs. | ||
| SBET (m2 g−1) | Va (cm3 g−1) | Pore diameter (nm) |
|---|---|---|
| 719 | 2.73 | 24.82 |
Fig. 4 shows the effect of solvent on the catalytic function. The reaction did not proceed in THF or in solvent-free condition and showed low conversion in ethanol and water. Acetonitrile was the best solvent for photocatalytic reactions in certain reports,41,42 and it also produced a high conversion of 83.9%. Methanol resulted in 99.9% conversion. Interestingly, the solvent exhibited a great effect on the photocatalytic activity of PbCrO4 NPs towards the oxidation of thioanisole. Thus, methanol was selected as a safe solvent.
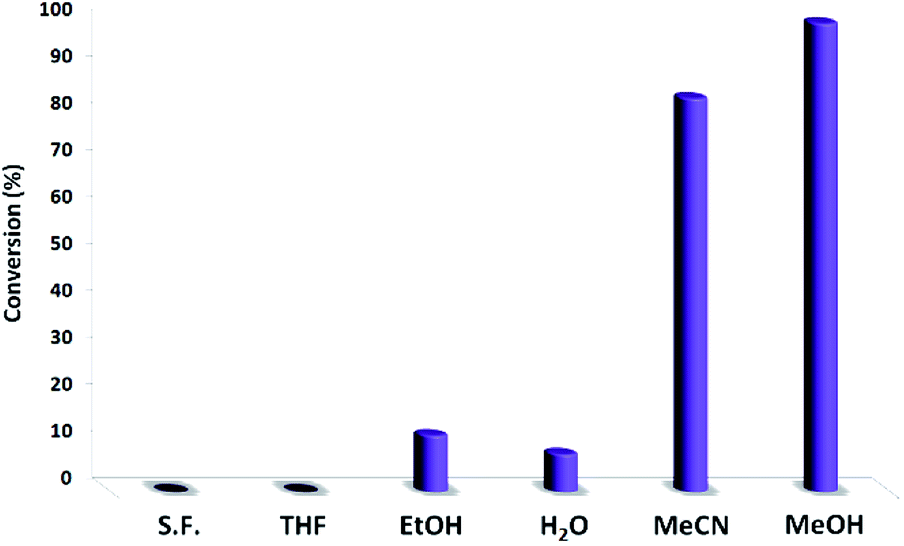 | ||
| Fig. 4 Photocatalytic oxidation of sulfides in different solvents. | ||
In the next step, we conducted aerobic sulfide oxidation with a variety of amines, which could be easily separated from the sulfoxide product (Fig. 5). The conversion of thioanisole without amine was very low under visible-light irradiation. Interestingly, with the introduction of amine into the reaction system, the progression of the reaction was significantly boosted. Primary amines can initiate the oxidation, and a decrease in the amount of amine leads to a decrease in the conversion of the product. As expected, the tertiary amines TMA and TEA remained stable during the oxidation process, ensuring higher conversions of thioanisole. Based on the data in Fig. 6, common oxidants such as H2O2, TBHP, UHP and oxone were weaker oxidants than O2 for this transformation. During the reaction process, the concentration of methyl phenyl sulfide gradually decreased, whereas the concentration of methyl phenyl sulfoxide gradually increased (Fig. 7). No intermediates or other by-products were observed during the reaction process. After light irradiation for 2 h, methyl phenyl sulfide was quantitatively converted into the target product of methyl phenyl sulfoxide.
 | ||
| Fig. 5 The influence of amine on the selective aerobic oxidation of thioanisole under visible-light irradiation. | ||
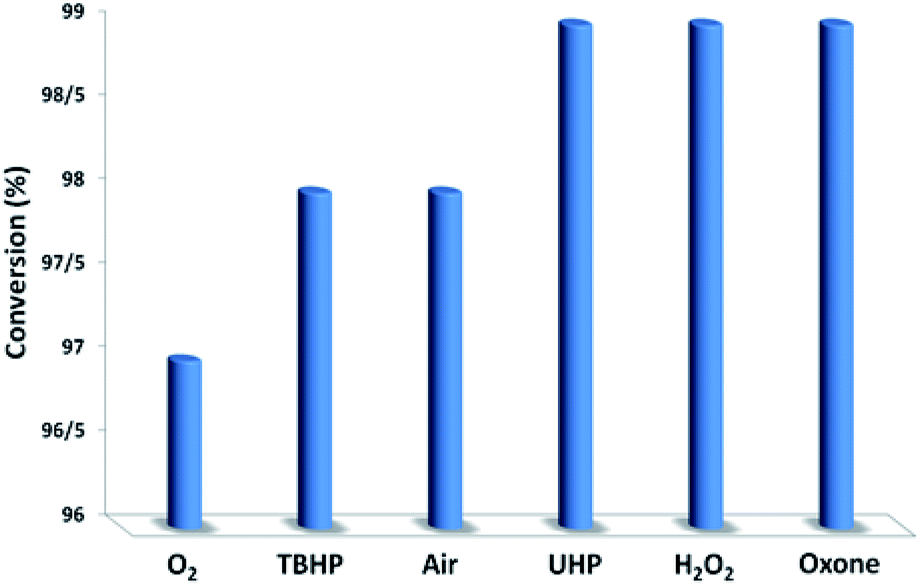 | ||
| Fig. 6 The screening of the oxidant nature. | ||
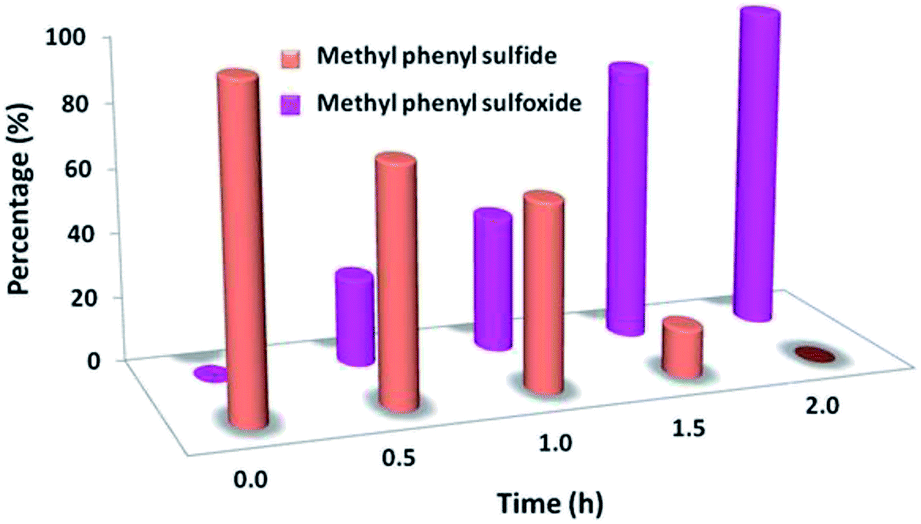 | ||
| Fig. 7 Time course of the product distribution for the photocatalytic oxidation of methyl phenyl sulfide into methyl phenyl sulfoxide. | ||
Moreover, the reaction shows light-wavelength-dependent behavior. Thus, a wide range of visible-light spectra can be used for this reaction, representing a giant step forward compared to the results obtained in previous reports, where only visible light of approximately 400 nm could be used (Fig. 8).
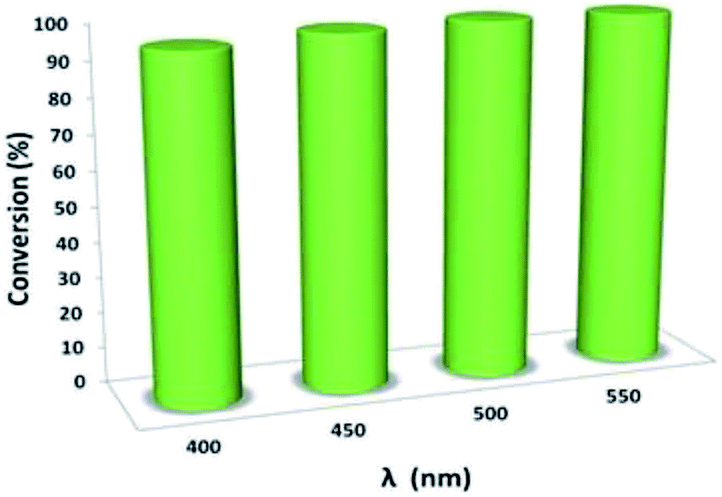 | ||
| Fig. 8 Control experiment for the synergistic photocatalytic oxidation of thioanisole. | ||
To further prove the catalytic nature of PbCrO4, we varied the amount of PbCrO4 used in the reaction to study its effect on the selective aerobic oxidation of thioanisole under visible-light irradiation; more details and results are summarized in Fig. 9. We discovered that decreasing the amount of PbCrO4 led to a drop in conversion to some extent in comparison with the starting result. Increasing PbCrO4 did not lead to an apparent increase in conversion. These combined results prove that PbCrO4 truly acts as a catalyst for the photoredox process.
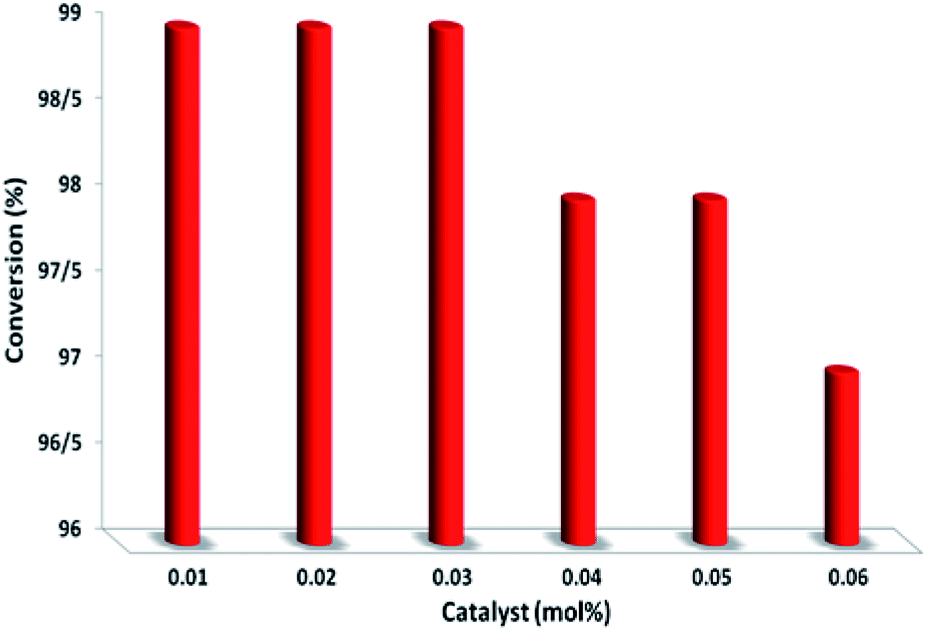 | ||
| Fig. 9 The effect of catalyst amount. | ||
In comparison to previously reported methods, our method is milder, greener from viewpoint of oxidant, support, and metal; it is also cost-effective with higher conversion and selectivity towards production of sulfoxide. This comparison clearly exhibits that our proposed catalyst is excellent (Table 2).
After the formulation of tentative reaction mechanism, we finally seek to establish the scope of the substrates for this visible-light photoredox catalysis, which is important for demonstrating the generality of this reaction scheme. The reaction results are summarized in Table 3. The sulfides were successfully oxidized into the corresponding sulfoxides with good to excellent yields. The electronic properties of the substituents in the aryl sulfides played an important role in the substrate activity. Generally, the substrates with electron-donating groups demonstrate higher activity than the substrates with electron-withdrawing groups; the latter require longer reaction times to give high yields of the corresponding sulfoxides. The substrates with electron-donating groups more easily donate electrons to sulfur atoms than the substrates with electron-withdrawing groups, which can be beneficial for the oxidation reactions.
| Entry | Substrate | Product | Conversion [%] |
|---|---|---|---|
| 1 | 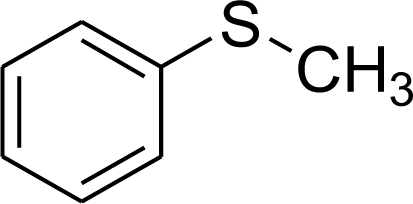 |
 |
99.9 |
| 2 | 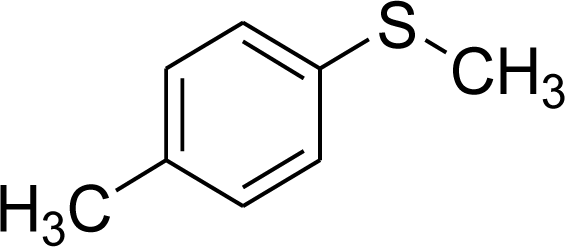 |
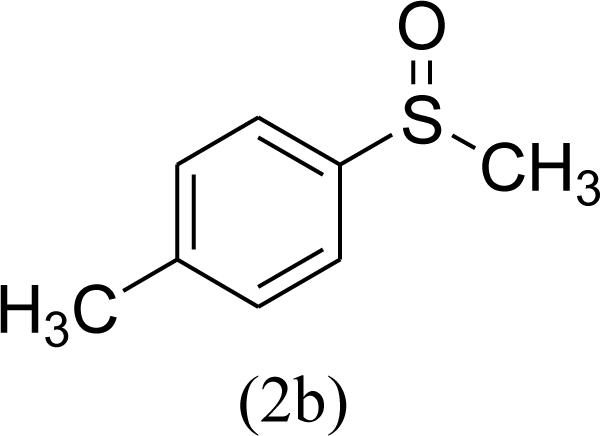 |
99.9 |
| 3 |  |
 |
99.9 |
| 4 | 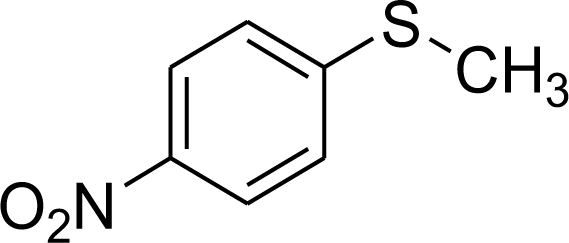 |
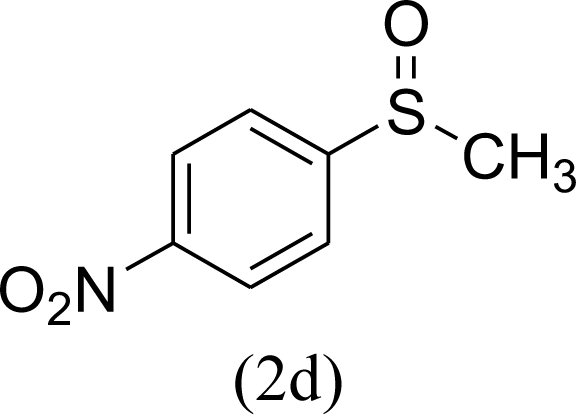 |
87.2 |
| 5 |  |
 |
83.9 |
| 6 |  |
 |
86.1 |
One of the most important merits of heterogeneous catalysts is that they can be recycled and reused. Thus, the recyclability of the PbCrO4 catalyst was investigated for the oxidation of thioanisole. After the first catalytic run, the catalyst was separated by centrifugation, leached and washed with water and methanol, and dried at 80 °C under vacuum. The used PbCrO4 catalyst was subjected to a next run under the same conditions. As shown in Fig. 10, the PbCrO4 catalyst demonstrated excellent recyclability. It was recycled six consecutive times with almost unaltered catalytic activity.
 | ||
| Fig. 10 The reusability of catalysts for the oxidation of thioanisole. | ||
The catalytic activity of nano-PbCrO4 depends on the size of the particles, and we observed that PbCrO4 nanoparticles with sizes are effective in catalyzing reactions. However, these nanoparticles generally lose their activity slowly during every reaction cycle. This loss in activity is observed because the nanoparticles grow in size through the Ostwald ripening process, in which small nanoparticles merge together to form large nanoparticles. In the case of the PbCrO4 catalyst system, we observed no appreciable Ostwald ripening, and the particle size and distribution remained the same even after the reaction, which in turn maintained the catalytic activity. Therefore, even after several reaction cycles, the size and distribution of PbCrO4 nanoparticles remain nearly the same (Fig. 11) and thus, the system maintains nearly the same catalytic activity.
 | ||
| Fig. 11 TEM images of PbCrO4 nano-catalysts after reaction (a). Particle size distribution before reaction (b) and after reaction (c). | ||
The tentative mechanism of visible-light photoredox catalysis by PbCrO4 with triethylamine as a redox mediator is rationalized in Scheme 1. PbCrO4 can absorb a wide range of visible light. Next, the electron travels through the conduction band of PbCrO4 to react with O2, thereby forming O2˙−. The S-centered free-radical cation prefers to react with O2˙− formed at the conduction band of PbCrO4, allowing the evolution of sulfide peroxide. In the protic solvent CH3OH, the final methyl phenyl sulfoxide is formed, accompanied by the side products of HCHO and H2O. At this stage, this proposed mechanism lacks direct evidence (Scheme 1).
 | ||
| Scheme 1 Possible mechanism for the visible-light-induced oxidation of sulfides with O2. | ||
Conclusions
In conclusion, PbCrO4 nanoparticles were successfully synthesized via a green method in the presence of gum of ferula assa-foetida as a novel nanocatalyst. SEM, TEM, XRD and EDX have been used to characterize the prepared nanocatalyst. Through the photocatalysis of PbCrO4 and TEA, the visible-light-induced selective oxidation of sulfide into sulfoxide with O2 was successfully conducted in CH3OH. The interaction between PbCrO4 and TEA gives rise to visible-light activity in the reaction system. This separable solid nanocatalyst can be prepared by a very simple procedure using inexpensive and commercially available precursors, and the catalyst can be reused for six cycles without any loss in its activity as well as composition. Thus, the procedure serves as a valuable alternative in the selective synthesis of sulfoxides.Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. C. Ling, G. M. Wang, J. Reddy, C. C. Wang, J. Z. Zhang and Y. Li, Angew. Chem., Int. Ed., 2012, 51, 4074–4079 CrossRef CAS.
- N. Dimitratos, J. A. Lopez-Sanchez and G. J. Hutchings, Chem. Sci., 2012, 3, 20–44 RSC.
- M. Bordeaux, A. Galarneau and J. Drone, Angew. Chem., 2012, 124, 10870–10881 CrossRef.
- S. R. Zhang, L. Nguyen, Y. Zhu, S. H. Zhan, C. K. Tsung and F. Tao, Acc. Chem. Res., 2013, 46, 1731–1739 CrossRef CAS PubMed.
- R. Blume, M. Hvecker, S. Zafeiratos, D. Techner, A. KnopGericke, R. Schlgl, L. Gregoratti, A. Barinov and M. Kiskinova, Nanostructured Catalysts: Selective Oxidations, The Royal Society of Chemistry, 2011, pp. 248–265 Search PubMed.
- J. Piera and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2008, 47, 3506–3523 CrossRef CAS PubMed.
- F. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., 2010, 122, 5642–5646 CrossRef.
- Z. Guo, B. Liu, Q. Zhang, W. Deng, Y. Wang and Y. Yang, Chem. Soc. Rev., 2014, 43, 3480–3524 RSC.
- T. Matsumoto, M. Ueno, N. Wang and S. Kobayashi, Chem.–Asian J., 2008, 3, 196–214 CrossRef CAS.
- P. S. Kulkarni and C. A. M. Afonso, Green Chem., 2010, 12, 1139–1149 RSC.
- M. V. Gómez, R. Caballero, E. Vázquez, A. Moreno, A. Hoz and Á. Díaz-Ortiz, Green Chem., 2007, 9, 331–336 RSC.
- B. Karimi, M. Ghoreishi-Nezhad and J. H. Clark, Org. Lett., 2005, 7, 625–628 CrossRef CAS PubMed.
- R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977–1986 RSC.
- K. Sato, M. Aoki and R. Noyori, Science, 1998, 281, 1646–1647 CrossRef CAS PubMed.
- K. Sato, M. Hyodo, M. Aoki, X.-Q. Zheng and R. Noyori, Tetrahedron, 2001, 57, 2469–2476 CrossRef CAS.
- P. Kowalski, K. Mitka, K. Ossowska and Z. Kolarska, Tetrahedron, 2005, 61, 1933–1953 CrossRef CAS.
- K. Kaczorowska, Z. Kolarska, K. Mitka and P. Kowalski, Tetrahedron, 2005, 61, 8315–8327 CrossRef CAS.
- B. M. Choudary, B. Bharathi, C. V. Reddy and M. L. Kantam, J. Chem. Soc., Perkin Trans. 1, 2002, 2069–2074 RSC.
- K. Jeyakumar, R. D. Chakravarthy and D. K. Chand, Catal. Commun., 2009, 10, 1948–1951 CrossRef CAS.
- C. M. Gordon, Appl. Catal., A, 2001, 222, 101–117 CrossRef CAS.
- S. Rostamnia, RSC Adv., 2015, 5, 97044–97065 RSC.
- B. Zhou, J. Song, Z. Zhang, Z. Jiang, P. Zhang and B. Han, Green Chem., 2017, 19, 1075–1081 RSC.
- W. Huang, B. C. Ma, H. Lu, R. Li, L. Wang, K. Landfester and K. Zhang, ACS Catal., 2017, 7, 5438–5442 CrossRef CAS.
- X. Lang, J. Zhao and X. Chen, Angew. Chem., Int. Ed., 2016, 55, 4697–4700 CrossRef CAS PubMed.
- X. Lang, W. Hao, W. R. Leow, S. Li, J. Zhao and X. Chen, Chem. Sci., 2015, 6, 5000–5005 RSC.
- X. Lang, W. R. Leow, J. Zhao and X. Chen, Chem. Sci., 2015, 6, 1075–1082 RSC.
- B. Zhang, J. Li, B. Zhang, R. Chong, R. Li, B. Yuan, S.-M. Lu and C. Li, J. Catal., 2015, 332, 95–100 CrossRef CAS.
- S. J. Tans, M. H. Devoret, H. Dai, A. Thess, R. E. Smalley, L. J. Geerligs and C. Dekker, Nature, 1997, 386, 474–476 CrossRef CAS.
- J. Wang, M. S. Gudiksen, X. Duan, Y. Cui and C. M. Lieber, Science, 2001, 293, 1455–1457 CrossRef CAS PubMed.
- H. Kind, H. Yan, B. Messer, M. Law and P. Yang, Adv. Mater., 2002, 14, 158–160 CrossRef CAS.
- J. Hu, T. W. Odom and C. M. Lieber, Acc. Chem. Res., 1999, 32, 435–445 CrossRef CAS.
- X. G. Peng, L. Manna, W. D. Yang, J. Wickham, E. Scher, A. Kadavanich and A. P. Alivisators, Nature, 2000, 404, 59–61 CrossRef CAS PubMed.
- K. S. Knight, Mineral. Mag., 2000, 64, 291–300 CrossRef CAS.
- S. Vaucher, M. Li and S. Mann, Angew. Chem., Int. Ed., 2000, 39, 1793–1796 CrossRef CAS.
- X. L. Hu and Y. J. Zhu, Chem. Lett., 2004, 33, 880–881 CrossRef CAS.
- L. J. H. Erkens, H. Hamers, R. J. M. Hermans, E. Claeys and M. Bijnens, Surf. Coat. Int., Part B, 2001, 84, 169–176 CrossRef CAS.
- K. A. Wishah and M. M. Abdul-Gader, Appl. Phys. A, 1998, 66, 229–234 CrossRef CAS.
- S. M. Sadeghzadeh, Appl. Organomet. Chem., 2016, 30, 835–842 CrossRef CAS.
- M. A. Nasseri and S. M. Sadeghzadeh, J. Iran. Chem. Soc., 2014, 11, 27–33 CrossRef CAS.
- S. M. Sadeghzadeh, Microporous Mesoporous Mater., 2016, 234, 310–316 CrossRef CAS.
- Z. Chai, T.-T. Zeng, Q. Li, L.-Q. Lu, W.-J. Xiao and D. Xu, J. Am. Chem. Soc., 2016, 138, 10128–10131 CrossRef CAS PubMed.
- F. Raza, J. H. Park, H.-R. Lee, H.-I. Kim, S.-J. Jeon and J.-H. Kim, ACS Catal., 2016, 6, 2754–2759 CrossRef CAS.
- K. Sato, M. Hyodo, M. Aoki, X.-Q. Zheng and R. Noyori, Tetrahedron, 2001, 57, 2469–2476 CrossRef CAS.
- N. Komatsu, M. Hashizume, T. Sugita and S. Uemura, J. Org. Chem., 1993, 58, 4529–4533 CrossRef CAS.
- B. M. Trost and D. P. Curran, Tetrahedron Lett., 1981, 22, 1287–1290 CrossRef CAS.
- B. Karimi, M. Ghoreishi-Nezhad and J. H. Clark, Org. Lett., 2005, 7, 625–628 CrossRef CAS PubMed.
- K. Nakajima, K. Kojima, M. Kojima and J. Fujita, Bull. Chem. Soc. Jpn., 1990, 63, 2620–2630 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |