DOI:
10.1039/C8RA08162J
(Paper)
RSC Adv., 2018,
8, 40974-40983
Synthesis and anti-glioblastoma effects of artemisinin-isothiocyanate derivatives†
Received
2nd October 2018
, Accepted 19th November 2018
First published on 7th December 2018
Abstract
A series of novel artemisinin (ART) derivatives containing an isothiocyanate (ITC) group were synthesized. All the compounds showed more potent anti-tumor effects than those of parent dihydroartemisinin (DHA) towards glioblastoma multiforme U87 in vitro. Among them, 5b had the strongest cytotoxic activity which exerted its effects in a concentration-dependent but not time-dependent manner (IC50 7.41 μM for 24 h, 7.35 μM for 72 h). Pyknosis was observed in 5b-treated U87 cells. Multiple intrinsic apoptotic pathways were induced by 5b including the upregulation of caspase 9, the release of cytochrome c, an increase of the proapoptotic protein Bax, a decrease of the anti-apoptotic protein Bcl 2, and the activation of execution pathways by the upregulation of caspase 3. In addition to apoptosis, an autophagic mechanism was also involved in 5b-induced cytotoxicity to human GBM U87 cells by upregulating the expression of LC3-II and downregulating p62. Furthermore, 5b also significantly attenuated the migration of U87 cells. Therefore, our results suggest that 5b may be a promising molecule for the further development of a novel drug for the treatment of glioblastoma.
Introduction
Glioblastoma multiforme (GBM) is the most prevalent and severe primary central nervous system tumor. Even when using conventional treatments such as surgery, radiation therapy and chemical treatment, the average survival time is less than 15 months.1 The average occurrence rate of GBM is 3.19/100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 population, and the median age of diagnosis is 64 years old. The occurrence is higher in men than in women.2 Therefore, the discovery of effective therapy for GBM is an area of interest.
000 population, and the median age of diagnosis is 64 years old. The occurrence is higher in men than in women.2 Therefore, the discovery of effective therapy for GBM is an area of interest.
Artemisinin (ART) and its derivatives (collectively termed as artemisinins (ARTs)) are sesquiterpene lactones isolated from sweet wormwood (Artemisia annua), which are used as a remedy for fevers and chills in Chinese Traditional Medicine.3 The endoperoxide bridge of ARTs is very important for their activities; they are still the first-line drug for malarial treatment. ARTs have not only antimalarial property but also anticancer effects.4,5 It has been confirmed that dihydroartemisinin (DHA), the active metabolite of ARTs, showed cytotoxic activity on ten human glioma cell lines through the induction of autophagy.6 DHA also exerted an anti-tumor role through induced ROS-mediated mitochondrion and ER stress apoptotic pathways as well as autophagic cell death in human GBM cells.7 Although ART and its derivatives are extremely effective in their antimalarial role, they are less potent as anticancer agents, especially as monotherapy drugs because of their mild activity and short elimination half-life.8 Therefore, to improve the clinical outcome of ARTs, future efforts should be focused on the combination of longer-acting drugs for maximum efficacy or the development of potent derivatives.9–11
Naturally occurring isothiocyanates (ITCs), which are generated by the enzymatic hydrolysis of glucosinolates, are abundant in cruciferous vegetables (Fig. 1).12–14 It has been proven that some natural and synthetic ITCs have anticancer activities with multiple targets and mechanisms. ITCs can enhance the antitumor efficacy of other drugs, or act as covalent inhibitors.15 ITCs alone are able to suppress the growth of various cancer cell lines such as non–small-cell lung cancer cells, human malignant astrocytoma cells, and HCT 116 human colon cancer cells.16–20 They exert their effects mainly by increasing the production of reactive oxygen species (ROS), induction of apoptosis, induction of cell cycle arrest and so on.18–23 However, the exact mechanisms of ITCs against tumors are still not clear. Some research uncovered that aryl isothiocyanates (e.g., benzyl isothiocyanate (BITC), phenyl isothiocyanate (PITC)) and allyl isothiocyanates (AITC) (Fig. 1) act as H2S donor agents.24,25 H2S as the third most important gasotransmitter is involved in the development of cancer by shortening the cell cycle and inducing proliferation.26,27
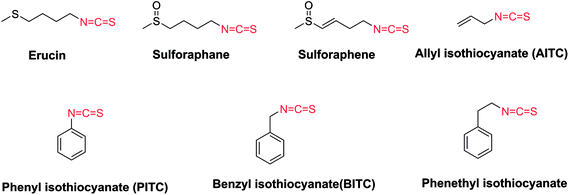 |
| | Fig. 1 The representatives of naturally occurring isothiocyanates. | |
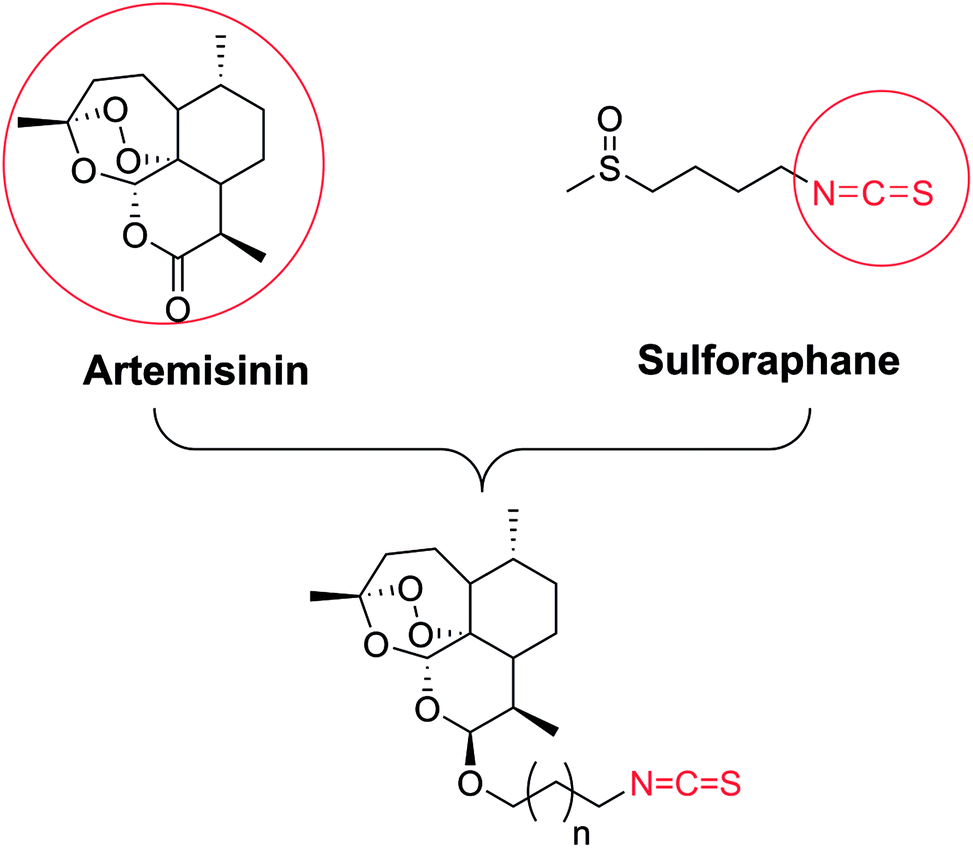
Based on those studies, we hypothesized that artemisinin-based isothiocyanates might be more efficacious compounds towards human GBM cells with multiple mechanisms. In this study, the synthesis, bio-evaluation and mechanism of artemisinin-based isothiocyanates against GBM in vitro have been carried out, and we found a novel DHA-ITC derivative 5b to have potent GBM cell toxicity.
Results and discussion
Chemistry
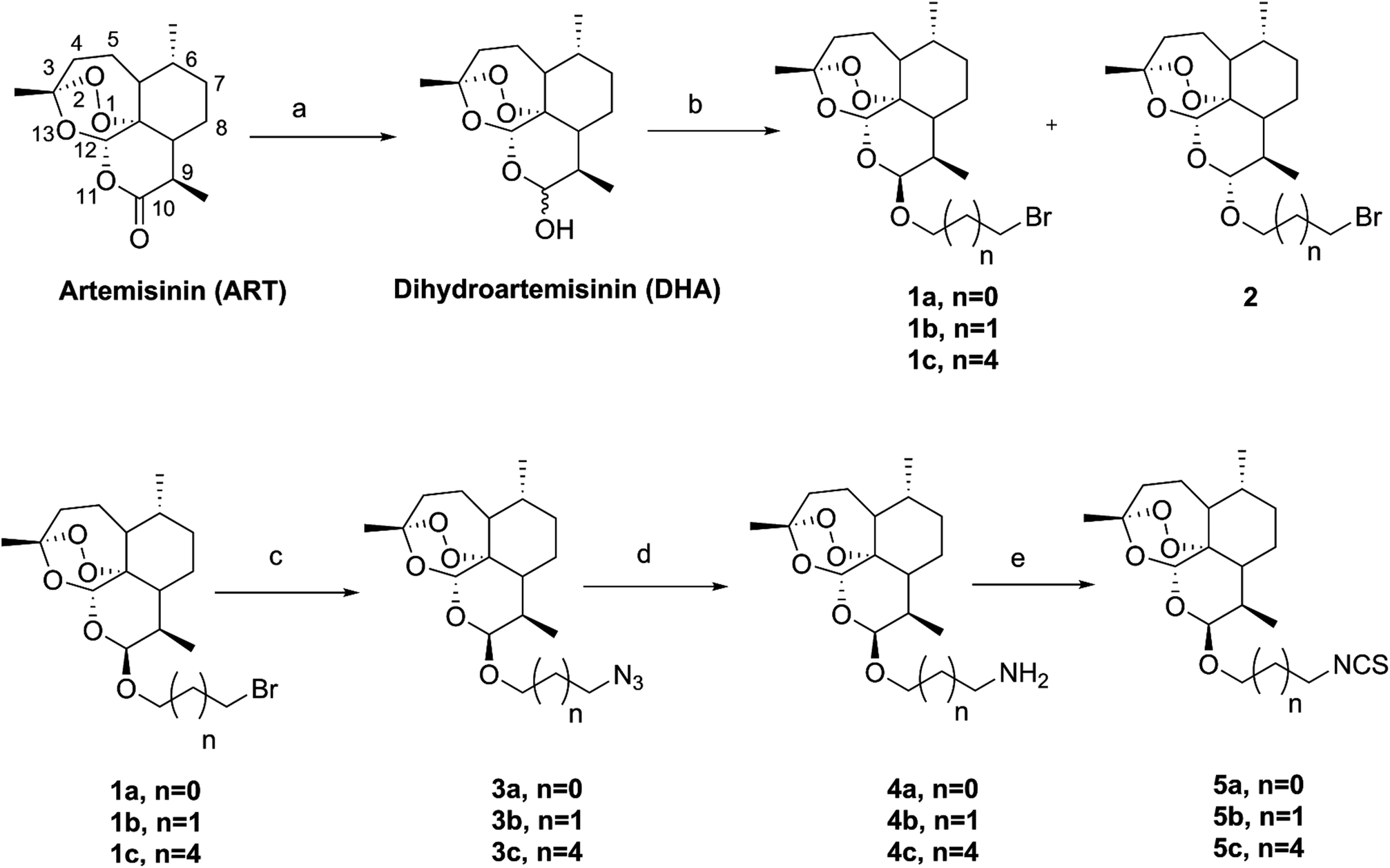
The synthetic strategy employed for the synthesis of the target compounds is depicted in Scheme 1. A series of novel hybrids were afforded by linking DHA at C-10 and an isothiocyanate group. The detailed synthetic route was described in Scheme 2. The DHA derivatives 4a–c were synthesized according to the literature.28,29 The reaction of DHA with the corresponding bromo-substituted alkanol in CH2Cl2 by using boron trifluoride ethyl ether (BF3·Et2O) as a catalyst afforded substituted ART enantiomers. 1a–c were obtained by silica gel column chromatography elution with petroleum ether/ethyl acetate (97:3) as the β configuration (indicated the coupling constants between 9-H and 10-H in 1H NMR, J9,10 = 3–4 Hz).30 1a–c were then converted to 4a–c via an azidation and reduction reaction. Finally, the intermediates 4a–c bearing an amino group reacted with carbon disulfide (CS2) in dried THF31 to afford the target compounds 5a–c. All the synthesized compounds were purified by silica gel column chromatography.
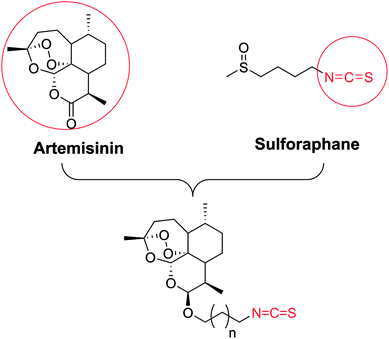 |
| | Scheme 1 The design strategy. | |
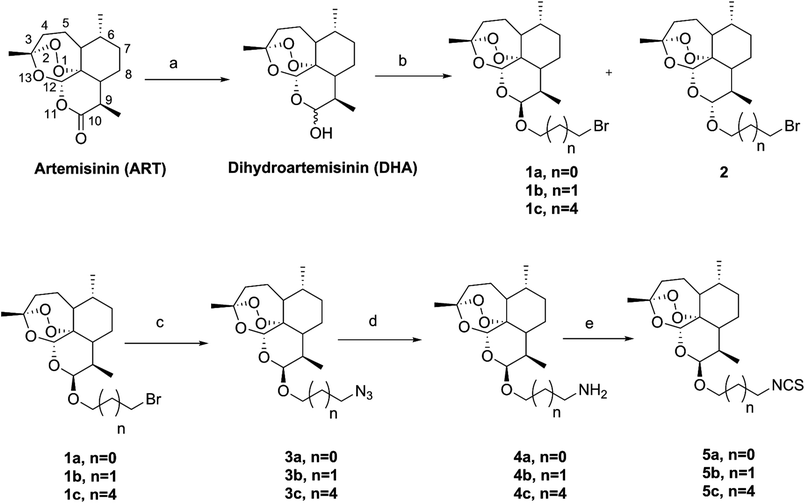 |
| | Scheme 2 The synthetic route of artemisinin-isothiocyanate derivatives 5a–c. Reagents and conditions:aNaBH4/MeOH, 0 °C, 2 h, 94%; bBF3·Et2O/CH2Cl2, bromo-substituted alkanol; cNaN3/NaI/DMF, 60 °C, 4 h, 85–98%; d(1) (Ph)3P/THF, 60 °C, 2 h; (2) H2O, 70 °C, 2 h, 65–91%; eCS2/THF/Et3N, 0 °C-r.t, 30 min, then AcCl, 0 °C-r.t, 30 min, 49–76%. | |
The structures of compounds 5a–c were elucidated on the basis of 1H NMR, 13C NMR and HR-ESI-MS spectra. In a typical example, the HR-ESI-MS of compound 5b displayed a peak at m/z 384.1802, corresponding to the quasimolecular ions [M + H]+, confirming the molecular formula of compound 5b as C19H29NO5S. In the 1H-NMR spectrum of 5b, the signals of one singlet at δ 1.42 and two doublets at δ 0.89 and 0.94 could be attributed to Me-14, Me-16 and Me-15, respectively. The doublet peak appearing at δ 4.78 (d, J = 3.2 Hz) indicated H-10 in the α configuration.30 In the 13C-NMR spectrum, there were 19 peaks appearing in the range from δ 12 to 130 ppm. Among them, a weak peak appearing at δ 129.5 could be ascribed to the carbon of the isothiocyanate (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CS). The other NMR characterizations have been assigned through the comparison of relative data in literature.32,33
CS). The other NMR characterizations have been assigned through the comparison of relative data in literature.32,33
Biological evaluation
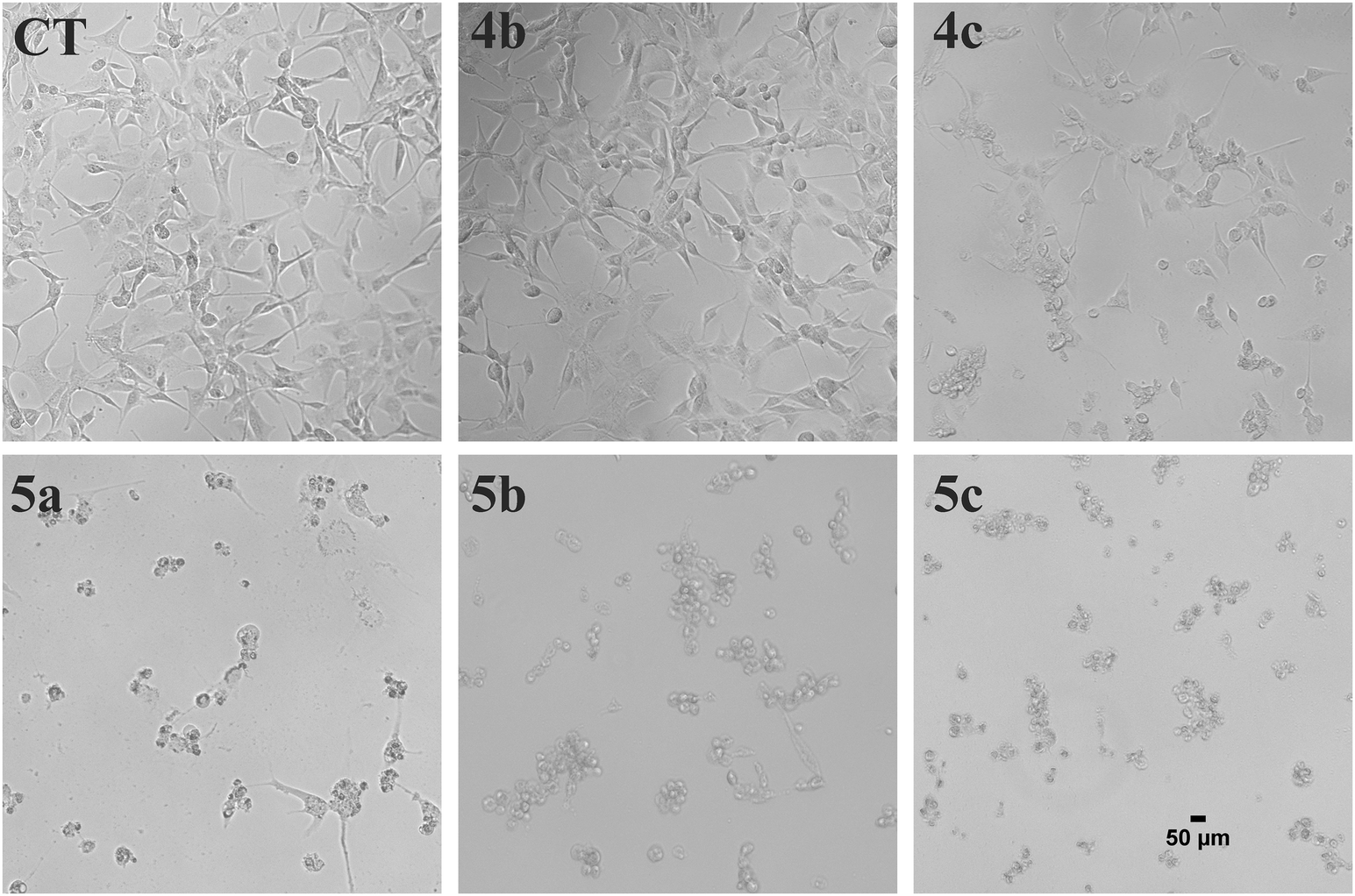
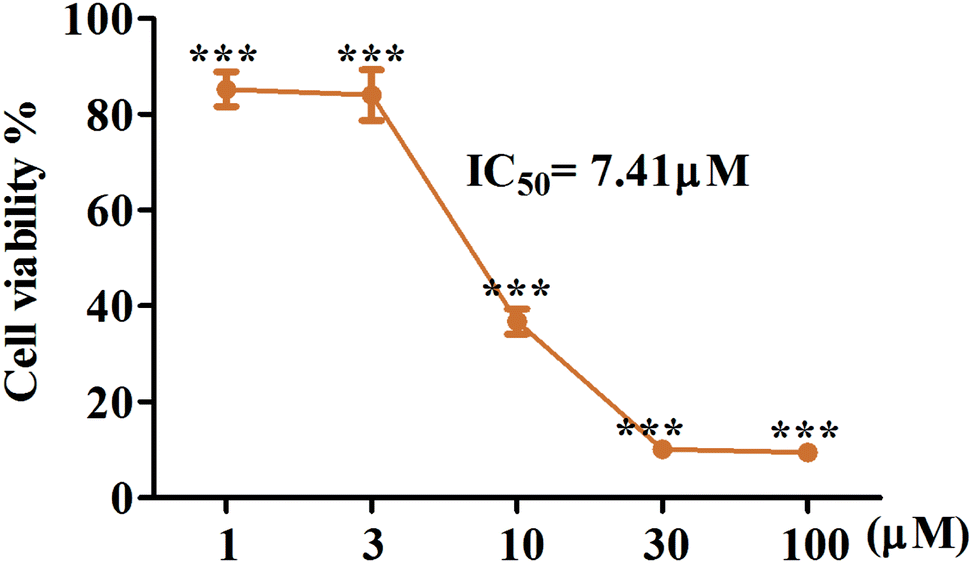
Artemisinin-based isothiocyanate derivatives induced cytotoxicity in glioblastoma U87 cells. The cytotoxicity of artemisinin-based isothiocyanate derivatives on human glioblastoma U87 cells was firstly tested by MTT assay. U87 cells were exposed to various concentrations of compounds for 24 h, and all compounds could induce cell death by MTT assay (Table 1). Cells shrunk and fragmented when exposed to compounds under the phase-contrast microscope (Fig. 2). Among all the compounds, 5b showed the most cytotoxic activity with an IC50value of 7.41 μM and was thus selected for further experiments. As shown in Fig. 3, 5b reduced the viability of GBM cells in a concentration-dependent manner. To evaluate the effects of 5b on the proliferative cancer cell line, U87 glioblastoma cells were exposed to various concentrations of 5b for different durations. 5b significantly suppressed the survival of U87 cells and the inhibitory efficiency was stronger than that of DHA (see Table 1 and Fig. 3).
Table 1 IC50 values (μM) against human U87 cells
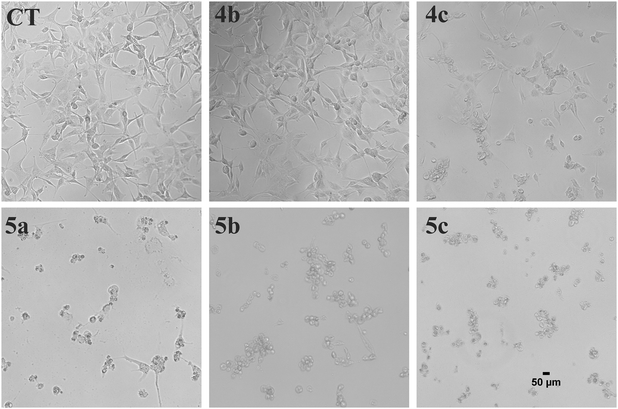 |
| | Fig. 2 Morphological examination of U87 cells treated with artemisinin-isothiocyanates (5a–c) and the key intermediates (4b, 4c). The cells were treated with 30 μM of the compounds for 24 h and the morphological changes were examined with phase contrast microscope. Scale bar = 50 μm. | |
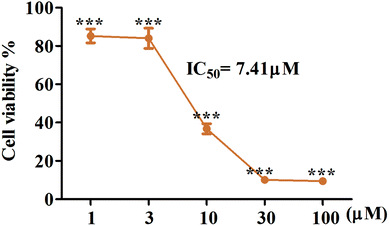 |
| | Fig. 3 5b possesses the cytotoxic activity on human GBM cell lines, U87. The cells were treated with indicated concentrations of 5b for 24 h. Cell viability was tested via MTT assay and IC50 value was calculated. 5b reduced the cell viability of U87 cells in a concentration dependent manner. All data were represented as mean ± SD from three independent experiments; ***P < 0.001 versus control. | |
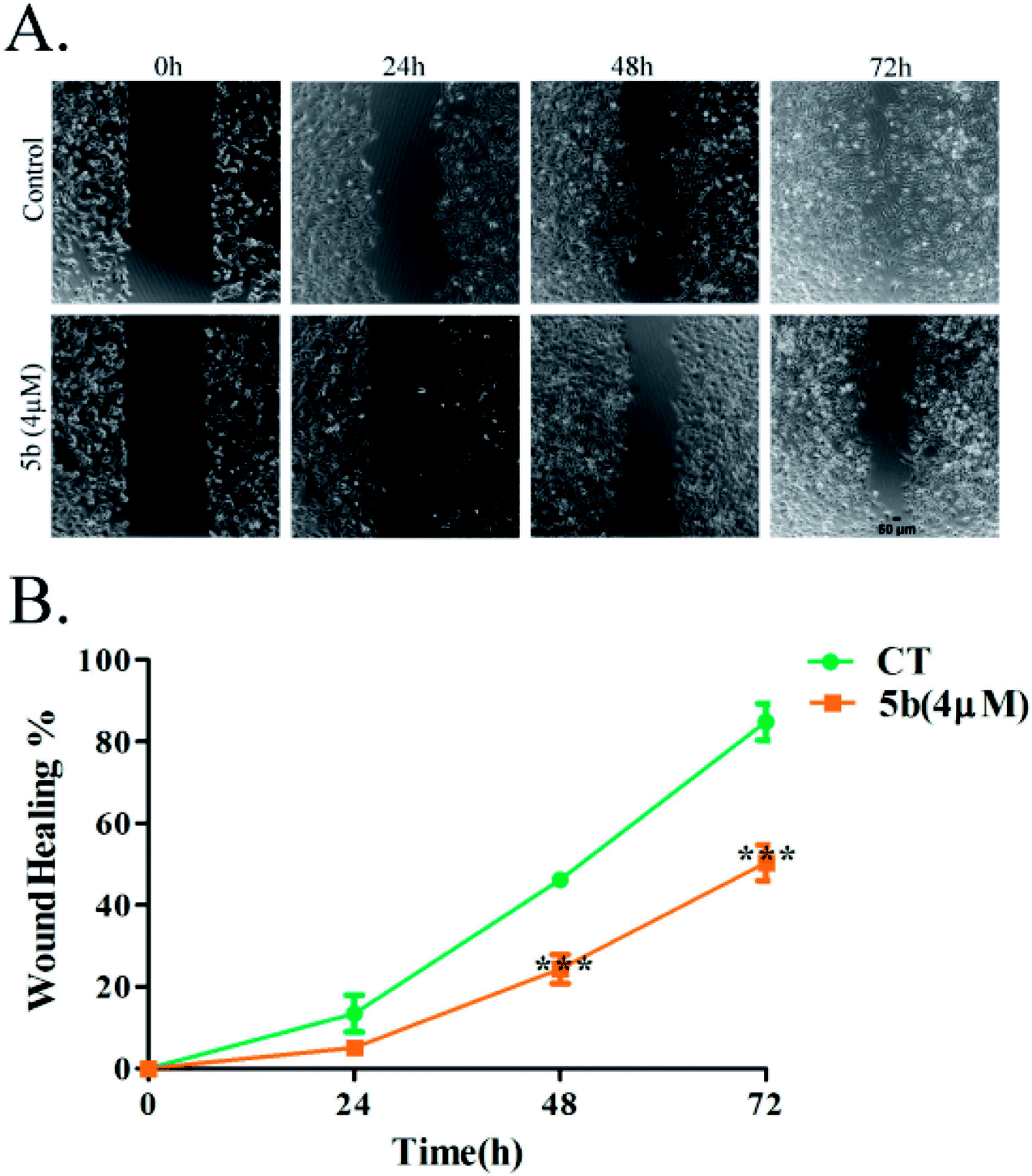
Artemisinin-based isothiocyanate derivative 5b inhibits cell migration. Cell migration is a landmark of cancer invasion and metastasis, immune responses, angiogenesis, wound repair, and embryonic morphogenesis.34 Wound-healing assay is a well-established test of cancer cell migration. To investigate the effects of 5b on wound-healing phenomena, cells were exposed to 4 μM (around half of the IC50) of 5b for 0–72 h.35 The migration of U87 cells was reduced significantly by 5b (Fig. 4).
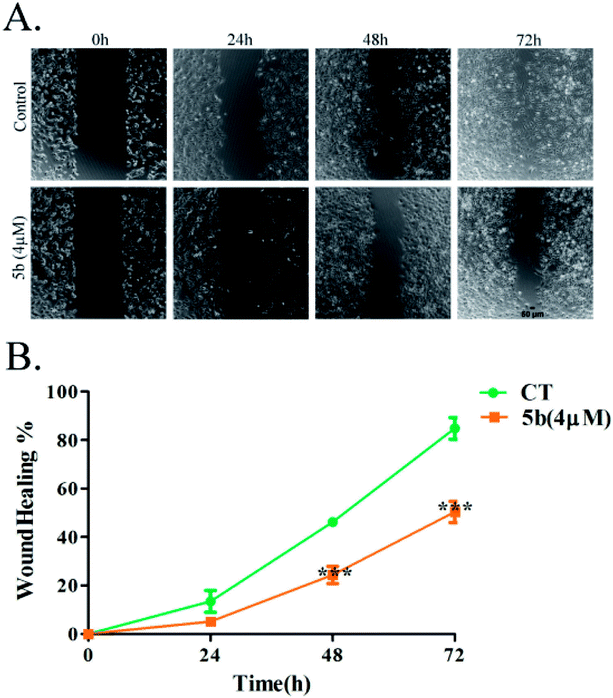 |
| | Fig. 4 5b inhibits the migration of human glioblastoma cells, U87. Data were expressed as means ± SD. ***P < 0.001 versus control. All experiments were performed in triplicates. | |
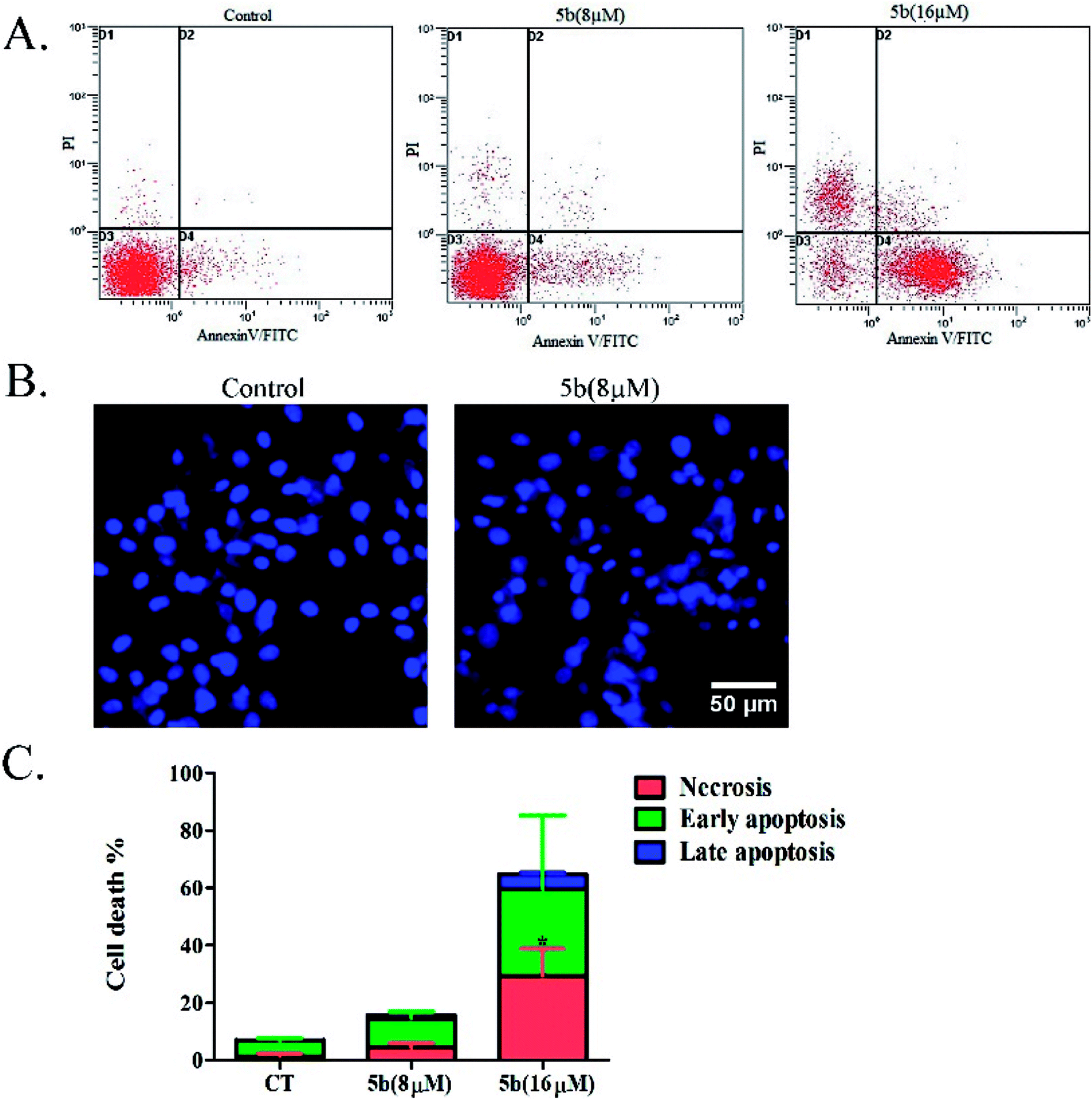
Artemisinin-based isothiocyanate derivative 5b induces apoptosis in U87 cells. To determine the cell death pathway, the U87 cells were treated with two doses (8 and 16 μM) of 5b, which were selected to determine the apoptotic effects of 5b in a concentration-dependent manner. After 16 h of incubation, apoptotic markers of treated cells were analyzed with fluorescein isothiocyanate (FITC)-conjugated annexin V and PI. Annexin V detects phosphatidylserine (PS) on the cell membrane during apoptosis. Propidium iodide (PI) stains the DNA of apoptotic and necrotic cells. As shown in Fig. 5A, 5b induced a higher proportion of early apoptotic and necrotic cells. The nuclear morphological changes were examined by DAPI staining. Pyknosis and nuclear shrinkage were observed in 5b-treated cells (Fig. 5B).
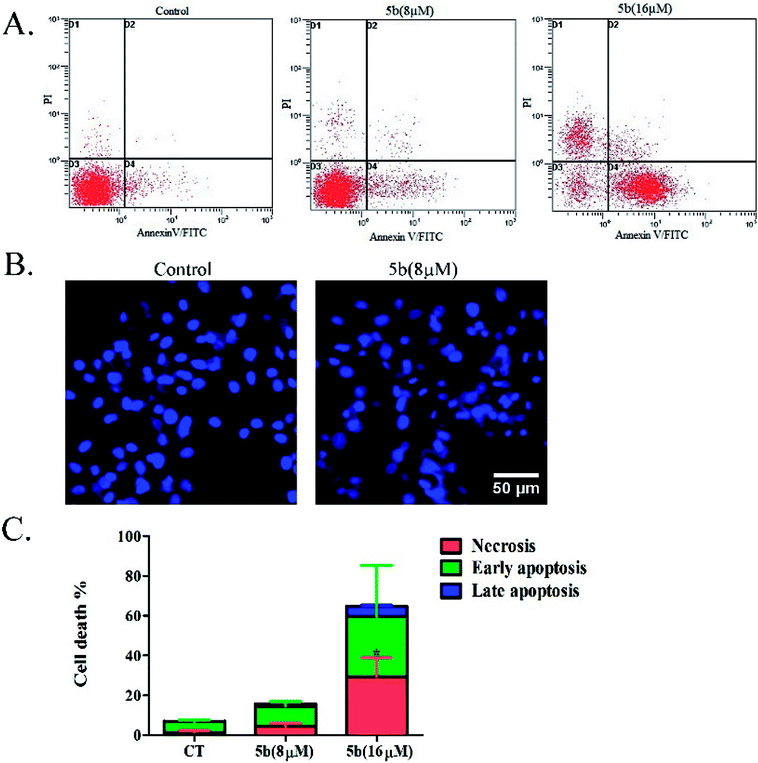 |
| | Fig. 5 Apoptotic analysis of 5b on U87 glioblastoma cells. (A) U87 cells were treated with DMSO or 8 and 16 μM concentrations of 5b for 16 h, and apoptotic rates were analyzed by flow cytometry after annexin V/PI staining. (B) U87 cells were treated with DMSO or 8 μM of 5b for 16 h and the nuclear morphological changes were observed using DAPI staining and fluorescence microscope. (C) Data are expressed as mean ± SD of three independent experiments presented in (A). *P < 0.05 versus control. | |
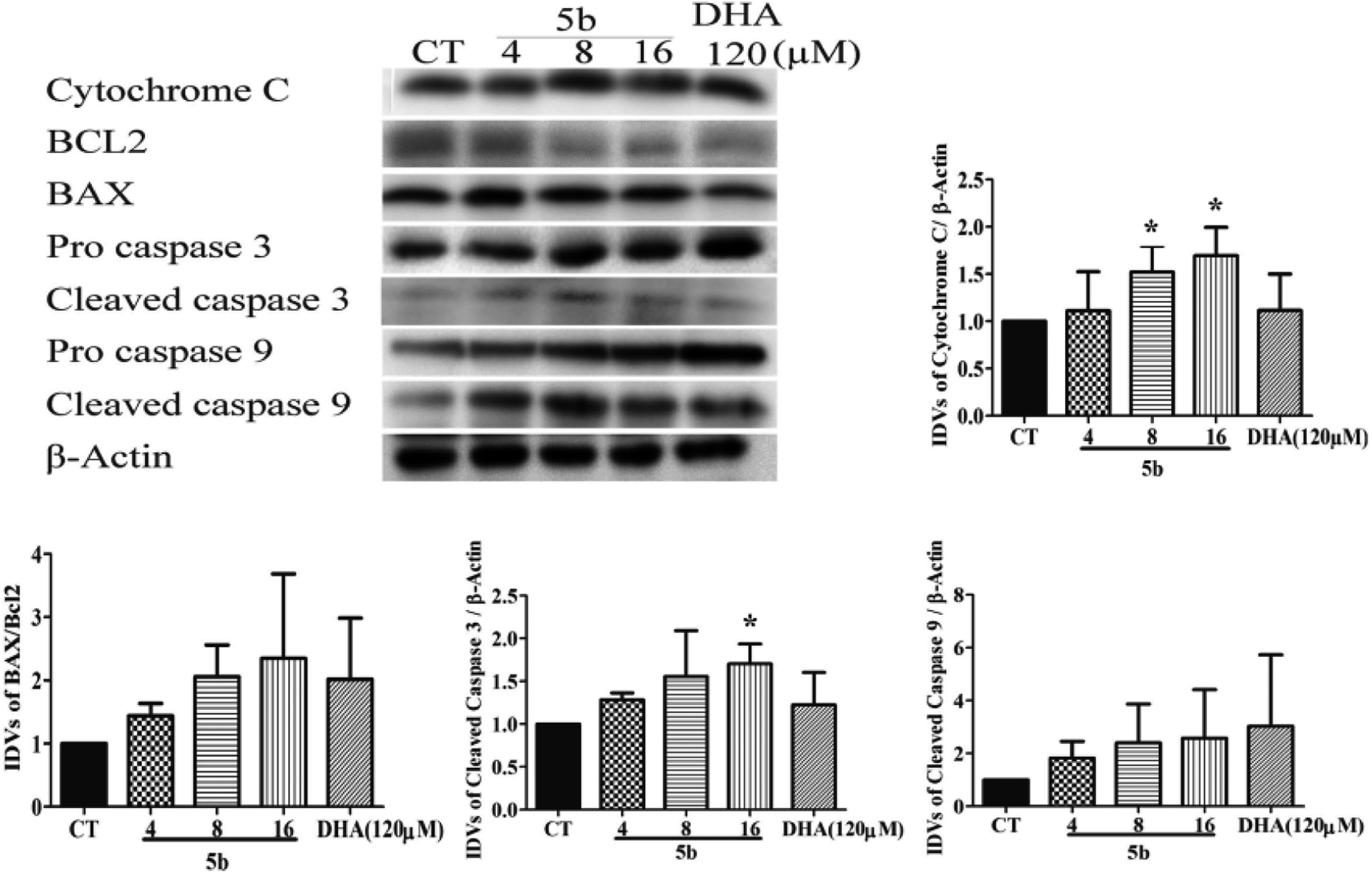
To support the fact that apoptosis was involved in the cytotoxic effects of 5b on GBM cells, we further investigated the caspase family apoptotic proteins and the Bcl2 family apoptotic proteins. As there are three classical apoptotic pathways, we investigated intrinsic and execution pathways. As shown in Fig. 6, the caspase family apoptotic proteins were induced in U87 cells treated with various concentrations of 5b (4–16 μM) and DHA (120 μM) for 16 h. Caspase 9 and cytochrome c were induced in U87 cells treated with 5b. It was assumed that release of cytochrome c activated the caspase-dependent mitochondrial pathway. Cytochrome c binds and activates Apaf1 as well as procaspase-9, forming an “apoptosome”. The clustering of procaspase-9 leads to caspase-9 activation.36,37 The anti-apoptotic protein Bcl-2 was downregulated and pro-apoptotic protein BAX was upregulated in 5b-treated cells. The Bcl-2 family of proteins governs mitochondrial membrane permeability via the regulation of cytochrome c released from the mitochondria.36 Caspase 3 is considered to be the most important of the executioner caspases in apoptotic cell death. Cleaved (active) caspase 3 was increased in 5b-treated cells (Fig. 6).
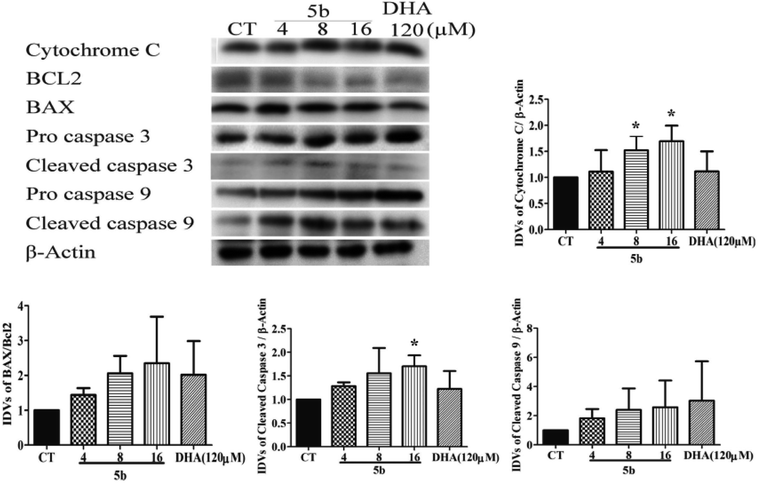 |
| | Fig. 6 5b induces apoptosis by regulating caspase family proteins and Bcl2 family proteins in human GBM cells. Cells were treated with the indicated concentrations of 5b and DHA for 16 h. The proteins were collected for western blot assay and β-actin was used as protein loading control. The result was obtained by three different independent experiments. | |
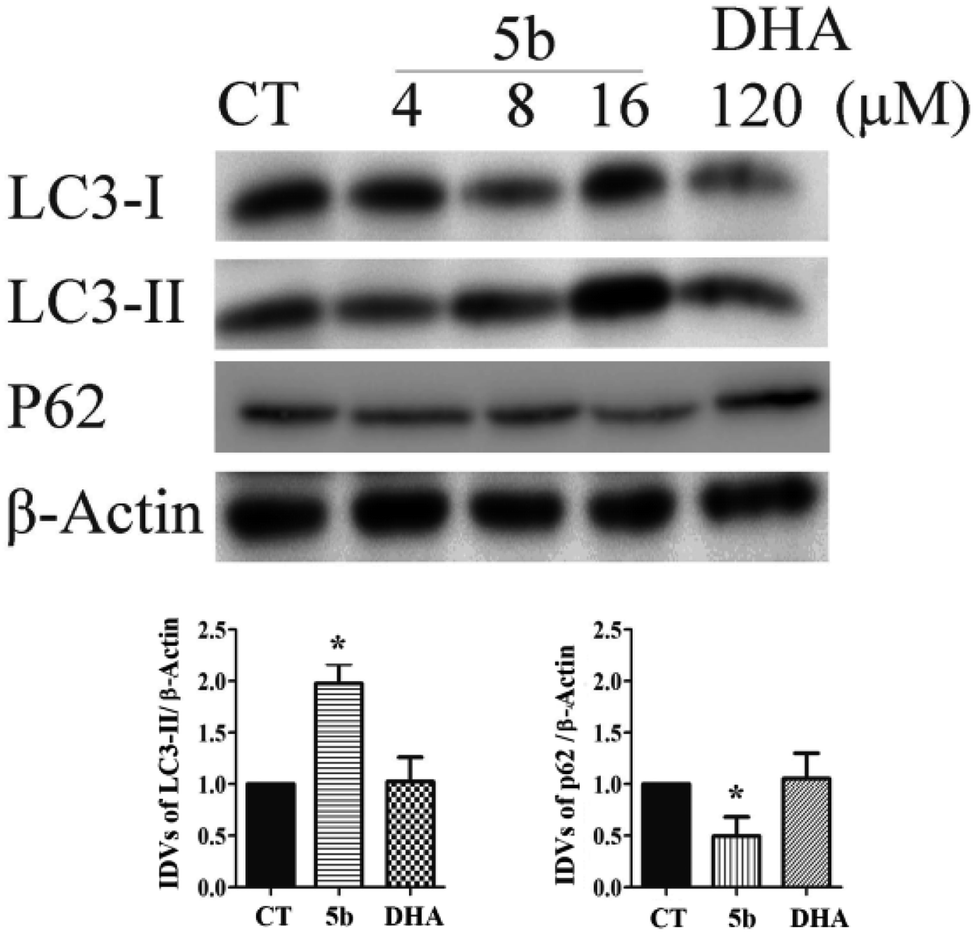
Artemisinin-based isothiocyanate derivative 5b induces autophagy in U87 cells. Autophagy assures cellular homeostasis and is gaining increasing importance in cancer treatment, where it impacts carcinogenesis as well as the propagation of the malignant phenotype and the development of resistance.38 To observe whether autophagy was also involved in 5b-induced cytotoxicity, autophagic markers, such as LC3-II, microtubule-associated proteins 1A/1B light chain 3B, and p62/SQSTM1, were investigated in U87 cells treated with 5b for 16 h. LC3-II expression increased in the 5b-treated group compared to the control groups. Moreover, the decrease of p62 was observed in the 5b-treated group (Fig. 7).
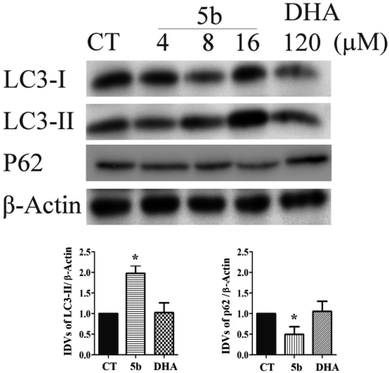 |
| | Fig. 7 5b induces autophagy in human GBM cells. Cells were treated with the indicated concentrations of 5b and DHA for 16 h. The proteins were collected for western blot assay with LC3B, p62 and β-actin antibodies. The result was obtained by three different independent experiments. | |
Conclusion
In conclusion, novel ART derivatives containing an ITC group have been synthesized. All the compounds showed more potent activity than that of the parent DHA towards glioblastoma multiforme U87 in vitro. The data in the present study demonstrated that 5b significantly induced cytotoxicity (IC50 7.41 μM) towards human GBM cells through inducing both apoptotic and autophagic cell death and also inhibited the migration of human GBM cells. It was a stronger inducer in GBM cells compared to DHA (about twenty times stronger). Therefore, it suggested that 5b may be a potential multifunctional small molecule for developing new drugs for the treatment of GBM.
Experimental
Chemistry
Melting points were measured on an XT-4 apparatus (TaikeCorp., Beijing, China) and were uncorrected. 1H and 13C NMR spectra were recorded on Bruker 400 MHz, and the data were recorded using CDCl3 as the solvent. Chemical shifts (δ) are reported in ppm downfield from an internal TMS standard. HR-ESI-MS spectra were determined on an AB SciexTripleTOF 5600+ apparatus. Flash column chromatography on silica gel (200–300 mesh) was used for the routine purification of the reaction products. Analytical TLC was performed on silica gel 60 F254 plates (Merck KGaA, Darmstadt, Germany). All solvents and chemical reagents were obtained from commercial sources and used without further purifications.
General procedure for the synthesis of compounds 1a–c
The DHA derivatives 1a–c were synthesized via the reaction of DHA with the corresponding bromo-substituted alkanol in CH2Cl2 using boron trifluoride ethyl ether (BF3·Et2O)28,29 and isolation in the β configuration by silica gel column chromatography elution with petroleum ether/ethyl acetate (97:3) (indicated are the coupling constants between 9-H and 10-H in 1H NMR, J9,10 = 3–4 Hz).30
The solution of DHA (3.0 mmol) and 3-bromo-1-propanol, or 2-bromo-1-ethanol, or 6-bromo-hexanol (4.6 mmol) in CH2Cl2 (20 mL) below 0 °C was added to BF3·Et2O (0.63 mL, 5.0 mmol). The mixture was stirred for about 50 min at the same temperature under nitrogen protection and monitored by TLC. At the end of the reaction, the organic layer was collected and washed with saturated NaHCO3 solution (1 × 20 mL), water (1 × 20 mL) and brine (1 × 20 mL), dried over anhydrous Na2SO4 and then concentrated in vacuo to give the crude product, which was purified by silica gel column chromatography (ether/ethyl acetate, 97:3,v/v) to afford compounds 1a–c.
1-Bromo-2-(10β-dihydroartemisinoxy)ethane (1a). Colorless solid; yield: 36%; mp 73–74 °C; 1H NMR (refer to literature 28); 13C NMR (100 MHz, CDCl3) δ 104.3(C-3), 102.2(C-10), 88.3(C-12), 81.3(C-12a), 68.4(–OCH2–), 52.8(C-5a), 44.5(C-8a), 37.6(C-6), 36.6(C-4), 34.9(C-7), 31.6(C-9), 31.1(BrCH2–), 26.4(C-14), 24.8(C-5), 24.6(C-8), 20.6(C-15), 13.2(C-16).
1-Bromo-3-(10β-dihydroartemisinoxy)propane (1b). Colorless solid; yield: 39%; mp: 69–71 °C;1H NMR (refer to literature 29);;13C NMR (100 MHz, CDCl3) δ 104.3(C-3), 102.3(C-10), 88.1(C-12), 81.2(C-12a), 65.8(–OCH2–), 52.7(C-5a), 44.5(C-8a), 37.6(C-6), 36.6(C-4), 34.8(C-7), 32.7(C-9), 31.1(BrCH2–), 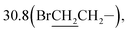 26.4(C-14), 24.8(C-5), 24.7(C-8), 20.5(C-15), 13.2(C-16).
26.4(C-14), 24.8(C-5), 24.7(C-8), 20.5(C-15), 13.2(C-16).
1-Bromo-6-(10β-dihydroartemisinoxy)hexane (1c). Colorless oil; yield: 40%; 1H NMR (400 MHz, CDCl3) δ 5.35 (s, 1H, H-12), 4.76 (d, J = 3.0 Hz, 1H, H-10), 3.98–3.85 (m, 1H, –OCH2–), 3.46–3.37 (m, 2H, –CH2Br), 3.34 (t, J = 6.7 Hz, 1H,–OCH2–), 2.68–2.50 (m, 1H, H-9), 2.34 (td, J = 14.1, 3.7 Hz, 1H, H-4), 2.01 (d, J = 14.3 Hz, 1H, H-4), 1.66–1.56 (m, 2H), 1.53–1.42 (m, 2H), 1.40 (s, 3H, CH3-14), 1.36–1.26 (m, 1H), 1.25–1.18 (m, 1H), 0.92 (d, J = 6.2 Hz, 3H, CH3-15), 0.87 (d, J = 7.2 Hz, 3H, CH3-16), 0.87 (m, 1H, H-7); 13C NMR (100 MHz, CDCl3) δ 104.3(C-3), 102.2(C-10), 88.1(C-12), 81.2(C-12a), 68.4(–OCH2–), 52.8(C-5a), 44.7(C-8a), 37.7(C-6), 36.7(C-4), 34.9(C-7), 34.0, 32.9(C-9), 31.1, 29.7, 28.1, 26.4(C-14), 25.7, 24.9(C-5), 24.7(C-8), 20.6(C-15), 13.1(C-16).
General procedure for the synthesis of compounds 3a–c
The intermediate 3a–c was synthesized from 1a–c according to the procedure previously reported.28,29 To the solution of 1a–c (0.89 mmol) and NaI (10.0 mg) in DMF (5.0 mL), NaN3 (2.7 mmol) was added. The reaction mixture was heated to 60 °C for 4 h. After the disappearance of the starting material, the solution was poured into ice water (20 mL), stirred for 1 h, and then extracted with CH2Cl2 (3 × 20 mL), dried over anhydrous Na2SO4 and concentrated to give a light yellow oil. The crude product was further purified by silica gel column chromatography (petroleum ether/ethyl acetate, 9:1, v/v) to provide 3a–c.
2-(10β-Dihydroartemisinoxy)ethyl azide (3a). Colorless solid; yield: 85%; mp: 86–87 °C; 1H NMR (refer to literature 28); 13C NMR (100 MHz, CDCl3) δ 104.34(C-3), 102.59(C-10), 88.14(C-12), 81.22(C-12a), 67.47(–OCH2–), 52.70(C-5a), 51.33(–CH2N3), 44.54(C-8a), 37.57(C-6), 36.56(C-4), 34.75(C-7), 30.93(C-9), 26.32(C-14), 24.84(C-5), 24.56(C-8), 20.53(C-15), 13.10(C-16).
3-(10β-Dihydroartemisinoxy)propyl azide (3b). Colorless solid; yield: 94%; mp: 80–82 °C; 1H NMR (400 MHz, CDCl3) δ 5.35 (s, 1H, H-12), 4.76 (d, J = 3.1 Hz, 1H, H-10), 3.97–3.84 (m, 1H, –OCH2–), 3.49–3.38 (m, 1H, –OCH2–), 3.34 (t, J = 6.7 Hz, 2H, –CH2N3), 2.68–2.53 (m, 1H, H-9), 2.34 (td, J = 14.0, 3.8 Hz, 1H, H-4), 2.00 (m, 1H, H-4), 1.83 (m, 3H), 1.77–1.68 (m, 2H), 1.66–1.56 (m, 2H), 1.54–1.42 (m, 2H), 1.40 (s, 3H, CH3-14), 1.35–1.24 (m, 1H), 1.24–1.12 (m, 1H), 0.92 (d, J = 6.2 Hz, 3H, CH3-15), 0.87 (d, J = 7.3 Hz, 3H, CH3-16), 0.87 (m, 1H, H-7); 13C NMR (100 MHz, CDCl3) δ 104.3(C-3), 102.3(C-10), 88.1(C-12), 81.2(C-12a), 65.2(–OCH2–), 52.7(C-5a), 48.8(–CH2N3), 44.5(C-8a), 37.6(C-6), 36.6(C-4), 34.8(C-7), 31.1(C-9), 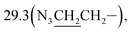 26.8(C-14), 24.8(C-5), 24.7(C-8), 20.5(C-15), 13.2(C-16).
26.8(C-14), 24.8(C-5), 24.7(C-8), 20.5(C-15), 13.2(C-16).
6-(10β-Dihydroartemisinoxy)hexyl azide (3c). Colorless oil; yield: 98%; 1H NMR (400 MHz, CDCl3) δ 5.36 (s, 1H, H-12), 4.75 (d, J = 3.0 Hz, 1H, H-10), 3.89–3.74 (m, 1H, –OCH2–), 3.40–3.30 (m, 1H, –OCH2–), 3.23 (t, J = 6.5 Hz, 2H, –CH2N3), 2.59 (m, 1H,H-9), 2.34 (t, J = 13.9 Hz, 1H, H-4), 2.00 (m, 1H, H-4), 1.80 (m, 3H), 1.51 (m, 5H), 1.41 (s, 3H, CH3-14), 1.38 (m, 7H), 1.26–1.12 (m, 3H),0.93 (d, J = 6.0 Hz, 3H, CH3-15), 0.87 (d, J = 6.6 Hz, 3H, CH3-16), 0.85–0.78 (m, 1H, H-7); 13C NMR (100 MHz, CDCl3) δ 104.3(C-3), 102.2(C-10), 88.1(C-12), 81.3(C-12a), 68.4(–OCH2–), 52.8(C-5a), 51.6(–CH2N3), 44.7(C-8a), 37.7(C-6), 36.7(C-4), 34.9(C-7), 31.1(C-9), 29.7, 29.0, 26.7, 26.4(C-14), 26.1, 24.9(C-5), 24.7(C-8), 20.6(C-15), 13.2(C-16).
General procedure for the synthesis of compounds 4a–c
The key intermediates 4a–c were prepared by the reduction of 3a–c according to literature.28,29 Triphenylphosphine (0.85 mmol) was added to the solution of 3a–c (0.71 mmol) in THF (5.0 mL). The solution was stirred at 55 °C for 2 h. Then, 1.0 mL of water was added, and stirred at room temperature for about 5 h. After removal of the solvent, 20 mL of water was added and then extracted with dichloromethane (3 × 20 mL). The organic layers were combined, dried over anhydrous Na2SO4, and concentrated to afford the crude product, which was further purified by silica gel column chromatography (methanol/dichloromethane, 5:95, v/v) to provide a colorless oil.
2-(10β-Dihydroartemisinoxy)ethylamine (4a). Colorless oil; yield: 65%; HR-ESI-MS (m/z) [M + H]+ calcd for: 328.2124, found: 328.2094;1H NMR (refer to literature 28); 13C NMR (100 MHz, CDCl3) δ 104.34(C-3), 102.39(C-10), 88.11(C-12), 81.27(C-12a), 71.08(–OCH2–), 52.76(C-5a), 44.60(C-8a), 42.26(–CH2NH2), 37.69(C-6), 36.62(C-4), 34.81(C-7), 31.15(C-9), 26.40(C-14), 24.88(C-5), 24.82(C-8), 20.55(C-15), 13.27(C-16).
3-(10β-Dihydroartemisinoxy)propylamine (4b). Colorless oil; yield: 91%; HR-ESI-MS (m/z) [M + H]+ calcd for: 342.2280, found: 342.2245; 1H NMR (400 MHz, CDCl3) δ 5.36 (s, 1H, H-12), 4.76 (d, J = 3.1 Hz, 1H, H-10), 3.96–3.84 (dt, J = 10.9, 6.0 Hz, 1H, –OCH2–), 3.42 (dt, J = 10.9, 6.0 Hz, 1H, –OCH2–), 2.77 (t, J = 6.9 Hz, 2H, –CH2NH2), 2.66–2.52 (m, 1H, H-9), 2.34 (td, J = 14.0, 3.7 Hz, 1H, H-4), 2.01 (m, 1H, H-4), 1.86 (m, 1H),1.79–1.66 (m, 4H), 1.66–1.60 (m, 1H), 1.53–1.42 (m, 2H),1.41 (s, 3H, CH3-14), 1.30 (m, 1H), 1.26–1.16 (m, 2H),0.92 (d, J = 6.1 Hz, 3H, CH3-15), 0.86 (d, J = 7.4 Hz, 3H, CH3-16), 0.86 (m, 1H, H-7); 13C NMR (100 MHz, CDCl3) δ 104.3(C-3), 102.3(C-10), 88.1(C-12), 81.3(C-12a), 66.6(–OCH2–), 52.8(C-5a), 44.6(C-8a), 39.9(–CH2NH2), 37.7(C-6), 36.6(C-4), 34.8(C-7), 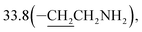 31.1(C-9), 26.4(C-14), 24.9(C-5), 24.7(C-8), 20.6(C-15), 13.2(C-16).
31.1(C-9), 26.4(C-14), 24.9(C-5), 24.7(C-8), 20.6(C-15), 13.2(C-16).
6-(10β-Dihydroartemisinoxy)hexylamine (4c). Colorless oil; yield: 86%; HR-ESI-MS (m/z) [M + H]+ calcd for: 384.2750, found: 384.2711; 1H NMR (400 MHz, CDCl3) δ 5.34 (s, 1H, H-12), 4.73 (d, J = 3.0 Hz, 1H, H-10), 3.78 (dt, J = 15.9, 6.6 Hz, 1H, –OCH2–), 3.41–3.24 (dt, J = 15.9, 6.6 Hz, 1H, –OCH2–), 2.64 (t, J = 7.0 Hz, 2H, –CH2NH2), 2.59–2.51 (m, 1H, H-9), 2.32 (td, J = 14.0, 3.7 Hz, 1H, H-4), 1.99 (m, 1H, H-4), 1.91–1.64 (m, 7H), 1.64–1.41 (m, 7H),1.40 (s, 3H,CH3-14), 1.38 (m, 1H), 1.29 (m, 6H), 1.24–1.11 (m, 2H), 0.91 (d, J = 6.2 Hz, 3H, CH3-15), 0.88 (m, 1H, H-7),0.85 (d, J = 7.2 Hz, 3H, CH3-16); 13C NMR (100 MHz, CDCl3) δ 104.2(C-3), 102.1(C-10), 88.1(C-12), 81.3(C-12a), 68.5(–OCH2–), 52.7(C-5a), 44.7(C-8a), 41.8(–CH2NH2), 37.7(C-6), 36.6(C-4), 34.8(C-7), 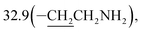 31.1(C-9), 29.8, 26.8, 26.4(C-14), 26.3, 24.9(C-5), 24.6(C-8), 20.6(C-15), 13.2(C-16).
31.1(C-9), 29.8, 26.8, 26.4(C-14), 26.3, 24.9(C-5), 24.6(C-8), 20.6(C-15), 13.2(C-16).
General procedure for the synthesis of compounds 5a–c
The desired compounds 5a–c were prepared according to the literature. CS2 (47 μL, 0.77 mmol) was added dropwise to a solution of compounds 4a–c (0.64 mmol) and Et3N (0.24 mL, 1.9 mmol) in anhydrous THF (5.0 mL) in an ice bath. Then, the solution was stirred for 30 min at r.t. and AcCl (54 μL, 0.77 mmol) was added into the solution at 0 °C. After 5 min at the same temperature, it was warmed to r.t and stirred for 15–30 min. Then, the reaction was quenched with 2.0 mL of 5% HCl (aq.) and extracted with ethyl acetate three times (3 × 10 mL). The organic layers were combined, dried over anhydrous Na2SO4, and concentrated to afford the crude product, which was purified by silica gel column chromatography (petroleum ether/ethyl acetate 97:3, v/v) to provide 5a–c as a colorless oil.
2-(10β-Dihydroartemisinoxy)ethyl isothiocyanate (5a). Colorless oil; yield: 49%; HR-ESI-MS (m/z) [M + H]+ calcd for: 370.1688, found: 370.1643; 1H NMR (400 MHz, CDCl3) δ 5.43 (s, 1H, H-12), 4.81 (d, J = 2.9 Hz, 1H, H-10), 4.00 (dt, J = 10.3, 5.1 Hz, 1H,–OCH2–), 3.65 (t, J = 5.1 Hz, 2H, –CH2NCS), 3.56 (dt, J = 9.7, 4.7 Hz, 1H, –OCH2–), 2.69–2.57 (m, 1H, H-9), 2.33 (td, J = 14.1, 3.7 Hz, 1H, H-4), 2.00 (d, J = 14.5 Hz, 1H, H-4), 1.90–1.79 (m, 1H), 1.75 (m, 2H), 1.64 (m, 1H), 1.45 (m, 3H), 1.40 (s, 3H, CH3-14), 1.28–1.14 (m, 2H), 0.91 (d, J = 6.4 Hz, 3H,CH3-15), 0.91 (d, J = 6.4 Hz, 3H,CH3-16), 0.88–0.79 (m, 1H, H-7); 13C NMR (100 MHz, CDCl3) δ 128.99(–NCS), 104.35(C-3), 102.57(C-10), 88.24(C-12), 81.21(C-12a), 66.65(–OCH2–), 52.68(C-5a), 45.65(–CH2NCS), 44.45(C-8a), 37.42(C-6), 36.55(C-4), 34.74(C-7), 30.88(C-9), 26.30(C-14), 24.82(C-5), 24.64(C-8), 20.50(C-15), 13.16(C-16).
3-(10β-Dihydroartemisinoxy)propyl isothiocyanate (5b). Colorless oil; yield: 53%; HR-ESI-MS (m/z) [M + H]+ calcd for: 384.1845, found: 384.1802; 1H NMR (400 MHz, CDCl3) δ 5.36 (s, 1H, H-12), 4.78 (d, J = 3.2 Hz, 1H, H-10), 4.02–3.88 (dt, J = 9.5, 6.4 Hz, 1H, –OCH2–), 3.60 (t, J = 6.6 Hz, 2H, –CH2NCS), 3.53–3.41 (dt, J = 9.5, 6.4 Hz, 1H,–OCH2–), 2.70–2.57 (m, 1H,H-9), 2.35 (td, J = 14.1, 3.8 Hz, 1H, H-4), 2.01 (m, 1H, H-4), 1.97–1.91 (m, 2H), 1.87 (m, 1H), 1.81–1.67 (m, 2H), 1.63 (m, 2H), 1.49 (m, 2H), 1.42 (s, 3H, CH3-14),0.94 (d, J = 6.3 Hz, 3H,CH3-15), 0.89 (d, J = 7.3 Hz, 3H, CH3-16), 0.87–0.80 (m, 1H, H-7); 13C NMR (100 MHz, CDCl3) δ 129.5(–NCS), 104.4(C-3), 102.4(C-10), 88.1(C-12), 81.2(C-12a), 64.8(–OCH2–), 52.8(C-5a), 44.5(C-8a), 42.5(–CH2NCS), 37.7(C-6), 36.6(C-4), 34.8(C-7), 31.0(C-9), 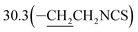 26.4(C-14), 24.9(C-5), 24.8(C-8), 20.5(C-15), 13.2(C-16).
26.4(C-14), 24.9(C-5), 24.8(C-8), 20.5(C-15), 13.2(C-16).
6-(10β-Dihydroartemisinoxy)hexyl isothiocyanate (5c). Colorless oil; yield: 76%; HR-ESI-MS (m/z) [M + H]+ calcd for: 426.2314, found: 426.2302; 1H NMR (400 MHz, CDCl3) δ 5.35 (s, 1H, H-12), 4.74 (d, J = 3.2 Hz, 1H, H-10), 3.80 (dt, J = 9.5, 6.4 Hz, 1H, –OCH2–), 3.48 (t, J = 6.6 Hz, 2H, –CH2NCS), 3.33 (dt, J = 9.5, 6.4 Hz, 1H, –OCH2–), 2.65–2.47 (m, 1H, H-9), 2.33 (td, J = 14.0, 3.8 Hz, 1H, H-4), 2.06–1.94 (m, 1H, H-4), 1.92–1.80 (m, 1H), 1.80–1.70 (m, 2H), 1.70–1.61 (m, 3H), 1.53 (m, 4H), 1.41 (s, 3H, CH3-14), 1.35 (m, 3H), 1.20 (m, 2H), 0.92 (d, J = 6.2 Hz, 3H, CH3-15), 0.89 (m, 1H, H-7), 0.86 (d, J = 7.2 Hz, 3H, CH3-16); 13C NMR (100 MHz, CDCl3) δ 129.8(–NCS), 104.2(C-3), 102.2(C-10), 88.1(C-12), 81.3(C-12a), 68.4(–OCH2–), 52.7(C-5a), 45.2(–CH2NCS), 44.6(C-8a), 37.7(C-6), 36.6(C-4), 34.8(C-7), 31.1(C-9), 30.1, 29.6, 26.5, 26.4(C-14), 25.7, 24.9(C-5), 24.7(C-8), 20.6(C-15), 13.2(C-16).
Cell lines and reagents
In the present study, a U87 cell line (from Prof. Wenbo Zhu, Zhongshan Medical School, Sun Yat-sen University) was used. The cells were maintained in Dulbecco's modified Eagle's medium (Gibo; Thermo Scientific, China) supplemented with 10% fetal bovine serum (PAN BIOTECH) in a humidified incubator at 37 °C with 5% CO2. Artemisinin-based isothiocyanate derivatives were dissolved in dimethyl sulfoxide and stored at −20 °C in the dark. Annexin V-FITC apoptosis detection kits were from Best Bio(China). The antibodies used are listed in Table 2.
Table 2 Information of antibodies for western blot
| Antibody name |
Manufacturer |
Catalog |
Dilutions |
| Primary antibody |
| Bax |
ABclonal |
A2211 |
1:1000 |
| BCL2 |
ABclonal |
A0208 |
1:1000 |
| Caspase 3 |
Cell signaling technology |
9665 |
1:1000 |
| Caspase 9 |
Cell signaling technology |
9508 |
1:1000 |
| Cytochrome C |
Cell signaling technology |
11940 |
1:1000 |
| LC3B |
Cell signaling technology |
3868 |
1:1000 |
| P62 |
MBL |
PM045 |
1:1000 |
| β-Actin |
Thermo Fisher |
MA5-11869 |
1:2500 |
|
| Secondary antibody |
| Anti-rabbit |
Beyotime |
A0208 |
1:1000 |
| Anti-mouse |
Beyotime |
A0216 |
1:1000 |
Cytotoxic assay
The cells were seeded in 96-well plates at a density of 1 × 104 cells/100 μL and treated with different concentrations of the compounds ranging from 1 to 250 μM. After 24 hours of incubation, MTT (Sigma-Aldrich) was added to cells following the treatment and incubated for 4 h at 37 °C. DMSO was subsequently added, and the absorbance was measured with a microplate reader after shaking for ∼30 s. The half maximal inhibitory concentration (IC50) was calculated. The cytotoxicity of 5b and DHA were investigated for the 24 and 72 h incubations.
Wound healing assay
The wound healing assay was performed in 6-well tissue culture plates. After the cells were allowed to attach and reach 80% confluence, a scratch in the cell monolayer was made using a sterilized pipette tip (10 μL). Thereafter, all wound-healing processes were performed in serum-free conditions. After washing with 0.01 M phosphate-buffered saline (PBS, pH 7.4), the plates were incubated with 5b or with DMSO, and images were captured at 0, 24, 48 and 72 h using an EVOS® Digital microscope. Cell death would affect the wound healing results. Thus, to avoid too much cell death, half of the IC50 (4 μM) of 5b was used. The distance of wound healing was calculated with the MRI wound healing tool of the Image J software (National Institutes of Health, MD, USA).
DAPI staining
Morphological changes of the nuclear chromatin of apoptotic cells were identified by staining with the DNA binding dye 4′,6-diamidino-2-phenylindole (DAPI, cat. no. D8417, Sigma Aldrich). Cells were grown in 12-well plates at a density of 1 × 105 cells per well followed by the desired treatment. After 16 h of incubation, the cells were washed with cold PBS, fixed with 4% paraformaldehyde for 30 min at room temperature, rewashed and then stained with DAPI solution (1:1000in PBS) at 37 °C for 5 min. After removing the staining solution, apoptotic cells were visualized using a fluorescence microscope.
Apoptotic analysis by flow cytometry
For this assay, U87 cancer cells were seeded at a density of 2 × 106 cells per mL and then incubated with 0, 8 and 16 μM of 5b for 16 h. The cells were harvested using trypsinization and then washed with ice-cold PBS and resuspended in binding buffer. After adding Annexin V-FITC and PI, the cells were incubated at 4 °C in the dark for 15 minutes according to the manufacturer instructions. The samples were analyzed by flow cytometer (Beckman Coulter, Epics XL) for the percentage of apoptotic and necrotic cells.
Western blot assay
Treated cells were washed with cold PBS and lysed in a radioimmunoprecipitation assay (RIPA) buffer supplemented with a proteinase inhibitor for extracting total proteins. Protein concentrations were determined by the bicinchoninic acid (BCA) protein assay. After denaturation, the proteins were separated in SDS polyacrylamide gel electrophoresis and transferred onto PVDF membranes. Nonspecific binding was blocked with 5% milk in TBST buffer for 2 h, followed by incubation with primary antibodies at 4 °C overnight and secondary antibodies at room temperature for 2 h. Blots were visualized using ECL detection reagents.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported by the Guangdong Provincial International Cooperation Project of Science & Technology (No. 2013B051000038), the National Natural Science Foundation of China (No. 31371070, No. 81671264), Grant (No. 201704020222, No. 201807010094) from Guangzhou Science, Technology and Innovation Commission and the Fundamental Research Funds for the Central Universities (No. 15ykjc08b) to R. Pi., the Science and Technology Project of Guangzhou City (201607010293) to X. He, (2014Y2-00500) to M. Li. The authors also thank the Talented Young Scientist Program supported by the China Science and Technology Exchange Center for the fellowship.
Notes and references
- R. Stupp, W. P. Mason, M. J. van den Bent, M. Weller, B. Fisher, M. J. Taphoorn, K. Belanger, A. A. Brandes, C. Marosi, U. Bogdahn, J. Curschmann, R. C. Janzer, S. K. Ludwin, T. Gorlia, A. Allgeier, D. Lacombe, J. G. Cairncross, E. Eisenhauer and R. O. Mirimanoff, N. Engl. J. Med., 2005, 352, 987–996 CrossRef CAS PubMed.
- J. P. Thakkar, T. A. Dolecek, C. Horbinski, Q. T. Ostrom, D. D. Lightner, J. S. Barnholtz-Sloan and J. L. Villano, Cancer Epidemiol., Biomarkers Prev., 2014, 23, 1985–1996 CrossRef CAS PubMed.
- M. A. van Agtmael, T. A. Eggelte and C. J. van Boxtel, Trends Pharmacol. Sci., 1999, 20, 199–205 CrossRef CAS PubMed.
- L. Gang, S. Song, X. Liu, A. Zhang, Z. Miao and C. Ding, RSC Adv., 2016, 6, 98975–98984 RSC.
- N. H. Zuma, F. J. Smit, C. de Kock, J. Combrinck, P. J. Smith and D. D. N'Da, Eur. J. Med. Chem., 2016, 122, 635–646 CrossRef CAS PubMed.
- Z. S. Zhang, J. Wang, Y. B. Shen, C. C. Guo, K. E. Sai, F. R. Chen, X. Mei, F. U. Han and Z. P. Chen, Oncol. Lett., 2015, 10, 379–383 CrossRef CAS PubMed.
- C. Qu, J. Ma, X. Liu, Y. Xue, J. Zheng, L. Liu, J. Liu, Z. Li, L. Zhang and Y. Liu, Front. Cell. Neurosci., 2017, 11, 310 CrossRef PubMed.
- Y. K. Wong, C. Xu, K. A. Kalesh, Y. He, Q. Lin, W. S. F. Wong, H. M. Shen and J. Wang, Med. Res. Rev., 2017, 37, 1492–1517 CrossRef CAS PubMed.
- X. Wu, Q. H. Zhou and K. Xu, Acta Pharmacol. Sin., 2009, 30, 501–512 CrossRef CAS PubMed.
- M. L. Abba, N. Patil, J. H. Leupold, M. E. M. Saeed, T. Efferth and H. Allgayer, Cancer Lett., 2018, 429, 11–18 CrossRef CAS PubMed.
- Y. Zhang, G. Xu, S. Zhang, D. Wang, P. Saravana Prabha and Z. Zuo, Nat. Prod. Bioprospect., 2018, 8, 303–319 CrossRef CAS PubMed.
- J. W. Fahey, A. T. Zalcmann and P. Talalay, Phytochemistry, 2001, 56, 5–51 CrossRef CAS.
- N. K. Amaglo, R. N. Bennett, R. B. L. Curto, E. A. Rosa, V. L. Turco, A. Giuffrida, A. L. Curto, F. Crea and G. M. Timpo, Food Chem., 2010, 122, 1047–1054 CrossRef CAS.
- C. Müller, J. Van Loon, S. Ruschioni, G. R. De Nicola, C. E. Olsen, R. Iori and N. Agerbirk, Phytochemistry, 2015, 118, 139–148 CrossRef PubMed.
- S. M. Lewis, Y. Li, M. J. Catalano, A. R. Laciak, H. Singh, D. R. Seiner, T. J. Reilly, J. J. Tanner and K. S. Gates, Bioorg. Med. Chem. Lett., 2015, 25, 4549–4552 CrossRef CAS PubMed.
- Y. J. Jeong, H. J. Cho, F. L. Chung, X. Wang, H. S. Hoe, K. K. Park, C. H. Kim, H. W. Chang, S. R. Lee and Y. C. Chang, Oncotarget, 2017, 8, 63949–63962 Search PubMed.
- Y. T. Yang, Y. Shi, M. Jay and A. J. Di Pasqua, Chem. Res. Toxicol., 2014, 27, 946–948 Search PubMed.
- T. S. Rajan, G. R. De Nicola, R. Iori, P. Rollin, P. Bramanti and E. Mazzon, Fitoterapia, 2016, 110, 1–7 CrossRef CAS PubMed.
- K. C. Liu, T. Y. Shih, C. L. Kuo, Y. S. Ma, J. L. Yang, P. P. Wu, Y. P. Huang, K. C. Lai and J. G. Chung, Am. J. Chin. Med., 2016, 44, 1289–1310 CrossRef CAS PubMed.
- R. K. Singh, T. S. Lange, K. Kim, Y. Zou, C. Lieb, G. L. Sholler and L. Brard, Bioorg. Med. Chem. Lett., 2007, 17, 5846–5852 CrossRef CAS PubMed.
- P. Sestili and C. Fimognari, BioMed Res. Int., 2015, 2015, 402386 Search PubMed.
- R. Grzywa, Ł. Winiarski, M. Psurski, A. Rudnicka, J. Wietrzyk, T. Gajda and J. Oleksyszyn, Bioorg. Med. Chem. Lett., 2016, 26, 667–671 CrossRef CAS PubMed.
- A. Minarini, A. Milelli, C. Fimognari, E. Simoni, E. Turrini and V. Tumiatti, Expert Opin. Drug Metab. Toxicol., 2014, 10, 25–38 CrossRef CAS PubMed.
- V. Citi, A. Martelli, L. Testai, A. Marino, M. C. Breschi and V. Calderone, Planta Med., 2014, 80, 610–613 CrossRef CAS PubMed.
- L. D. C. Mannelli, E. Lucarini, L. Micheli, I. Mosca, P. Ambrosino, M. V. Soldovieri, A. Martelli, L. Testai, M. Taglialatela, V. Calderone and C. Ghelardini, Neuropharmacology, 2017, 121, 49–59 CrossRef PubMed.
- S. Huang, J. H. Chua, W. S. Yew, J. Sivaraman, P. K. Moore, C. H. Tan and L. W. Deng, J. Mol. Biol., 2010, 396, 708–718 CrossRef CAS PubMed.
- Z. Ma, Q. Bi and Y. Wang, Oral Dis., 2015, 21, 156–162 CrossRef CAS PubMed.
- Y. Liu, Z. Liu, J. Shi, H. Chen, B. Mi, P. Li and P. Gong, Molecules, 2013, 18, 2864–2877 CrossRef CAS PubMed.
- Y. Tian, Z. Liang, H. Xu, Y. Mou and C. Guo, Molecules, 2016, 21, 758 CrossRef PubMed.
- J. M. Wu, F. Shan, G. S. Wu, Y. Li, J. Ding, D. Xiao, J. X. Han, G. Atassi, S. Leonce, D. H. Caignard and P. Renard, Eur. J. Med. Chem., 2001, 36, 469–479 CrossRef CAS PubMed.
- B. Luo, J. Wang, X. Li, W. Lu, J. Yang, Y. Hu, P. Huang and S. Wen, Molecules, 2017, 22, 773 CrossRef PubMed.
- A. K. Pathak, D. U. Jain and R. F. Sharma, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1995, 34B, 992–993 CAS.
- A. R. Butler, L. Conforti, P. Hulme, L. M. Renton and T. J. Rutherford, J. Chem. Soc., Perkin Trans. 1, 1999, 2, 2089–2092 RSC.
- A. Grada, M. Otero-Vinas, F. Prieto-Castrillo, Z. Obagi and V. Falanga, J. Invest. Dermatol., 2017, 137, e11–e16 CrossRef CAS PubMed.
- L. Jia, Q. Song, C. Zhou, X. Li, L. Pi, X. Ma, H. Li, X. Lu and Y. Shen, PLoS One, 2016, 11, e0147157 CrossRef PubMed.
- S. Elmore, Toxicol. Pathol., 2007, 35, 495–516 CrossRef CAS PubMed.
- N. Morishima, K. Nakanishi, H. Takenouchi, T. Shibata and Y. Yasuhiko, J. Biol. Chem., 2002, 277, 34287–34294 CrossRef CAS PubMed.
- A. M. Schläfli, S. Berezowska, O. Adams, R. Langer and M. P. Tschan, Eur. J. Histochem., 2015, 59, 2481 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08162j |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ef,
Mingtao Lid,
Ying Ou-yangg,
Rongbiao Pi*bd and
Xixin He
ef,
Mingtao Lid,
Ying Ou-yangg,
Rongbiao Pi*bd and
Xixin He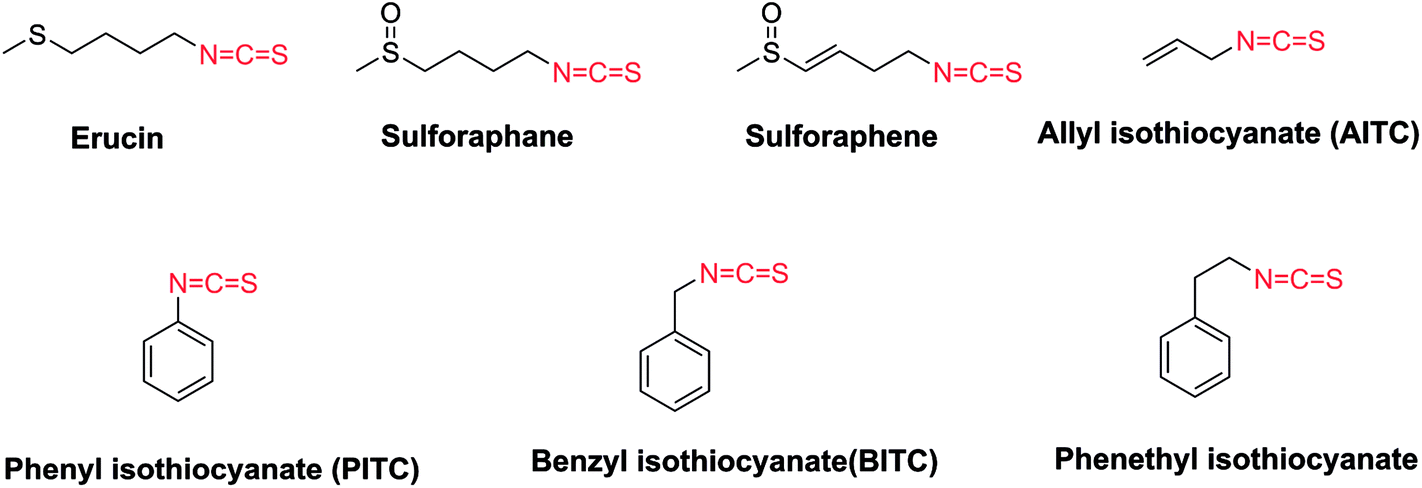
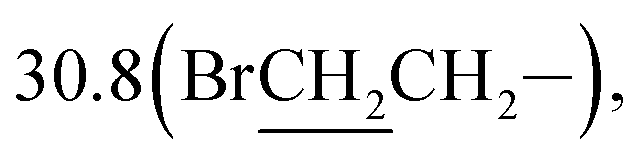 26.4(C-14), 24.8(C-5), 24.7(C-8), 20.5(C-15), 13.2(C-16).
26.4(C-14), 24.8(C-5), 24.7(C-8), 20.5(C-15), 13.2(C-16).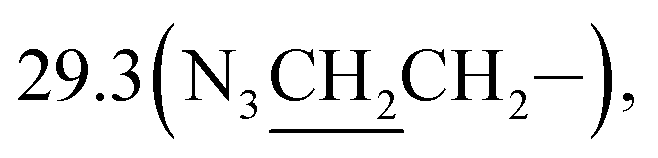 26.8(C-14), 24.8(C-5), 24.7(C-8), 20.5(C-15), 13.2(C-16).
26.8(C-14), 24.8(C-5), 24.7(C-8), 20.5(C-15), 13.2(C-16).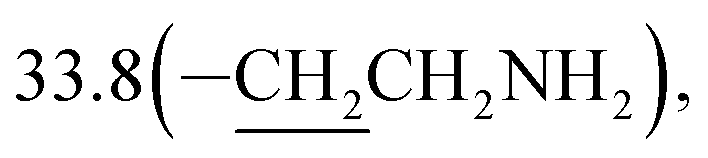 31.1(C-9), 26.4(C-14), 24.9(C-5), 24.7(C-8), 20.6(C-15), 13.2(C-16).
31.1(C-9), 26.4(C-14), 24.9(C-5), 24.7(C-8), 20.6(C-15), 13.2(C-16).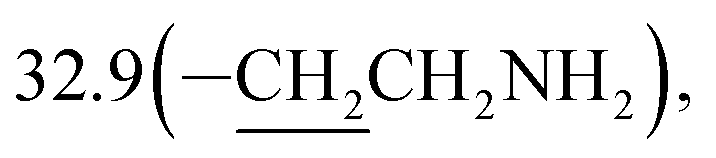 31.1(C-9), 29.8, 26.8, 26.4(C-14), 26.3, 24.9(C-5), 24.6(C-8), 20.6(C-15), 13.2(C-16).
31.1(C-9), 29.8, 26.8, 26.4(C-14), 26.3, 24.9(C-5), 24.6(C-8), 20.6(C-15), 13.2(C-16).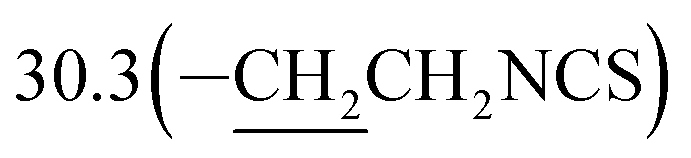 26.4(C-14), 24.9(C-5), 24.8(C-8), 20.5(C-15), 13.2(C-16).
26.4(C-14), 24.9(C-5), 24.8(C-8), 20.5(C-15), 13.2(C-16).