
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA convenient one-pot synthesis of N-substituted amidoximes and their application toward 1,2,4-oxadiazol-5-ones†
Wong Phakhodee *ab,
Chuthamat Duangkamola,
Nitaya Wiriyaa and
Mookda Pattarawarapanab
*ab,
Chuthamat Duangkamola,
Nitaya Wiriyaa and
Mookda Pattarawarapanab
aDepartment of Chemistry, Faculty of Science, Chiang Mai University, Chiang Mai 50200, Thailand. E-mail: wongp2577@gmail.com
bResearch Center on Chemistry for Development of Health Promoting Products from Northern Resources, Chiang Mai University, Chiang Mai, 50200, Thailand
First published on 14th November 2018
Abstract
The first direct one-pot approach for the synthesis of N-substituted amidoximes from secondary amides or the intermediate amides has been developed. Through the Ph3P–I2-mediated dehydrative condensation, a variety of N-aryl and N-alkyl amidoximes (R1(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NOH)NHR2, where R1 or R2 = aryl, alkyl, or benzyl) were readily afforded under mild conditions and short reaction times. The synthetic application of the obtained amidoximes has also been demonstrated through the formation of 1,2,4-oxadiazolones via base-mediated carbonylative cyclization with 1,1′-carbonyldiimidazole.
NOH)NHR2, where R1 or R2 = aryl, alkyl, or benzyl) were readily afforded under mild conditions and short reaction times. The synthetic application of the obtained amidoximes has also been demonstrated through the formation of 1,2,4-oxadiazolones via base-mediated carbonylative cyclization with 1,1′-carbonyldiimidazole.
Introduction
N-Substituted amidoximes are privileged structures that have been used extensively as versatile building blocks in the synthesis of various heterocycles such as benzimidazoles,1 4-aminoquinazolines,2 1-aminoisoquinolines,3 oxadiazoles,4 oxadiazolones (thiones),5 and triazoles.1c They also serve as the key intermediates in the synthesis of amidines as well as metal ion chelating ligands in coordination chemistry.6 In drug development, various N-substituted amidoxime derivatives have been introduced as prodrug candidates to achieve good cell permeability and oral bioavailability.6a The representative examples are shown in Fig. 1. Epacadostat (A) is currently being studied in a phase 3 clinical trial in patients with unresectable or metastatic melanoma.7 B is a novel inhibitor of indoleamine 2,3-dioxygenase.8 C and D are potent inhibitors of Escherichia coli RNA polymerase.9 Oseltamivir prodrug E has been developed as an anti-influenza agent with favourable pharmacokinetics.10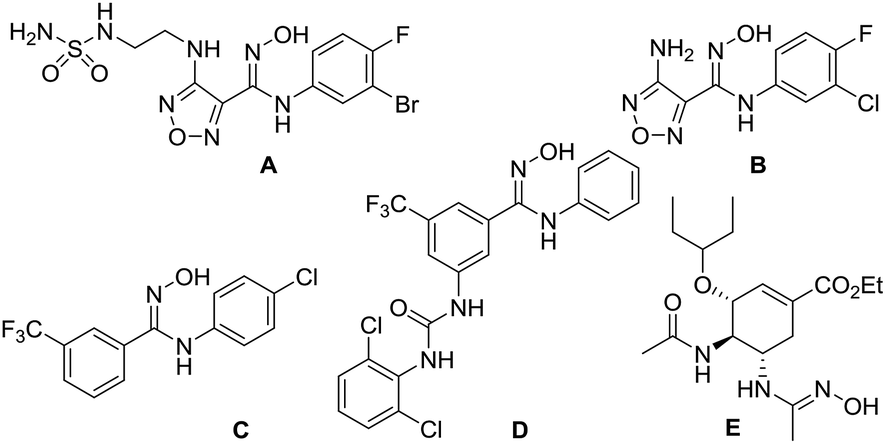 | ||
| Fig. 1 Examples of bioactive N-substituted amidoximes. | ||
Despite their essential applications, only a limited number of synthetic routes for N-substituted amidoximes has been reported in the literature. Whereas the synthesis of unsubstituted amidoximes starting from benzonitriles is well-established,6a the synthesis of N-substituted amidoximes is much less straightforward requiring laborious multistep procedures.
One of the most commonly used methods involves the reaction of amines with N-hydroxyimidoyl chloride (I).11 However, I is not readily available and has to be prepared in two steps starting from condensation of aldehydes with hydroxylamine hydrochloride, followed by chlorination of the formed oximes with N-chlorosuccinimide12 or chlorine gas.5c Moreover, the protocol is ineffective for preparing aliphatic imidoyl chlorides as the starting aldehydes are less reactive. Aliphatic imidoyl chlorides are also highly unstable and are often obtained in low yields.12,13
Alternatively, N-substituted amidoximes could be synthesized via the addition of hydroxylamine or its analogs to the activated amide derivatives such as imidoyl chloride (II) derived from dehydrative chlorination of secondary amide with PCl5 or P2O5,14 thioamide (III) from treatment of the starting amides with Lawesson's reagent (p-methoxyphenylthionophosphine sulphide dimer),15 or imidoylbenzotriazole (IV) from the reaction of an amide with oxalyl chloride and benzotriazole.16 Other methods include hydrolysis of 1,2,4-oxadiazolones (V),17 or the reaction of primary nitroalkanes (VI) with amines18 or lithium amides.19 However, these approaches suffer from various limitations such as the use of highly toxic reagents, multistep synthesis with tedious work-up and purification, harsh reaction conditions, long reaction times, low yields, and limit substrate scope. Therefore, the development of general and practical one-pot approaches to access a variety of amidoximes derivatives from readily available precursors is still desirable.
In the recent years, iodine-mediated organic synthesis has attracted considerable attention.20 This could be attributed to its inexpensiveness, green nature, and high efficiency in promoting a range of reactions. In particular, the combination of iodine with triphenylphosphine (Ph3P) provides a highly effective dehydrating agent leading to rapid and high yielding synthesis under mild reaction conditions.21 In a continuation of our interest in developing facile one-pot methods using the Ph3P–I2 combination,22 we have designed a one-pot approach for the synthesis of N-substituted amidoximes which enables the use of inexpensive and commercially available secondary amides, acid chlorides or carboxylic acids as the key precursors (Scheme 1). Herein, we wish to report our detailed study in this aspect.
 | ||
| Scheme 1 Synthetic approaches toward N-substituted amidoximes. | ||
Results and discussion
We started our investigations by optimizing the reaction of N-phenylbenzamide (1a) with hydroxylamine hydrochloride. Typically, the reaction was carried out by addition of N-phenylbenzamide (1 equiv.) into a mixture of Ph3P (1.5 equiv.) and halogenated additive (1.5 equiv.) in freshly dry dichloromethane, followed by addition of base (5 equiv.) and hydroxylamine hydrochloride (1.5 equiv.).As shown in Table 1, among the tested organic bases, only triethylamine gave rise to the formation of the desired product 2a in high yield (entries 1–6). The reaction in the presence of the commonly used imidazole gave the product in low yield (entry 4). Using diisopropylethylamine (DIPEA) although led to increase conversion to the product (entry 5), the reaction was incomplete even after 24 h. According to entries 7–9, the reaction did not proceed when replacing iodine with other halogenated additives such as N-chlorosuccinimide (NCS), N-bromosuccinimide (NBS), or carbon tetrabromide (CBr4). In addition, no conversion was observed when the reaction was carried out in the absence of Ph3P (entry 10) indicating that phosphonium iodide or triphenylphosphoranediiodide are acting as the key activating species. It should be noted also that the reaction did not proceed when using oxalyl chloride or thionyl chloride as the dehydrating agent.
|
|||
|---|---|---|---|
| Entry | Reagent | Base | Yield (%) |
| a Reaction conditions: N-phenylbenzamide (0.28 mmol), hydroxylamine hydrochloride (0.42 mmol), Ph3P (0.42 mmol), additive (0.42 mmol), base (1.4 mmol), CH2Cl2 (2 mL), 0 °C-RT, 2 h. nr = no reaction. | |||
| 1 | I2/Ph3P | DABCO | nr |
| 2 | I2/Ph3P | DBU | Trace |
| 3 | I2/Ph3P | NMM | 10 |
| 4 | I2/Ph3P | Imidazole | 20 |
| 5 | I2/Ph3P | DIPEA | 45 |
| 6 | I2/Ph3P | Et3N | 85 |
| 7 | NCS/Ph3P | Et3N | nr |
| 8 | NBS/Ph3P | Et3N | nr |
| 9 | CBr4/Ph3P | Et3N | nr |
| 10 | I2 | Et3N | nr |

With the optimized reaction conditions in hand, the scope and generality of the reaction were studied. For this purpose, the reactions of diverse N-substituted aromatic and aliphatic amides were investigated, and the results are shown in Scheme 2. N-Aryl substituted secondary amides bearing either an electron-donating group (EDG), such as methyl or methoxy, or an electron-withdrawing group (EWG), such as chloro, bromo, or nitro on the N-phenyl ring underwent smooth conversion to give 2a–2e in high yields. However, the electronic effect of the 4-nitro group in the aromatic ring of R1 of amide 1f seems to lower the yield of 2f. The condition is applicable to substrate bearing heterocyclic ring although slightly lower yield of the product 2g was obtained due to the difficulty in the product isolation.
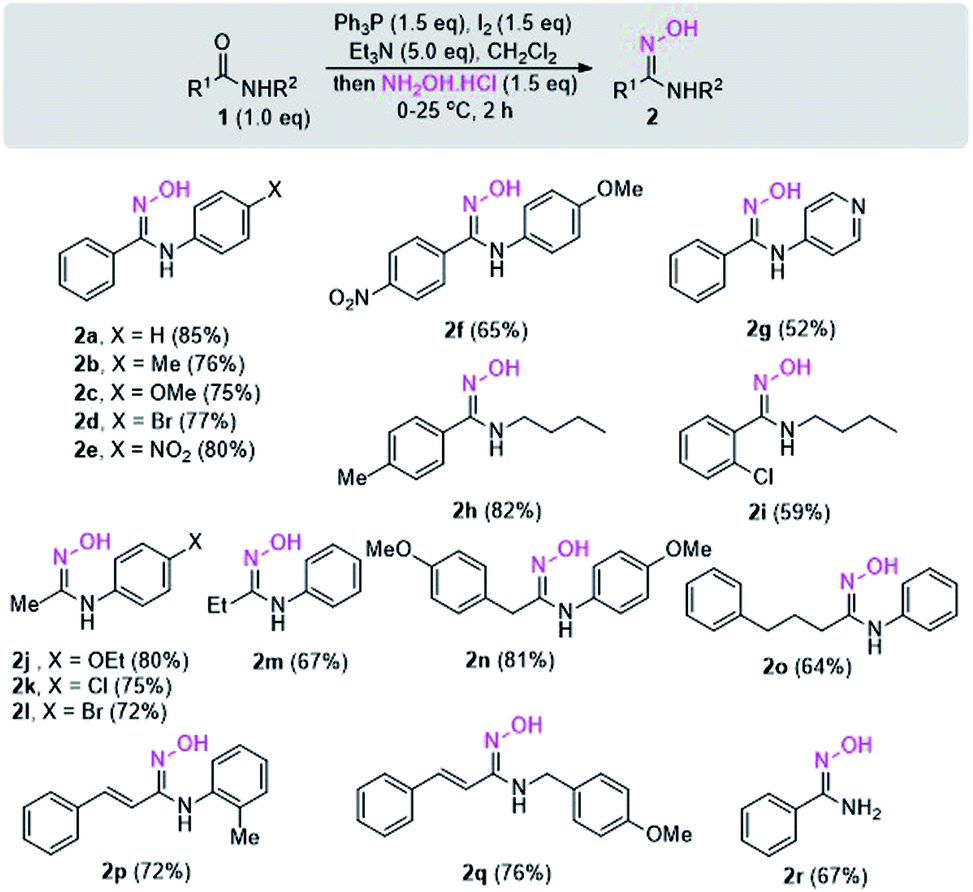 | ||
| Scheme 2 Synthesis of N-substituted amidoximes 2 starting from amides. | ||
N-Alkyl substituted amides were also converted into the corresponding amidoximes 2h–2i in satisfactory yields. Unlike other reported procedures which are not suitable for preparation of amidoximes with R1 = alkyl,12,13 our conditions enable the synthesis of various acetimidamide derivatives such as 2j–2l from N-arylacetamide substrates without any difficulty. Other secondary amides having R1 = ethyl, benzyl, or even long alkyl chain also provided the products 2m–2o in moderate to high yields. Additionally, the reaction condition is applicable with amides bearing α,β-unsaturated moiety. The exclusive formation of amidoximes 2p and 2q indicated no complication due to Michael addition to the α,β-unsaturated system. The presence of steric ortho-substituted methyl group did not affect the reaction (see compound 2p). It should be noted that primary amide is also a viable substrate as illustrated in the synthesis of compound 2r. However, the reactions with tertiary amides such as N,N-dimethylbenzamide or N-benzoylpiperidine failed to give the amidoxime products (data not shown).
With the success in the formation of N-substituted amidoximes from amide substrates, we thus further investigated a direct one-pot reaction toward amidoximes using acid chlorides or carboxylic acids as the amide precursors. According to Scheme 3, condensation of amines with acid chlorides led to an in situ formation of amides which could then be activated with Ph3P–I2 before treatment with hydroxylamine hydrochloride. The reaction using benzoyl chloride gave rise to various amidoximes having N-aryl or N-alkyl substituent in moderate to good yields.
 | ||
| Scheme 3 Synthesis of N-substituted amidoximes through in situ formation of amides. | ||
Due to the remarkable difference in the reactivity of carboxylic acid and amide, it was envisioned that a direct one-pot synthesis of amidoximes from carboxylic acids via intermediate amides would be possible through a sequential addition of an amine, then hydroxylamine hydrochloride to a carboxylic acid in the presence of an excessive amount of dehydrating agent.
To our delight, the procedure enables the synthesis of various N-substituted amidoximes in moderate to good yields (Scheme 3). No complication from the formation of amidine side-product was observed. This one-pot three-component coupling led to an improve in the efficiency of the reaction avoiding laborious separation and purification of the amide intermediate.
Based on the obtained results and our previous experiences in the Ph3P–I2-mediated synthesis, the mechanism for the formation of N-substituted amidoximes was proposed as shown in Scheme 4. The combination of Ph3P with I2 provides triphenylphosphoranediiodide I and triphenylphosphonium iodide II. Upon additon of an amide and base, phosphorylation at the oxygen atom of the amide then leads to the formation of imidinium intermediate III. This species could be converted into another reactive intermediate, imidoyl iodide IV. Displacement of III or IV with hydroxylamine hydrochloride then gives rise to N-substituted amidoxime.
 | ||
| Scheme 4 Proposed mechanism for the Ph3P–I2 mediated synthesis of N-substituted amidoxime. | ||
To obtain scientific evidences regarding the mechanism of the process, 31P{1H} NMR spectroscopy was used to monitor the progress of the reaction of N-phenylbenzamide. As shown in Scheme 4 and Fig. S1 (ESI†), addition of Ph3P to the deuterated chloroform solution of I2 resulted in an appearance of a resonance peak at −19.8 ppm corresponded to phosphoranediiodide species I.23 No significant change in the 31P{1H} NMR spectrum was observed after adding N-phenylbenzamide except that a small signal appeared at 42.8 ppm. This signal could be attributed to the presence of phosphonium salt II. Upon adding Et3N, a signal of Ph3PO rapidly appeared at 29.16 ppm as a major peak along with some other minor phosphorus species. Unfortunately, the signal arises from phosphonium salt intermediate III could not be observed. This data suggests a rapid conversion of III to IV leading to a release of Ph3PO at this stage. Addition of hydroxylamine hydrochloride finally led to a complete disappearance of the species I, while the signal of Ph3PO appeared in a greater amount.
To our delight, when monitoring the reaction between benzoyl chloride and aniline, a signal at 65.38 ppm which could be attributed to the amide phosphonium salt III was detected upon adding Ph3P and I2 to the preformed N-phenylbenzamide (Fig. S1, ESI†). This data is consistent with the observed chemical shift for the nucleoside phosphonium salt (δ = 66.2 ppm) in the synthesis of O6-(benzotriazol-1-yl)inosine nucleosides promoted by the same reagent combination.24
To further demonstrate the synthetic application of the developed methodology, the conversion of 2 into 1,2,4-oxadiazol-5(4H)-one 3 was investigated. These structures are known to be valuable heterocycles as amidine precursors25 or as bioisosteric replacement for the carboxylic acid, amide or ester.26 Additionally, compounds bearing oxadiazolone pharmacophore have been shown to possess a range of pharmacological and biological activities.14b,27 The reported methods for the preparation of 1,2,4-oxadiazolones from N-substituted amidoximes often require multiple steps, harsh reaction conditions, and long reaction times.5,14b,28
Through a screening for the best reaction conditions, it was found that, by treatment of amidoximes 2 with 1,1′-carbonyldiimidazole (CDI) in the presence of finely ground potassium carbonate, carbonylative cyclization proceeded rapidly to afford products 3 in good to excellent yields (Scheme 5). The reaction took place within 10–20 min at room temperature rather than several hours under the reported refluxing conditions.14b The method is also applicable with a range of amidoxime substrates containing N-aryl, N-alkyl, and N-benzyl groups.
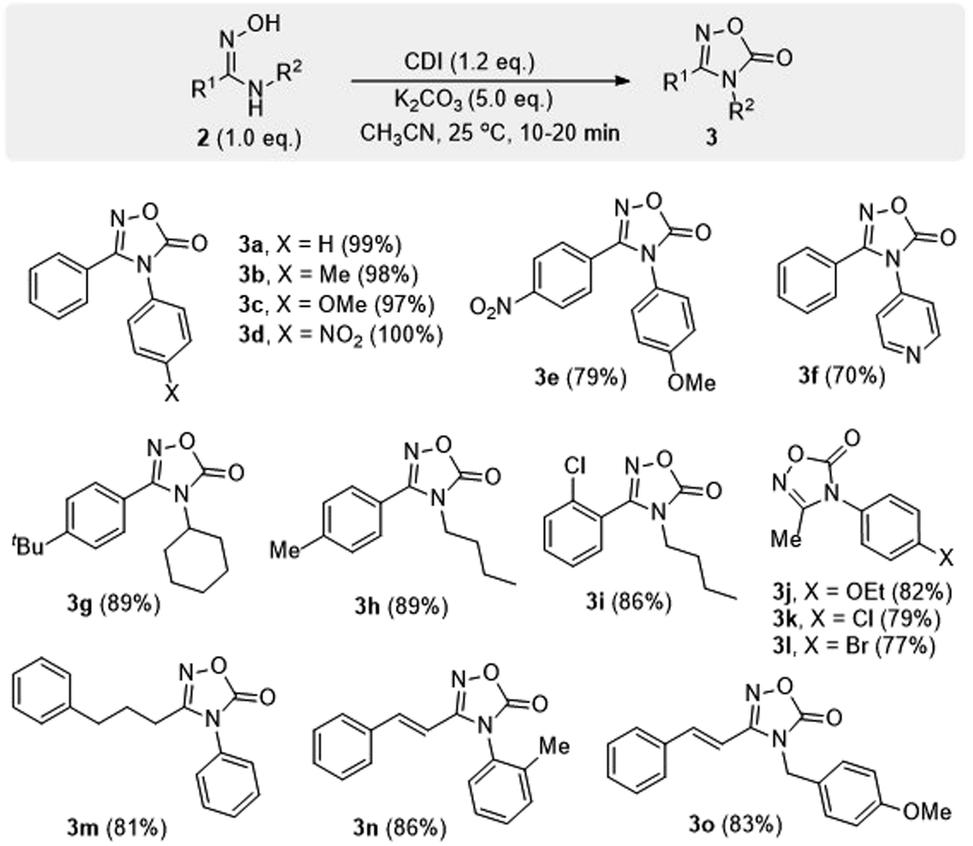 | ||
| Scheme 5 Synthesis of 1,2,4-oxadiazol-5(4H)-ones 3. | ||
Conclusions
In summary, we have developed a convenient one-pot approach for the preparation of N-substituted amidoximes from amides, or the requisite acid chlorides, or carboxylic acids. The protocol represents a fast, efficient, and economic alternative to the earlier reported multistep methods. The mild nature of the procedure enables a range of starting materials accessible. One application of amidoximes as building blocks in the synthesis of 1,2,4-oxadiazol-5(4H)-ones via base-mediated carbonylative cyclization has been demonstrated. Further utility of these amidoximes in organic synthesis of other important pharmacophores is anticipated.Experimental
General information
All reagents were purchased from Sigma-Aldrich Co., USA, and used without further purification. Dichloromethane was freshly distilled over CaH2 before use. The reaction was monitored by thin-layer chromatography carried out on silica gel plates (60F254, MERCK, Germany) and visualized under UV light (254 nm). Melting points were determined using Mettler Toledo DSC equipment at a heating rate of 6 °C min−1 and are uncorrected. NMR spectra were determined using a Bruker AVANCE™ (400 MHz for 1H). Chemical shifts were reported in parts per million (ppm, δ) downfield from TMS. High resolution mass spectra (HRMS) were recorded using the LC-DAD-ESI-MS/MS system consisted of a Waters Alliance 2695 LC-DAD and a Q-TOF 2 (quadrupole mass filter-time-of-fiight) mass spectrometer with a Z-spray ES source.General procedure for the synthesis of amidoximes from amides or acid chloride
To a solution of iodine (0.1904 g, 0.75 mmol) and triphenylphosphine (0.1967 g, 0.75 mmol) in dry dichloromethane (4 mL) was added amide (0.50 mmol), triethylamine (0.35 mL, 2.50 mmol), and hydroxylamine hydrochloride (0.0521 g, 0.75 mmol) at 0 °C. The reaction mixture was then warm up to room temperature and stirred until completion of the reaction (typically within 2 h). The crude mixture was concentrated under reduced pressure then purified by column chromatography using 30–70% ethyl acetate in hexane. For the synthesis of amidoximes from acid chloride, a mixture of acid chloride (0.50 mmol), amine (0.50 mmol), and triethylamine (0.10 mL, 0.75 mmol) in dry dichloromethane (4 mL) was stirred at 0 °C to room temperature until complete disappearance of the acid chloride. The in situ generated amide was then subjected to the reaction with hydroxylamine hydrochloride as described above.General procedure for the synthesis of amidoximes from carboxylic acids
To a solution of iodine (0.3807 g, 1.5 mmol) and triphenylphosphine (0.3935 g, 1.5 mmol) in dry dichloromethane (5 mL) was added in one portion with carboxylic acid derivative (0.50 mmol), and amine (0.50 mmol), followed by triethylamine (0.45 mL, 3.25 mmol) at 0 °C. The resulting mixture was continuously stirred at room temperature for 1 h. Hydroxylamine hydrochloride (0.0521 g, 0.75 mmol) was then added and the reaction mixture was allowed to stir until completion of the reaction (typically within 2 h). The crude mixture was concentrated under reduced pressure then purified by column chromatography using 30–70% ethyl acetate in hexane.General procedure for the synthesis of 1,2,4-oxadiazol-5(4H)-one
To a solution of N-substituted amidoxime 2 (0.30 mmol) dissolved in acetonitrile (2.0 mL) was added 1,1′-carbonyldiimidazole (0.0584 g, 0.36 mmol), followed by finely grounded K2CO3 (0.2073 g, 1.50 mmol). The resulting mixture was stirred at room temperature for 10–20 min. The crude mixture was concentrated under reduced pressure and then purified by short column chromatography using 10–40% ethyl acetate in hexane to give the corresponding product.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from The Thailand Research Fund through the Royal Golden Jubilee PhD Program to C. D. (Grant No. PHD/0086/2557) and N. W. (Grant No. PHD/0072/2559) are gratefully acknowledged. This research work was also partially supported by Chiang Mai University. Special appreciation to Chulabhorn Research Institute (CRI), Thailand for the ESI-MS analysis.Notes and references
-
(a) Y. Yamamoto, T. Tsuritani and T. Mase, Tetrahedron Lett., 2008, 49, 876 CrossRef CAS
; (b) A.-A. Gaber and L. Taib, J. Chem. Sci., 2016, 128, 745 CrossRef CAS
- S. S. Patil, P. C. Mhaske, S. V. Patil and V. D. Bobade, J. Heterocycl. Chem., 2011, 48, 652 CrossRef CAS
- K. Muralirajan, R. Kuppusamy, S. Prakash and C. H. Cheng, Adv. Synth. Catal., 2016, 358, 774 CrossRef CAS
-
(a) F.-L. Zhang, Y.-F. Wang and S. Chiba, Org. Biomol. Chem., 2013, 11, 6003 RSC
-
(a) Y. Durust, C. Altug and F. Kilic, Phosphorus, Sulfur Silicon Relat. Elem., 2007, 182, 299 CrossRef CAS
-
(a) K. C. Fylaktakidou, D. J. Hadjipavlou-Litina, K. E. Litinas, E. A. Varella and D. N. Nicolaides, Curr. Pharm. Des., 2008, 14, 1001 CrossRef CAS PubMed
- E. W. Yue, R. Sparks, P. Polam, D. Modi, B. Douty, B. Wayland, B. Glass, A. Takvorian, J. Glenn, W. Zhu, M. Bower, X. Liu, L. Leffet, Q. Wang, K. J. Bowman, M. J. Hansbury, M. Wei, Y. Li, R. Wynn, T. C. Burn, H. K. Koblish, J. S. Fridman, T. Emm, P. A. Scherle, B. Metcalf and A. P. Combs, ACS Med. Chem. Lett., 2017, 8, 486 CrossRef CAS PubMed
- E. W. Yue, B. Douty, B. Wayland, M. Bower, X. Liu, L. Leffet, Q. Wang, K. J. Bowman, M. J. Hansbury, C. Liu, M. Wei, Y. Li, R. Wynn, T. C. Burn, H. K. Koblish, J. S. Fridman, B. Metcalf, P. A. Scherle and A. P. Combs, J. Med. Chem., 2009, 52, 7364 CrossRef CAS PubMed
-
(a) I. Artsimovitch, C. Chu, A. S. Lynch and R. Landick, Science, 2003, 302, 650 CrossRef CAS PubMed
- D. Schade, J. Kotthaus, L. Riebling, J. Kotthaus, H. Mueller-Fielitz, W. Raasch, O. Koch, N. Seidel, M. Schmidtke and B. Clement, J. Med. Chem., 2014, 57, 759 CrossRef CAS PubMed
-
(a) F. Fang, J. Zhang, L. Cao, S. Shen, Y. Guo, Z. He and H. Hu, Tetrahedron, 2016, 72, 2476 CrossRef CAS
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210 CrossRef CAS PubMed
- D. F. Bushey and F. C. Hoover, J. Org. Chem., 1980, 45, 4198 CrossRef CAS
-
(a) H. C. Shen, F.-X. Ding, Q. Deng, S. Xu, X. Tong, X. Zhang, Y. Chen, G. Zhou, L.-Y. Pai, M. Alonso-Galicia, S. Roy, B. Zhang, J. R. Tata, J. P. Berger and S. L. Colletti, Bioorg. Med. Chem. Lett., 2009, 19, 5716 CrossRef CAS PubMed
-
(a) B. Ganem, Y. Dong, Y. F. Zheng and G. D. Prestwich, J. Org. Chem., 1999, 64, 5441 CrossRef CAS PubMed
- A. R. Katritzky, N. M. Khashab, N. Kirichenko and A. Singh, J. Org. Chem., 2006, 71, 9051 CrossRef CAS PubMed
-
(a) G. D'Alo and P. Grunanger, Farmaco, Ed. Sci., 1966, 21, 346 Search PubMed
- A. V. Aksenov, A. N. Smirnov, N. A. Aksenov, A. S. Bijieva, I. V. Aksenova and M. Rubin, Org. Biomol. Chem., 2015, 13, 4289 RSC
- G. Sanguineti, H. V. Le and B. Ganem, Tetrahedron, 2011, 67, 10208 CrossRef CAS PubMed
-
(a) F. C. Kuepper, M. C. Feiters, B. Olofsson, T. Kaiho, S. Yanagida, M. B. Zimmermann, L. J. Carpenter, G. W. Luther, Z. Lu, M. Jonsson and L. Kloo, Angew. Chem., Int. Ed., 2011, 50, 11598 CrossRef CAS PubMed
- J.-R. Dormoy and B. Castro, Triphenylphosphine-Iodine, Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2009, DOI:10.1002/047084289X.rt376.pub2
-
(a) W. Phakhodee, S. Wangngae and M. Pattarawarapan, J. Org. Chem., 2017, 82, 8058 CrossRef CAS PubMed
- S. Wangngae, C. Duangkamol, M. Pattarawarapan and W. Phakhodee, RSC Adv., 2015, 5, 25789 RSC
- S. Bae and M. K. Lakshman, J. Org. Chem., 2008, 73, 1311 CrossRef CAS PubMed
- R. E. Bolton, S. J. Coote, H. Finch, A. Lowdon, N. Pegg and M. V. Vinader, Tetrahedron Lett., 1995, 36, 4471 CrossRef CAS
-
(a) J. E. Mangette, M. R. Johnson, V.-D. Le, R. A. Shenoy, H. Roark, M. Stier, T. Belliotti, T. Capiris and P. R. Guzzo, Tetrahedron, 2009, 65, 9536 CrossRef CAS
-
(a) R. Budriesi, B. Cosimelli, P. Ioan, C. Z. Lanza, D. Spinelli and A. Chiarini, J. Med. Chem., 2002, 45, 3475 CrossRef CAS PubMed
-
(a) T. L. Gilchrist, C. J. Moody and C. W. Rees, J. Chem. Soc., Perkin Trans. 1, 1979, 1871 RSC
- F. Minisci, R. Galli and A. Quilico, Tetrahedron Lett., 1963, 4, 785 CrossRef
- A. E.-A. M. Gaber and H. McNab, J. Anal. Appl. Pyrolysis, 2009, 86, 369 CrossRef CAS
- Y. S. Kara, Spectrochim. Acta, Part A, 2015, 149, 920 CrossRef CAS PubMed
- T. Bacchetti and A. Alemagna, Atti Accad. Naz. Lincei, Cl. Sci. Fis., Mat. Nat., Rend., 1960, 28, 824 CAS
- C.-C. Lin, T.-H. Hsieh, P.-Y. Liao, Z.-Y. Liao, C.-W. Chang, Y.-C. Shih, W.-H. Yeh and T.-C. Chien, Org. Lett., 2014, 16, 892 CrossRef CAS PubMed
- A. Renodon-Cornière, S. Dijols, C. Perollier, D. Lefevre-Groboillot, J.-L. Boucher, R. Attias, M.-A. Sari, D. Stuehr and D. Mansuy, J. Med. Chem., 2002, 45, 944 CrossRef
- J. K. Augustine, V. Akabote, S. G. Hegde and P. Alagarsamy, J. Org. Chem., 2009, 74, 5640 CrossRef CAS PubMed
- M. W. Partridge and H. A. Turner, J. Chem. Soc., 1958, 2086 RSC
- H. Ağirbaş and Y. Kara, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 1435 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08207c |
| This journal is © The Royal Society of Chemistry 2018 |