
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGram-scale synthesis of porphycenes through acid-catalyzed oxidative macrocyclizations of E/Z-mixed 5,6-diaryldipyrroethenes†
Toshikazu Ono *ab,
Ning Xua,
Daiki Kogaa,
Toshihiro Ideoc,
Manabu Sugimotoc and
Yoshio Hisaeda*a
*ab,
Ning Xua,
Daiki Kogaa,
Toshihiro Ideoc,
Manabu Sugimotoc and
Yoshio Hisaeda*a
aDepartment of Chemistry and Biochemistry, Graduate School of Engineering, Center for Molecular Systems (CMS), Kyushu University, Fukuoka 819-0395, Japan. E-mail: tono@mail.cstm.kyushu-u.ac.jp; yhisatcm@mail.cstm.kyushu-u.ac.jp
bPRESTO, Japan Science and Technology Agency (JST), 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
cFaculty of Advanced Science and Technology, Kumamoto University, 2-39-1 Kurokami, Chuo-ku, Kumamoto 860-8555, Japan
First published on 26th November 2018
Abstract
The gram-scale production of porphycene derivatives is reported. This has been achieved by acid-catalyzed ring closure of an E/Z-mixture of 5,6-diaryldipyrroethenes, resulting in the formation of meso-tetraarylporphycenes in yields of up to 80%. E/Z-isomerization of the 5,6-diaryldipyrroethenes under acidic conditions was key to proceed the effective macrocyclization.
Porphycene, the first structural isomer of porphyrin, was reported in 1986 (ref. 1) and has since been the subject of extensive study.2 Porphycene derivatives have attracted considerable attention in photodynamic therapy,3 protein mimicry,4–7 catalysis,8,9 tautomerism,10–12 and materials chemistry.13,14 Various porphycenes with meso- and β-substituents and their metal complexes have been reported to have tunable functionalities, crystallinities and solubilities.15–31
However, the main obstacle to the widespread application of porphycenes is the lack of efficient and economical methods for their production. Therefore, porphycene chemistry has advanced more slowly than that of the parent isomer porphyrin. The gram-scale synthesis of porphycenes has yet to be reported, despite reports of gram-scale synthesis of other tetrapyrrole macrocycles, such as porphyrin,32,33 corrole,34 and norcorrole.35 The first reported synthetic approach (Fig. 1, Method a) proposed by Vogel in 1986, is recognized as the standard preparation method.1 This method is based on the McMurry reductive cyclization of two 5,5′-diacyl-2,2′-bipyrroles. However, this synthesis is very difficult owing to the susceptibility of pyrrole intermediates to rapid oxidation in air and the unimpressive yields obtained. The final McMurry cyclization is difficult to scale up and usually gives very low yields. An alternative approach (Fig. 1, Method b) was proposed by Srinivasan in 2008.36 This synthesis of 9,10,19,20-tetraarylporphycenes involves the formation of two 2,2′-linkages followed by aromatization. It also includes an oxidative ring closure reaction of 5,6-diaryldipyrroethanes with p-toluenesulfonic acid (p-TSA) as acid catalyst, followed by oxidation with 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ). In 2014, Ravikanth optimized the same methodology using 5,6-diaryldipyrroethenes as precursors with a wider scope of aryl substituents.37 However, these alternative systems have not been explored further, probably due to the last oxidative macrocyclization step giving a low yield of 3–7%.36,37
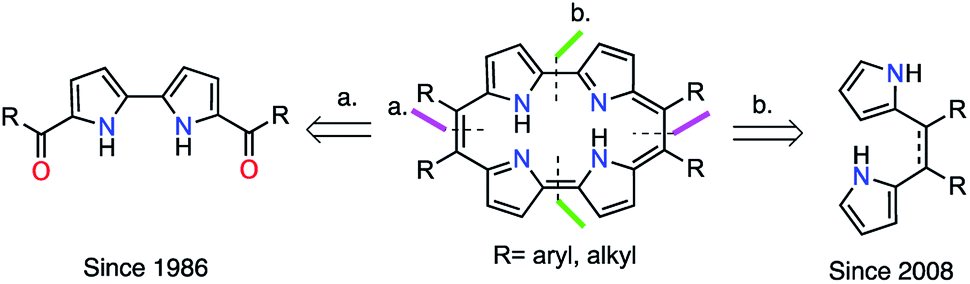 | ||
| Fig. 1 Retrosynthetic analysis of porphycenes. | ||
Inspired by these reports, we have recently demonstrated the syntheses of 9,10,19,20-tetralkylporphycenes through the oxidative macrocyclization of 5,6-dialkyldipyrroethenes as precursors.38 The coupling reaction of 5,6-dialkyldipyrroloethenes with p-TSA as acid catalyst, followed by oxidation with DDQ, was unsuccessful. In contrast, the coupling reaction of 5,6-dialkyldipyrroloethene with hypervalent iodine(III) reagent, [bis(trifluoroacetoxyiodo)]benzene (PIFA), as oxidant, followed by autooxidation in air, was successful, affording 9,10,19,20-tetralkylporphycenes in 3–13% yields.38 These findings indicated that careful selection of the macrocyclization/oxidation conditions was important in the alternative strategy (Fig. 1, Method b). From this background, we now report a highly efficient and gram-scale production of porphycene derivatives based on this alternative strategy. Notably, yields of the oxidative macrocyclization of 5,6-diaryldipyrroethenes were dramatically increased to up to 80%, and the gram-scale production of novel porphycenes was demonstrated. To our knowledge, such a high-yielding scalable synthesis of porphycenes has not previously been reported.2
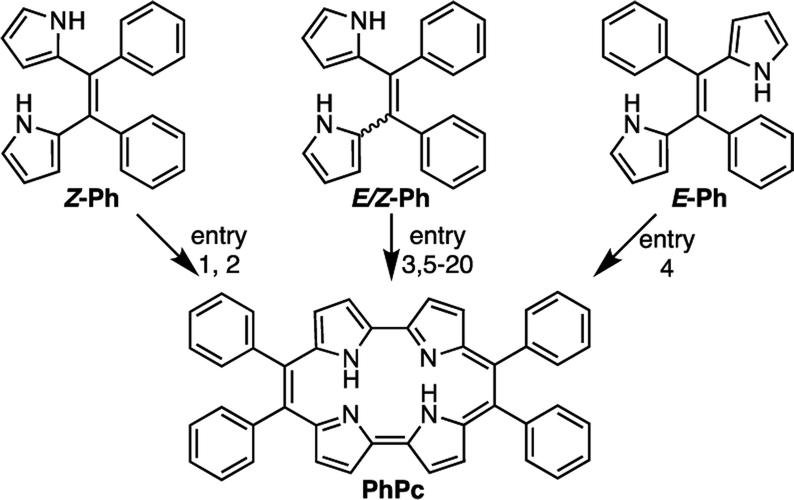
The E/Z mixture of 5,6-diphenyldipyrroloethene (Ph) was synthesized from commercially available 2-benzoylpyrrole (1) using a McMurry coupling in the presence of Zn and TiCl4 in THF. The E/Z isomer ratio of the mixture was determined to be 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by integrating the pyrrole α-H proton signals in the 1H NMR spectra. We successfully separated the E- and Z-isomers from the mixture by a silica gel column chromatography. The (E)-5,6-diaryldipyrroethanes (E-Ph) and (Z)-5,6-diaryldipyrroethanes (Z-Ph) were characterized according to the previous reports.39
:1 by integrating the pyrrole α-H proton signals in the 1H NMR spectra. We successfully separated the E- and Z-isomers from the mixture by a silica gel column chromatography. The (E)-5,6-diaryldipyrroethanes (E-Ph) and (Z)-5,6-diaryldipyrroethanes (Z-Ph) were characterized according to the previous reports.39
First, we investigated optimizing the oxidative macrocyclizations using the synthesis of 9,10,19,20-tetrphenylporphycenes (PhPc) as the model reaction (Table 1). According to the previous reports, the coupling reaction of Z-Ph with p-TSA as acid catalyst, followed by oxidation with DDQ was performed to give PhPc in 5% yield (Table 1, entry 1). This result was consistent with previous results.37 In contrast, the coupling reactions of Z-Ph with p-TSA followed by oxidation with p-chloranil produced PhPc in 45% yield (entry 2). The coupling reaction of E/Z-Ph with p-TSA followed by oxidation with p-chroranil produced PhPc in 35% yield (entry 3), while that of E-Ph with p-TSA followed by oxidation with p-chloranil produced the target porphycene in 30% yield (entry 4). In porphycene synthesis, coupling two E-Ph molecules is expected not to result in macrocycles, but linear polymers such as polypyrroles. However, as shown in entries 3 and 4, the efficient synthesis of porphycenes proceeded from E-Ph. The 1H NMR spectra of CD2Cl2 solutions of E-Ph, Z-Ph, and E/Z-Ph showed negligible E/Z-isomerization at room temperature in the dark after 1 day (Fig. S1–S3†). Therefore, we surmised that the isomers interconverted in the presence of p-TSA in CH2Cl2. Indeed, it has been reported that compounds containing alkene in its structure undergoes E/Z-isomerization by proton in situ.40 E/Z-isomerization of Ph under acidic condition was also supported by density-functional theory calculations (Fig. S34 and Table S5, see detail in ESI†). A plausible mechanism for formation of porphycenes are shown in Fig. 2. The acid (proton) plays two important roles: E/Z isomerization and oxidative ring closure reaction of 5,6-diaryldipyrroloethene.
|
|||||
|---|---|---|---|---|---|
| Entry | Pyrrole | Acidb | Oxidant | Solvent | Yieldc (%) |
| a Reaction conditions: [pyrrole] = 1.6 × 10−3 M; [acid] = 0.5 eq. to pyrrole; [Oxidant] = 3 eq. to pyrrole; room temperature.b Abbreviations: p-TSA, p-toluenesulfonic acid monohydrate; TfOH trifluoromethane sulfonic acid; TFA, trifluoroacetic acid.c Yield of isolated product.d [Acid] = 1.0 eq. to pyrrole.e [Acid] = 2.0 eq. to pyrrole.f [Acid] = 10 eq. to pyrrole. | |||||
| 1 | Z-Ph | p-TSA | DDQ | CH2Cl2 | 5 |
| 2 | Z-Ph | p-TSA | p-Chloranil | CH2Cl2 | 45 |
| 3 | E/Z-Ph | p-TSA | p-Chloranil | CH2Cl2 | 35 |
| 4 | E-Ph | p-TSA | p-Chloranil | CH2Cl2 | 30 |
| 5 | E/Z-Ph | p-TSA | Quinone | CH2Cl2 | 14 |
| 6 | E/Z-Ph | p-TSA | Bromanil | CH2Cl2 | 42 |
| 7 | E/Z-Ph | p-TSA | Fluoranil | CH2Cl2 | 32 |
| 8 | E/Z-Ph | p-TSA | o-Chloranil | CH2Cl2 | 17 |
| 9 | E/Z-Ph | p-TSA | p-Chloranil | CHCl3 | 32 |
| 10 | E/Z-Ph | p-TSA | p-Chloranil | Toluene | 6 |
| 11 | E/Z-Ph | p-TSAd | p-Chloranil | CH2Cl2 | 50 |
| 12 | E/Z-Ph | p-TSAe | p-Chloranil | CH2Cl2 | 65 |
| 13 | E/Z-Ph | p-TSAf | p-Chloranil | CH2Cl2 | 8 |
| 14 | E/Z-Ph | TfOH | p-Chloranil | CH2Cl2 | 62 |
| 15 | E/Z-Ph | TfOHe | p-Chloranil | CH2Cl2 | 6 |
| 16 | E/Z-Ph | TFA | p-Chloranil | CH2Cl2 | 6 |
| 17 | E/Z-Ph | TFAe | p-Chloranil | CH2Cl2 | 18 |
| 18 | E/Z-Ph | BF3·Et2O | p-Chloranil | CH2Cl2 | 30 |
| 19 | E/Z-Ph | BF3·Et2Oe | p-Chloranil | CH2Cl2 | 8 |
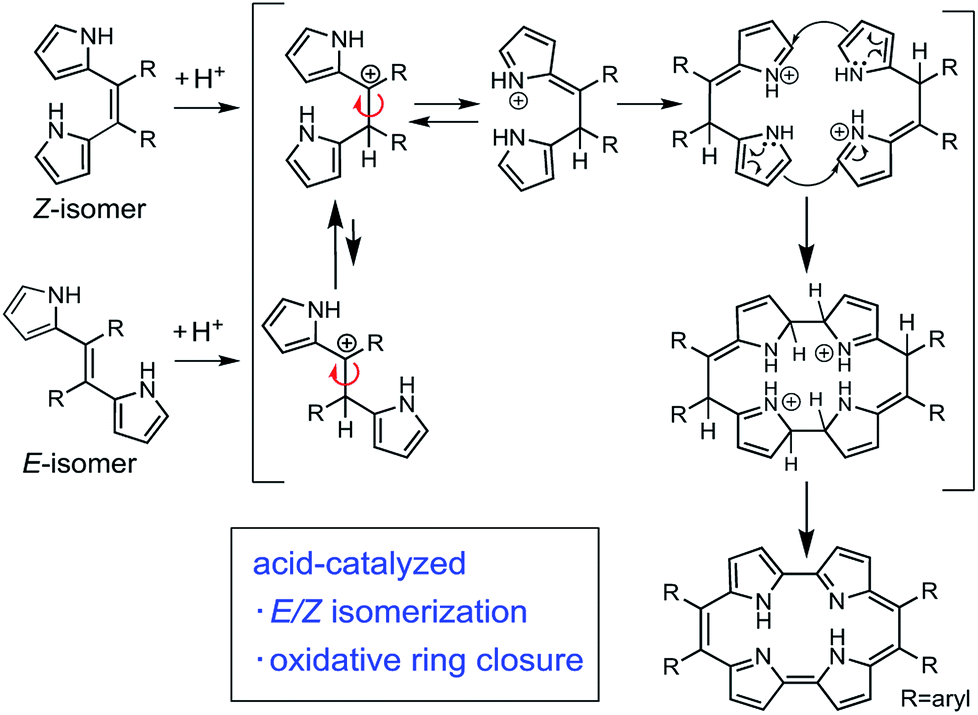 | ||
| Fig. 2 Plausible mechanism for the formation of porphycenes. | ||
To further enhance the yield of PhPc, the oxidants, solvents, and acid catalysts were optimized. First, the trial experiments with E/Z-Ph were conducted with p-TSA as acid catalyst in CH2Cl2 followed by treatment with different oxidants (Table 1, entry 3 and 5–8). Oxidants p-chloranil, quinone, bromanil, fluoranil, and o-chloranil gave PhPc yields of 35%, 14%, 42%, 32%, and 17%, respectively, when using p-TSA (0.5 eq.) as catalyst. The results indicated oxidation required a moderate oxidant, with too strong or too weak oxidants decreasing the PhPc yield. However, when toluene was used as solvent, PhPc was afforded with a significantly decreased yield of 6% (entry 10). Furthermore, when the amount of p-TSA was increased from 0.5 eq. to 1.0 eq., 2.0 eq., and 10 eq., the yields of PhPc clearly fluctuated from 35% to 50%, 65%, and 8%, respectively (entries 11–13). Furthermore, using a stronger Brønsted acid, trifluoromethane sulfonic acid (TfOH; 0.5 eq.), as catalyst gave the PhPc in a reasonably good yield of 62% (entry 14). However, the yield decreased to 6% when 2.0 eq. of TfOH was used (entry 15). A weaker Brønsted acid, trifluoroacetic acid (TFA), was also tested, affording PhPc in lower yields of 6% (0.5 eq.) or 18% (2.0 eq.) (entries 16 and 17). Lewis acid catalyst BF3·Et2O (0.5 eq.) gave PhPc in a moderate yield of 30% (entry 18). The yield was decreased to 8% when the amount of BF3·Et2O was increased to 2.0 eq. (entry 19). The coupling reaction was also performed using PIFA as oxidant, which produced PhPc in 1% yield (see ESI†). Based on these results, we selected p-TSA as acid catalyst, p-chloranil as oxidant, and CH2Cl2 as solvent as the optimal reaction conditions.
Next, we attempted to broaden the reaction scope to the synthesis of meso-tetraarylporphycenes with various aryl-substituents. Considering the solubility and electronic control of the porphycene framework, we attempted to synthesize a set of new meso-tetraaryl substituted porphycenes bearing 3,5-bis(trifluoromethy)phenyl (CF3Pc) and 3,5-difluorophenyl groups (FPc) as electron-withdrawing groups, and 3,5-dimethyphenyl groups (CH3Pc) as electron-donating groups (Fig. 3(a)). The starting materials (1–4) were obtained in 54–68% yields. Compounds 1–4 were then subjected to the McMurry coupling conditions. A conventional workup gave E/Z mixtures of the corresponding dipyrroethenes (E/Z-Ph, E/Z-CF3, E/Z-F, and E/Z-CH3) in yields of 42–67%. Finally, optimizations of oxidative macrocyclizations afforded porphycenes (PhPc, CF3Pc, FPc, and CH3Pc) in yields of 46–80%. Notably, the yields of CF3Pc and FPc were 80% and 72%, respectively. Therefore, we had succeeded in preparing PhPc, CF3Pc, and FPc with isolated yields of 0.90 g, 1.06 g, and 1.42 g. The overall yield of FPc from pyrrole and 3,5-difluorobenzoyl chloride was 27%. This is an unprecedented result that enables the gram-scale synthesis of porphycene derivatives in high yields. These compounds were characterized by 1H, 13C, and 19F NMR spectroscopy and high-resolution mass analysis (Fig. S4–S30†).
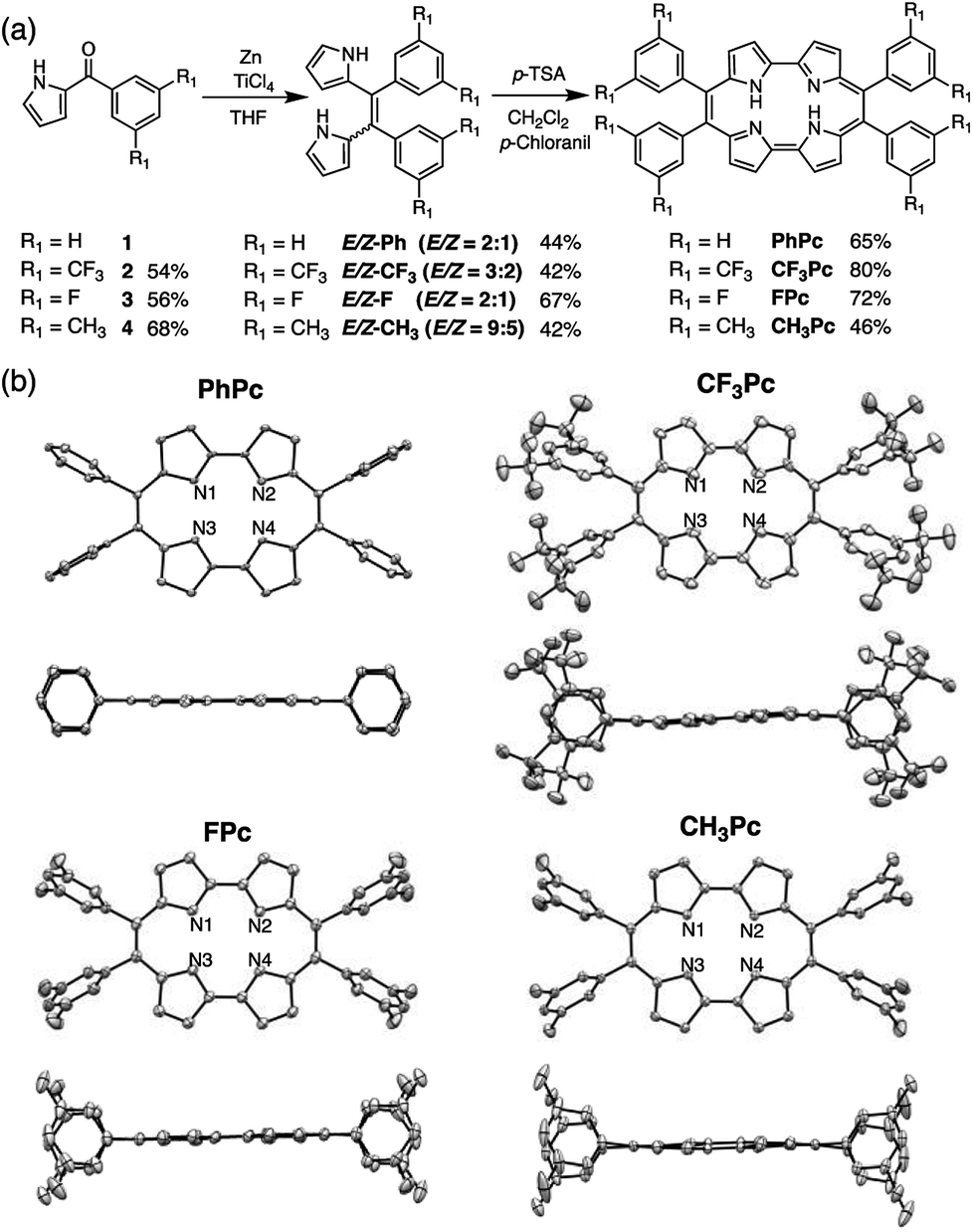 | ||
| Fig. 3 (a) Synthesis of meso-tetraarylporphycenes, (b) single-crystal X-ray structures of PhPc, CF3Pc, FPc, and CH3Pc (above) top view and (below) side view. Thermal ellipsoids are drawn at the 50% probability level. For clarity, only N atoms are numbered and hydrogen atoms are omitted. | ||
Single crystals of porphycenes were fully characterized (Fig. 3(b) and Tables S1–S4†).‡ The molecular structures exhibited perfect rectangular cores in PhPc (N1⋯N2 2.874(5) Å; N1⋯N3 2.547(6) Å), CF3Pc (N1⋯N2 2.894(8) Å; N1⋯N3 2.552(8) Å), FPc (N1⋯N2 2.866(3) Å; N1⋯N3 2.562(4) Å), and CH3Pc (N1⋯N2 2.873(2) Å; N1⋯N3 2.568(2) Å). Side views of the porphycene derivatives showed that PhPc and FPc had planar geometries, while CF3Pc and CH3Pc showed slightly distorted structures due to steric repulsion between the bulky 3,5-di(trifluoromethyl) or 3,5-dimethylphenyl substituents in the crystals.
The optoelectronic properties of the porphycenes were also investigated (Table 2). The porphycenes generally showed a strong Soret band at approximately 380 nm and three weak Q bands in the region of 578–654 nm (Fig. S31†). The introduction of electron-withdrawing groups (CF3Pc, FPc) resulted in a hypochromic shift in the absorption spectra, while introducing electron-rich groups (CH3Pc) resulted in a bathochromic shift. The compounds were reasonably fluorescent, with a strong fluorescence band at approximately 660 nm and a shoulder band at approximately 720 nm. The quantum yields of porphycenes in CH2Cl2 were in the range 16.0–29.2%, and the lifetimes were in the range of 2.4–5.6 ns (Fig. S32†). The electrochemical properties of the porphycenes were characterized by cyclic voltammetry and differential pulse voltammetry in CH2Cl2 vs. Ag/AgCl (Fig. S33†). All porphycenes displayed the typical two reversible one-electron reductions and one reversible and/or quasi-reversible one-electron oxidation. Owing to the presence of electron-withdrawing groups for CF3Pc and FPc, the first oxidation and first reduction potentials were more positive and less negative, respectively, compared with those of PhPc. Conversely, owing to the presence of electron-rich groups for CH3Pc, the first oxidation and first reduction potentials were less negative and more positive over PhPc. The HOMO–LUMO energy gaps of porphycene derivatives (ΔE = Eox1 − Ered1) were 2.10, 1.92, 1.87, and 1.82 V for CF3Pc, FPc, PhPc, and CH3Pc, respectively. The optoelectronic properties were supported by DFT calculations (Tables S6–S8, Fig. S35†).
| Soret banda [nm] | Q bandsa [nm] | λem [nm] | ΦPLb [%] | τc [ns] | Reduction E1/2/V vs. Ag/AgCl | Oxidation E1/2/V vs. Ag/AgCl | HOMO–LUMO gap | ||
|---|---|---|---|---|---|---|---|---|---|
| Ered [V] I | Ered [V] II | Eox [V] | ΔEred–ox [V] | ||||||
| a Values parentheses correspond to log.b Absolute photoluminescence quantum yields.c Fluorescence lifetime. | |||||||||
| PhPc | 381 (5.1) | 584 (4.2), 625 (4.4), 653 (4.6) | 667, 727 | 16.0 | 2.6 | −0.73 | −0.98 | +1.14 | 1.87 |
| CH3Pc | 384 (5.1) | 578 (4.3), 619 (4.4), 648 (4.6) | 655, 721 | 29.2 | 5.6 | −0.51 | −0.81 | +1.59 | 2.10 |
| FPc | 381 (5.1) | 578 (4.3), 619 (4.4), 647 (4.5) | 655, 720 | 17.5 | 3.4 | −0.59 | −0.88 | +1.33 | 1.92 |
| CH3Pc | 383 (5.1) | 586 (4.3), 627 (4.5), 654 (4.6) | 655, 727 | 15.2 | 2.4 | −0.76 | −1.08 | +1.06 | 1.82 |
In conclusion, meso-tetraarylporphycenes were synthesized on a gram-scale in a few steps with high overall yields using an optimized acid-catalyzed oxidative macrocyclization of E/Z-mixtures of 5,6-diaryldipyrroethene. E/Z-isomerization of the 5,6-diaryldipyrroethenes under acidic conditions was key to effective macrocyclization, as supported by both experimental and theoretical observations. The straightforward production of porphycenes will enable practical applications of porphycene derivatives.
Conflicts of interest
There were no conflicts to declare.Acknowledgements
This work was supported by JST PRESTO Grant Number JPMJPR1414 and JSPS KAKENHI Grant Numbers JP17H04875 (Grant-in-Aid for Young Scientists (A) for T. O.), JP17H05161 (π-System Figuration for T. O.), JP26102015 (π-System Figuration for M. S.), JP16H04119 (Grant-in-Aid for Scientific Research for Y. H.), JP16H01035, JP18H04265 (Precisely Designed Catalysts with Customized Scaffolding for Y. H.), and Nissan Chemical Corporation. We thank Dr Kenji Yoza (Bruker AXS) for X-ray crystallographic analyses.Notes and references
- E. Vogel, M. Köcher, H. Schmickler and J. Lex, Angew. Chem., Int. Ed., 1986, 25, 257–259 CrossRef.
- G. Anguera and D. Sanchez-Garcia, Chem. Rev., 2017, 117, 2481–2516 CrossRef CAS PubMed.
- J. C. Stockert, M. Canete, A. Juarranz, A. Villanueva, R. W. Horobin, J. Borrell, J. Teixido and S. Nonell, Curr. Med. Chem., 2007, 14, 997–1026 CrossRef CAS PubMed.
- T. Hayashi, H. Dejima, T. Matsuo, H. Sato, D. Murata and Y. Hisaeda, J. Am. Chem. Soc., 2002, 124, 11226–11227 CrossRef CAS PubMed.
- K. Oohora, Y. Kihira, E. Mizohata, T. Inoue and T. Hayashi, J. Am. Chem. Soc., 2013, 135, 17282–17285 CrossRef CAS PubMed.
- K. Oohora, H. Meichin, Y. Kihira, H. Sugimoto, Y. Shiro and T. Hayashi, J. Am. Chem. Soc., 2017, 139, 18460–18463 CrossRef CAS PubMed.
- K. Oohora, H. Meichin, L. Zhao, M. W. Wolf, A. Nakayama, J. Y. Hasegawa, N. Lehnert and T. Hayashi, J. Am. Chem. Soc., 2017, 139, 17265–17268 CrossRef CAS PubMed.
- W.-C. Lo, C.-M. Che, K.-F. Cheng and T. C. Mak, Chem. Commun., 1997, 1205–1206 RSC.
- T. Koide, I. Aritome, T. Saeki, Y. Morita, Y. Shiota, K. Yoshizawa, H. Shimakoshi and Y. Hisaeda, ACS Omega, 2018, 3, 4027–4034 CrossRef CAS.
- J. Waluk, Chem. Rev., 2017, 117, 2447–2480 CrossRef CAS PubMed.
- T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Nat. Chem., 2013, 6, 41–46 CrossRef PubMed.
- J. N. Ladenthin, T. Frederiksen, M. Persson, J. C. Sharp, S. Gawinkowski, J. Waluk and T. Kumagai, Nat. Chem., 2016, 8, 935–940 CrossRef CAS PubMed.
- W. Brenner, J. Malig, R. D. Costa, D. M. Guldi and N. Jux, Adv. Mater., 2013, 25, 2314–2318 CrossRef CAS PubMed.
- R. D. Costa, J. Malig, W. Brenner, N. Jux and D. M. Guldi, Adv. Mater., 2013, 25, 2600–2605 CrossRef CAS PubMed.
- E. Vogel, M. Balci, K. Pramod, P. Koch, J. Lex and O. Ermer, Angew. Chem., Int. Ed., 1987, 26, 928–931 CrossRef.
- E. Vogel, M. Köcher, J. Lex and O. Ermer, Isr. J. Chem., 1989, 29, 257–266 CrossRef CAS.
- E. Vogel, P. Koch, X. L. Hou, J. Lex, M. Lausmann, M. Kisters, M. A. Aukauloo, P. Richard and R. Guilard, Angew. Chem., Int. Ed., 1993, 32, 1600–1604 CrossRef.
- D. Kuzuhara, J. Mack, H. Yamada, T. Okujima, N. Ono and N. Kobayashi, Chem.–Eur. J., 2009, 15, 10060–10069 CrossRef CAS PubMed.
- T. Sarma, P. K. Panda, P. T. Anusha and S. V. Rao, Org. Lett., 2011, 13, 188–191 CrossRef CAS PubMed.
- A. Rana and P. K. Panda, Org. Lett., 2014, 16, 78–81 CrossRef CAS PubMed.
- A. Rana, S. Lee, D. Kim and P. K. Panda, Chem. Commun., 2015, 51, 7705–7708 RSC.
- A. Rana and P. K. Panda, Chem. Commun., 2015, 51, 12239–12242 RSC.
- M. Duran-Frigola, R. Tejedor-Estrada, D. Sanchez-Garcia and S. Nonell, Phys. Chem. Chem. Phys., 2011, 13, 10326–10332 RSC.
- K. Oohora, A. Ogawa, T. Fukuda, A. Onoda, J. Y. Hasegawa and T. Hayashi, Angew. Chem., Int. Ed., 2015, 54, 6227–6230 CrossRef CAS PubMed.
- O. Planas, D. Fernandez-Llaneza, I. Nieves, R. Ruiz-Gonzalez, E. Lemp, A. L. Zanocco and S. Nonell, Phys. Chem. Chem. Phys., 2017, 19, 25537–25543 RSC.
- D. Kuzuhara, M. Sakaguchi, W. Furukawa, T. Okabe, N. Aratani and H. Yamada, Molecules, 2017, 22, 908 CrossRef PubMed.
- T. Ono, D. Koga, K. Yoza and Y. Hisaeda, Chem. Commun., 2017, 53, 12258–12261 RSC.
- V. Roznyatovskiy, V. Lynch and J. L. Sessler, Org. Lett., 2010, 12, 4424–4427 CrossRef CAS PubMed.
- L. Cuesta, E. Karnas, V. M. Lynch, P. Chen, J. Shen, K. M. Kadish, K. Ohkubo, S. Fukuzumi and J. L. Sessler, J. Am. Chem. Soc., 2009, 131, 13538–13547 CrossRef CAS PubMed.
- M. Abe, H. Futagawa, T. Ono, T. Yamada, N. Kimizuka and Y. Hisaeda, Inorg. Chem., 2015, 54, 11061–11063 CrossRef CAS PubMed.
- T. Sarma, B. S. Kumar and P. K. Panda, Angew. Chem., Int. Ed., 2015, 54, 14835–14839 CrossRef CAS PubMed.
- A. D. Adler, F. R. Longo, J. D. Finarelli, J. Goldmach, J. Assour and L. Korsakoff, J. Org. Chem., 1967, 32, 476 CrossRef CAS.
- J. S. Lindsey, H. C. Hsu and I. C. Schreiman, Tetrahedron Lett., 1986, 27, 4969–4970 CrossRef CAS.
- B. Koszarna and D. T. Gryko, J. Org. Chem., 2006, 71, 3707–3717 CrossRef CAS PubMed.
- T. Ito, Y. Hayashi, S. Shimizu, J. Y. Shin, N. Kobayashi and H. Shinokubo, Angew. Chem., Int. Ed., 2012, 51, 8542–8545 CrossRef CAS PubMed.
- K. Anju, S. Ramakrishnan, A. P. Thomas, E. Suresh and A. Srinivasan, Org. Lett., 2008, 10, 5545–5548 CrossRef CAS PubMed.
- E. Ganapathi, T. Chatterjee and M. Ravikanth, Eur. J. Org. Chem., 2014, 2014, 6701–6706 CrossRef CAS.
- T. Ono, D. Koga and Y. Hisaeda, Chem. Lett., 2017, 46, 260–262 CrossRef CAS.
- K. Garg, E. Ganapathi, P. Rajakannu and M. Ravikanth, Phys. Chem. Chem. Phys., 2015, 17, 19465–19473 RSC.
- A. Souizi, A. Robert, P. Batail and L. Ouahab, J. Org. Chem., 1987, 52, 1610–1611 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of experimental procedure, general information for measurements and materials. CCDC 1868292–1868294 and 1868296. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra09040h |
‡ Crystal data for PhPc (from CHCl3/methanol): C44H30N4·2(CHCl3), Mw = 853.46, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 9.235(16), b = 9.638(17), c = 11.86(2) Å, α = 78.93, β = 76.86(5), γ = 73.21(4)°, V = 975(3) Å3, Z = 1, T = 100 K, Dc = 1.158 g cm−3, GOF = 1.107, R1 = 0.0618 and wR2 = 0.1604 for all data, CCDC 1868296. Crystal data for CF3Pc (from CH2Cl2/methanol): C52H22F24N4, Mw = 1158.74, monoclinic, C2/c, a = 35.48(3), b = 14.931(11), c = 8.852(6) Å, β = 99.42(2)°, V = 4626(6) Å3, Z = 4, T = 103 K, Dc = 1.664 g cm−3, GOF = 1.031, R1 = 0.0988 and wR2 = 0.2659 for all data, CCDC 1868292. Crystal data for FPc (from C2H4Cl2/methanol): C24H22F8N4·0.793(C2H4Cl2), Mw = 837.12, monoclinic, P21/c, a = 8.4081(6), b = 13.0237(9), c = 17.9996(12) Å, β = 103.022(2)°, V = 1920.4(2) Å3, Z = 2, T = 103 K, Dc = 1.448 g cm−3, GOF = 1.073, R1 = 0.0566 and wR2 = 0.1716 for all data, CCDC 1868294. Crystal data for CH3Pc (from THF/methanol): C52H46N4, Mw = 726.93, monoclinic, P21/n, a = 8.7406(11), b = 15.369(2), c = 16.406(2) Å, β = 103.303(4)°, V = 2144.8(5) Å3, Z = 2, T = 103 K, Dc = 1.126 g cm−3, GOF = 1.095, R1 = 0.0611 and wR2 = 0.1848 for all data, CCDC 1868293. , a = 9.235(16), b = 9.638(17), c = 11.86(2) Å, α = 78.93, β = 76.86(5), γ = 73.21(4)°, V = 975(3) Å3, Z = 1, T = 100 K, Dc = 1.158 g cm−3, GOF = 1.107, R1 = 0.0618 and wR2 = 0.1604 for all data, CCDC 1868296. Crystal data for CF3Pc (from CH2Cl2/methanol): C52H22F24N4, Mw = 1158.74, monoclinic, C2/c, a = 35.48(3), b = 14.931(11), c = 8.852(6) Å, β = 99.42(2)°, V = 4626(6) Å3, Z = 4, T = 103 K, Dc = 1.664 g cm−3, GOF = 1.031, R1 = 0.0988 and wR2 = 0.2659 for all data, CCDC 1868292. Crystal data for FPc (from C2H4Cl2/methanol): C24H22F8N4·0.793(C2H4Cl2), Mw = 837.12, monoclinic, P21/c, a = 8.4081(6), b = 13.0237(9), c = 17.9996(12) Å, β = 103.022(2)°, V = 1920.4(2) Å3, Z = 2, T = 103 K, Dc = 1.448 g cm−3, GOF = 1.073, R1 = 0.0566 and wR2 = 0.1716 for all data, CCDC 1868294. Crystal data for CH3Pc (from THF/methanol): C52H46N4, Mw = 726.93, monoclinic, P21/n, a = 8.7406(11), b = 15.369(2), c = 16.406(2) Å, β = 103.303(4)°, V = 2144.8(5) Å3, Z = 2, T = 103 K, Dc = 1.126 g cm−3, GOF = 1.095, R1 = 0.0611 and wR2 = 0.1848 for all data, CCDC 1868293. |
| This journal is © The Royal Society of Chemistry 2018 |