
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLinking lignin source with structural and electrochemical properties of lignin-derived carbon materials†
Wenqi Lia,
Yan Zhangb,
Lalitendu Dasa,
Yikai Wangc,
Mi Lide,
Namal Wanninayakeb,
Yunqiao Pud,
Doo Young Kim b,
Yang-Tse Chengc,
Arthur J. Ragauskasdef and
Jian Shi*a
b,
Yang-Tse Chengc,
Arthur J. Ragauskasdef and
Jian Shi*a
aBiosystems and Agricultural Engineering, University of Kentucky, Lexington, KY 40506, USA. E-mail: j.shi@uky.edu; Fax: +1 859 257 5671; Tel: +1 859 218 4321
bDepartment of Chemistry, University of Kentucky, Lexington, KY 40506, USA
cDepartment of Chemical and Materials Engineering, University of Kentucky, Lexington, KY 40506, USA
dJoint Institute of Biological Science, Biosciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
eDepartment of Chemical and Biomolecular Engineering, University of Tennessee, Knoxville, TN 37996, USA
fDepartment of Forestry, Wildlife, and Fisheries, Center for Renewable Carbon, University of Tennessee Institute of Agriculture, Knoxville, TN 37996, USA
First published on 19th November 2018
Abstract
Valorization of lignin to high-value chemicals and products along with biofuel production is generally acknowledged as a technology platform that could significantly improve the economic viability of biorefinery operations. With a growing demand for electrical energy storage materials, lignin-derived activated carbon (AC) materials have received increasing attention in recent years. However, there is an apparent gap in our understanding of the impact of the lignin precursors (i.e., lignin structure, composition and inter-unit linkages) on the structural and electrochemical properties of the derived ACs. In the present study, lignin-derived ACs were prepared under identical conditions from two different lignin sources: alkaline pretreated poplar and pine. The lignin precursors were characterized using composition analysis, size exclusion chromatography, and 2D HSQC nuclear magnetic resonance (NMR). Distinctive distributions of numerous micro-, meso- and macro-porous channels were observed in the two lignin-derived ACs. Poplar lignin-derived ACs exhibited a larger BET surface area and total mesopore volume than pine lignin-derived AC, which contributed to a larger electrochemical capacitance over a range of scan rates. X-ray photoelectron spectroscopic analysis (XPS) results revealed the presence of oxygen-containing functional groups in all lignin-derived ACs, which participated in redox reactions and thus contributed to an additional pseudo-capacitance. A possible process mechanism was proposed to explain the effects of lignin structure and composition on lignin-derived AC pore structure during thermochemical conversion. This study provides insight into how the lignin composition and structure affect the derived ACs for energy storage applications.
1. Introduction
For the past two centuries, the extensive use of fossil fuels has brought human-kind to an age of unprecedented prosperity and rapid development. However, there is a growing consensus that the introduction of sustainable technologies needs to be developed to address environmental challenges, energy security, and rural development.1 How we respond to these challenges will remarkably affect both current and future generations and Earth's eco-systems. Biofuels provide a potential and promising alternative to traditional fossil fuels and help mitigate the global warming trend,2 which can be broadly categorized as first and second-generation biofuels. First-generation biofuels are produced from starch-based crops, such as corn and sucrose from sugarcane. Second-generation biofuels utilize lignocellulose biomass, such as agriculture residues and woody resources. Lignocellulosic biomass is a composite consisting primarily of cellulose, hemicellulose and lignin, in which cellulose and a portion of the hemicellulose are currently used to produce biofuels, while lignin is underutilized under current biorefinery configurations.Lignin is the second most abundant terrestrial aromatic polymer on Earth.3 As projected by the 2009 Renewable Fuel Standard (RFS2) on the basis of the Energy Independence and Security Act of 2007, the US alone will generate approximately 60 million dry tons of lignin annually as a byproduct from the cellulosic biorefineries by year 2022.4 This will add to the existing ∼100 million tons of lignin from paper and pulping industry.5 Despite its great potential as a feedstock for a variety of high-value chemicals and materials, lignin valorization is challenging, due to the complex structure and compositional heterogeneity of lignin.4 Today, lignin is usually burnt for heat and power, which provides a low-value product and does not fully capture the value of this biopolymer.6 Therefore, other options are needed to unlock the full potential of lignin.4 Utilization of lignin or lignin derived materials as an electrochemical electrode is well suited to meet the demand in the fast-growing energy storage market, such as batteries and supercapacitors.7
Supercapacitor, also known as electrical double layer capacitor (EDLC), is used as an energy storage device. Compared to batteries, supercapacitors have an advantage with high power density and long lifespan, which are suitable for short-term energy storage and burst power delivery. They are capable of bridging the gap between traditional dielectric capacitors and batteries or fuel cells.8 Porous carbon materials, which have high surface areas for charge storage, are the most popular electrode materials for supercapacitors.9 Lignin has been considered as a preferred precursor for activated carbon materials because its high carbon content, highly branched and cross-linked structure, and low feedstock cost. Despite many publications on lignin-derived activated carbon materials for supercapacitor applications, the results reported by different groups vary significantly, as shown in Table 1. In addition to inconsistent processing conditions, we suspect that the heterogeneity in structure and composition of lignin precursors could contribute to the discrepancy in the supercapacitance performances.
| Feedstock | Target materials | Carbonization | Activation | SSA (m2 g−1) | Electrolyte | Capacitance (F g−1) | Reference |
|---|---|---|---|---|---|---|---|
| Kraft lignin | AC | Pyrolysis (1000 °C, 15 min) | KOH (1000 °C, 35 min) | 1148 | 6 M KOH | 91.7 @ 2 mV s−1 | Saha et al., 201447 |
| Hardwood lignin | AC | Pyrolysis | KOH (700 °C, 2h) | 907 | 1 M H2SO4 | 165@ 50 mV s−1 | Zhang et al., 201544 |
| Corn stover lignin | AC | Hydrothermal (180 °C, 18h) | KOH (800 °C, 3h) | 1660 | 6 M KOH | 420 @ 0.1 A g−1 | Guo et al., 201746 |
| Alkali lignin | ACF | Pyrolysis (850 °C, 0.5 h) | KOH (850 °C, 0.5h) | — | 6 M KOH | 344 @ 10 mV s−1 | Hu et al., 201451 |
| Poplar lignin | AC | Hydrothermal (200 °C, 24h) | KOH (800 °C, 1h) | 2218 | 6 M KOH | 312 @ 1 A g−1 | Zhang et al., 201630 |
| Acid washed lignin | AC | Pyrolysis (900 °C, 15 min) | Template | 803 | 6 M KOH | 152.5 @ 1 mV s−1 | Li et al., 201652 |
| Black liquor lignin | AC | Pyrolysis (900 °C, 2h) | KOH (900 °C, 2h) | 1406 | 1.5 M NEt4BF4/ACN | 87 @ 5 mV s−1 | Adriana et al., 201453 |
| Alkali lignin | AC | Pyrolysis (500 °C, 1h) | KOH (800 °C, 1h) | 3775 | 6 M KOH | 286.7 @ 0.2 A g−1 | Zhang et al., 201543 |
| Alkali lignin | ECNF mats | Pyrolysis (1200, 1h) | — | 583 | 6 M KOH | 64 @ 0.4 A g−1 | Lai et al., 201454 |
| Kraft lignin, ethanol extracted lignin, alkali lignin | AC | — | — | 1092, 519, 126 | 1 M H2SO4 | 91, 35, 53 @ 0.5 A g−1 | Jeon et al., 201555 |
| Softwood kraft lignin | AC | — | KOH (800 °C, 1h) | 1800 | EMIMBF4 | 200 @ 10 A g−1 | Klose et al., 201756 |
Properties of lignin, including composition, structure and reactivity, vary significantly depending on the source of biomass feedstocks and isolation methodology. Lignin is mostly composed of three major monomeric units, namely guaiacyl (G), syringyl (S), and p-hydroxyphenyl (H).10 Different types of biomass have different specific ratios of G/S/H. For instance, softwood is composed over 95% G lignin with minor amount of H lignin, while hardwood contains approximately equal percentages of G and S lignins.11 The high ratio of S-units impedes the formation β-5′ and/or 5-5 linkages among lignin monomers (i.e., fewer branches) due to structural hindrance. As a result, hardwood lignin has the structure of more linear polymer chains than softwood lignin.12 Softwood lignins with relatively higher numbers of C–C linkages compared to ether linkages usually contain more condensed structures. Lignins with condensed structures are typically more rigid and less prone to degradation, since the bond dissociation energy13 required to break C–C linkages such as β-5′ are higher than that to break ether linkages such as β-O-4′.14 Furthermore, the chemical reactivities of lignin linkages are different. It was found that the cleavage of β-O-4′ linkage within H and S lignin was easier than G lignin; while the reactivities (as calculated by electrophilicity) of the linkages between H and S unit were higher than those of G lignin.15
In addition to the lignin sources, biomass pretreatment, during which lignin is fractionated from biomass feedstocks are well known to influence the properties of the extracted lignin. A number of pretreatment methods have been developed, such as acidic, alkali, organic solvents, and ionic liquids, all proven to overcome or reduce the natural recalcitrance of lignocellulosic biomass thus improving sugar yield from enzymatic hydrolysis. Due to the differences in pretreatment chemistry and processing condition, various pretreatment approaches remarkably affect the structure and chemical properties of the extracted lignin. Acid pretreatment methods will generally eliminate hemicellulose, while leaving cellulose fraction unaffected. Although little lignin is dissolved, the acidic conditions are known to catalyze the rearrangement of lignin during pretreatment.16 Alkaline-based pretreatments, which include ammonia and lime pretreatment, can effectively remove lignin in biomass by hydrolyzing lignin-carbohydrate ester linkages and some ether bonds.17 Compared to NaOH pretreatment, the lignin isolated from ammonia pretreatment yields a lignin with a lower oxygen and higher aromatic content.18 Organosolv pretreatment removes and modifies lignin via organic solvents, such as methanol, ethanol and acetone, with either acid or base catalysts. The organosolv pretreatment resulted in a significant deconstruction of lignin by the cleavage of inter-unit linkages and condensation of S and G units in comparison with dilute acid and ammonia pretreatment.19 For example, lignin were isolated, migrated and redistributed during organosolv pretreatment of Liriodendron tulipifera with the reformed droplets found on the surface of pretreated biomass.20
Based on the above discussion, there is a gap in our understanding on the determining factors of lignin source, structure and composition on the electrochemical performances of the derived activated carbons. The non-uniformity in lignin structure, composition and reactivity of linkages of diverse lignin sources could lead to different activated carbon materials and thus influence the supercapacitor behavior. In the present study, lignin-derived activated carbon (AC) materials were prepared under identical conditions from two different lignin sources: poplar and pine derived lignin. Electrochemical properties and capacitance behavior of the derived ACs were examined for supercapacitor application to better understand the impact of lignin source. A possible process mechanism was also proposed to explain the effects of lignin structure and composition on lignin-derived AC pore structure during thermochemical conversion.
2. Experimental
2.1 Materials
All biomass feedstocks, including debarked hybrid poplar and lodge pole pine, were obtained from the Idaho National Laboratory. The received biomass was ground by a Thomas Model 4 Wiley® Mill (Thomas Scientific, NJ, USA) to 1 mm particles. Then the ground biomass was sieved via a Ro-Tap® testing sieve shaker (Model B, W. S. Tyler Industrial Group, Mentor, OH, USA) to acquire a particle size range of 0.25 to 0.425 mm for lignin isolation. KOH solution was purchased from Sigma-Aldrich (St. Louis, MO, USA); while NaOH and HCl were purchased from Fisher Scientific (Hampton, NH, USA). Hemicellulase enzyme mixture Cellic HTec2 was provided by Novozymes North America (Franklinton, NC, USA).2.2 Lignin isolation
Biomass samples were pretreated in NaOH at 140 °C for 60 min in a 500 ml Parr reactor. Specifically, 40 g of biomass was mixed with 360 ml of 2 wt% NaOH to obtain a 10 wt% biomass loading. A 300 mesh nylon filter was used to separate the solid and liquid phases in the pretreated biomass slurry. The solid fraction was kept and stored at 4 °C for composition analysis. Lignin was precipitated from the liquid fraction by adjusting the pH value to 1.5–2.0 with 1 M HCl. The recovered lignin was washed 4 times with hot water to reach a neutral pH effluent. To remove carbohydrate impurities from isolated lignin, a dose of hemicellulose enzymes (Cellic HTec2 loading of 0.34 mg protein per g starting biomass) was added to the recovered lignin and incubated at 50 °C in a pH 4.8, 0.05 M citrate buffer solution for 72 h on an orbital shaker (Forma 435, Thermo Fisher Scientific Inc., Waltham, MA, USA). The purified lignin was washed 4 times with 35 ml of hot DI water and then freeze-dried (Freezone Model 77530, Labconco, Kansas City, MO, USA) and collected for carbonization.2.3 Preparation of activated carbons
Lignin derived activated carbon was prepared in an alumina crucible and was placed in a tube furnace (GSL-1500X-OTF, MTI Corporation, Richmond, CA, USA) for carbonization. 2 grams of lignin sample was heated under an argon environment from room temperature to 700 °C ramping at 2 °C min−1 and held for 1 h at 700 °C before cooling down to room temperature. The collected biochar was ground using a pestle and mortar and then dispersed in the KOH solution with a biochar to KOH mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 to form a slurry. The slurry was placed on a hot plate at 80 °C and was stirred by a magnetic stirrer during drying. The dried biochar KOH composite was transferred to a crucible and put into a tube furnace for activation. The furnace temperature was set to 700 °C and held for 1 h. After activation, the activated carbon was neutralized with 0.1 M HCl and then washed with deionized water until the effluent pH becomes 7.0. Finally, the activated carbon was collected and dried in a convection oven (Heratherm, Fisher Scientific Inc., Waltham, MA, USA) at 80 °C.
:3 to form a slurry. The slurry was placed on a hot plate at 80 °C and was stirred by a magnetic stirrer during drying. The dried biochar KOH composite was transferred to a crucible and put into a tube furnace for activation. The furnace temperature was set to 700 °C and held for 1 h. After activation, the activated carbon was neutralized with 0.1 M HCl and then washed with deionized water until the effluent pH becomes 7.0. Finally, the activated carbon was collected and dried in a convection oven (Heratherm, Fisher Scientific Inc., Waltham, MA, USA) at 80 °C.
2.4 Lignin characterization
:8 (v/v) anhydrous acetic acid and acetyl bromide mixture and stirred at 50 °C for 2 h. The acetic acid and excess acetyl bromide solvent were dried in flowing N2. The acetylated lignin was immediately dissolved in tetrahydrofuran (THF). The molecular weight distribution of the lignin samples was determined by an HPLC system (Ultimate 3000, Dionex Corporation, Sunnyvale, CA, USA) equipped with an ultraviolet detector and an Agilent Mixed-D PLgel 5 μm 300 × 7.5 mm column (Agilent Technologies, Santa Clara, CA, USA) using THF as mobile phase at a flow rate of 0.5 ml min−1. The materials eluting from the column were monitored by recording absorbance at 290 nm. The chromatograms were calibrated using low molecular weight polystyrene standards (Product no. 48937, Sigma-Aldrich).2.5 Physical and chemical properties characterization
2.6 Electrochemical characterization
Electrochemical characterization of all lignin-derived carbon materials was carried out using a three-electrode, single-compartment system. A platinum wire was used as the counter electrode, a commercial Ag/AgCl electrode as the reference electrode, and the lignin-derived activated carbon as the working electrode. Lignin-derived activated carbons were first dispersed in DI water (10 μg ml−1) and sonicated for 30 min to make a homogeneous dispersion. Afterwards, a 10 μL droplet of the dispersion was placed on a freshly polished glass carbon disk. After drying in an oven at 40 °C for 1 h, a thin and smooth electrode film (1.4 mg cm−2) was coated on the glass carbon disk and ready to be tested. The electrolyte was 1 M H2SO4 in an aqueous solution. Cyclic voltammetric curves (CV) and galvanostatic charge–discharge curves (GCD) were recorded with a potentiostat (CHI 760, CH Instruments, Austin, TX, USA).3. Results and discussion
3.1 Lignin isolation and characterization
The alkaline treatment using dilute NaOH or lime is a common biomass pretreatment method which results in delignification.25 During an alkaline treatment process, the ester bonds between lignin and xylan are typically cleaved along with the breakdown of the β-O-4′ lignin interunit linkages, leading to dissolution of lignin fragments in the liquid phase and generation of a cellulose and hemicellulose enriched solid suitable for biofuel production.26 The dissolved lignin can then be precipitated from the alkaline solution by adjusting the pH to acidic. Since hemicellulose will bind and precipitate with lignin, the main impurities of the isolated lignin are the 5-carbon sugars. To remove xylan, the pretreated lignin samples were subject to enzymatic hydrolysis.27 The purities of pine and poplar derived lignin samples were determined using the two-step acid hydrolysis procedure.22 As shown in Table 2, most of xylan was eliminated and high purity lignin (94–98%) was acquired after enzymatic saccharification. The ash content is the measure of the mineral content and other inorganic matter in biomasses,28 and the isolated lignin also contains a small amounts of ash, depending on the biomass feedstock, pretreatment, and isolation method used.29 The low ash content of the isolated lignins, shown in Table 2, may be attributed to the acid adjustment, enzymatic hydrolysis and multiple hot water washing, which removed most of ash within lignin.| Lignin sources | Glucan | Xylan | Ash (%) | Mw (Da) | Mn (Da) | PDI |
|---|---|---|---|---|---|---|
| Pine | 0.76 ± 0.05 | 1.18 ± 0.02 | 0.50 ± 0.05 | 2847.73 | 1671.53 | 1.70 |
| Poplar | 1.08 ± 0.01 | 4.95 ± 0.08 | 0.19 ± 0.05 | 2799.78 | 1656.28 | 1.69 |
The weight average molecular weight (Mw), number average molecular weight (Mn) and polydispersity index (PDI) of pine and poplar derived lignin are shown in Table 2. The Mw of pine and poplar were 2847.7 and 2799.8 dalton, respectively. The lower molecular weight might result from the depolymerization of native plant lignin during alkaline pretreatment.30 The PDI value was acquired from the ratio of Mw/Mn, which represents the heterogeneity of the size distribution of the isolated lignin samples. The PDI values were below 2 for both poplar and pine derived lignin samples, indicating increased uniformity of molecular weight distribution.
FT-IR spectra of the three lignin are shown in Fig. S1.† All lignin samples exhibited a broad absorption band at 3400 cm−1, which corresponds to the O–H stretching vibrations in phenolic and aliphatic O–H groups.31 The bands between 2920 and 2840 cm−1 represent C–H vibrations of CH2 and CH3 groups.32 All lignin samples have a peak at 1740 cm−1, representing a stretching of carbonyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in carboxylic acid or ketone and aldehydes.33 The bands at 1600 cm−1 and 1510 cm−1 are attributed to aromatic ring stretch vibrations (CC).34 The broad bands ranged from 1470 to 1330 cm−1 are assigned to C–H aromatic ring vibrations and the deformation vibration of O–H in CH2 and CH3 groups.34 The bands at 1220, 1110 cm−1 are believed to associated with guaiacyl (G) and syringyl (S) units of lignin, respectively.34,35 Compared to poplar lignin, pine lignin has an intense peak at the band of 1220 cm−1, indicating C–C, C–O, and CO stretching (G), which agrees with an earlier report.11
O) in carboxylic acid or ketone and aldehydes.33 The bands at 1600 cm−1 and 1510 cm−1 are attributed to aromatic ring stretch vibrations (CC).34 The broad bands ranged from 1470 to 1330 cm−1 are assigned to C–H aromatic ring vibrations and the deformation vibration of O–H in CH2 and CH3 groups.34 The bands at 1220, 1110 cm−1 are believed to associated with guaiacyl (G) and syringyl (S) units of lignin, respectively.34,35 Compared to poplar lignin, pine lignin has an intense peak at the band of 1220 cm−1, indicating C–C, C–O, and CO stretching (G), which agrees with an earlier report.11
To examine the lignin chemistry and chemical structure of the alkaline extracted lignin, 2D 13C–1H HSQC NMR was applied to characterize the pine and poplar derived lignins. The spectra of the aromatic region between 6.0–8.0/100–150 ppm of the lignins, revealing the lignin aromatic structural subunits, were shown in the Fig. 1A. On a basis of total aromatic amount, pine lignin was almost composed of G unit (S: 0%; G: >99%; H: <0.5%), while poplar was composed of S and G unit (S: 75.2%; G: 24.8%; H: 0%). The aliphatic region between 2.5–6.0/50–90 ppm of the lignins, revealing the lignin inter-units and side chains, can be seen in the Fig. 1B. Both pine and poplar lignins were found to be dominated by β-O-4′ linkages accompanying with a small amount of β-5′ and β-β′ linkages (pine derived lignin: β-O-4′, 85.6%; β-5′, 11.6%; β-β′, 2.8%; poplar derived lignin: β-O-4′, 87.7%; β-5′, 2.5%; β-β′, 9.8%). The poplar lignin contained more oxygen-containing functionalities (i.e., OCH3 groups) than pine due to significantly higher S-unit, which likely contributed to more pore structures after carbonization, and thus result in a higher capacitance as discussed in the Electrochemical characterization section. Comparing to untreated native structure of lignin, the S unit of poplar was increased ~21% after alkaline pretreatment, indicating that pretreatment also altered the structure of lignin which might affect the lignin-derived AC supercapacitor performance.
 | ||
| Fig. 1 13C–1H (HSQC) spectra of aromatic regions (left) and aliphatic regions (right) of alkaline lignin from pine (PI lignin) and poplar (POP lignin). *The structures of lignin compositional units were coded with colors corresponding to the cross peaks in the spectra of aromatic regions while the structures of side-chain linkages were coded with colors corresponding to the cross peaks in the spectra alkyl regions. | ||
3.2 Lignin carbonization and biochar activation
The weight loss of lignin sample after carbonization is attributed to the loss of H2O, CO, CO2, methane, and phenolic volatiles depolymerized during pyrolysis.36 The reaction between carbon and KOH and a series of intermediate products contribute to the weight loss during the activation process.37 The mass yield during carbonization and activation is summarized in Table S1.†38 On basis of 100 g of lignin sample, pine and poplar lignin yielded 41.3 g and 29.4 g of biochar, respectively, although their yields of activated carbon are similar, 22.2 g for pine lignin and 23.2 g for poplar. It is speculated that the different S/G ratio and inter-lignin linkages affect the carbonization and biochar activation of pine and poplar derived lignins. The less amount of methoxy group in softwood lignin favors a highly branched and condensed structure,39 and thus has more C–C bond and less ether bond than hardwood lignin. As the C–C bond has stronger bond dissociation energy than ether bond, softwood lignin may require more energy to break down.40 On the other hand, more methoxy groups mean higher oxygen content in lignin; the oxygen creates active sites during carbonization that are readily activated. Since the reactivity of the linkage between S and S unit is higher than G lignin, poplar lignin is probably more reactive than pine lignin due to its relatively higher S-lignin content.15 The premature biochar may significantly affect subsequent activation due to fewer active sites generated during carbonization which can affect pore structure and specific surface area (SSA).3.3 Morphology, pore structure of carbon materials
Fig. 2 shows the SEM images of the pine and poplar lignins, the carbonized biochar and the KOH activated carbon from those lignin samples. As can be seen in Fig. 2a and b, the raw lignin samples are 20–40 μm particles with high surface roughness. During pyrolysis, lignin starts to melt at approximately 200 °C;41 from that moment, melting and aggregation between lignin particles occur simultaneously, as shown in Fig. 2c and d, forming a 3-dimensional biochar structure after carbonization. KOH chemical activation of these biochar resulted in the lignin-derived activated carbon (AC) samples. These AC samples exhibited a 3D structure with a large number of pores in different sizes, as shown in Fig. 2e and f. The pores connected through channels have important roles in promoting electrode surface areas available for the adsorption of ions and electrolytes and thus enhancing electrochemical capacitance in supercapacitors. | ||
| Fig. 2 SEM images of lignin, biochar and activated carbon samples: (a) pine lignin, (b) poplar lignin, (c) pine lignin-derived biochar, (d) poplar lignin-derived biochar, (e) pine lignin-derived AC, (f) poplar lignin-derived AC. | ||
XRD spectra of lignin-derived ACs are shown in Fig. S2.† All three lignin-derived ACs exhibited similar features with two broad peaks at approximately 20 and 40 degree. While the positions of the two peaks correspond to (002) and (100) crystalline planes of graphitic carbon materials, the peaks are broad, indicating their amorphous structure. The XRD results show that the derived ACs have a turbostratic structure (between graphite and amorphous carbon).42
To examine the pore structure of lignin-derived ACs, the BET specific surface area and pore size distribution were determined by N2 adsorption and desorption analyses. The adsorption isotherm plots are shown in Fig. 3a. While the pore size distribution, which was calculated by BJH method, is shown in Fig. 3b. All ACs revealed the mixed character of types I and IV. Such a behavior reflects the coexistence of micropores, mesopores, and macropores.43,44 However, the pine lignin-derived AC represented a very low N2 adsorption volume and a flat adsorption curve, indicating the low porosity content.
 | ||
| Fig. 3 (a) N2 adsorption–desorption isotherms and (b) calculated pore size distribution of pine and poplar lignin derived activated carbons (LAC). | ||
BET specific surface area (SSA) and the volume of micropores and mesopores of the lignin-derived ACs were shown in Table 3. Compared with the low SSA of 314.95 m2 g−1 for pine derived lignin-derived AC, poplar derived lignin-derived ACs had much higher SSA of 621.25 m2 g−1. Poplar lignin-derived AC had relatively lower micropores volume as compared with its mesopore volume, likely attributing to the structurally fragmented lignin when going through alkali extraction and reprecipitation. In addition to lower SSABET, mesopore volume of pine lignin-derived AC was also significantly lower than that of the poplar lignin-derived ACs. Poplar lignin-derived AC exhibited a high mesopore ratio of 44.4% as compared to 16.7% of pine lignin-derived AC. Taking together, SSA and pore size distribution results illustrate that the lignin source greatly influences the porosity and structure of lignin-derived ACs which can be correlated to the structure, S/G ratio, and interunit linkages of the lignin precursors.
| Lignin | SBET (m2 g−1) | Vmicro (cm3 g−1) | Vmeso (cm3 g−1) | Vtotal (cm3 g−1) | Vmeso/Vmicro (%) |
|---|---|---|---|---|---|
| Pine | 314.95 | 0.12 | 0.02 | 0.14 | 16.7 |
| Poplar | 621.25 | 0.18 | 0.08 | 0.27 | 44.4 |
3.4 Chemical composition analysis of lignin-derived activated carbon materials
X-ray photoelectron spectroscopy (XPS) was applied for the elemental and chemical composition analyses of the lignin-derived ACs. The high resolution XPS C1s and O1s spectra of the lignin-derived ACs are shown in Fig. 4. The deconvolution of the original C1s peaks revealed four individual component peaks at 284.4, 285.5, 286.4, 287.5, 288.8, 291.0 eV including C-sp2, C-sp3, C–O, CO, O–CO, π–π, respectively. The content of each functional group in the AC samples are summarized in Table 4. The surface C and O elemental percentages were determined with EDX. Compared to pine lignin-derived AC of 7.6 atom% surface oxygen content, poplar lignin-derived ACs had higher surface oxygen content as indicated by the atomic percentage of 11.5%. The higher surface oxygen contents of poplar lignin-derived ACs are likely caused by the greater extent of activation occurring during carbonization and KOH activation processes than pine lignin-derived AC.44 Oxygen-containing functional groups can improve the wettability of electrode in aqueous electrolyte and provide an additional pseudocapacitive current through reversible faradaic reactions.45,46 One example of such reversible faradaic reactions for oxygen functional group is a redox process between quinone and hydroquinone.
 | ||
| Fig. 4 Survey spectra of (a) poplar; (b) pine and high resolution C1s of XPS spectra: (c) poplar; (d) pine lignin-derived ACs. | ||
| Lignin | sp2, % | sp3, % | C–O, % | CO, % |
O–CO, % |
|
|---|---|---|---|---|---|---|
| Pine (7.6 at%) | 71.8 | 8.6 | 8.6 | 5.0 | 5.7 | |
| Poplar (11.5 at%) | 68.1 | 11.6 | 8.2 | 5.5 | 6.1 |
3.5 Electrochemical characterization
The electrochemical behaviors of the lignin-derived ACs were examined in a single-compartment electrochemical cell in the potential window from −0.6 to 0.6 V in 1 M H2SO4. The cyclic voltammetry (CV) curves are shown in Fig. 5a and b. The quasi-rectangular shape CV curves, representing a double layer capacitor character, were observed for all the lignin-derived ACs. In addition, peaks at 0.2 to 0.4 V indicated pseudo-capacitance, which was caused by redox reactions of surface oxygen containing functional groups as depicted by the XPS results (Table 4).44 The specific capacitances of the lignin-derived ACs were determined at different sweep rates from CV curves as shown in Fig. 5c. Compared with pine, poplar lignin-derived AC showed a better current response at each scan rate, which is attributed to the significantly larger SSA and mesopore volume. | ||
| Fig. 5 Cyclic voltammetry (CV) curves of (a) poplar lignin-derived AC, (b) pine lignin-derived AC at scan rates ranging from 10 to 50 mV s−1, and (c) capacitive performance of lignin-derived ACs in 1 M H2SO4 at scan rates ranging from 10 to 50 mV s−1. | ||
To further investigate the electrochemical properties of lignin-derived ACs, galvanostatic charge–discharge (GCD) curves were recorded. Fig. 6 gives the GCD character of the lignin-derived ACs at current densities range from 0.5 to 2 A g−1 and all of the GCD curves exhibits a typical triangular shape. As expected, pine lignin-derived ACs represented the shortest one, which demonstrate the same capacitive performance trend as the specific capacitance evaluated from CV curves, seen in Fig. 6a and b. At current density of 0.5 A g−1, the specific capacitance of pine lignin-derived AC was 48.3 F g−1, while poplar lignin-derived ACs achieved 86.7 F g−1, as shown in Fig. 6c. Consistent with the CV curves, the GCD curves of pine lignin-derived AC exhibited higher resistivity than poplar. Note that no carbon black was blended when preparing the electrode. The high resistivity of pine lignin derived AC could be attributed to the incomplete carbonization during pyrolysis, a hypothesis supported by previous studies showing less efficient pyrolysis of G-lignin type softwood lignin than S-lignin dominant hardwood lignin.49,50
 | ||
| Fig. 6 Galvanostatic charge–discharge (GCD) curves of (a) poplar lignin-derived AC, (b) pine lignin-derived AC at current density ranging from 0.5 to 2 A g−1, and (c) comparative GCD curves of pine and poplar lignin-derived ACs at a current density of 0.5 A g−1. | ||
Compared to the data reported in literature shown in Table 1, both SSA and supercapacitor capacitance presented herein are on the same order of magnitude. The difference may come from dissimilar lignin feedstocks and process conditions. It is worth noting that the main goal of this study is to understand the impact of lignin sources on lignin-derived AC pore structure and thus supercapacitor capacitance from a perspective of lignin composition and structure rather than synthesis a new material with super high performance. Therefore, the process conditions of both carbonization and activation are not optimized43 and no other additives, such as carbon black, and binder were blended during lignin-derived ACs synthesis so as to enhance conductivity, as reported elsewhere.44,46,47
3.6 Possible mechanisms of the formation pathway of lignin-derived ACs
Reaction mechanism of lignin pyrolysis is only partly understood due to the complex product composition and possible multiple reaction phases involving complicit prime and secondary reactions.37,48 Based on the results from present study and other literature, we propose a possible pathway for generating the lignin-derived ACs that may help understand the different lignin-derived AC's pore structure between hardwood and softwood lignin, as shown in Scheme 1. During pyrolysis, the depolymerization of lignin starts with the cleavage of ether bonds, such as β-O-4′ linkages at low temperature and produce primarily guaiacol-type and syringol-type monomers and a variety of oligomers.49,50 It is believed that the primary reaction of lignin pyrolysis involves free radical reactions and the monomer products are presented as free radicals.48 Since free radical reactions can initiate chain reactions, it would not terminate as long as the free radicals are present. Hence, the originally volatilized guaiacol and syringol-typed free radicals formed from ether linkages cleavage would subsequently go through repolymerization and condensation into oligomers and finally form solid fractions, namely char and coke. A portion of the monomers capture hydrogen from other compounds, entering into liquid fraction;48 while the other portion of monomers further decompose into gases, such as CH4, CO, CO2 and H2.50 As the reactions continue, the chain reactions within char and coke are forced to terminate after devolatilization and depletion of hydrogen, leaving some free radicals to serve as active sites for the activation of lignin-derived ACs. As discussed previously, softwood lignin, such as pine lignin, is dominated by G lignin, while hardwood lignin has an almost equal amount of G and S lignin unit. It is a crucial difference to the free radical reaction of lignin pyrolysis, since hardwood lignin would have significantly more free radicals acquired from ether linkages cleavage which serve as precursors to propagate the chain reactions and eventually have more chance to maintain free radicals reactive site available for activation. This hypothesis is supported by more efficient devolatilization of hardwood lignin pyrolysis than softwood.49,50 Under the presence of a chemical agent, such as KOH, and pyrolysis, the free radical chain reactions are able to resume and release vapors, such as K, H2, CO and H2O, which contribute to the growth of porosity of carbon materials.37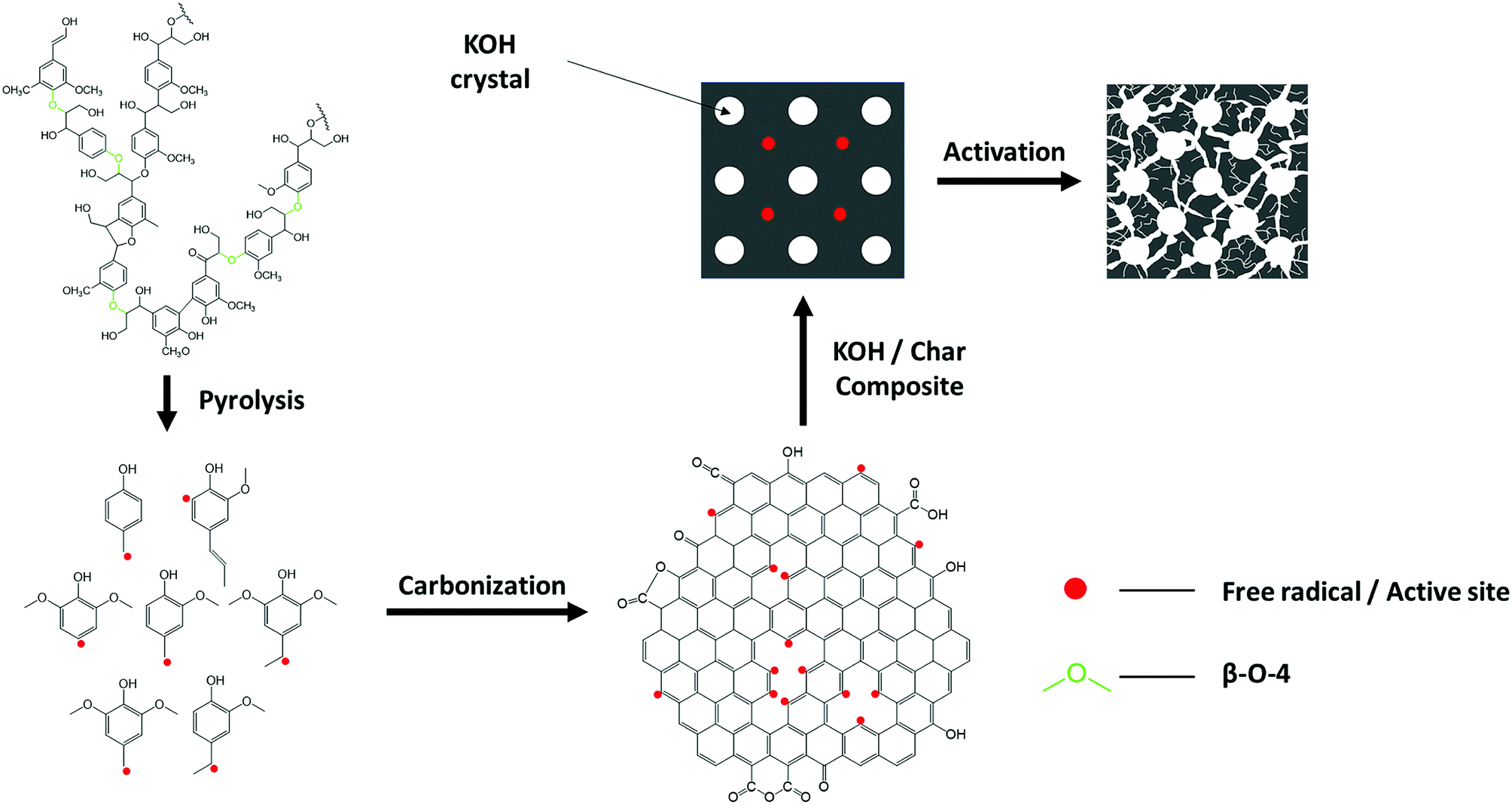 | ||
| Scheme 1 A schematic of possible mechanisms diagram of the formation pathway of lignin-derived AC. | ||
4. Conclusions
Lignin-derived activated carbon materials from alkali pretreated hardwood and softwood were synthesized and characterized. Each lignin-derived AC exhibited a three-dimensional pore structure with numerous micro-, meso- and macro-porous channels. Comparing with pine (softwood) lignin-derived AC, poplar (hardwood) lignin-derived AC showed a higher level of specific surface area and volume of both mesopores and micropores. When applied as supercapacitor electrodes, the poplar lignin-derived AC had a higher value of specific capacitance at each current scan rate than the softwood lignin-derived AC. X-ray photoelectron spectroscopic (XPS) revealed the presence of oxygen-containing functional groups in all lignin-derived ACs, which contributed additional pseudo-capacitance to the total capacitance. By delineating the carbonization and activation parameters, results from this study suggest that lignin structure and composition are important factors determining the pore structure and electrochemical properties of the derived carbon materials. Further investigation is yet needed to understand the kinetics of carbonization and crystal growth during lignin thermochemical conversion.Author contribution
JS, YTC, and DYK conceived the experiments and analyzed the results. WL, YZ, LD, YW, NW conducted experiments and analyzed data. ML, YP, and AJR did NMR analysis and analyzed data. All authors involved writing or revising the manuscript. All authors reviewed and approved the manuscript.Conflicts of interest
The authors declare no conflicts of interest in the present work.Acknowledgements
This work is supported by the USDA National Institute of Food and Agriculture under project accession no. 1015068 and the National Science Foundation under Cooperative Agreements 1355438. Oak Ridge National Laboratory is managed by UT-Battelle, LLC under Contract DE-AC05-00OR22725 with the U.S. Department of Energy (DOE). The views and opinions of the authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, expressed or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. We thank Novozymes for providing enzyme samples and the Idaho National Laboratory for providing plant materials. YZ, NW, and DYK appreciate support from National Science Foundation under Cooperative Agreement No. 1355438 and Kentucky Science & Engineering Foundation grant (KSEF-3884-RDE-020).References
- A. J. McMichael, Climate change and human health: Risks and responses, World Health Organization, Geneva, Switzerland, 2003 Search PubMed.
- N. Panwar, S. Kaushik and S. Kothari, Renewable Sustainable Energy Rev., 2011, 15, 1513–1524 CrossRef.
- V. K. Thakur, M. K. Thakur, P. Raghavan and M. R. Kessler, ACS Sustainable Chem. Eng., 2014, 2, 1072–1092 CrossRef CAS.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 709–719 CrossRef CAS PubMed.
- F. S. Chakar and A. J. Ragauskas, Ind. Crops Prod., 2004, 20, 131–141 CrossRef CAS.
- Z. Strassberger, S. Tanase and G. Rothenberg, RSC Adv., 2014, 4, 25310–25318 RSC.
- W. E. Tenhaeff, O. Rios, K. More and M. A. McGuire, Adv. Funct. Mater., 2014, 24, 86–94 CrossRef CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774–1785 RSC.
- A. Brandt, L. Chen, B. E. van Dongen, T. Welton and J. P. Hallett, Green Chem., 2015, 17, 5019–5034 RSC.
- Bioenergy Feedstock Library, Idaho National Laborotory, U.S. Department of Energy, https://bioenergylibrary.inl.gov/, (accessed October 2018).
- W.-J. Liu, H. Jiang and H.-Q. Yu, Green Chem., 2015, 17, 4888–4907 RSC.
- A. Elmouwahidi, Z. Zapata-Benabithe, F. Carrasco-Marín and C. Moreno-Castilla, Bioresour. Technol., 2012, 111, 185–190 CrossRef CAS PubMed.
- S. Kubo and J. F. Kadla, Biomacromolecules, 2005, 6, 2815–2821 CrossRef CAS PubMed.
- J. Shi, S. Pattathil, R. Parthasarathi, N. A. Anderson, J. Im Kim, S. Venketachalam, M. G. Hahn, C. Chapple, B. A. Simmons and S. Singh, Green Chem., 2016, 18, 4884–4895 RSC.
- B. Yang and C. E. Wyman, Biotechnol. Bioeng., 2004, 86, 88–98 CrossRef CAS PubMed.
- V. Chaturvedi and P. Verma, 3 Biotech, 2013, 3, 415–431 CrossRef PubMed.
- R. Gupta and Y. Y. Lee, Bioresour. Technol., 2010, 101, 8185–8191 CrossRef CAS PubMed.
- C. G. Yoo, M. Li, X. Meng, Y. Pu and A. J. Ragauskas, Green Chem., 2017, 19, 2006–2016 RSC.
- B.-W. Koo, B.-C. Min, K.-S. Gwak, S.-M. Lee, J.-W. Choi, H. Yeo and I.-G. Choi, Biomass Bioenergy, 2012, 42, 24–32 CrossRef CAS.
- F. Lu and J. Ralph, J. Agric. Food Chem., 1997, 45, 2590–2592 CrossRef CAS.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Laboratory analytical procedure, 2008, Vol. 1617, pp. 1–16 Search PubMed.
- C. G. Yoo, Y. Pu, M. Li and A. J. Ragauskas, ChemSusChem, 2016, 9, 1090–1095 CrossRef CAS PubMed.
- S.-L. Sun, J.-L. Wen, M.-G. Ma and R.-C. Sun, J. Agric. Food Chem., 2014, 62, 8120–8128 CrossRef CAS PubMed.
- Y. Sun and J. Cheng, Bioresour. Technol., 2002, 83, 1–11 CrossRef CAS PubMed.
- R. A. Silverstein, Y. Chen, R. R. Sharma-Shivappa, M. D. Boyette and J. Osborne, Bioresour. Technol., 2007, 98, 3000–3011 CrossRef CAS PubMed.
- M. Selig, N. Weiss and Y. Ji, Enzymatic Saccharification of Lignocellulosic Biomass: Laboratory Analytical Procedure (LAP): Issue Date, 3/21/2008, National Renewable Energy Laboratory, 2008 Search PubMed.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter and D. Templeton, Biomass Compositional Analysis Laboratory Procedures, National Renewable Energy Laboratory, Golden, 2004 Search PubMed.
- S. Nakagame, R. P. Chandra and J. N. Saddler, Biotechnol. Bioeng., 2010, 105, 871–879 CAS.
- L. Zhang, T. You, T. Zhou, X. Zhou and F. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 13918–13925 CrossRef CAS PubMed.
- O. Faix, Holzforschung, 1991, 45, 21–28 CrossRef CAS.
- R. García, C. Pizarro, A. G. Lavín and J. L. Bueno, Fuel, 2014, 117, 1139–1147 CrossRef.
- L. A. M. Nevárez, L. B. Casarrubias, A. Celzard, V. Fierro, V. T. Muñoz, A. C. Davila, J. R. T. Lubian and G. G. Sánchez, Sci. Technol. Adv. Mater, 2011, 12, 045006 CrossRef PubMed.
- O. Gordobil, R. Moriana, L. Zhang, J. Labidi and O. Sevastyanova, Ind. Crops Prod., 2016, 83, 155–165 CrossRef CAS.
- A. García, X. Erdocia, M. G. Alriols and J. Labidi, Chem. Eng. Process., 2012, 62, 150–158 CrossRef.
- S. Wang, K. Wang, Q. Liu, Y. Gu, Z. Luo, K. Cen and T. Fransson, Biotechnol. Adv., 2009, 27, 562–567 CrossRef CAS PubMed.
- W. J. Liu, H. Jiang and H. Q. Yu, Green Chem., 2015, 17, 4888–4907 RSC.
- S. Mukkamala, M. C. Wheeler, A. R. P. van Heiningen and W. J. DeSisto, Energy Fuels, 2012, 26, 1380–1384 CrossRef CAS.
- A. E. Imel, A. K. Naskar and M. D. Dadmun, ACS Appl. Mater. Interfaces, 2016, 8, 3200–3207 CrossRef CAS PubMed.
- R. Parthasarathi, R. A. Romero, A. Redondo and S. Gnanakaran, J. Phys. Chem. Lett., 2011, 2, 2660–2666 CrossRef CAS.
- D. A. Baker, N. C. Gallego and F. S. Baker, J. Appl. Polym. Sci., 2012, 124, 227–234 CrossRef CAS.
- S.-W. Woo, K. Dokko, H. Nakano and K. Kanamura, J. Mater. Chem., 2008, 18, 1674–1680 RSC.
- W. Zhang, M. Zhao, R. Liu, X. Wang and H. Lin, Colloids Surf., A, 2015, 484, 518–527 CrossRef CAS.
- W. L. C. -Zhang, H. B. Lin, Z. Q. Lin, J. Yin, H. Y. Lu, D. C. Liu and M. Z. Zhao, ChemSusChem, 2015, 8, 2114–2122 CrossRef CAS PubMed.
- Y. J. Oh, J. J. Yoo, Y. I. Kim, J. K. Yoon, H. N. Yoon, J.-H. Kim and S. B. Park, Electrochim. Acta, 2014, 116, 118–128 CrossRef CAS.
- N. Guo, M. Li, X. Sun, F. Wang and R. Yang, Green Chem., 2017, 19, 2595–2602 RSC.
- D. Saha, Y. C. Li, Z. H. Bi, J. H. Chen, J. K. Keum, D. K. Hensley, H. A. Grappe, H. M. Meyer, S. Dai, M. P. Paranthaman and A. K. Naskar, Langmuir, 2014, 30, 900–910 CrossRef CAS PubMed.
- W. Li, S. Zhou, Y. Xue, Y. J. Lee, R. Smith and X. Bai, ACS Sustainable Chem. Eng., 2017, 5, 8939–8950 CrossRef CAS.
- J. Zhao, W. Xiuwen, J. Hu, Q. Liu, D. Shen and R. Xiao, Polym. Degrad. Stab., 2014, 108, 133–138 CrossRef CAS.
- M. Asmadi, H. Kawamoto and S. Saka, J. Anal. Appl. Pyrolysis, 2011, 92, 417–425 CrossRef CAS.
- S. Hu, S. Zhang, N. Pan and Y.-L. Hsieh, J. Power Sources, 2014, 270, 106–112 CrossRef CAS.
- H. Li, D. Yuan, C. Tang, S. Wang, J. Sun, Z. Li, T. Tang, F. Wang, H. Gong and C. He, Carbon, 2016, 100, 151–157 CrossRef CAS.
- A. M. Navarro-Suárez, J. Carretero-González, V. Roddatis, E. Goikolea, J. Ségalini, E. Redondo, T. Rojo and R. Mysyk, RSC Adv., 2014, 4, 48336–48343 RSC.
- C. Lai, Z. Zhou, L. Zhang, X. Wang, Q. Zhou, Y. Zhao, Y. Wang, X.-F. Wu, Z. Zhu and H. Fong, J. Power Sources, 2014, 247, 134–141 CrossRef CAS.
- J. W. Jeon, L. Zhang, J. L. Lutkenhaus, D. D. Laskar, J. P. Lemmon, D. Choi, M. I. Nandasiri, A. Hashmi, J. Xu and R. K. Motkuri, ChemSusChem, 2015, 8, 428–432 CrossRef CAS PubMed.
- M. Klose, R. Reinhold, F. Logsch, F. Wolke, J. Linnemann, U. Stoeck, S. Oswald, M. Uhlemann, J. Balach and J. Markowski, ACS Sustainable Chem. Eng., 2017, 5, 4094–4102 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08539k |
| This journal is © The Royal Society of Chemistry 2018 |