
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient hydrogen evolution from water electrolysis using nanocrystalline transition metal phosphide catalysts†
Weiming Wu,
Xiao-Yuan Wu ,
Sa-Sa Wang and
Can-Zhong Lu*
,
Sa-Sa Wang and
Can-Zhong Lu*
CAS Key Laboratory of Design and Assembly of Functional Nanostructures, Fujian Provincial Key Laboratory of Nanomaterials, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China. E-mail: czlu@fjirsm.ac.cn; Fax: +86 591 63173355; Tel: +86 591 63173355
First published on 26th November 2018
Abstract
Nanocrystalline transition metal phosphides (CoP and Ni2P) were successfully synthesized by a simple calcination method by using transition metal hydroxides and NaH2PO2 as raw materials. Their catalytic activities for the hydrogen evolution from water electrolysis were evaluated with silicotungstic acid as an electron-coupled-proton buffer, whereby hydrogen and oxygen could be produced separately. It was found that the CoP sample showed higher catalytic activity (32 mmol min−1 g−1) and good stability (12 h) as compared to the Ni2P sample, and its catalytic activity could rival that of the commercial Pt/C catalyst. The electrochemical results revealed that CoP had high cathodic current and small charge transfer resistance, which further suggested it was indeed an efficient noble metal-free catalyst for hydrogen evolution from water electrolysis.
1. Introduction
Hydrogen is considered as an ideal clean green fuel.1,2 Its large-scale application can solve the energy and environment issues at a global level. Water electrolysis is an important method for hydrogen production on a large scale. Therefore, this technology has received considerable attention in the field of chemistry. Generally, water and energy (in form of electricity) are the only required inputs for the electrolysis of water.3,4 The separation of hydrogen and oxygen is an important process in this technology, because these gases will diffuse in electrolysis devices and can give rise to hazardous O2/H2 mixtures. The proton exchange membrane electrolysis is the most mature technology used for mitigating the gas crossover, but it still a challenge to prevent such gas crossover.5,6If hydrogen and oxygen are produced separately in the electrolyzer systems, this annoying problem will be rooted out. Cronin group has introduced a concept of the electron-coupled proton buffer (ECPB), which can decouple electrolytic H2 and O2 production, producing these gases separately.7–9 In the reported systems, silicotungstic acid is one attractive material as a candidate of the ECPBs for the electrolysis of water due to its high solubility and reversible redox ability in water.9 Silicotungstic acid can accept two electrons to from a 2e-reduced species in water. Furthermore, its 2e-reduced species can reduce H+ ions, which come from the oxidation of water (2H2O = 4H+ + O2), into H2 gas directly in the present of catalysts (such as Pt/C). This may not only separate H2 and O2 gases, but also give the catalysts freedom from the limitation of the low work surfaces of electrodes. However, nobel metal Pt is often used as a catalyst for the hydrogen evolution in these studies. For real wide-spread application, catalysts made from elementally abundant and less expensive materials are urgently required.
Earth-abundant transition metal phosphides have received special attention in recent years, because these phosphide nanocrystals can efficiently split water into H2 by utilizing electricity.10–21 Nevertheless, very few reports describe the H2 evolution from water electrolysis using these phosphide nanomaterials in the ECPBs systems. Herein, nanocrystalline transition metal phosphides (CoP and Ni2P) were successfully synthesized by a simple calcination method, and their catalytic activities for the hydrogen evolution from water electrolysis were evaluated with silicotungstic acid as an ECPB. The highly efficient catalytic activity of CoP nanocrystal was confirmed via various electrochemical techniques. Our results may allow us to highlight the promising application of transition metal phosphides in the highly efficient hydrogen evolution from water electrolysis.
2. Results and discussion
2.1. Preparation and characterization
The crystal structures and morphologies of the as-prepared transition metal phosphides have been characterized by X-ray diffraction (XRD) and transmission electron microscope (TEM) techniques, respectively. Fig. 1a shows the XRD pattern of the CoP sample. All diffraction peaks of the pattern are well indexed to orthorhombic cobalt phosphide (CoP, JCPDS card no. 29-0497). The main peaks at 2θ values about 31.6, 32.0, 35.3, 36.3, 36.7, 45.1, 46.2, 48.1, 48.4, 52.3, 56.0, 56.4 and 56.8° can be assigned to the diffraction peaks of the (011), (002), (200), (111), (102), (210), (112), (211), (202), (103), (020), (212) and (301) planes, respectively. Furthermore, as shown in Fig. 1d, the nickel phosphide sample is mainly composed of hexagonal Ni2P (JCPDS card no. 03-0953), and contains a small amount of hexagonal Ni5P4 (JCPDS card no. 18-0883). The TEM images of the as-prepared samples are showed in Fig. 1b and e, respectively. It is found that the cobalt phosphide sample consists of CoP nanoparticles with a diameter of 10–20 nm, while the particle size of the Ni2P sample ranges from 20 to 30 nm. Moreover, HRTEM images show clear lattice fringes for the as-prepared samples. Note that the d = 0.189 and 0.283 nm of lattice fringes in Fig. 1c match those of the (211) and (011) crystallographic planes of CoP, respectively. And the lattice spacing of d = 0.221 nm in Fig. 1f corresponds to the (111) plane of Ni2P. In addition, the morphology of the CoP sample has been confirmed by scanning electron microscope (SEM) (see Fig. S1†). Surface chemical compositions of the as-prepared sample have also been studied by X-ray photoelectron spectroscopy (XPS). Fig. S2† shows the high-resolution XPS spectra of Co 2p and P 2p for the CoP sample. The peaks at 778.6, 793.6, 129.3 and 130.1 eV can be attributed to the Co 2p3/2, Co 2p1/2, P 2p3/2 and P 2p1/2 in CoP, respectively.12,22 Two satellite peaks at 786.1 and 802.9 eV are observed for the Co 2p in the CoP sample.22 The additional peaks at 781.6 and 797.7 eV in the high-resolution XPS spectrum of Co 2p and the broad peak at 133.5 eV in the high-resolution XPS spectrum of P 2p may come from the cobalt phosphate formed on the surface of CoP due to the exposure of the sample to air.12,22,23 These results suggest that nanocrystalline transition metal phosphides can be obtained in this work by a simple calcination method via using transition metal hydroxides and NaH2PO2 as raw materials.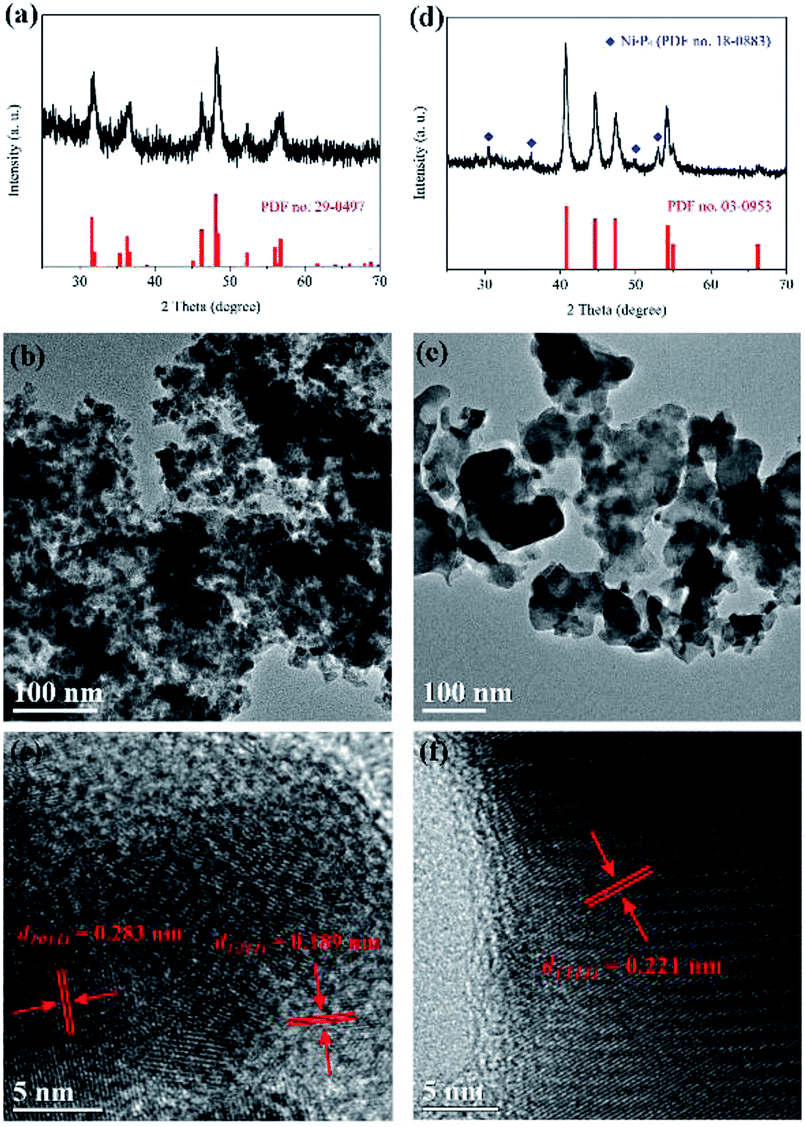 | ||
| Fig. 1 XRD patterns, TEM images and high-resolution TEM images of the as-prepared samples. (a)–(c) for the CoP sample and (d)–(f) for the Ni2P sample. | ||
2.2. Catalytic performance for H2 evolution from water electrolysis
The catalytic activities of the as-prepared transition metal phosphides for the hydrogen evolution from water electrolysis have been evaluated with silicotungstic acid as an ECPB, whereby hydrogen and oxygen can be produced separately. In the present system, H4SiW12O40 is firstly reduced to H6SiW12O40 in a two-compartment home-made H-cell by using a classical two-electrode configuration (see Fig. S3†), and water are oxidized to molecular oxygen at the same time. Then, the 2e-reduced silicotungstic acid will reduce H+ ions to molecular hydrogen in water with the obtained samples as the catalysts. Fig. 2 shows the hydrogen yield as a function of reaction time in the presence of the as-prepared catalysts. It is found that the as-prepared samples show excellent activities for the H2 evolution from water electrolysis, and the catalytic activity of the CoP sample is much higher than that of the Ni2P sample. For the CoP sample, the initial rate of the H2 evolution is 32 mmol min−1 g−1, corresponding to a turnover frequency of 2.9 min−1. In theory, 74.6 mL (3.3 mmol) of H2 can be obtained, because the 2e-reduced silicotungstic acid can provide 6.7 mmol of electrons to take part in the hydrogen evolution. However, only a half of the hydrogen yield (∼37 mL) is collected in the present system. This can be ascribed to the low reducing ability of the 1e-reduced silicotungstic acid (E (H5SiW12O40/H4SiW12O40) = 0.01 V vs. RHE).9 As a part of this work, the catalytic activity of the commercial Pt/C (5 wt%, Aladdin Co.) has been investigated under same conditions (see Fig. 2). Note that the catalytic activity the CoP sample can rival that of the commercial Pt/C catalyst. These results may highlight the promising application of earth-abundant transition metal phosphides in the highly efficient hydrogen evolution from water electrolysis.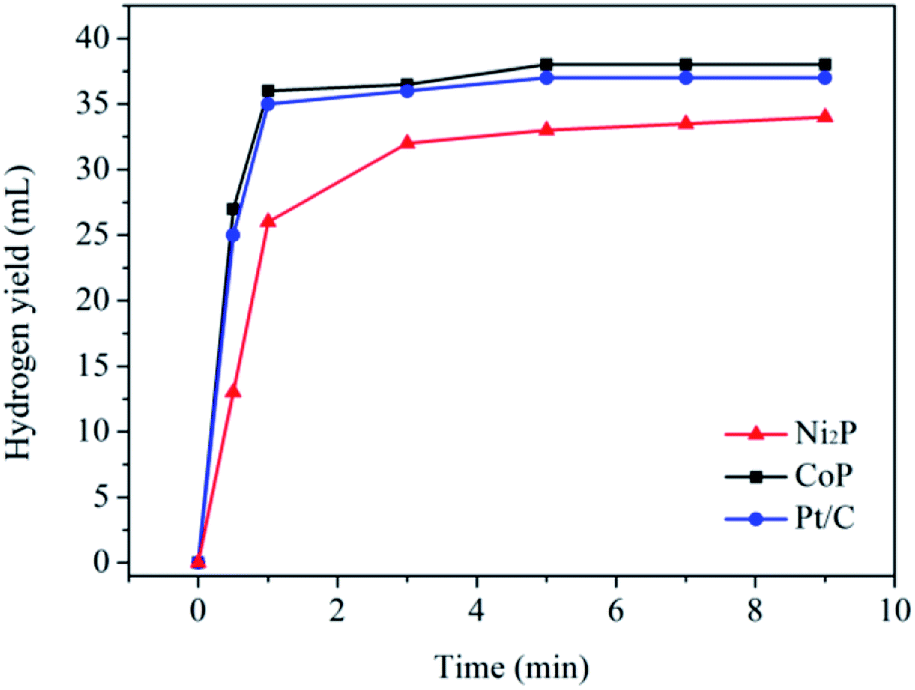 | ||
| Fig. 2 Hydrogen yield as a function of reaction time in the presence of the as-prepared catalysts. | ||
2.3. Stability test
It is widely regarded that the stability of a catalyst is a very important factor for its practical applications. Therefore, the stability of the CoP sample for the hydrogen evolution from water electrolysis has been carried out. For the sake of simplicity, the CoP sample is directly added into the one compartment of the two-compartment H-cell containing 1e-reduced silicotungstic acid (H5SiW12O40). After a potential of 8.3 V is set between the working electrodes, the evolving H2 and O2 gas are captured in two 100 mL measuring cylinder filled with water, respectively. As shown in Fig. 3 and S4,† H2 and O2 evolutions proceed continuously in a molar ratio of H2/O2 of 2.1 (∼49 mL h−1 for H2 and ∼23 mL h−1 for O2), almost equal to the theoretical value of 2 for overall water splitting. Moreover, it can be seen that the production rates of hydrogen and oxygen do not obviously decrease after 12 h with a faradaic efficiency of nearly 100%. The low rate of the H2 evolution for the initial phase can be attributed to the introduction of air during the addition of the CoP sample. The crystal structure, morphology and surface chemical compositions of the as-prepared sample before and after the reaction have also been studied by XRD, TEM and XPS (see Fig. S5–S7†). There are no obvious changes in the crystal structure, morphology and surface chemical compositions after the reaction for the CoP sample. Furthermore, the Co2+ concentration measured by inductively coupled plasma-atomic emission spectrometry (ICP-AES) is only 40 ppm after the reaction. The Co2+ ions in the present system may come from the cobalt phosphate formed on the surface of the CoP particles due to the exposure of the sample to air.10,12,13,17,22,23 These results suggest that the CoP catalyst exhibits good stability in the present system.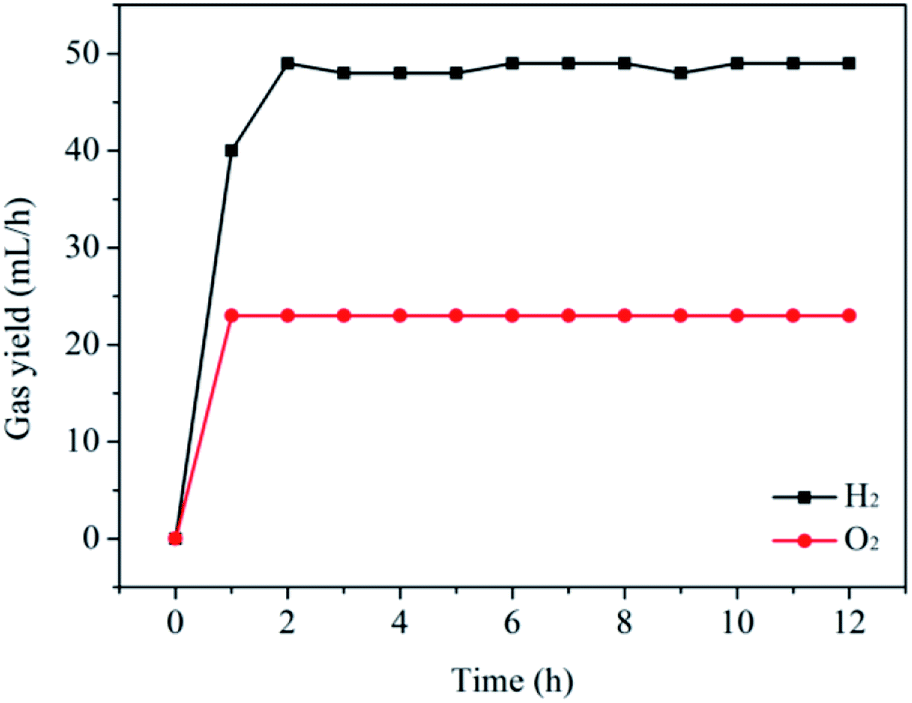 | ||
| Fig. 3 Production rates of hydrogen and oxygen during the durability experiment for the CoP sample. | ||
2.4. Roles of ECPB and catalyst
Production rate of H2 in the two-electrode system have been investigated under different conditions (see Table 1). It is found that the production of H2 is negligible in the present of H4SiW12O40. However, after reducing H4SiW12O40 to H5SiW12O40 and adding the CoP sample, H2 can be detected with a rate of 48 mL h−1. Control experiments exhibit only a half of H2 is obtained without the ECPB, and the adding of the catalyst can't improve the production of H2 (see entry 3 and 4 in Table 1). These results confirm H4SiW12O40 and CoP are the efficient ECPB and catalyst, respectively. Furthermore, H2 evolution from such a system is no longer directly coupled to the rate of water oxidation, and the activity of the catalyst don't limit by the electrode's work surface.| Entry | Componentsb | Current (mA) | Production rate of H2 (mL h−1) |
|---|---|---|---|
| a Conditions: a two-compartment H-cell separated by a piece of 0.18 mm-thick Nafion N-117 membrane; platinum mesh working electrode (1 × 1 cm2), 0.9 mL of H3PO4 and 60 mL of H2O in the left compartment; carbon felt working electrode (3 × 2 cm2) and various components in the right compartment; Ar atmosphere; reaction time, 4 h.b Dosages: H4SiW12O40, 10 mmol; H3PO4, 0.9 mL (13.3 mmol); CoP, 50 mg. | |||
| 1 | H4SiW12O40 | 100 | No detected |
| 2 | H4SiW12O40 + CoP | 100 | 48 |
| 3 | H3PO4 | 50 | 26 |
| 4 | H3PO4 + CoP | 50 | 28 |
2.5. Electrochemical properties of transition metal phosphides
Electrochemical measurements have been conducted in a typical three-electrode cell to get further insight into the hydrogen evolution reaction process of transition metal phosphides. Fig. 4a shows the polarization curves of the as-prepared samples loaded on glassy carbon electrodes, which are measured in the N2-saturated 0.5 M H2SO4 with a scan rate of 100 mV s−1. The Pt/C electrode prepared with a similar content has also been examined for comparison. The overpotential required for different electrocatalysts to produce a cathodic current density of 10 mA cm−2 (η10) is usually compared in the literature to assess their catalytic activities, and the lower overpotentials are helpful for the hydrogen evolution from water electrolysis.11–14 The η10 of the CoP and Ni2P sample shown in Fig. 4a are 65 and 150 mV, respectively. Although the η10 of the CoP sample is higher than that of the Pt/C sample (η10 = 28 mV), the CoP sample shows a high cathodic current in the range of −0.1 to −0.2 V vs. RHE. It only needs ∼130 mV to afford a current density of 100 mA cm−2, which is lower than that of the Pt/C sample (η100 > 200 mV). This may explain the efficient catalytic activity of the CoP sample at a low redox potential (E(H6SiW12O40/H5SiW12O40) = −0.49 V vs. RHE), which can rival that of the commercial Pt/C catalyst.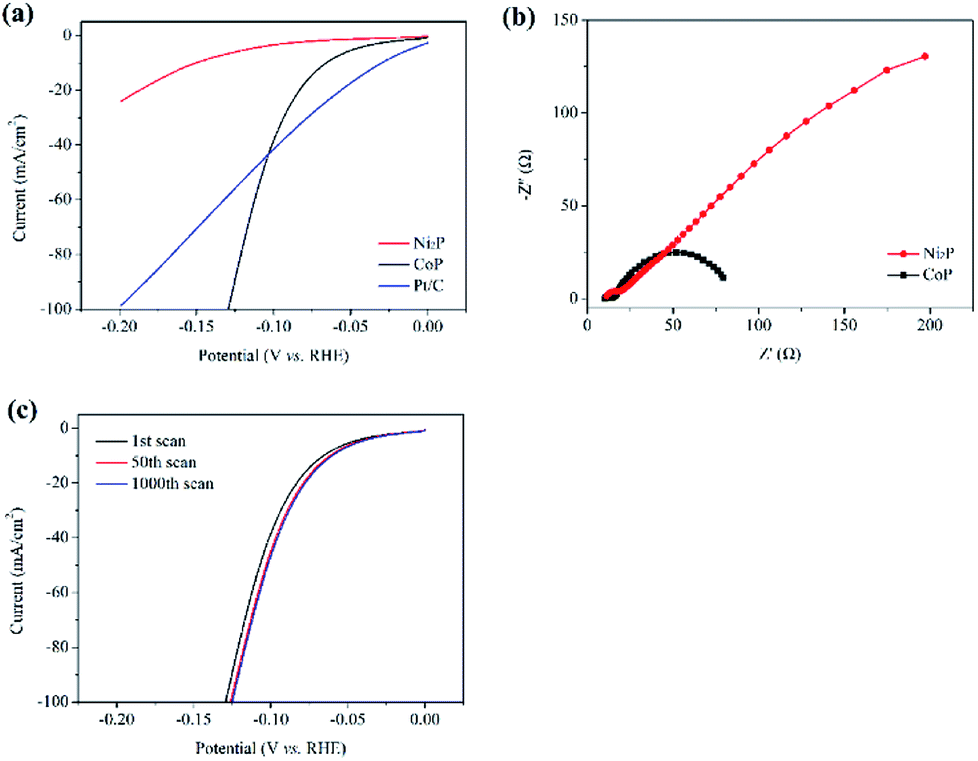 | ||
| Fig. 4 (a) Polarization curves, (b) Nyquist plots for different samples and (c) polarization curves for the CoP sample before and after 1000 cycles. | ||
Furthermore, the charge transfer rate has been studied by electrochemical impedance spectroscopy (EIS) and the expected semicircular Nyquist plots for the transition metal phosphides (Fig. 4b), with a significantly decreased diameter for the CoP sample, have been obtained. It is generally accepted that a small diameter means a low impedance and gives rise to fast charge transfer kinetics.18,24 These results imply that the CoP sample indeed is an efficient catalyst for the H2 evolution from water electrolysis in this work. The polarization curves that are measured for the CoP sample before and after 50 cyclic voltammetry cycles at a scan rate of 100 mV s−1 are shown in Fig. 4c. At the end of the cycling experiment, a significant improvement of current density has occurred, which can be attributed to the elimination of the oxidation layer on the CoP sample. While increasing the cycling times to 1000, the curve exhibits negligible loss in current density compared to that of the 50th scan. This further suggests the excellent stability of the CoP sample for the H2 evolution from water electrolysis in acid condition.
3. Conclusions
Nanocrystalline transition metal phosphides (CoP and Ni2P) with a diameter of 10–30 nm were successfully synthesized by a simple calcination method via using transition metal hydroxides and NaH2PO2 as raw materials. They were used as the catalysts for the hydrogen evolution from water electrolysis with silicotungstic acid as an ECPB, whereby hydrogen and oxygen could be produced separately. It was found that the CoP sample showed higher catalytic activity (32 mmol min−1 g−1) as compared to the Ni2P sample, and its catalytic activity could rival that of the commercial Pt/C catalyst. The stability tests demonstrated that the production rate of hydrogen did not obviously decrease after 12 h. And the analysis results of the XRD, TEM, XPS and ICP showed that the crystal structure, morphology and surface chemical compositions of the as-prepared CoP sample had no obvious change. These results revealed that it exhibited good stability in the present system. The electrochemical results indicated that CoP had high cathodic current and small charge transfer resistance, which further suggested it was indeed a noble metal-free catalyst for efficient hydrogen evolution. Our results may allow us to highlight the promising application of transition metal phosphides in the highly efficient hydrogen evolution from water electrolysis.4. Experimental section
4.1. Preparation
5 mmol of transition metal chlorides (NiCl2·6H2O or CoCl2·6H2O, Sinopharm Chemical Reagent Co. (SCRC)) was dissolved in 40 mL of deionized water. Then, 20 mL of 1 M NaOH aqueous solution (SCRC) was dropped under stirring (200 rpm). Transition metal hydroxides were collected and were washed with deionized water two times after 30 min of stirring, and then were dried in air at 60 °C overnight.50 mg of the obtained hydroxides and 250 mg of NaH2PO2 (Aladdin Co.) were mixed well in an agate mortar. These mixtures were heated to 300 °C with a ramping rate of 2 °C min−1, and kept at 300 °C for 60 min. This process was carried out in Ar flow (50 mL min−1) all through. After naturally cooled to room temperature under Ar atmosphere, the as-prepared phosphides were soaked in 1 M HCl aqueous solution (SCRC) for 36 h to remove any unstable species. Then, the transition metal phosphides were washed with deionized water and absolute ethyl alcohol three times, and dried in vacuum at 60 °C for 1 h.
4.2. Characterization
XRD patterns were collected on a Miniflex II X-ray diffractometer (Rigaku Co.) with Cu Kα radiation. The data were recorded in a 2θ range of 25–70°. TEM images were obtained using a FEI Tecnai 20 transmission electron microscope at an accelerating voltage of 200 kV. Samples for TEM imaging were prepared by placing a drop of the sample ethanol suspension on a copper grid coated with carbon film and dried in the atmosphere. SEM image was obtained on a JEOL JSM-7500F field emission scanning electron microscope at an accelerating voltage of 5 kV. XPS measurement was carried out by using a Escalab 250Xi X-ray photoelectron spectrometer (Thermo Fisher scientific Co.) equipped with Al Kα X-ray source. The binding energy calibration of the spectra was referred to C 1s peak located at 284.6 eV for the analysis. Electrochemical experiments were taken on a CHI660e workstation (CH Instruments, Inc.). The electrochemical measurements were performed in a typical three electrode cell, using Pt wire and saturated Ag/AgCl electrode as counter electrode and reference electrode, respectively. Glassy carbon electrode (3 mm in diameter) coated with the as-prepared samples were used as the working electrodes. In the fabrication of the working electrode, the catalyst ink was prepared by dispersing 5.0 mg of the catalyst into a mixed solvent containing 0.98 mL of deionized water and 20 μL of 5 wt% Nafion solution (D521, Alfa Aesar Co.), and then the mixture was sonicated for 30 min to form a homogeneous ink. After that, 5 μL of the catalyst ink was loaded onto glassy carbon electrode and dried at room temperature. The electrolyte was a 0.5 M H2SO4 aqueous solution (SCRC) and was purged with nitrogen gas for 30 min prior to the measurements. In all measurements, the saturated Ag/AgCl reference electrode was calibrated with respect to reversible hydrogen electrode (RHE). In 0.5 M H2SO4, E(RHE) = E(Ag/AgCl) + 0.198 V.4.3. H2 evolution from water electrolysis
The electrochemical reduction of silicotungstic acid (H4SiW12O40) was carried out by a modified method:8,9 10 mmol of H4SiW12O40 (SCRC) and 60 mL of deionized water were added into one compartment of a two-compartment H-cell, and 0.9 mL H3PO4 (13.3 mmol, SCRC) and 60 mL of deionized water were placed into the other compartment. The compartments of the H-cells were separated by a piece of 0.18 mm-thick Nafion N-117 membrane (Alfa Aesar Co.). All electrochemical experiments were performed by using a classical two-electrode configuration (TPR6010S regulated power supply, ATTEN instruments Co.). The H4SiW12O40 compartment was equipped with a large area carbon felt working electrode (3 × 2 cm2), and the other compartment of the cell was equipped with a large area platinum mesh working electrode (1 × 1 cm2). To fully reduce H4SiW12O40 to H6SiW12O40 by two electrons, a potential of 8.3 V was set between the working electrodes and 1931 C of charge was passed at this potential (working current: 0.1 A, working time: 5.4 h). Prior to the experiment, the H4SiW12O40 solution was bubbled with argon (60 mL min−1) for 60 min, stirred vigorously and kept under an argon atmosphere throughout the experiment.50 mg of the as-prepared sample was dispersed in 5 mL of deionized water in a 50 mL three-necked round-bottomed flask. After thoroughly flushed with argon, the freshly produced H6SiW12O40 (20 mL) was added to the sample aqueous dispersion under Ar atmosphere and stirred vigorously via a pressure-equalizing dropping funnel. The evolving H2 gas was captured in a 100 mL measuring cylinder filled with water. Furthermore, the evolving H2 gas was identified by gas chromatography (GC2014C, Shimazu Co., TCD, molecular sieve 5A column, argon carrier).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB20000000), the Key Program of Frontier Science, CAS (QYZDJ-SSW-SLH033), the National Natural Science Foundation of China (21521061 and 21701169), the Natural Science Foundation of Fujian Province (2006L2005), and the China Postdoctoral Science Foundation (2015M570561).Notes and references
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- A. L. Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Métayé, A. Fihri, S. Palacin and M. Fontecave, Science, 2009, 326, 1384 CrossRef PubMed.
- J. D. Holladay, J. Hu, D. L. King and Y. Wang, Catal. Today, 2009, 139, 244 CrossRef CAS.
- G. Ou, P. Fan, H. Zhang, K. Huang, C. Yang, W. Yu, H. Wei, M. Zhong, H. Wu and Y. Li, Nano Energy, 2017, 35, 207 CrossRef CAS.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901 CrossRef CAS.
- M. Paidar, V. Fateev and K. Bouzek, Electrochim. Acta, 2016, 209, 737 CrossRef CAS.
- B. Rausch, M. D. Symes and L. Cronin, J. Am. Chem. Soc., 2013, 135, 13656 CrossRef CAS PubMed.
- M. D. Symes and L. Cronin, Nat. Chem., 2013, 5, 403 CrossRef CAS PubMed.
- B. Rausch, M. D. Symes, G. Chisholm and L. Cronin, Science, 2014, 345, 1326 CrossRef CAS PubMed.
- Z. Huang, Z. Chen, Z. Chen, C. Lv, H. Meng and C. Zhang, ACS Nano, 2014, 8, 8121 CrossRef CAS PubMed.
- P. Jiang, Q. Liu, Y. Liu, J. Tian, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2014, 53, 12855 CrossRef CAS PubMed.
- Q. Liu, J. Tian, W. Cui, P. Jiang, N. Cheng, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2014, 53, 6710 CrossRef CAS PubMed.
- X. Li, Y. Fang, F. Li, M. Tian, X. Long, J. Jin and J. Ma, J. Mater. Chem. A, 2016, 4, 15501 RSC.
- C. Tang, L. Gan, R. Zhang, W. Lu, X. Jiang, A. M. Asiri, X. Sun, J. Wang and L. Chen, Nano Lett., 2016, 16, 6617 CrossRef CAS PubMed.
- L. Zhou, M. Shao, J. Li, S. Jiang, M. Wei and X. Duan, Nano Energy, 2017, 41, 583 CrossRef CAS.
- S. Cao, C. J. Wang, W. F. Fu and Y. Chen, ChemSusChem, 2017, 10, 4306 CrossRef CAS PubMed.
- W. Wu, X. Yue, X. Y. Wu and C. Z. Lu, RSC Adv., 2016, 6, 24361 RSC.
- X. Wang, Y. V. Kolen'ko, X. Q. Bao, K. Kovnir and L. Liu, Angew. Chem., Int. Ed., 2015, 54, 8188 CrossRef CAS PubMed.
- X. Wang, Y. V. Kolen'ko and L. Liu, Chem. Commun., 2015, 51, 6738 RSC.
- X. Wang, W. Li, D. Xiong, D. Y. Petrovykh and L. Liu, Adv. Funct. Mater., 2016, 26, 4067 CrossRef CAS.
- W. Li, X. Gao, D. Xiong, F. Xia, J. Liu, W. Song, J. Xu, S. M. Thalluri, M. F. Cerqueira, X. Fu and L. Liu, Chem. Sci., 2017, 8, 2952 RSC.
- X. Yang, A. Lu, Y. Zhu, M. N. Hedhili, S. Min, K. Huang, Y. Han and L. Li, Nano Energy, 2015, 15, 634 CrossRef CAS.
- Y. Zhu, Y. Liu, T. Ren and Z. Yuan, Adv. Funct. Mater., 2015, 25, 7337 CrossRef CAS.
- Y. Hou, A. B. Laursen, J. Zhang, G. Zhang, Y. Zhu, X. Wang, S. Dahl and I. Chorkendorff, Angew. Chem., Int. Ed., 2013, 52, 3621 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Electrochemical reduction of H4SiW12O40 by a two-compartment home-made H-cell by using a classical two-electrode configuration; production of hydrogen and oxygen during the durability experiment for the CoP sample; XRD patterns for the CoP sample before and after the catalytic test. See DOI: 10.1039/c8ra07195k |
| This journal is © The Royal Society of Chemistry 2018 |