DOI:
10.1039/C8RA06709K
(Paper)
RSC Adv., 2018,
8, 38733-38744
Characterization of reactive intermediates formation in dacomitinib metabolism and bioactivation pathways elucidation by LC-MS/MS: in vitro phase I metabolic investigation
Received
9th August 2018
, Accepted 11th November 2018
First published on 19th November 2018
Abstract
Dacomitinib (DCB) is a second generation irreversible tyrosine kinase inhibitor (TKI) that is claimed to overcome the disadvantages of the resistance developed by the first line epidermal growth factor receptor (EGFR) TKIs. In the current study, metabolites of phase I for DCB were systematically explored. DCB reactive metabolites were also investigated in rat liver microsomes in presence of potassium cyanide or methoxylamine that were employed as capturing agents for iminium reactive intermediates and aldehyde, respectively, to form stable complexes which can be detected by LC-MS/MS. As a result, four in vitro phase I metabolites were observed with major pathway of piperidine ring hydroxylation. Additionally, two potentially reactive intermediates, one aldehyde and one iminium ions were characterized. Two different pathways of bioactivation were ultimately proposed.
1. Introduction
Lung carcinoma is the principal cause of death among all cancer types. Non-small cell lung cancer (NSCLC) represents the most widespread (around 90%) of all lung cancer in patients.1–5 During the past few years, the epidermal growth factor receptor (EGFR) signaling pathway as a therapeutic target in NSCLC has gained more recognition.6 Inhibitors of the tyrosine kinase (TKIs) that affect the epidermal growth factor receptor (EGFR) are very effective against most EGFR mutations with a good therapeutic window. First line EGFR TKIs (e.g. erlotinib and gefitinib) had a very good response against these active mutations at the beginning.7,8 Unfortunately, acquired resistance (around 60%) and toxicities that occurred in treatment9,10 reduce their therapeutic efficacies.11,12
DCB is a second generation irreversible EGFR TKIs that overcomes the disadvantages of the acquired resistance first line EGFR TKIs.13–15 When compared with gefitinib, DCB improved progression-free survival in the management of patients with positive EGFR mutation NSCLC. DCB is regarded as a new way of treatment for this medical case.16 The U.S. Food and Drug Administration (FDA) accepted Pfizer's application for DCB as new drug and granted priority review for the first-line management of patients with locally advanced or metastatic NSCLC with EGFR-activating mutations. On September 27th 2018 FDA approved DCB tablets (VIZIMPRO™), for the first-line treatment of individuals suffering from metastatic NSCLC with EGFR exon 19 deletion or exon 21 L858R substitution mutations.17 A marketing authorization application for DCB was also accepted by the EMA (European Medicines Agency) for the same indication18
The most common severe (grade 3) adverse reactions of DCB were acne (in 14% of patients) and diarrhea (in 8% of patients). In spite of its increased adverse reactions particularly in the skin and gastrointestinal tract, the potent activity of DCB allowed for consideration of this effective therapy in EGFR-positive NSCLC.19–21 Proteins in the body can be modified by covalent binding to reactive metabolites that is regarded as the primary step in organ toxicities.22,23 In most cases, such reactive intermediates are produced through reactions of phase I metabolism. The reactive intermediates in drug metabolism can cause many side effects. As a result of their transient nature, a trapping agent is always used to trap reactive intermediates to facilitate the formation of stable adducts. The formed adducts can be isolated from the incubation mixture, detected and characterized by LC-MS/MS.24,25
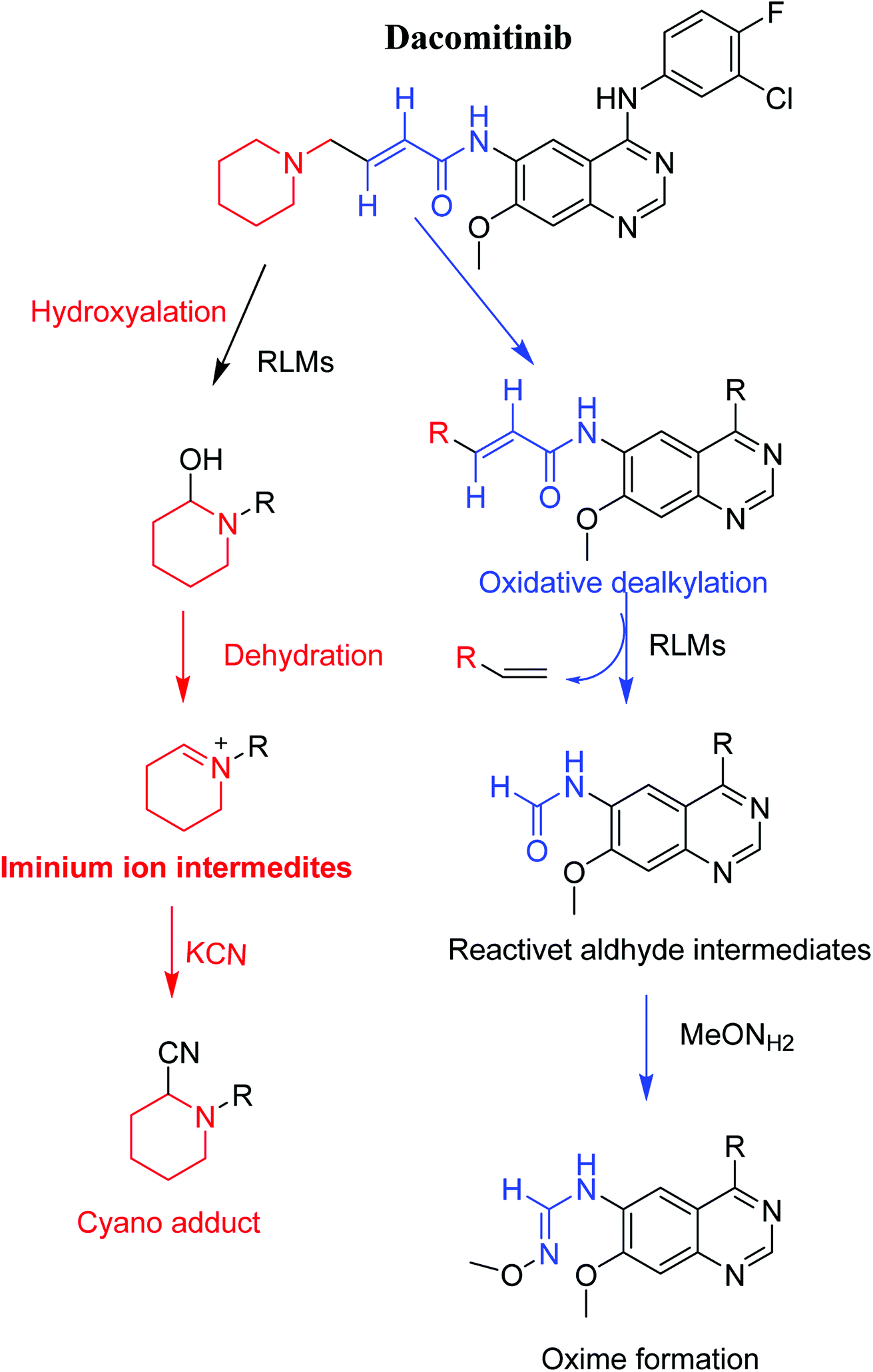
Chemically the structure of DCB encompasses a piperidine ring and a butenamide group. Drugs contains piperidine ring are subjected to bioactivation that forms iminium ions intermediates which may be stabilized by KCN forming cyano adducts. Butenamide group undergoes bioactivation through oxidative dealkylation developing an unstable reactive aldehyde that can be stabilized by reaction with methoxylamine forming oxime.26–28 These stabilized adducts may be extracted, separated and identified using LC-MS/MS.24–26,29,30
Upon literature review, one article was found that focused primarily on the pharmacokinetics of dacomitinib in human.31 Such study was aimed at characterizing the principal elimination routes of dacomitinib in humans. The study did not involve complete metabolic profiling with structural elucidation of dacomitinib metabolites. In contrast, the main target of the present work is reactive metabolites screening to provide a possible reason for toxicity of DCB via in vitro metabolic studies. Reactive metabolites cannot be determined in vivo because as soon as they are formed, they will bind to endogenous materials as DNA or proteins that cannot be detected by mass spectrometry. The basic outcome of this work is identification and characterization of two possible reactive metabolites: one aldehyde and one iminium ion with description of structure details. Bioactivation pathways were also proposed. Four in vitro phase I dacomitinib metabolites were also identified. The presence of such reactive intermediates in DCB metabolism may be the reason of its side effects as they are regarded the first step in drug-induced organ toxicities.32
2. Chemicals and methods
2.1. Chemicals
HPLC grade solvents were utilized throughout the entire study. Dacomitinib reference standard was acquired from Med Chem. Express (Princeton, NJ, USA). Acetonitrile (ACN), ammonium formate (NH4COOH), potassium cyanide (KCN), methoxyl amine (MeONH2) and formic acid (HCOOH) were procured from Sigma-Aldrich (USA). Water (ultrapure) was taken from in-house Milli-Q plus purification system (Millipore, Midford, MA USA). Rat liver microsomes (RLMs) were acquired in-house using Sprague Dawley rats.33–36 The used rats were attained from the experimental animal care center at college of Pharmacy, King Saud University (KSA). The University's Ethics Review Committee approved the animal experimental design.
2.2. Chromatography
The optimized LC-MS/MS chromatographic parameters for the separation and elucidation of incubation mixture extract components are listed in Table 1.
Table 1 Adjusted parameters of the proposed LC-MS/MS methodology
| LC parameters |
MS/MS parameters |
| HPLC |
Agilent 1200 |
Mass spectrometer |
Agilent 6410 QqQ |
| Gradient mobile phase |
A: 10 mM NH4COOH in H2O |
Ionization source |
Positive ESI |
| B: ACN |
Drying gas: N2 gas |
| Flow rate: 0.4 mL min−1 |
Flow rate (12 L min−1) |
| Run time: 75 min |
Pressure (60 psi) |
| Agilent eclipse plus C18 column |
Length |
150 mm |
ESI temperature: 350 °C |
| Internal diameter |
2.1 mm |
Capillary voltage: 4000 V |
| Particle size |
3.5 µm |
Collision gas |
High purity N2 |
| Temperature: |
22 ± 1 °C |
Modes |
Mass scan and product ion (PI) |
| Gradient system |
Time |
% B |
Analyte |
DCB and its reactive metabolites |
| 0 |
5 |
| 5 |
5 |
Mass parameters |
Fragmentor voltage: 140 V |
| 40 |
60 |
| 70 |
90 |
| 75 |
5 |
Collision energy of 20 eV |
2.3. RLMs incubations
Metabolic reactions were done by incubation 30 µM of DCB with 1.0 mg mL−1 RLMs in addition to 50 mM Na/K phosphate buffer (pH 7.4) that contains 3.3 mM MgCl2. Incubation was done at 37 °C for 2 h in a temperature-controlled rocking water bath. The addition of 1.0 mM NADPH started the metabolic reactions while the addition of 2 mL ice-cold ACN was utilized to quench those reaction.
2.4. Extraction and purification method of DCB RLMs incubations
Protein precipitation method by ACN was performed to extract and purify DCB, its in vitro phase I metabolites and its adduct from RLMs incubation mixtures. Removal of proteins was done by centrifugation of metabolic mixtures at 9000g for 15 min at 4 °C. Supernatants were transferred to clean tubes. Evaporation of the supernatants was done under nitrogen stream. The mobile phase was used to reconstitute the residues and 1 mL from each was put in HPLC vial. Fifteen µL were injected into LC system.36–38
2.5. Characterization of DCB bioactive intermediates
The same metabolic incubation of DCB with RLMs was repeated in the presence of 2.5 mM methoxyl amine and 1.0 mM KCN to seize the aldehyde intermediates and reactive iminium intermediates, respectively. Each experiment was repeated three times to verify the outcomes. Three controls were prepared to confirm the results. Control 1 (C1) contains all incubation ingredients including DCB and RLMs except NADH and nucleophiles to confirm in vitro phase I metabolites. C2 contains all incubation ingredients including RLMs except NADH and DCB to confirm the absence of any interference from indigenous materials. C3 contains all incubation ingredients including NADH and DCB and nucleophiles (KCN or MeONH2) except RLMs to further proof that the reactive metabolites formation occurred only through metabolic reaction.
2.6. Identification of DCB reactive metabolites
Full mass scan and extracted ion chromatograms were utilized to annotate metabolites in the incubation mixtures, while product ion (PI) was employed to identify DCB in vitro metabolites, adducts and reactive intermediates formed in DCB metabolism. Locating metabolites in metabolic mixture extract chromatogram was performed by EIC of m/z of the proposed DCB metabolites.
3. Results and discussion
3.1. PI study of DCB
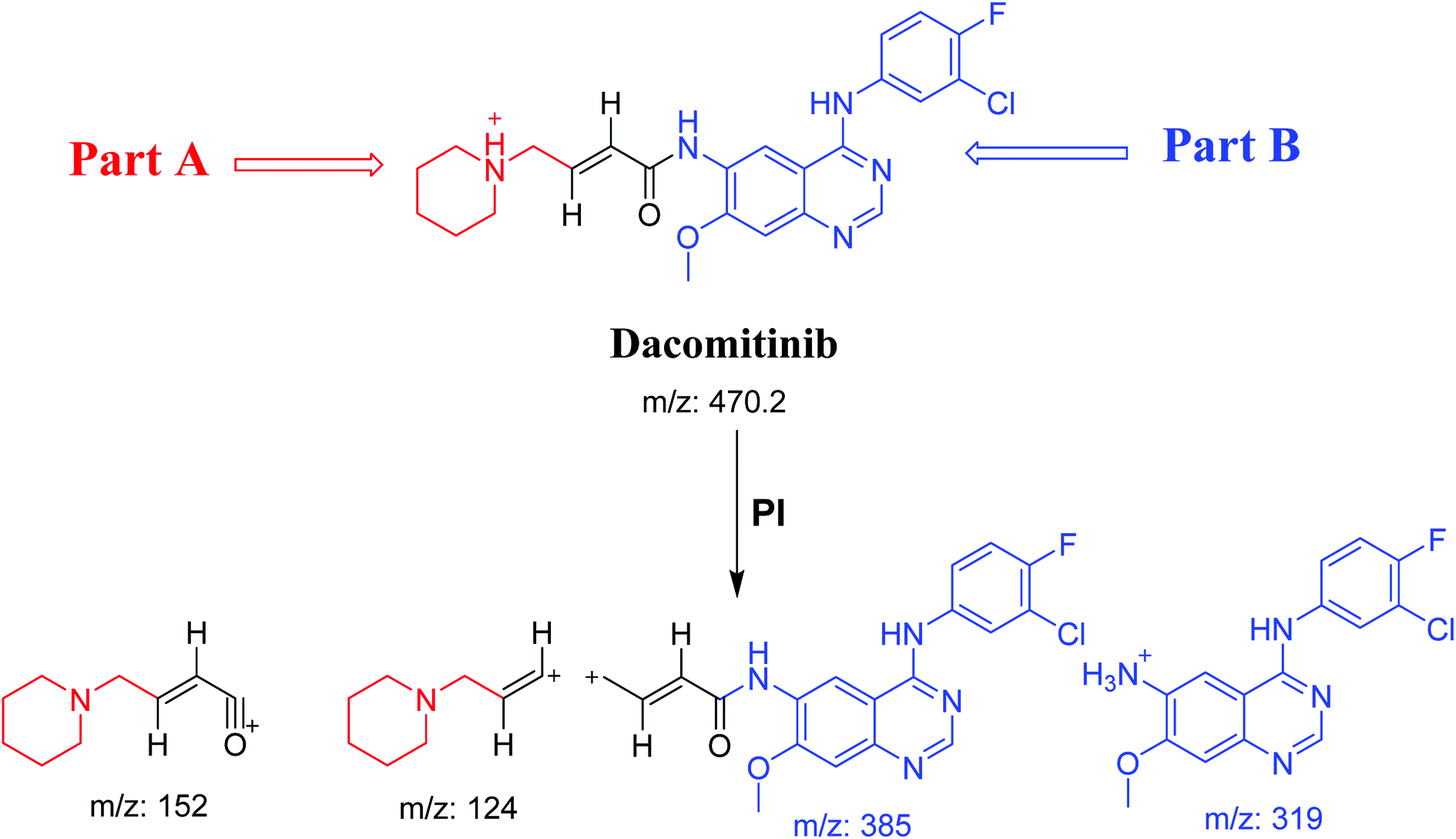
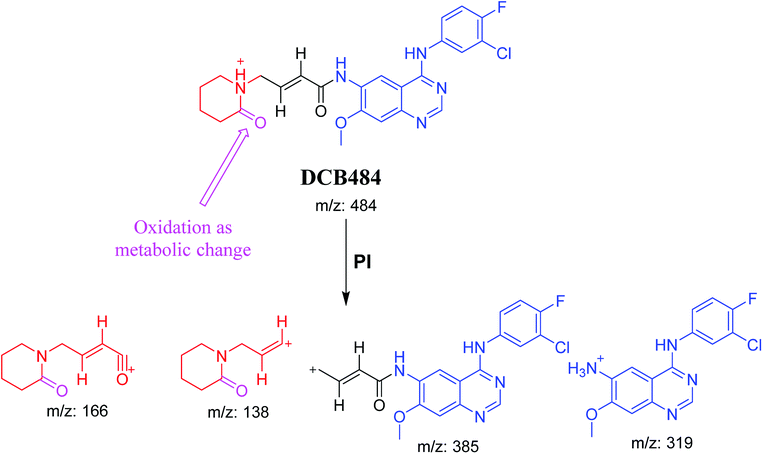
DCB chromatographic peak appears at 33.5 min in PI chromatogram (Fig. 1A). Collision induced dissociation (CID) of molecular ion at m/z 470 generates four characteristic fragment ions at m/z 385, m/z 319, m/z 152 and m/z 124 (Fig. 1B). Fragment ions at m/z 152 and m/z 124 were used as indicator for any metabolic change in part A of DCB chemical structure while fragment ions at m/z 385 and m/z 319 were used as indicator for any metabolic change in part B of DCB chemical structure (Scheme 1).
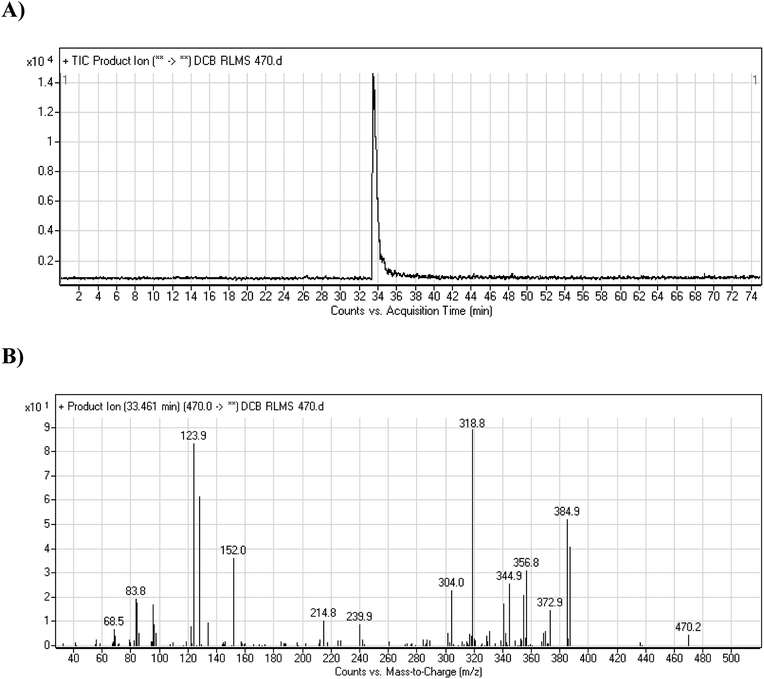 |
| | Fig. 1 PI chromatogram of ion at m/z 470 showing DCB peak at 33.5 min (A), PIs of DCB (B). | |
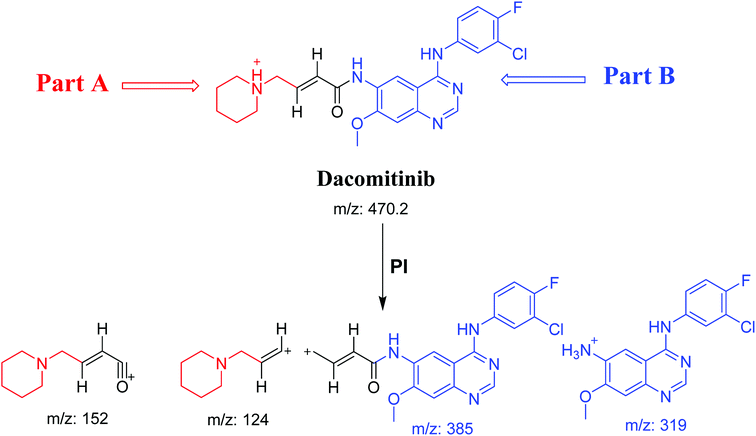 |
| | Scheme 1 PIs of DCB. | |
3.2. Identification of in vitro DCB metabolites
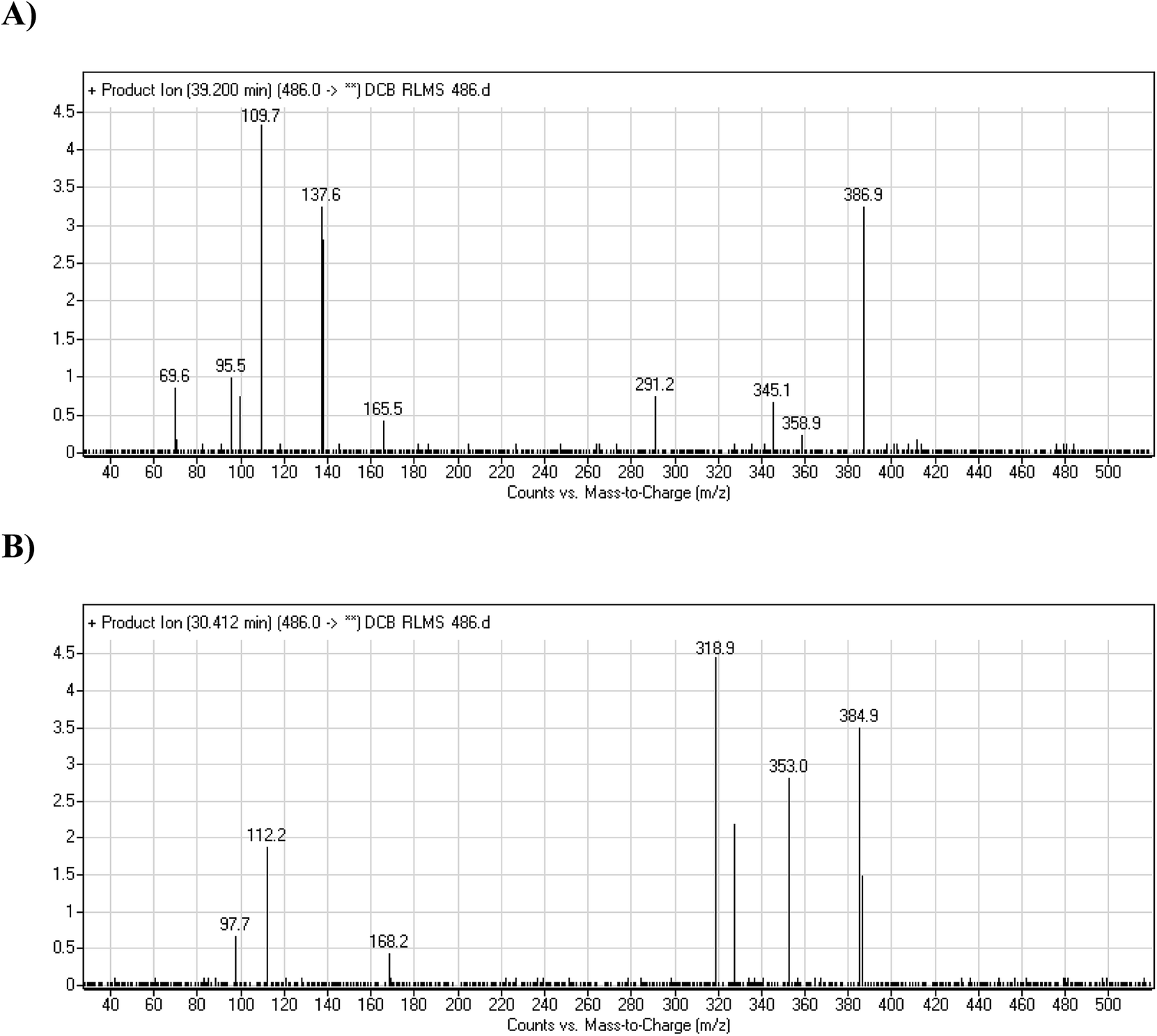
After extraction and purification of RLMs incubations, fifteen µL were injected into LC-MS/MS instrument. Major metabolic phase I pathway for DCB was piperidine ring hydroxylation. One methoxyl amine conjugate and one cyano adduct were characterized after incubation with RLMs with 2.5 mM methoxylamine and 1.0 mM KCN, respectively (Table 2).
Table 2 In vitro phase I and reactive metabolites of DCB
| |
MS scan |
Most abundant fragment ions |
Rt. (min) |
Metabolic reaction |
| DCB |
470 |
385, 319, 152, 124 |
33.5 |
Parent drug |
| DCB484 |
484 |
385, 319, 166, 144 |
39.5 |
α Oxidation at piperidine ring |
| DCB472 |
472 |
387, 321, 152, 124 |
32.2 |
Reduction at quinazoline ring |
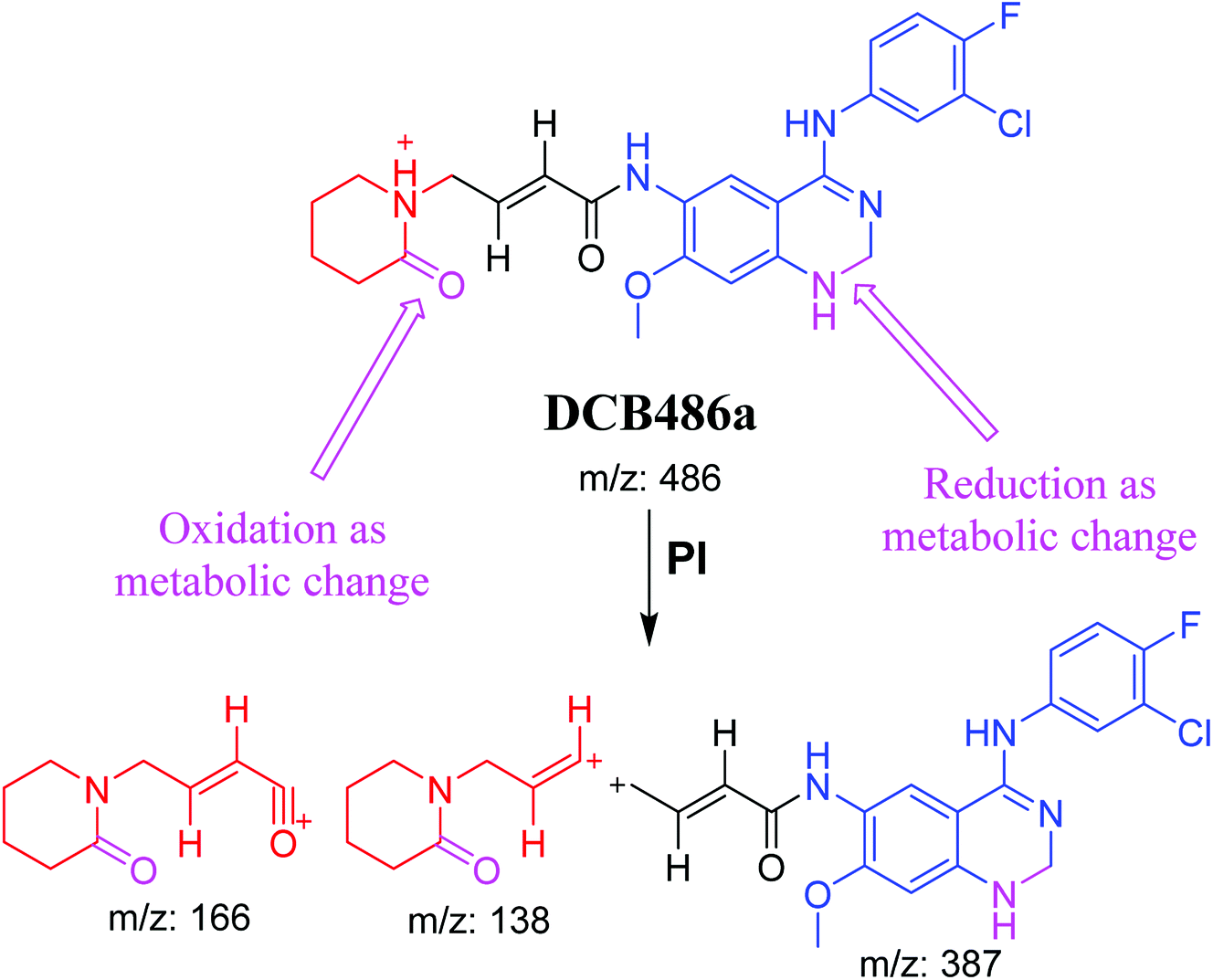
| DCB486a |
486 |
387, 166, 138 |
39.2 |
α Oxidation at piperidine ring and reduction at quinazoline ring |
| DCB486b |
486 |
385, 319, 168 |
30.4 |
Hydroxylation at piperidine ring |
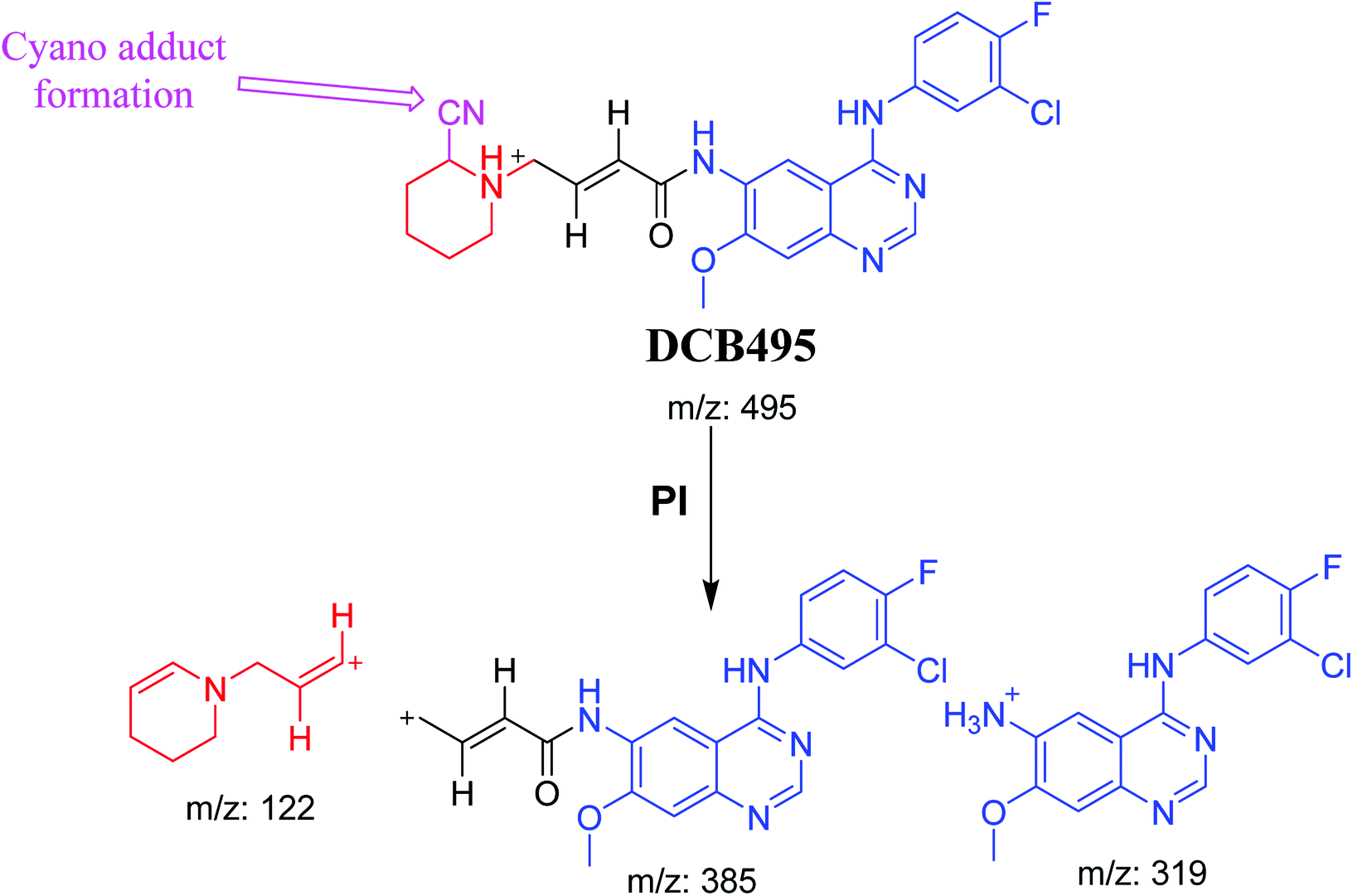
| DCB495 |
495 |
385, 319, 122 |
52.5 |
Cyano adduct |
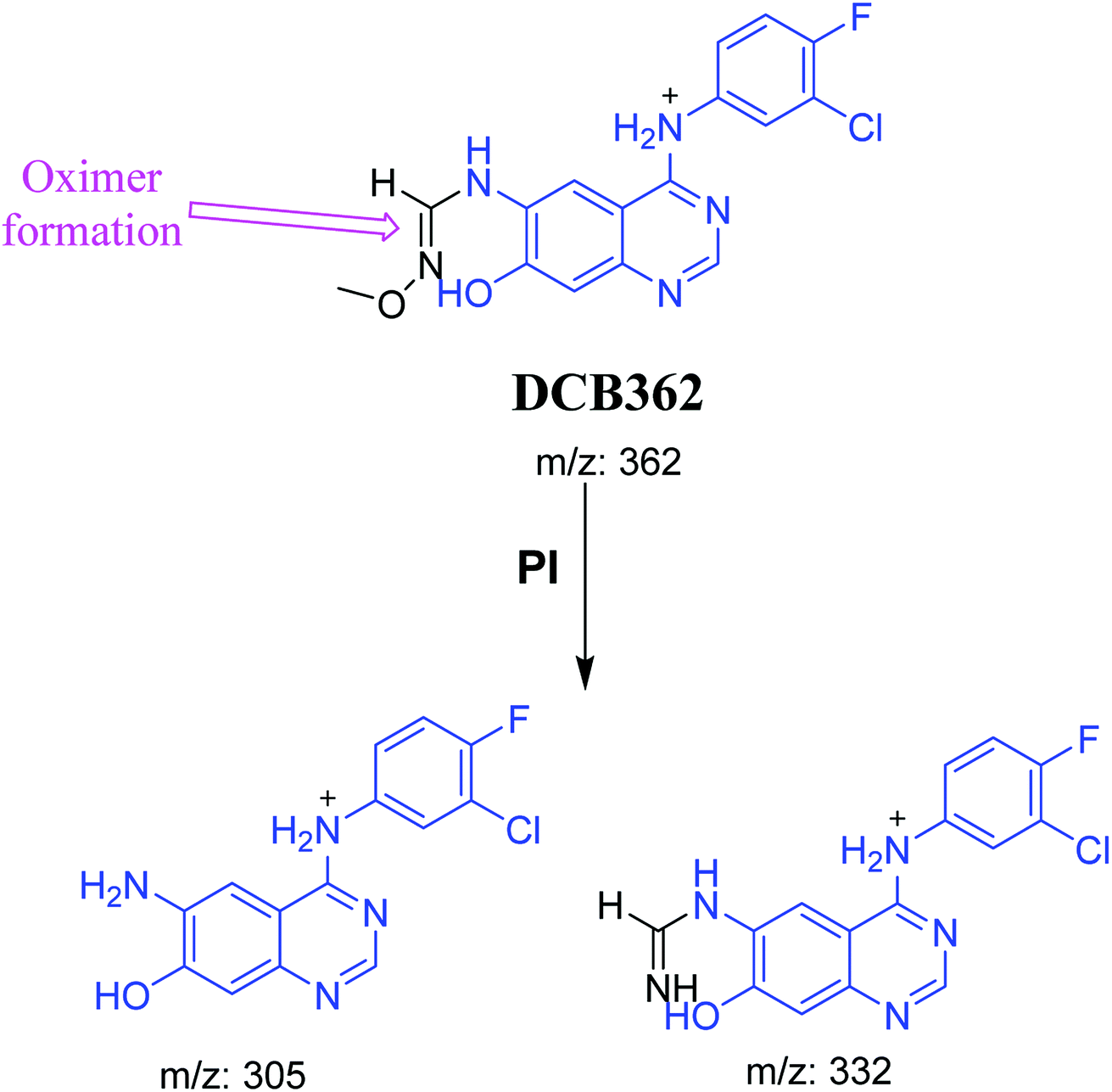
| DCB362 |
362 |
332, 305 |
38.7 |
Methoxyl amine conjugate (oxime) |
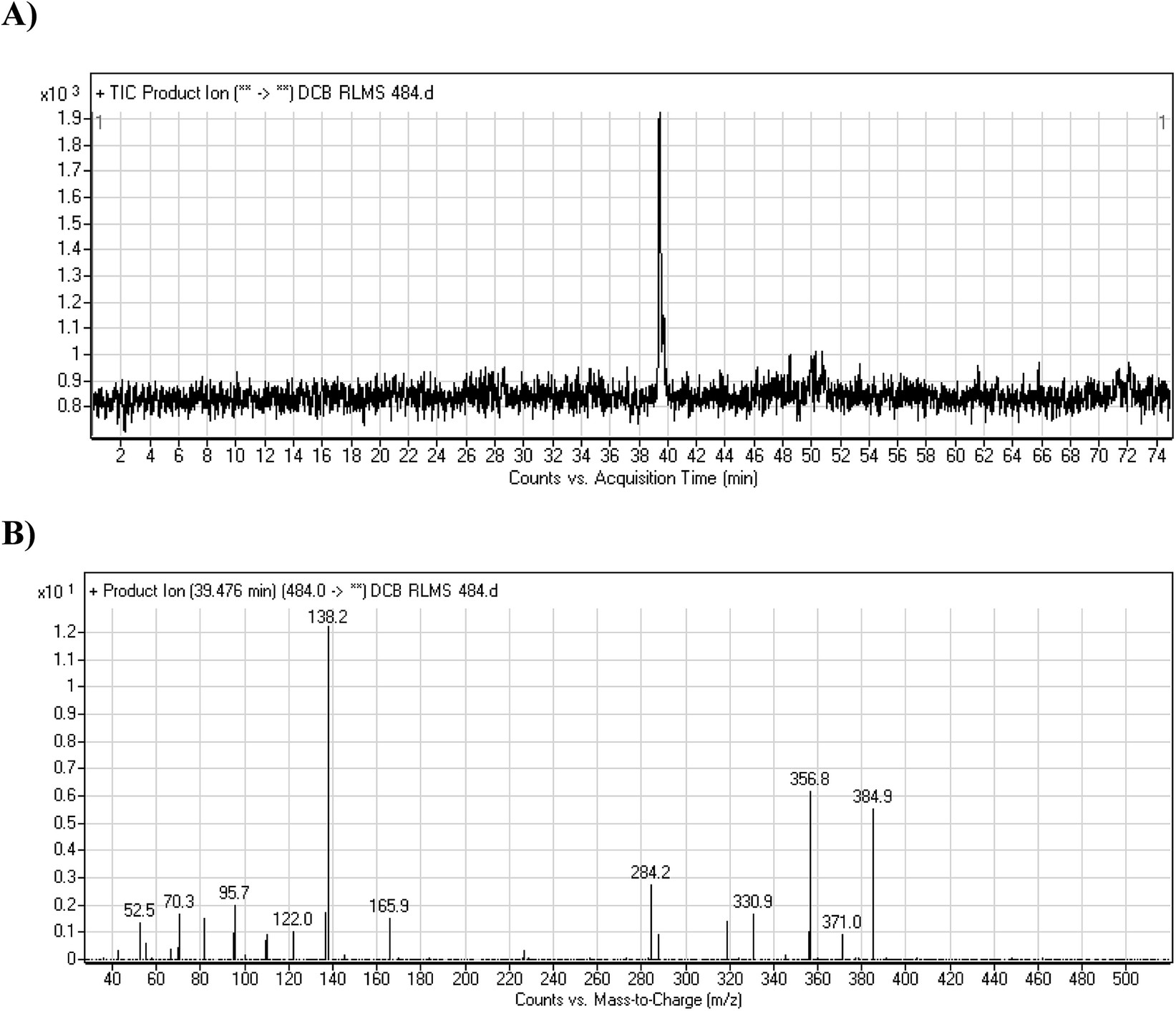
3.2.1. DCB484 phase I metabolite. DCB484 chromatographic peak appears at 39.5 min in PI chromatogram (Fig. 2A). CID of molecular ion at m/z 484 generates four characteristic fragment ions at m/z 385, m/z 319, m/z 166 and m/z 138 (Fig. 2B). In contrast to DCB fragmentation, fragment ions at m/z 166 and m/z 138 indicated that metabolic alteration took place in part A of DCB chemical structure while fragment ions at m/z 385 and m/z 319 indicated that no metabolic change occurred in part B of DCB chemical structure. α-Oxidation metabolic reaction was hypothesized to take place in piperidine ring (Scheme 2).
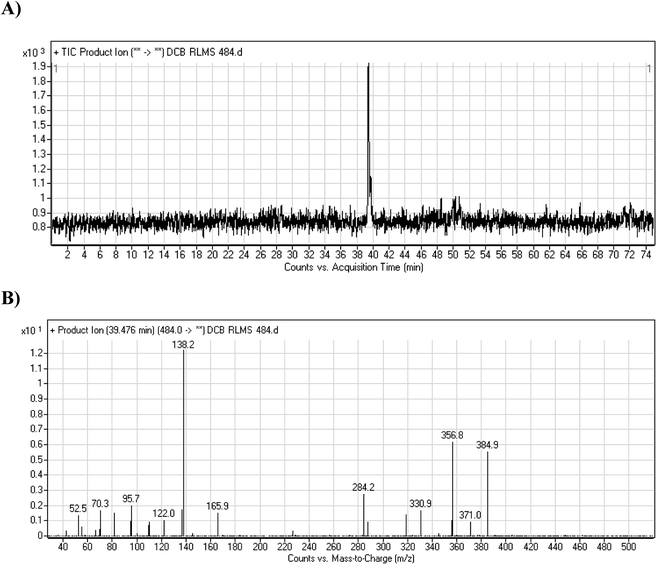 |
| | Fig. 2 PI chromatogram of ion at m/z 484 showing DCB484 peak at 39.5 min (A), PIs of DCB484 (B). | |
 |
| | Scheme 2 PIs of DCB484. | |
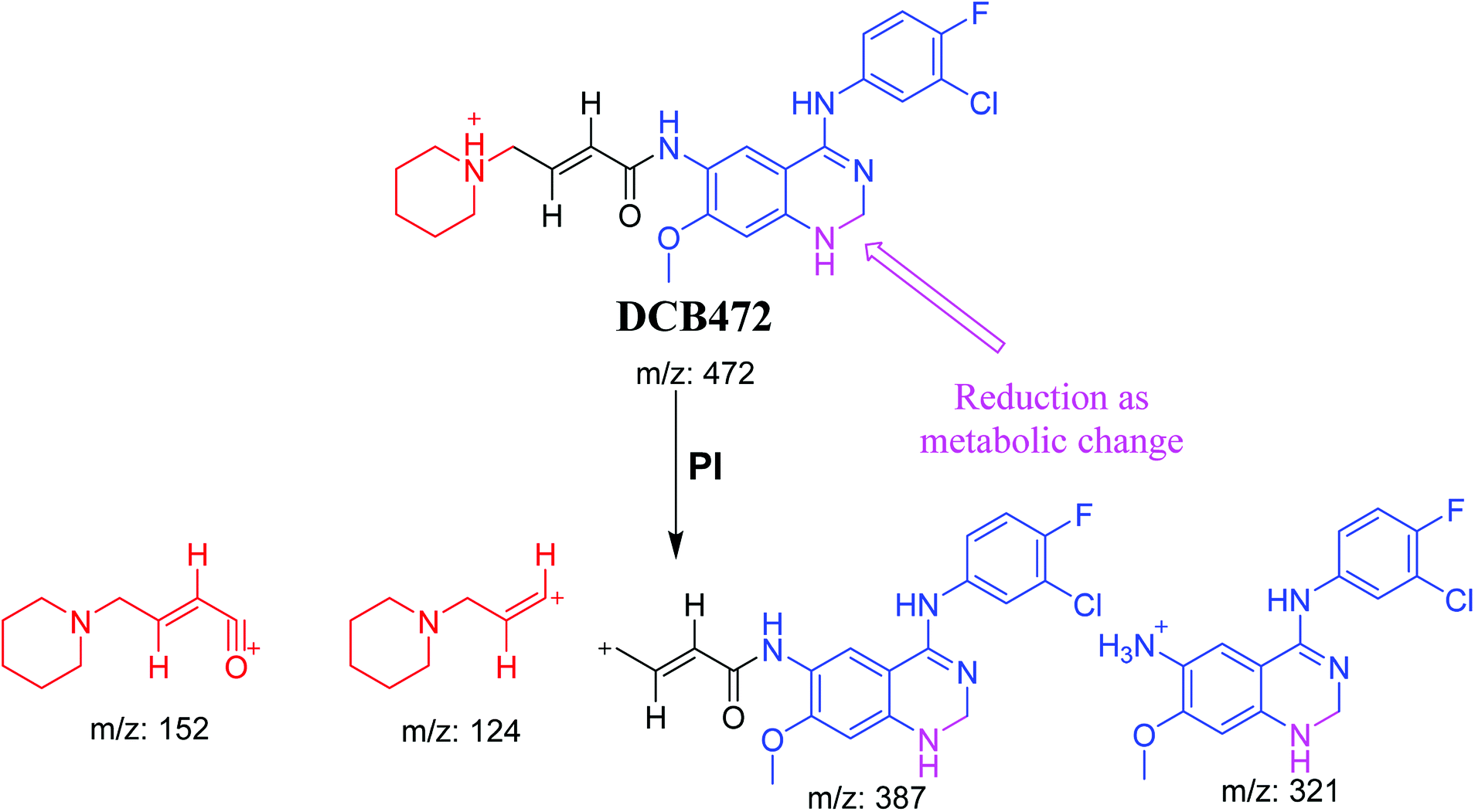
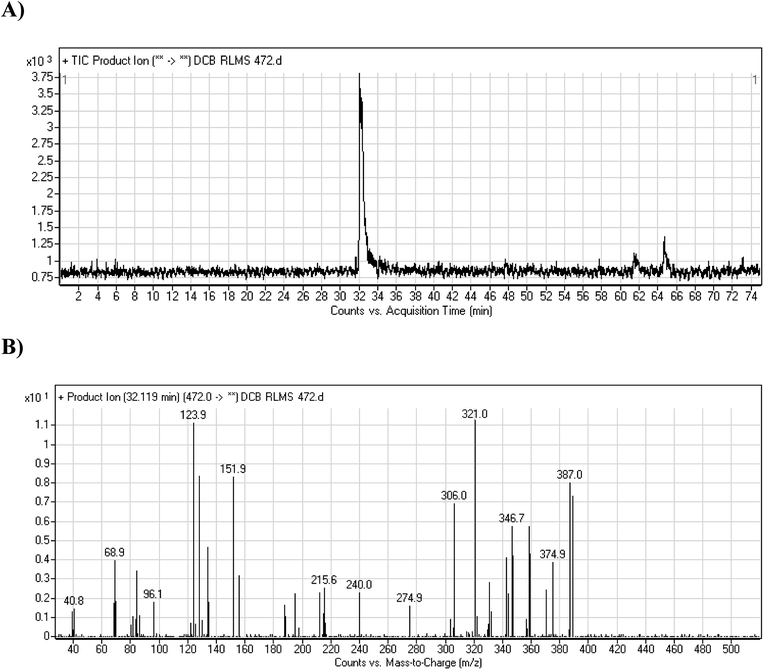
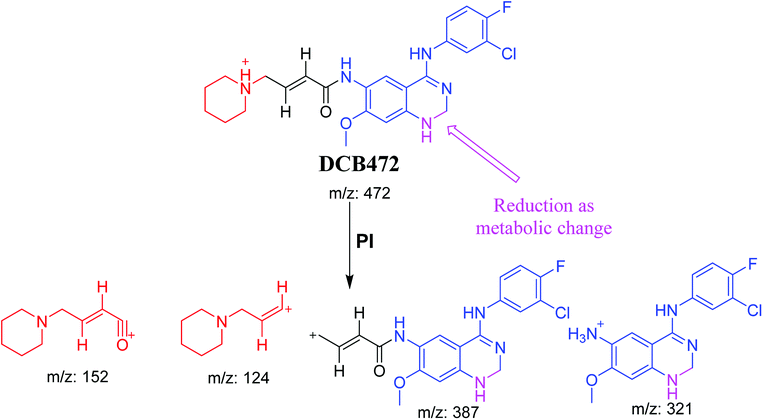
3.2.2. DCB472 phase I metabolite. DCB472 chromatographic peak appears at 32.1 min in PI chromatogram (Fig. 3A). CID of molecular ion at m/z 472 generates four characteristic fragment ions at m/z 387, m/z 321, m/z 152 and m/z 124 (Fig. 3B). In contrast to DCB fragmentation, fragment ions at m/z 152 and m/z 124 indicated that no metabolic modification took place in part A of DCB chemical structure while fragment ions at m/z 387 and m/z 321 indicated that there was a metabolic change in part B of the DCB chemical structure. Reduction metabolic reaction was suggested to take place in quinazoline ring (Scheme 3).
 |
| | Fig. 3 PI chromatogram of ion at m/z 472 showing DCB472 peak at 32.1 min (A), PIs of DCB472 (B). | |
 |
| | Scheme 3 PIs of DCB472. | |
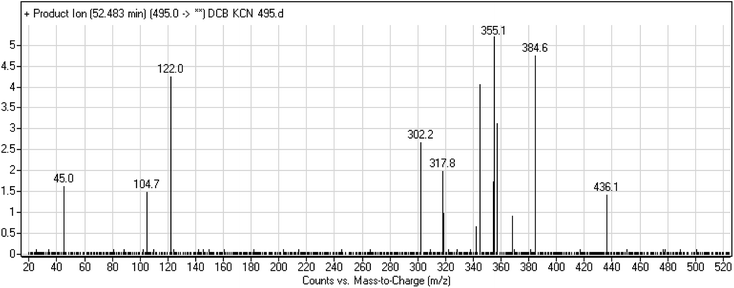
3.2.4. DCB495 cyano adduct. DCB495 chromatographic peak appears at 52.5 min in PI chromatogram. CID of molecular ion at m/z 495 generates three characteristic fragment ions at m/z 385, m/z 319 and m/z 122 (Fig. 5). In contrast to DCB fragmentation, fragment ions at m/z 385 and m/z 319 indicated that no metabolic effect on part A of the DCB chemical structure while fragment ions at m/z 122 indicated that a metabolic modification took place in part B of DCB chemical structure that signified a loss of HCN. In bioactivation of similar piperidine containing drugs,39,40 piperidine ring carbons was hypothesized to undergo bioactivation followed by attack by cyanide ion as explained in Scheme 6.
 |
| | Fig. 5 PI mass spectrum of DCB495. | |
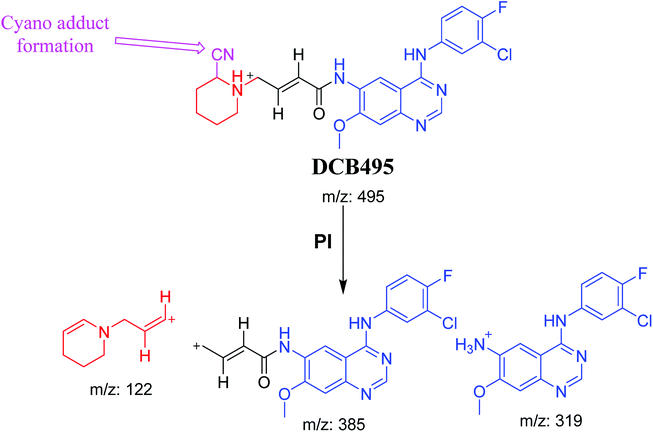 |
| | Scheme 6 PIs of DCB495. | |
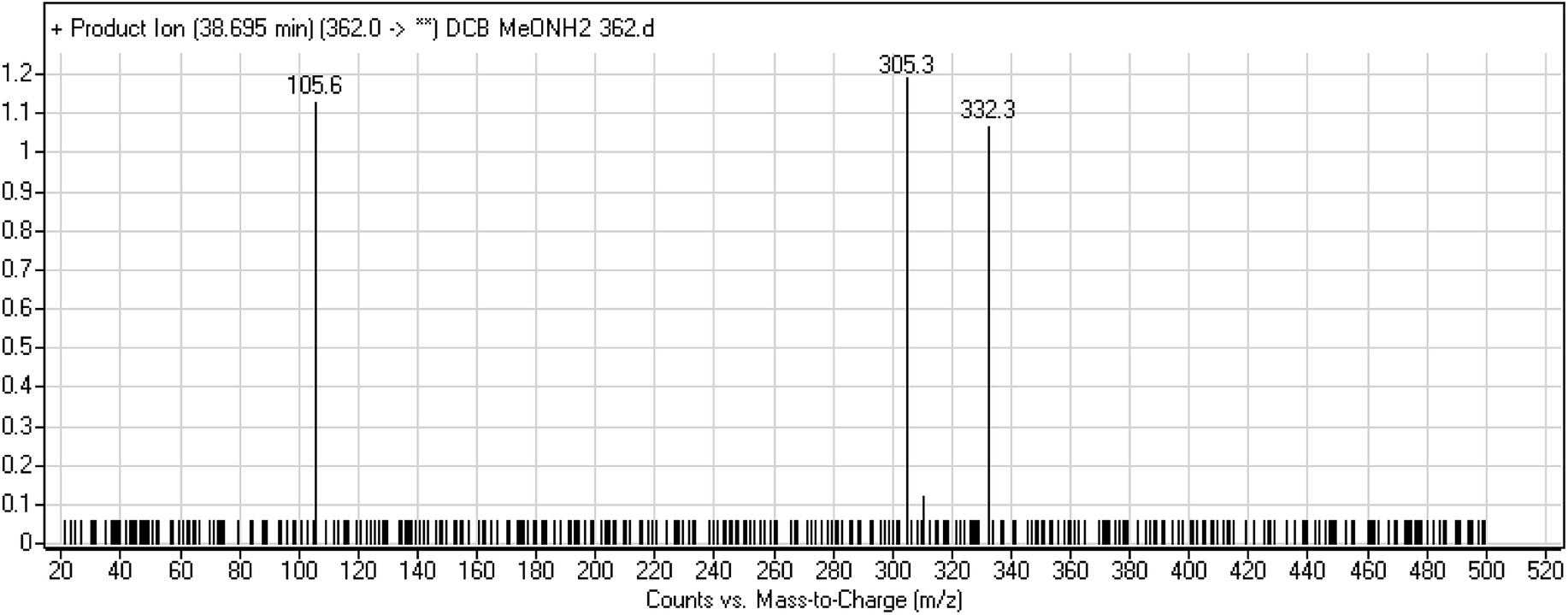
3.2.5. DCB362 oxime adduct of avitinib. DCB362 chromatographic peak appears at 38.69 min in PI chromatogram. CID of the molecular ion at m/z 362 generates three characteristic fragment ions at m/z 332, m/z 305 and m/z 106 (Fig. 6). In comparison to DCB fragmentation, fragment ions at m/z 332 and m/z 305 indicated that no metabolic change occurred in part B of DCB chemical structure contains butenamide group undergoes bioactivation through oxidative dealkylation to form unstable reactive aldehyde that can be stabilized by reaction with methoxylamine forming oxime. The formation of DCB362 oxime indicated that aldehyde intermediates were formed in the metabolism of DCB (Scheme 7).
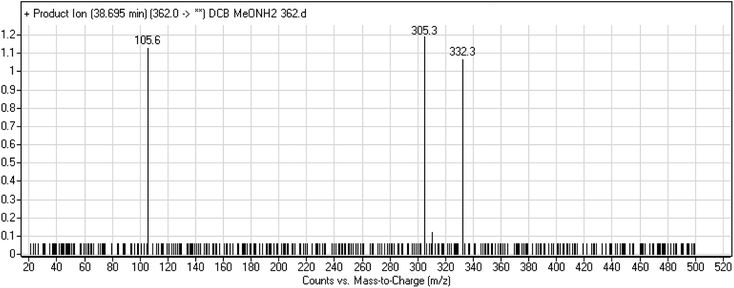 |
| | Fig. 6 PI mass spectrum of DCB362. | |
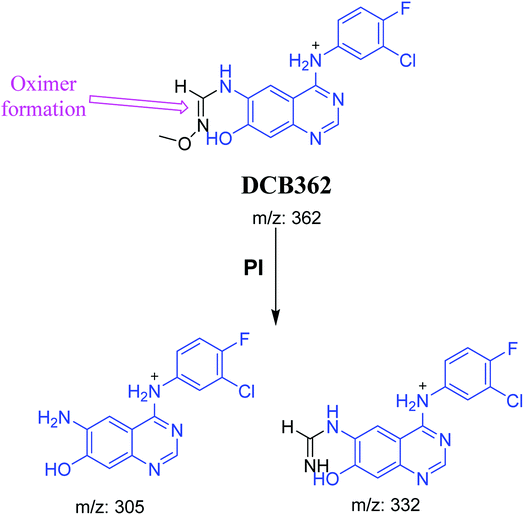 |
| | Scheme 7 PIs of DCB362. | |
3.3. Proposed pathways of bioactivation of DCB
Scheme 8 showed the different pathways for bioactivation of DCB. DCB495 cyanide adduct confirmed iminium intermediate formation in piperidine ring metabolism. Hydroxylation of piperidine ring in DCB followed by dehydration led to generation of unstable and reactive iminium ions intermediates that may be captured by cyanide to form stable adduct which can be detected in LC-MS/MS. The formation pathway of iminium intermediate and bioactivation mechanism of DCB was described earlier with similar drugs encompassing cyclic tertiary amine.36,37 The aldehyde (DCB362) was formed by oxidative dealkylation of butenamide group and captured by methoxylamine to form a stable oxime containing compound (Scheme 8).
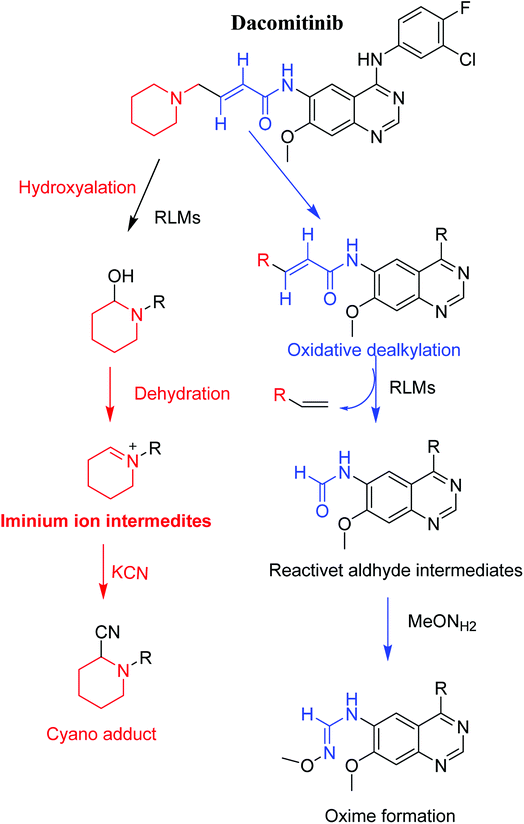 |
| | Scheme 8 Proposed pathways for DCB bioactivation. | |
4. Conclusions
Four DCB metabolites were isolated and detected by LC-MS/MS in vitro resulting from phase I metabolic reactions. Two possible reactive metabolites: one aldehyde and one iminium ion were detected and the bio-activation pathways were proposed (Fig. 7). These reactive intermediates in DCB metabolism may be the reason of its side effects as they are considered the primary step in drug-induced organ toxicities. The results obtained in the present study paves the way for the development of new drugs with more safety profile.
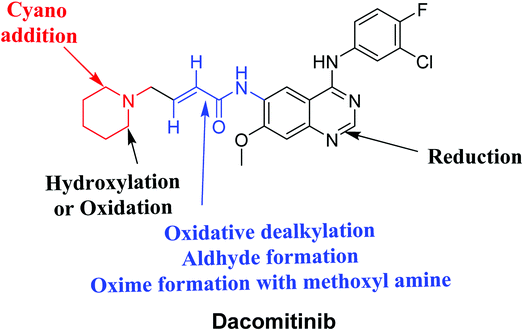 |
| | Fig. 7 Chemical structure of DCB showing bioactivation pathways including iminium and aldehyde formation. | |
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
“The authors would like to extend their sincere appreciation to the Deanship of Scientific Research at the King Saud University for funding this work through the Research Group Project No. RG-1435-025.”
References
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2016, 66, 7–30 CrossRef PubMed.
- S. M. Haghgoo, A. Allameh, E. Mortaz, J. Garssen, G. Folkerts, P. J. Barnes and I. M. Adcock, Eur. J. Pharmacol., 2015, 754, 82–91 CrossRef CAS PubMed.
- I. Abubakar, T. Tillmann and A. Banerjee, Lancet, 2015, 385, 117–171 CrossRef.
- D. S. Ettinger, W. Akerley, G. Bepler, M. G. Blum, A. Chang, R. T. Cheney, L. R. Chirieac, T. A. D'Amico, T. L. Demmy and A. K. P. Ganti, J. Natl. Compr. Cancer Network, 2010, 8, 740–801 CrossRef CAS.
- J. E. Larsen, T. Cascone, D. E. Gerber, J. V. Heymach and J. D. Minna, Cancer J., 2011, 17, 512 CrossRef CAS PubMed.
- D. B. Costa and S. S. Kobayashi, Transl. Lung Cancer Res., 2015, 4, 809 CAS.
- C. Gridelli, A. Rossi, D. P. Carbone, J. Guarize, N. Karachaliou, T. Mok, F. Petrella, L. Spaggiari and R. Rosell, Nat. Rev. Dis. Primers, 2015, 1, 15009 CrossRef PubMed.
- S. Peters, S. Zimmermann and A. A. Adjei, Cancer Treat. Rev., 2014, 40, 917–926 CrossRef CAS PubMed.
- G. Metro and L. Crinò, Transl. Lung Cancer Res., 2012, 1, 5 Search PubMed.
- M. G. Denis, A. Vallée and S. Théoleyre, Clin. Chim. Acta, 2015, 444, 81–85 CrossRef CAS PubMed.
- S. Jorge, S. Kobayashi and D. Costa, Braz. J. Med. Biol. Res., 2014, 47, 929–939 CrossRef CAS PubMed.
- M. R. Finlay, M. Anderton, S. Ashton, P. Ballard, P. A. Bethel, M. R. Box, R. H. Bradbury, S. J. Brown, S. Butterworth, A. Campbell, C. Chorley, N. Colclough, D. A. Cross, G. S. Currie, M. Grist, L. Hassall, G. B. Hill, D. James, M. James, P. Kemmitt, T. Klinowska, G. Lamont, S. G. Lamont, N. Martin, H. L. McFarland, M. J. Mellor, J. P. Orme, D. Perkins, P. Perkins, G. Richmond, P. Smith, R. A. Ward, M. J. Waring, D. Whittaker, S. Wells and G. L. Wrigley, J. Med Chem., 2014, 57, 8249–8267 CrossRef CAS PubMed.
- C.-S. Tan, D. Gilligan and S. Pacey, Lancet Oncol., 2015, 16, e447–e459 CrossRef CAS PubMed.
- B.-C. Liao, C.-C. Lin and J. C.-H. Yang, Curr. Opin. Oncol., 2015, 27, 94–101 CrossRef CAS PubMed.
- P. A. Jänne, J. C.-H. Yang, D.-W. Kim, D. Planchard, Y. Ohe, S. S. Ramalingam, M.-J. Ahn, S.-W. Kim, W.-C. Su and L. Horn, N. Engl. J. Med., 2015, 372, 1689–1699 CrossRef PubMed.
- Y. L. Wu, Y. Cheng, X. Zhou, K. H. Lee, K. Nakagawa, S. Niho, F. Tsuji, R. Linke, R. Rosell, J. Corral, M. R. Migliorino, A. Pluzanski, E. I. Sbar, T. Wang, J. L. White, S. Nadanaciva, R. Sandin and T. S. Mok, Lancet Oncol., 2017, 18, 1454–1466 CrossRef CAS PubMed.
- U.S. Food and Drug Administration (FDA), Vizimpro® (dacomitinib) tablets, 27th September 2018.
- F. Barrón, Z. L. Zatarain-Barrón, A. F. Cardona and O. Arrieta, J. Thorac. Dis., 2018, 10, 1265 CrossRef PubMed.
- C. Brzezniak, C. A. Carter and G. Giaccone, Expert Opin. Pharmacother., 2013, 14, 247–253 CrossRef CAS PubMed.
- K. Sanderson, Nat. Rev. Drug Discovery, 2013, 12, 649–651 CrossRef CAS PubMed.
- K. L. Reckamp, G. Giaccone, D. R. Camidge, S. M. Gadgeel, F. R. Khuri, J. A. Engelman, M. Koczywas, A. Rajan, A. K. Campbell and D. Gernhardt, Cancer, 2014, 120, 1145–1154 CrossRef CAS PubMed.
- S. R. Knowles, J. Uetrecht and N. H. Shear, Lancet, 2000, 356, 1587–1591 CrossRef CAS.
- C. Ju and J. Uetrecht, Curr. Drug Metab., 2002, 3, 367–377 CrossRef CAS PubMed.
- S. Ma and M. Zhu, Chem.-Biol. Interact., 2009, 179, 25–37 CrossRef CAS PubMed.
- A. F. Stepan, D. P. Walker, J. Bauman, D. A. Price, T. A. Baillie, A. S. Kalgutkar and M. D. Aleo, Chem. Res. Toxicol., 2011, 24, 1345–1410 Search PubMed.
- L. P. Masic, Curr. Drug Metab., 2011, 12, 35–50 Search PubMed.
- Z. Zhang, Q. Chen, Y. Li, G. A. Doss, B. J. Dean, J. S. Ngui, M. Silva Elipe, S. Kim, J. Y. Wu, F. Dininno, M. L. Hammond, R. A. Stearns, D. C. Evans, T. A. Baillie and W. Tang, Chem. Res. Toxicol., 2005, 18, 675–685 Search PubMed.
- B. K. Park, A. Boobis, S. Clarke, C. E. Goldring, D. Jones, J. G. Kenna, C. Lambert, H. G. Laverty, D. J. Naisbitt and S. Nelson, Nat. Rev. Drug Discovery, 2011, 10, 292–306 CrossRef CAS PubMed.
- S. Ma and R. Subramanian, J. Mass Spectrom., 2006, 41, 1121–1139 CrossRef CAS PubMed.
- A. Tolonen, M. Turpeinen and O. Pelkonen, Drug discovery today, 2009, 14, 120–133 CrossRef CAS PubMed.
- C. L. Bello, E. Smith, A. Ruiz-Garcia, G. Ni, C. Alvey and C.-M. Loi, Cancer Chemother. Pharmacol., 2013, 72, 379–385 CrossRef CAS PubMed.
- X. Xu, L. Mao, W. Xu, W. Tang, X. Zhang, B. Xi, R. Xu, X. Fang, J. Liu and C. Fang, Molecular cancer therapeutics, 2016 Search PubMed.
- R. von Jagow, H. Kampffmeyer and M. Kiese, Naunyn-Schmiedebergs Arch. Exp. Pathol. Pharmakol., 1965, 251, 73–87 CrossRef CAS PubMed.
- A. A. Kadi, M. Attwa and H. W. Darwish, RSC Adv., 2018, 8, 1182–1190 RSC.
- S. M. Amer, A. A. Kadi, H. W. Darwish and M. W. Attwa, RSC Adv., 2017, 7 CAS.
- A. A. Kadi, H. W. Darwish, M. W. Attwa and S. M. Amer, RSC Adv., 2016, 6, 72575–72585 RSC.
- S. M. Amer, A. A. Kadi, H. W. Darwish and M. W. Attwa, RSC Adv., 2017, 7, 4479–4491 RSC.
- A. A. Kadi, A. S. Abdelhameed, H. W. Darwish, M. W. Attwa and N. S. Al‐Shakliah, Biomed. Chromatogr., 2016, 30, 1248–1255 CrossRef CAS PubMed.
- L. P. Masic, Curr. Drug Metab., 2011, 12, 35–50 CrossRef CAS PubMed.
- N. Castagnoli Jr, J. M. Rimoldi, J. Bloomquist and K. P. Castagnoli, Chem. Res. Toxicol., 1997, 10, 924–940 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Adnan A. Kadi and
Ali S. Abdelhameed
*,
Adnan A. Kadi and
Ali S. Abdelhameed