
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterization of nano-crystalized HfC based on an aqueous solution-derived precursor
Youlin Jiangabc,
Dewei Ni *ab,
Qi Dingabcd,
Bowen Chenabc,
Xiaowu Chenab,
Yanmei Kanab and
Shaoming Dongab
*ab,
Qi Dingabcd,
Bowen Chenabc,
Xiaowu Chenab,
Yanmei Kanab and
Shaoming Dongab
aState Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, China. E-mail: deweini@mail.sic.ac.cn
bStructural Ceramics and Composites Engineering Research Center, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, China. E-mail: smdong@mail.sic.ac.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
dShanghaiTech University, Shanghai 201210, China
First published on 26th November 2018
Abstract
Nano-crystalized HfC was successfully synthesized based on an aqueous solution-derived precursor using hafnium tetrachloride and sucrose as raw materials. The precursor can be converted to pure phase HfC after pyrolysis at ∼600 °C and subsequent carbothermal reduction reaction at ∼1600 °C, with a ceramic yield of around 46.3%. Reaction mechanisms of the synthesis process are revealed based on FT-IR, TG-DSC, TEM analysis and thermodynamic calculations, etc. The obtained HfC nanoparticles possess an equiaxial shape with an average particle size of ∼73 nm. Oxygen content of the as-synthesized HfC powders is as low as 0.64 wt%, which exists in the form of an amorphous Hf–O–C thin layer covering the surface of the HfC particles. This feasible and promising method for HfC particle synthesis is believed to be suitable for the fabrication of continuous fibers reinforced HfC ceramic matrix composites.
Introduction
As a member of the ultrahigh temperature ceramics (UHTCs), hafnium carbide (HfC) has attracted extensive attention due to its extremely high melting point (3890 °C), low oxygen diffusion coefficient, good mechanical properties and excellent chemical stability.1–4 This makes HfC a promising candidate for ultra-high temperature and extreme environment applications such as hypersonic vehicle thermal structural components and rocket engine combustion chambers, etc.5Conventionally, HfC can be synthesized by various methods including solid state combustion via direct combination of the elements at elevated temperatures, chemical vapor deposition, carbothermal reduction of metal oxides, sol–gel route, laser and plasma chemical synthesis, etc.6–10 Solution-based processing possesses the advantages of forming a homogenous distribution of all elements on a molecular level, which can reduce the kinetic barriers and lower the reaction temperatures, thus leading to fine size of the synthesized particles.11–13 Moreover, it has great potential to be used as facile precursor for the fabrication of continuous fibers reinforced HfC ceramic matrix composites.14–20 Consequently, synthesis of HfC powders by solution-based processing has drawn much attention.
For the synthesis of HfC nanoparticles via solution-based processing, organic reactants and/or organic solvent were generally used, which is expensive and not eco-friendly. Sucrose is a non-toxic, inexpensive and easily accessible polyol sugar. As an environment friendly organic substance, it is an excellent candidate for eco-friendly applications. With several hydroxyl groups in its molecular structure, sucrose can be used as a chelating agent to form metal complexes with Hf ions and as a carbon source for subsequent carbothermal reduction.21 A plurality of hydroxyl groups on sucrose molecule might allow absorption of multiple Hf ions as well as the formation of a metal complex consists of several Hf ions and sucrose molecules.
Herein, we report a facile and promising aqueous solution-derived precursor approach for the synthesis of HfC nanoparticles by using hafnium tetrachloride and sucrose as raw materials. Reaction mechanisms of the synthesis process from the pre-ceramic precursor to HfC powders were revealed based on FT-IR, TG-DSC, TEM analysis and thermodynamic calculations, etc.
Experimental
Materials and processing
Commercial hafnium tetrachloride (HfCl4, purity 99.5%, Suzhou Chuanmao Metal Material Co. Ltd, Suzhou, China), sucrose (C12H22O11, AR grade, Aladdin industrial Corporation, Shanghai, China) and polyvinylpyrrolidone (PVP, Beijing HWRK Chemistry Co. Ltd, Beijing, China) were used as raw materials for the synthesis of HfC precursor.| HfO2 + 3C → HfC + CO(g) | (1) |
According to reaction corresponding amount of hafnium tetrachloride (HfCl4) and sucrose (C12H22O11) were used. 64.1 g hafnium tetrachloride was dissolved in 100 mL deionized water. Then 34.2 g sucrose and 2 g PVP were added to the solution. The molar ratio between HfCl4 and sucrose added was set as 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. PVP addition was fixed at around 2 wt% of the solution. After being magnetic stirred for 6 hours, a homogenous aqueous solution precursor was formed.
:1. PVP addition was fixed at around 2 wt% of the solution. After being magnetic stirred for 6 hours, a homogenous aqueous solution precursor was formed.
The obtained precursor was cross-linked and cured at 100 °C for 2 hours, during which it was gelatinized to form a jelly followed by dehydration. The cured precursor was then pyrolyzed at 600 °C for 2 hours under flowing argon with a heating rate of 5 °C min−1. To investigate the carbothermal reduction process comprehensively, the pyrolyzed precursor was further heat treated at 1400–1650 °C in vacuum and pure phase HfC powders were obtained at temperatures higher than 1600 °C.
Characterization
Fourier transform infrared (FTIR) spectra of starting materials and precursor were measured by NICOLET Is 10 spectrometer (Thermo Scientific, USA). Thermal gravity (TG) and differential scanning calorimetry (DSC) analysis were performed by a Netzsch STA 499 F3 Jupiter® under flowing argon atmosphere at a heating rate of 10 °C min−1 in the temperature range of 40–1400 °C.Crystal structures and phase composition of the as-synthesized powders were determined by X-ray diffraction (XRD) with a Rigaku Ultima IV diffractometer using Cu Kα radiation (λ = 1.540598 Å) in the angle range of 10–80°, operated at 40 kV, 40 mA with a step width of 3° min−1. Morphologies of the synthesized powders were observed by field emission scanning electron microscope (FESEM, Hitachi SU8220, Japan) along with energy dispersive spectroscopy (EDS) for chemical analysis. The particle sizes were determined by post processing of the SEM images with free software ImageJ®. To reduce the error in the determination of particle size, at least ten images with hundreds of particles were taken in random regions of the samples and used for the statistical analysis. Characterization of the powders were also performed by transmission electron microscope (TEM, JEOL JEM-2100F, Japan) along with selected area electron diffraction (SAED) and EDS. Oxygen content of the as-synthesized HfC powders was determined by nitrogen/oxygen determinator (LECO instrument LD, USA).
Results and discussion
The FTIR spectra of the raw materials (HfCl4, C12H22O11 and PVP) and the synthesized precursor (Fig. 1) were obtained to understand the molecular structure evolution and the principal interactions between the reactants. In the FTIR spectra for the HfCl4 (Fig. 1a), the vibration bands at 3431 cm−1 and 2353 cm−1 are associated with the ν(O–H) stretching of water molecules (adsorbed water molecule), while the band at 1634 cm−1 corresponds to the bending vibration of water molecules coordinated to Hf cations.11 For C12H22O11 (Fig. 1b), the band at 2918 cm−1 corresponds to the symmetric stretching vibration of ν(C–H), while the band at 1444 cm−1 is associated with the antisymmetric deformation peak of ν(–CH2). The band at 1064 cm−1 can be attributed to the stretching peak of ν(C–O), while the peak at 920 cm−1 corresponds to the stretching vibration of ν(C–O–C). For PVP (Fig. 1c), the respective assignments of the peaks are 3442 (ν(O–H)), 1643 (ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)) and 1444 (ν(–CH2)) cm−1. For precursor after heated and cured (Fig. 1d), the bands at 3431 (ν(O–H)), 2353 (ν(O–H)), 1641 (ν(O–H) + ν(CO)) and 1444 (ν(–CH2)) cm−1 can also be observed, while the peak at 1295 cm−1 corresponds to ν(C–N).17 The peak at 1091 cm−1 can be attributed to the stretching peak of ν(C–O), while the peak at 1026 cm−1 corresponds to the stretching vibration of ν(C–O–C). No obvious peak of (ν(Hf–O)) at around 750–500 cm−1 is observed in Fig. 1d, implying that no Hf–O bonding exists in the precursor. The slight difference of peak position of (ν(C–O)) between sucrose and precursor indicates the chemical environment change for C–O groups, which should results from the adsorption of Hf ions. It is conjectured that a complex of Hf ion, sucrose and PVP was formed. The hydrolysis of sucrose and the possible transesterification reaction between sucrose and PVP leads to the change of (ν(C–O–C)) peak. The precursor after pyrolysis at 600 °C was also characterized with FTIR, as shown in Fig. 1e. Most peaks of organo-functional groups disappear except the ones at around 3431 cm−1 and 1634 cm−1 which are related to ν(O–H) from adsorbed water, indicating the complete of pyrolysis reaction at 600 °C. Furthermore, tiny peaks of (ν(Hf–O)) at 750–500 cm−1 appear,10,22 which indicates the formation of HfO2 during pyrolysis process.
O)) and 1444 (ν(–CH2)) cm−1. For precursor after heated and cured (Fig. 1d), the bands at 3431 (ν(O–H)), 2353 (ν(O–H)), 1641 (ν(O–H) + ν(CO)) and 1444 (ν(–CH2)) cm−1 can also be observed, while the peak at 1295 cm−1 corresponds to ν(C–N).17 The peak at 1091 cm−1 can be attributed to the stretching peak of ν(C–O), while the peak at 1026 cm−1 corresponds to the stretching vibration of ν(C–O–C). No obvious peak of (ν(Hf–O)) at around 750–500 cm−1 is observed in Fig. 1d, implying that no Hf–O bonding exists in the precursor. The slight difference of peak position of (ν(C–O)) between sucrose and precursor indicates the chemical environment change for C–O groups, which should results from the adsorption of Hf ions. It is conjectured that a complex of Hf ion, sucrose and PVP was formed. The hydrolysis of sucrose and the possible transesterification reaction between sucrose and PVP leads to the change of (ν(C–O–C)) peak. The precursor after pyrolysis at 600 °C was also characterized with FTIR, as shown in Fig. 1e. Most peaks of organo-functional groups disappear except the ones at around 3431 cm−1 and 1634 cm−1 which are related to ν(O–H) from adsorbed water, indicating the complete of pyrolysis reaction at 600 °C. Furthermore, tiny peaks of (ν(Hf–O)) at 750–500 cm−1 appear,10,22 which indicates the formation of HfO2 during pyrolysis process.
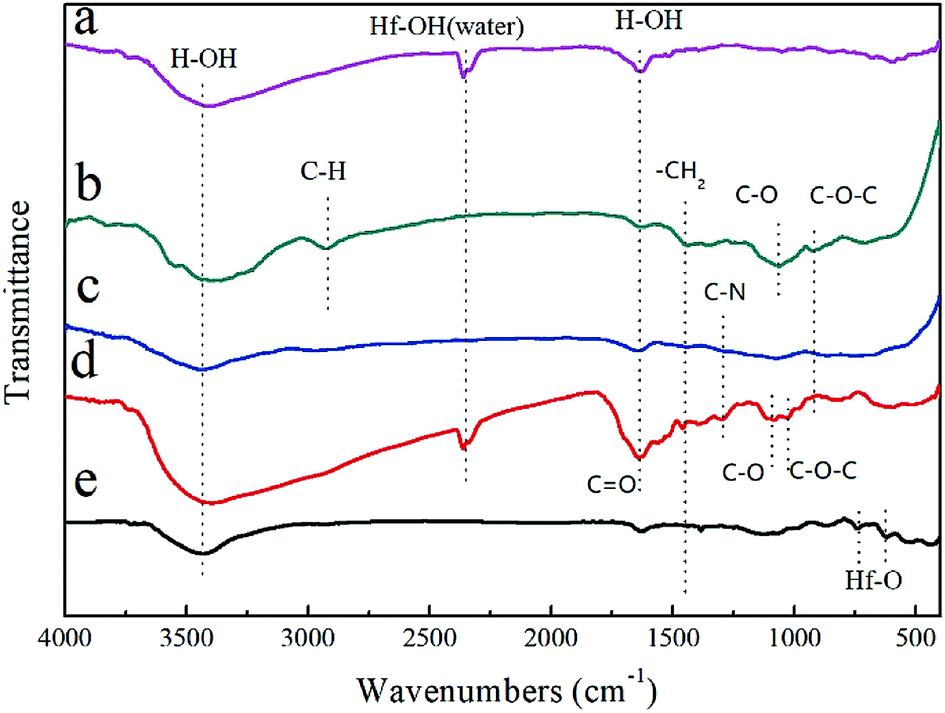 | ||
| Fig. 1 FTIR spectra of the (a) HfCl4, (b) C12H22O11, (c) PVP, (d) precursor after curing, and (e) precursor after pyrolysis at 600 °C. | ||
The pyrolysis process of the precursor was studied by TG-DSC analysis as illustrated in Fig. 2. The pyrolysis process of the precursor can be divided into approximately 3 stages. A weight loss of 9.8% at the first stage from starting temperature to around 200 °C is mainly contributed from the evaporation of free water and separation of adsorbed water. The process can last till up to 300 °C according to the endothermic peak on DSC curve. Together with evaporation of constitution water and small molecules produced by decomposition of organic matters, a weight loss of 26.4% between 200 and 600 °C can be observed. After 600 °C, a gradual descending on TG curve occurs probably due to the continuous carbonization of incompletely decomposed organic compounds. Carbothermal reduction reaction of HfO2 is initiated at ∼1400 °C based on the drop on TG curve. In accordance with the result of TG-DSC analysis, the pyrolysis temperature of precursor was set as 600 °C and the subsequent carbothermal reduction temperature was set in the temperature range of 1400–1650 °C. Meanwhile a dwelling time of 2 hours was set for both processes to ensure a complete reaction of each step.23
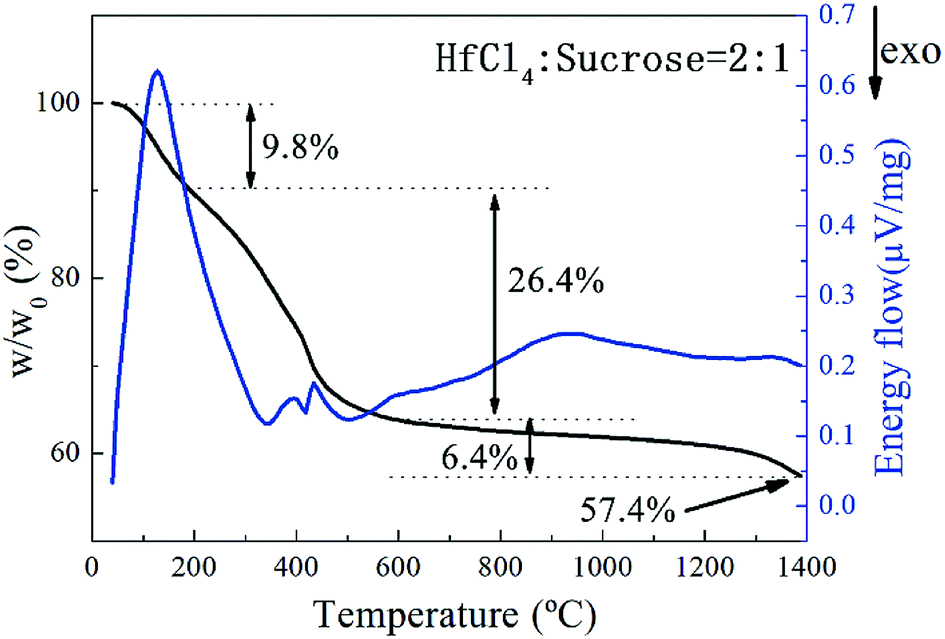 | ||
| Fig. 2 TG-DSC curves of the prepared HfC precursor after curing. | ||
Variation of phase composition during heat treatment was studied using XRD. The results are shown in Fig. 3. The original precursor is amorphous substance with no obvious crystallization peaks on the XRD pattern (Fig. 3a). After pyrolysis at 600 °C, it converts to monoclinic HfO2 and possible amorphous carbon, as can be observed in Fig. 3b. The main crystalline phase is still monoclinic HfO2 and a small amount of HfC appears after heat treatment at 1400 °C (Fig. 3c), indicating the initiation of carbothermal reduction reaction. As heat treatment temperature rising, the peaks of monoclinic HfO2 gradually decrease and HfC becomes the main crystalline phase as can be seen in Fig. 3d and e. Only diffraction peaks of cubic HfC can be found in Fig. 3e, indicating the complete of carbothermal reduction at 1600 °C. However, oxygen content of the as synthesized HfC powder at 1600 °C is still as high as 1.58 wt%. A further heat treatment at a higher temperature (1650 °C) was carried out, where the oxygen content is reduced to 0.64 wt%. Based on the results above, 1650 °C was used for the final carbothermal reduction. A shift of HfC characteristic peaks to higher angles is revealed, as shown in the red square frame of Fig. 3. This signifies the probable formation of non-stoichiometric HfC.
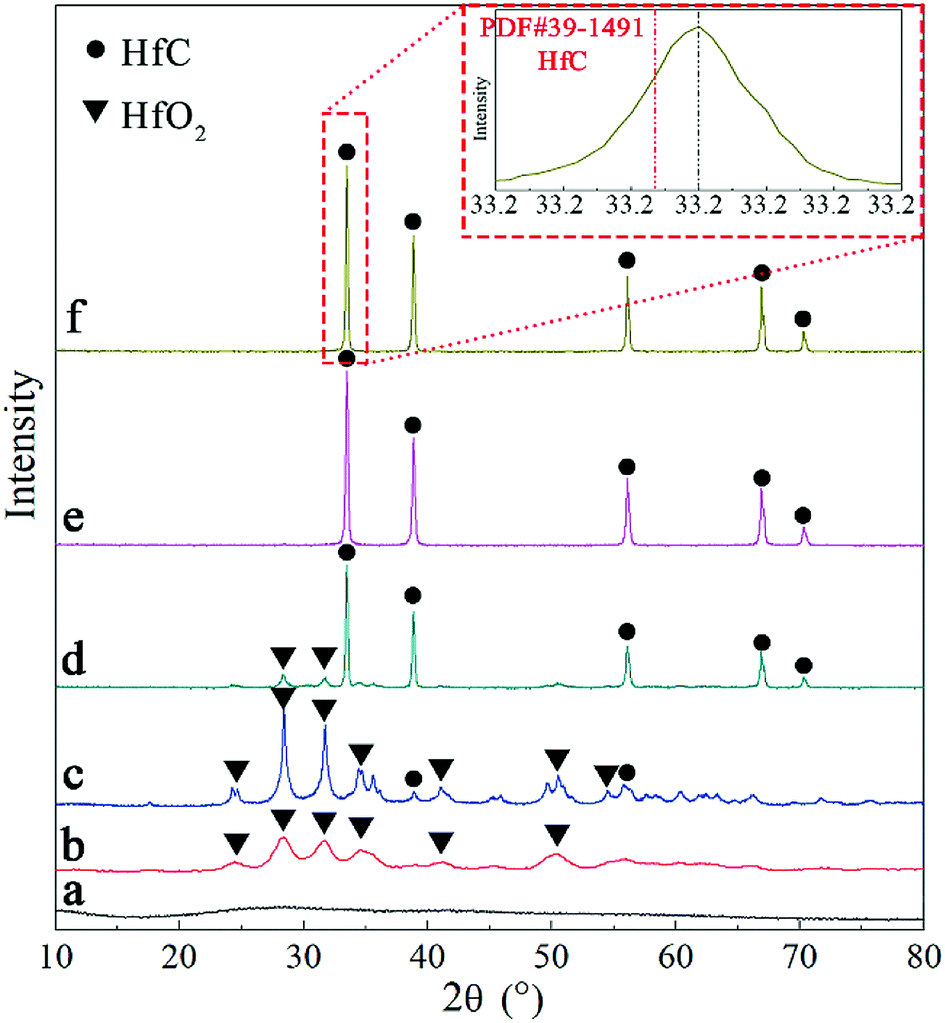 | ||
| Fig. 3 XRD patterns of (a) precursor, (b) product after pyrolysis at 600 °C and products after heat treated at (c) 1400 °C (d) 1500 °C (e) 1600 °C and (f) 1650 °C. | ||
Morphology of the as-synthesized HfC powders was observed by SEM, as shown in Fig. 4a. It can be seen that the obtained HfC particles present an equiaxial shape with some extent of agglomeration. In addition, floccule matter appears surrounding the HfC particles, which is proved to be residual carbon indicated by EDS analysis (not shown here), implying an existence of small amount of residual carbon. EDS analysis was used for an elements distribution map as shown in Fig. 4b–d. Carbon and hafnium are well distributed in the powder, indicating that the precursor has almost completely been transformed into homogenous HfC powder with residual carbon mixed. It is in strong agreement with XRD analysis. Oxygen makes up a very low share of the powder component, which is in line with the result of oxygen content test.
 | ||
| Fig. 4 (a) SEM image of synthesized powder and EDS mapping of (b) oxygen (c) carbon and (d) hafnium. | ||
Analysis of particle size distribution was performed by post processing of the SEM images using the freeware software ImageJ® and the results are shown in Fig. 5. As can be seen, the size of the HfC particles is mainly located between 30–100 nm and the average particle size is 73 ± 37 nm (Fig. 5a). Research has shown that the existence of residual carbon can limit grain growth by physically separating the particles.7 It is believed that the existence of residual free carbon in this work reduces the HfC particles mutual contact, thus preventing the coarsening and leading to the fine particle size. This has also been further confirmed in this work. Another HfC precursor with 25% excessive source was prepared. And consequently, a smaller average particle size of 57 ± 33 nm is achieved for the synthesized HfC powders, as shown in Fig. 5b.
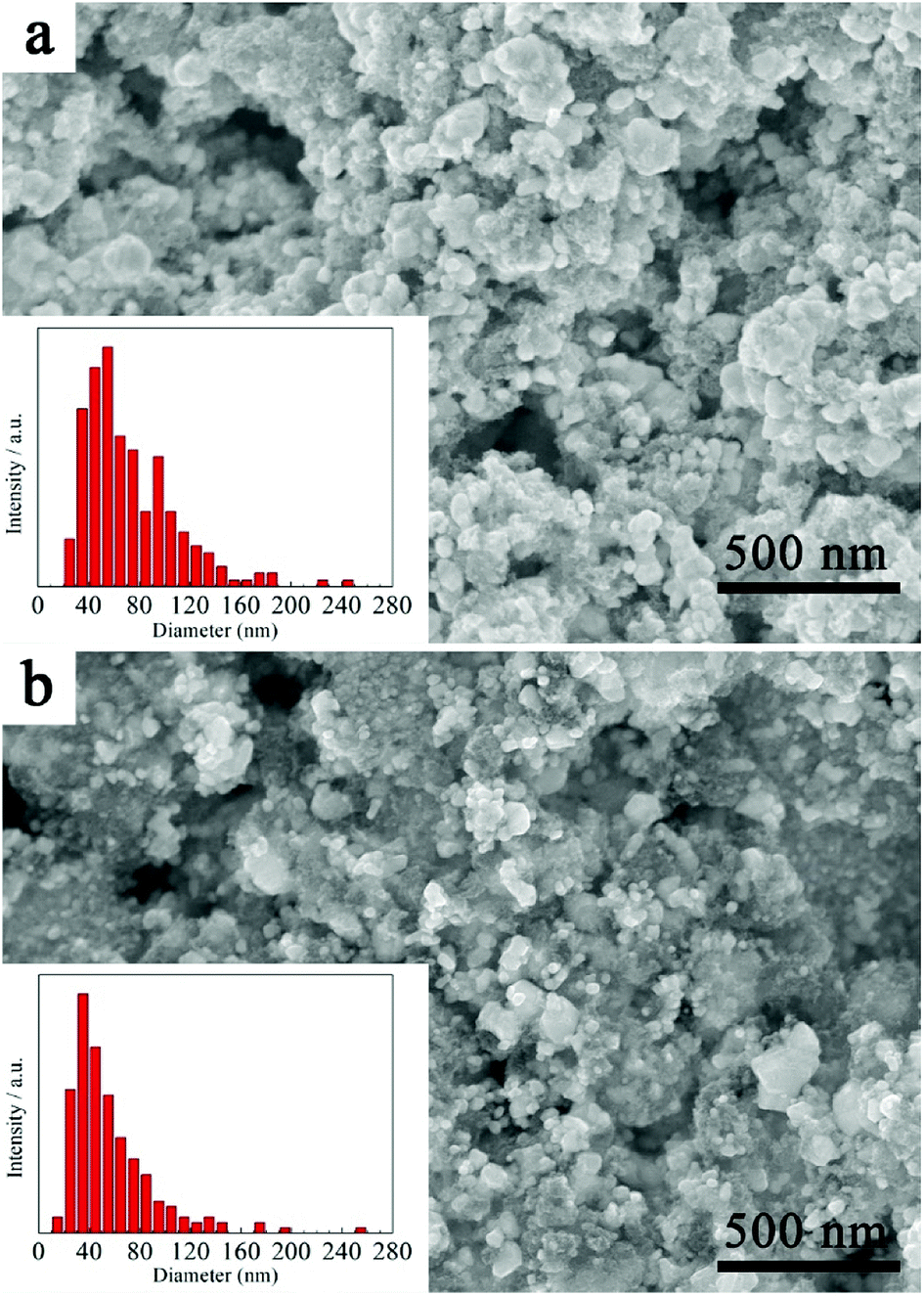 | ||
| Fig. 5 SEM images and particle size distribution of (a) as-synthesized HfC powder and (b) HfC powder synthesized from precursor with 25% excessive carbon source. | ||
Further analysis of the synthesized HfC powders was performed by TEM and HRTEM, as shown in Fig. 6. SAED patterns inserted in TEM images reveal the crystallized nature of the synthesized HfC particles and a face-centered cubic structure can be easily identified. The layer fringes with the interlayer distance are confirmed in the HRTEM images as can be seen in Fig. 6b. The interlayer distances of the synthesized HfC particles are 0.2680 nm corresponded to the (111) face and 0.2324 nm corresponded to (200) face, which are a little bit larger than d(111) = 0.26776 nm and d(200) = 0.23189 nm for standard HfC of PDF#39-1491. This reveals the presence of lattice distortion, which should result from the formation of non-stoichiometric Hf1−aC. With the increase of sucrose content in the precursor (Fig. 6c and d), larger lattice distortion (d(111) = 0.2687 nm and d(200) = 0.2346 nm) was achieved in the synthesized HfC powders. This confirms that more non-stoichiometric Hf1−aC is formed due to more residual carbon existed in the lattice. This is in good agreement with the XRD result.
 | ||
| Fig. 6 (a) TEM image and (b) HRTEM image of synthesized particles; (c) TEM image and (d) HRTEM image of particles synthesized with 25% excessive carbon source. The SEAD patterns of the HfC particles are inserted in (a) and (c) at the * position, respectively. | ||
Obviously, the HfC particles present a core–shell structure as shown in the HRTEM images. The core is composed of HfC crystal structure while the shell is in form of an amorphous gangue. To get a full understand of the components of the core–shell structure, element analysis was performed for the variation trend of C and O along the direction of the red arrow signed in Fig. 7. As can be observed, the carbon content keeps rising and then becomes stable in the inner core. However, the oxygen content rises till the outer edge of the amorphous shell, followed by a decrease at the border of the core and the shell, and in turn becomes stable in the core. Therefore, it can be concluded that the dominant proportion of oxygen content in the synthesized powders exists in the form of amorphous Hf–O–C thin layer covering on the surface of HfC particles. With the increase of sucrose content in the precursor, no evident decrease of the thickness of the amorphous Hf–O–C thin layer is observed (in average 3.29 nm and 3.27 nm, respectively). The amorphous Hf–O–C thin layer covered on the fine HfC particles is probably related to the mechanism of HfO2–C carbothermal reduction or surface oxidation during post-treatment.
| HfO2 → HfO(g) + 1/2O2(g) | (2) |
| C + 1/2O2(g) → CO(g) | (3) |
| HfO2 + CO(g) → HfO(g) + CO2(g) | (4) |
| C + CO2(g) → 2CO(g) | (5) |
| HfO(g) + CO(g) → HfCxOy + zCO2(g) | (6) |
| HfCxOy + CO(g) → Hf1−aC + CO2(g) | (7) |
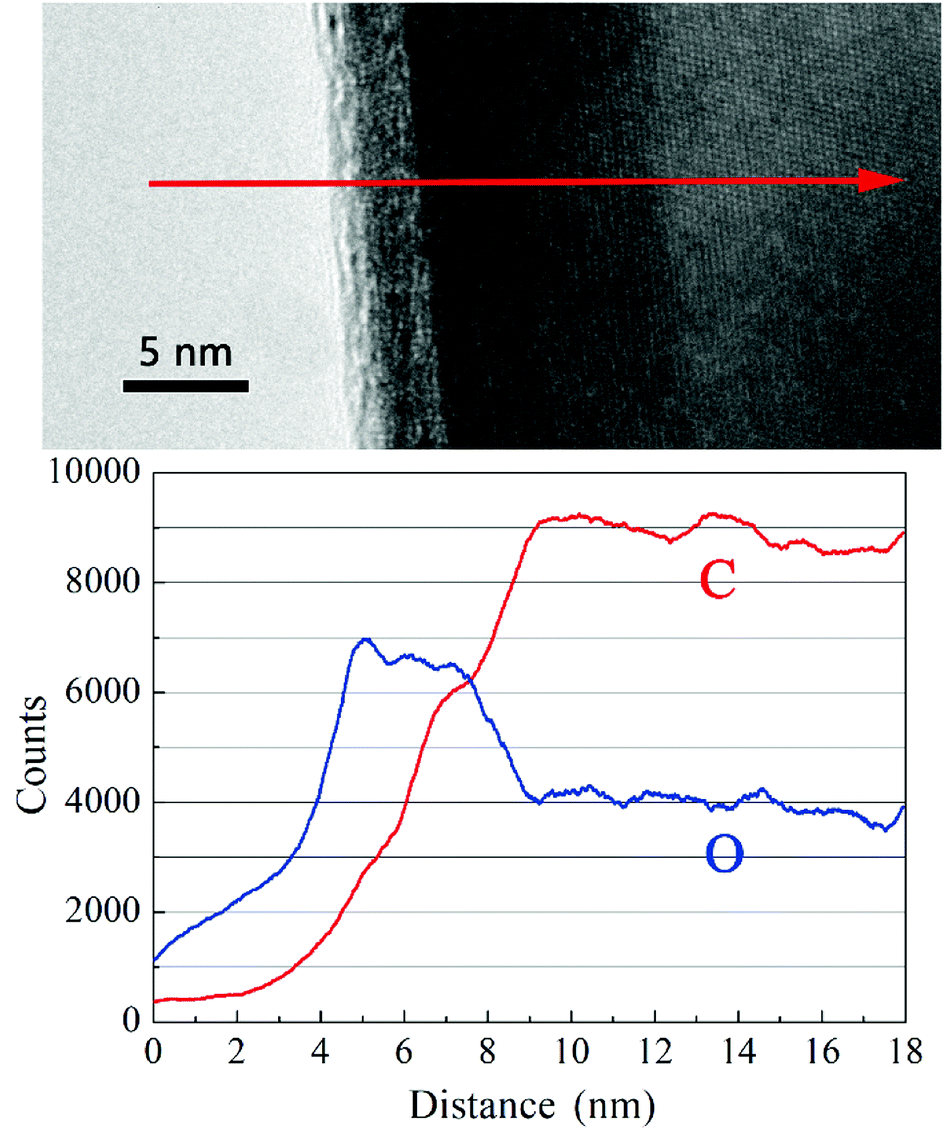 | ||
| Fig. 7 Variation of C and O contents via EDS lining scanning. | ||
The mechanisms of HfO2 carbothermal reduction has been widely discussed in literature.23,24 The formation of crystallized core and amorphous shell can be explained with the theory of gas–solid reaction.23 A proposed model is shown in Fig. 8. Carbon particles and HfO2 particles are produced from the precursor during pyrolysis. As shown in Fig. 8a, with temperature rising, HfO2 is destabilized, producing HfO(g) and O2(g) as described by eqn (2) at the beginning of the process. With the existence of O2(g), carbon particles is destabilized and CO(g) is emitted by eqn (3). Further destabilization of HfO2 carries out with the participation of CO(g) as described by eqn (4), as shown in Fig. 8b. The produce of CO2(g) in turn takes part in reaction of eqn (5), producing CO(g). The continuous reaction of eqn (3) and eqn (5) keeps providing CO(g). The condensation phenomenon of produced HfO(g) and CO(g) then accounts for the nucleation of HfCxOy as revealed in eqn (6) and Fig. 8c. The abrupt condensation of gaseous species gives an amorphous cladding layer around the HfCxOy oxycarbide crystal. Then the formation of oxycarbide phase is achieved through a solid state diffusion process within the amorphous layer. Researches25,26 have suggested that the destabilization of carbon might be the rate limiting step of the reaction progresses expressed in Fig. 8a–c, that is, the removal of CO(g) is slower while the release of HfO(g) within gaseous medium is comparably enhanced. Consequently, a preferential nucleation of oxycarbide in the carbon area is achieved. With an increased concentration of sucrose in the precursor, a rather low concentration of HfO(g) results in smaller particles. Fig. 8d shows the further reduction of the oxycarbide substance. With oxygen bumped out and consumed by CO(g), as described by eqn (7), the oxycarbide substance is transformed into ∼HfC. With excessive carbon diffusing into lattice, the product forms a matter with a non-stoichiometric Hf1−aC. The thin oxygen-rich outer layer covering the core observed in Fig. 6 and 7 might be resulted from the relatively short time for complete carbothermal reduction or surface oxidation caused by post-treatment.
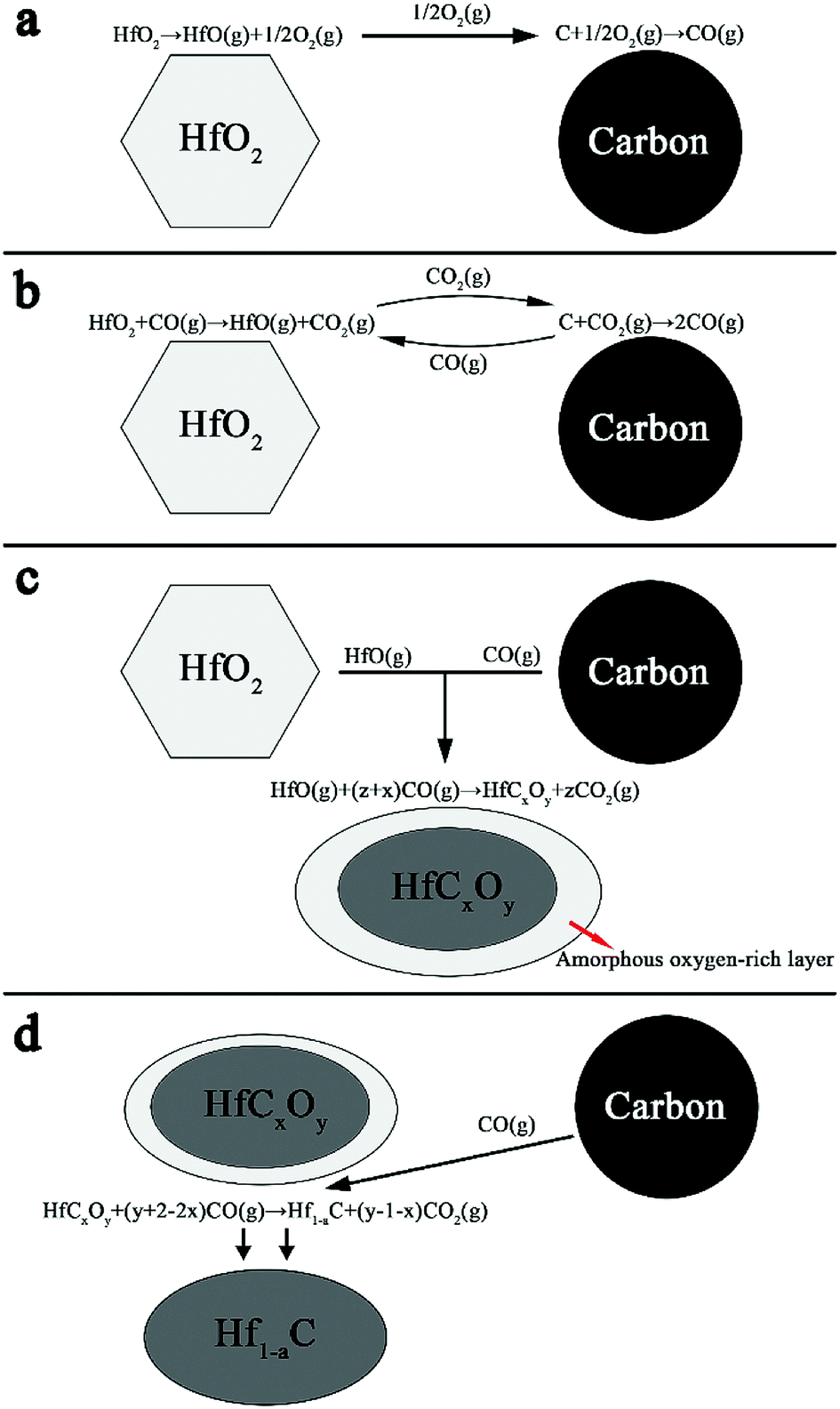 | ||
| Fig. 8 Schematic diagram of the HfO2–C system carbothermal reduction reaction process. | ||
Conclusions
Based on an aqueous solution-derived precursor, nano-crystalized HfC was successfully synthesized. The precursor was prepared using hafnium tetrachloride and sucrose as raw materials. The transformation process of the precursor during heat treatment was studied using FT-IR, TG-DSC, XRD, SEM and TEM, etc. The precursor can be converted to pure phase HfC after pyrolysis at ∼600 °C and subsequent carbothermal reduction reaction at ∼1600 °C, with a ceramic yield of around 46.3%. The obtained HfC nanoparticles possess an equiaxial shape with an average particle size of ∼73 nm. Oxygen content of the as-synthesized HfC powders is as low as 0.64 wt%, which existed in the form of amorphous Hf–O–C thin layer covering on the surface of HfC particles. This might be due do incomplete carbothermal reduction or surface oxidation caused by post-treatment. Carbothermal reduction reaction mechanisms of the synthesis process are revealed based on TG-DSC, HRTEM and thermodynamic calculations, etc. The theory of gas–solid reaction can be applied to explain the variation of substances during carbothermal reduction process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the “The National Key Research and Development Program of China” (2016YFB0700202), “Chinese Academy of Sciences Innovative Funding” (CXJJ-17-M169), “CAS Pioneer Hundred Talents Program” and “National Natural Science Foundation of China” (No. 51702341) are greatly acknowledged.References
- O. Cedillos-Barraza, D. Manara, K. Boboridis, T. Watkins, S. Grasso, D. D. Jayaseelan, R. J. Konings, M. J. Reece and W. E. Lee, Sci. Rep., 2016, 6, 37962 CrossRef CAS PubMed.
- M. M. Opeka, I. G. Talmy, E. J. Wuchina, J. A. Zaykoski and S. J. Causey, J. Eur. Ceram. Soc., 1999, 19, 2405–2414 CrossRef CAS.
- R. Savino, M. De Stefano Fumo, L. Silvestroni and D. Sciti, J. Eur. Ceram. Soc., 2008, 28, 1899–1907 CrossRef CAS.
- L. Silvestroni, A. Bellosi, C. Melandri, D. Sciti, J. X. Liu and G. J. Zhang, J. Eur. Ceram. Soc., 2011, 31, 619–627 CrossRef CAS.
- N. P. Padture, Nat. Mater., 2016, 15, 804–809 CrossRef CAS PubMed.
- B. B. Bokhonov and D. V. Dudina, Ceram. Int., 2017, 43, 14529–14532 CrossRef CAS.
- J. Liu, Y. Kan and G. Zhang, J. Am. Ceram. Soc., 2010, 93, 980–986 CrossRef CAS.
- D. Lu, W. Wang, H. Wang, J. Zhang, Y. Wang, F. Zhang and Z. Fu, Ceram. Int., 2016, 42, 8108–8114 CrossRef CAS.
- B. Matovic, B. Babic, D. Bucevac, M. Cebela, V. Maksimovic, J. Pantic and M. Miljkovic, Ceram. Int., 2013, 39, 719–723 CrossRef CAS.
- M. D. Sacks, C. an Wang, Z. Yang and A. Jain, J. Mater. Sci., 2004, 39, 6057–6066 CrossRef CAS.
- N. Patra, N. Al Nasiri, D. D. Jayaseelan and W. E. Lee, Ceram. Int., 2016, 42, 1959–1963 CrossRef CAS.
- T. Cai, D. Liu, W. Qiu, W. Han and T. Zhao, J. Am. Ceram. Soc., 2017, 101, 20–24 CrossRef.
- Z. Mei, X. Su, G. Hou, F. Guo and Y. Cheng, Adv. Mater. Res., 2012, 457–458, 283–286 CAS.
- A. Paul, S. Venugopal, J. G. P. Binner, B. Vaidhyanathan, A. C. J. Heaton and P. M. Brown, J. Eur. Ceram. Soc., 2013, 33, 423–432 CrossRef CAS.
- Y. Chang, W. Sun, X. Xiang, Z. Chen, Y. Wang, Z. Hao and Y. Xu, Ceram. Int., 2016, 42, 16906–16915 CrossRef CAS.
- M. Kütemeyer, L. Schomer, T. Helmreich, S. Rosiwal and D. Koch, J. Eur. Ceram. Soc., 2016, 36, 3647–3655 CrossRef.
- J. Cheng, J. Wang, X. Wang and H. Wang, Ceram. Int., 2017, 43, 7159–7165 CrossRef CAS.
- L. Duan, X. Zhao and Y. Wang, Ceram. Int., 2017, 43, 16114–16120 CrossRef CAS.
- D. Zhao, C. Zhang, H. Hu and Y. Zhang, Ceram. Int., 2011, 37, 2089–2093 CrossRef CAS.
- C. Yan, R. Liu, C. Zhang, Y. Cao and Y. Wang, J. Eur. Ceram. Soc., 2017, 37, 2343–2351 CrossRef CAS.
- N. Patra, D. D. Jayaseelan, W. E. Lee and T. Besmann, J. Am. Ceram. Soc., 2015, 98, 71–77 CrossRef CAS.
- Z. Mei, X. Su, G. Hou, F. Guo and Y. Zhang, Adv. Mater. Res., 2010, 148–149, 1453–1457 Search PubMed.
- F. Rejasse, G. Trolliard, O. Rapaud, A. Maitre and J. David, RSC Adv., 2015, 5, 45341–45350 RSC.
- A. Sondhi, C. Morandi, R. F. Reidy and T. W. Scharf, Ceram. Int., 2013, 39, 4489–4497 CrossRef CAS.
- J. David, G. Trolliard and A. Maitre, Acta Mater., 2013, 61, 5414–5428 CrossRef CAS.
- J. David, G. Trolliard, M. Gendre and A. Maître, J. Eur. Ceram. Soc., 2013, 33, 165–179 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |