DOI:
10.1039/C8RA07940D
(Paper)
RSC Adv., 2018,
8, 38166-38174
Preparation of 3-hydroxyquinolines from direct oxidation of dihydroquinolinium salts†
Received
25th September 2018
, Accepted 5th November 2018
First published on 14th November 2018
Abstract
A series of functionalized 3,4-dihydroquinolinium salts were prepared from the reaction of aryldiazonium salt with alkene in a nitrile solution. Further oxidation yielding either 3-hydroxyquinoline or quinoline products was investigated. A one-pot process from aryldiazonium salts, alkenes and nitriles leading to 3-hydroxyquinolines was also developed. Furthermore, an intramolecular trapping of an N-arylnitrilium ion with a vinyl group at the ortho position leading to 2-substituted quinolines was revealed.
Introduction
Functionalized quinolines are highly privileged structural units due to their widespread occurrence in various natural products and functional materials.1 The quinoline core is known to be a versatile building block in the preparation of small molecules with diverse therapeutic properties.2 Among the various quinoline derivatives, 3-hydroxyquinolines (quinoline-3-ols) are privileged (Fig. 1).3 Typically, Talnetant is a potential neurokinin 3 receptor antagonist primarily used in irritable bowel syndrome and psychotic disorders,3a whereas PSI-697 is utilized as an orally active effective P-selectin inhibitor.3b These ubiquitous cores have also been studied as selective inhibitors HIV-1 reverse transcriptase,4 antiallergic agents (TA-270),5 antioxidant3c and D-amino acid oxidase inhibitors (DAAO).6 In addition, these frameworks have been used as building blocks in functional materials.7 Notably, naturally occurring anti-HIV fungal metabolites such as viridicatol, viridication and 3-O-methylviridicatin are derivatives of 3-hydroxyquinolin-2(1H)-ones.8
 |
| | Fig. 1 Selected 3-hydroxyquinoline derivatives. | |
Despite the excellent significance, synthetic efforts towards this ubiquitous core remain largely underdeveloped. Classical routes leading to 3-hydroxyquinoline derivatives are Friedlander type condensations,9 Pfitzinger synthesis,10 Danheiser type annulation of 2-iodostyrenes with acetylenes,11 Knoevenagel condensation followed by decyanative epoxide-arene cyclization,12 and Cromwell's approach of epoxide opening/condensation of o-aminobenazalacetophenone oxides.13 In recent years, Jamison et al. demonstrated a convenient synthesis of 3-hydroxyquinolines via aerobic oxidation of corresponding aryl Grignard reagents under a continuous flow method.14 In 2017, Yadav's group described a NBS mediated deacylative formation of 3-hydroxyquinolines from 3-acetylquinolines via an epoxide intermediate.15 More recently, Mamedov et al. developed an in situ Meinwald rearrangement-intramolecular reductive cyclization of o-nitroarylacetophenone oxides under mild conditions.16 Nevertheless, the above-mentioned routes require pre-functionalized substrates, metal catalysts and/or multistep synthetic protocol with a limited functionality presented in the molecule. Thus, there is still a need for the development of a convenient method from easily available precursors with a diversity of functionality.
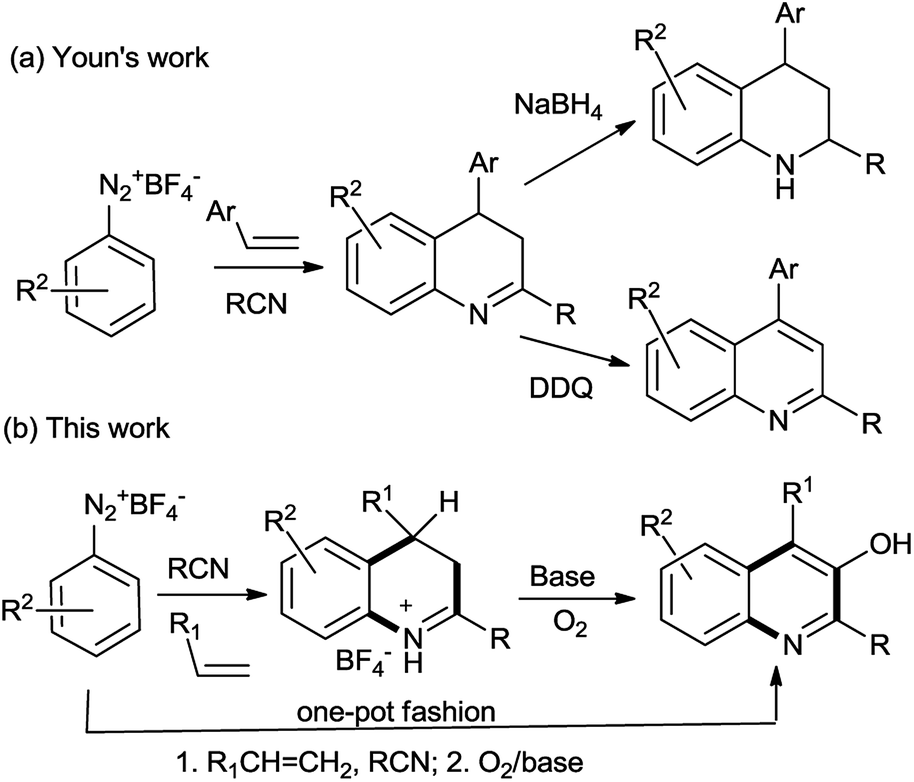
In early this year, Youn's group reported the reaction of aryldiazonium salts with nitrile followed by the cyclization with alkenes to achieve 3,4-dihydroquinolines, which was subsequently either oxidized or reduced to yield the corresponding quinolines or tetrahydroquinolines, respectively (Scheme 1a).17 In this work, we adopted the similar method to prepare 3,4-dihydroquinolines followed by a further oxidation at C-3 position leading to 3-hydroxyquinolines (Scheme 1b).
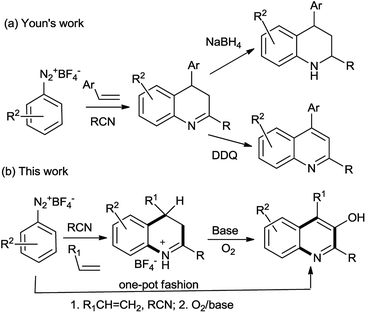 |
| | Scheme 1 Strategies leading to quinolone derivatives. | |
Results and discussion
Preparation of dihydroquinolinium salts
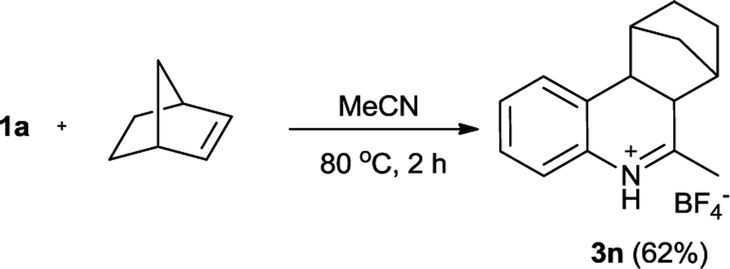
According to Youn's procedure, we have prepared a series of 4-aryl-dihydroquinolines by reaction of aryldiazonium tetrafluoroborate with styrenes in a nitrile solution (Table 1). Interestingly, when arylnitrile was employed, the corresponding 2,4-diaryl substituted quinolinium salt (3h) was obtained as the major products, but accompanied with quinazolines (4) as the side product (Scheme 2).18c Similarly, when α-methylstyrene was used as the substrate, a 4,4-disubstituted dihydroquinoline 3m was isolated in 49% yield. However, reaction of α-bromostyrene with 1a in acetonitrile gave quinoline 5 as the single product presumably due the elimination of HBr (Scheme 3). Instead of styrene, norbornylene smoothly participated in the reaction leading to fused-tricyclic quinolinium salts 3n in 62% yield (eqn. (1)).| |
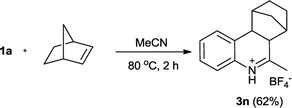 | (1) |
Table 1 Isolation of dihydroquinolinesa

|
| Entry |
Substituents |
Product 3 (yield) |
| Reaction conditions: a mixture of 1a (0.52 mmol), 2a (1.04 mmol) and solvent (2 mL) in reaction tube. Quinazoline 4 as the side product (18%). |
| 1 |
R1 = H, Ar = Ph, R = Me |
3a, (81%) |
| 2 |
R1 = 4-Me, Ar = Ph, R = Me |
3b, R1 = 6-Me (72%) |
| 3 |
R1 = 4-Me, Ar = Ph, R = Ph |
3c, R1 = 6-Me (54%) |
| 4 |
R1 = 4-(t-Bu), Ar = Ph, R = Me |
3d, R1 = 6 t-Bu (68%) |
| 5 |
R1 = H, Ar = Ph, R = Pr |
3e, (63%) |
| 6 |
R1 = H, Ar = Ph, R = i-Pr |
3f, (57%) |
| 7 |
R1 = H, Ar = Ph, R = c-C3H5 |
3g, (66%) |
| 8 |
R1 = H, Ar = Ph, R = Ph |
3h, (67%)b |
| 9 |
R1 = H, Ar = p-BrC6H4, R = Me |
3i, (80%) |
| 10 |
R1 = H, Ar = p-ClC6H4, R = Me |
3j, (62%) |
| 11 |
R1 = H, Ar = p-ClCH2C6H4, R = Me |
3k, (76%) |
| 12 |
R1 = H, Ar = 2,5-Me2C6H3, R = i-Pr |
3l, (39%) |
 |
| | Scheme 2 Reactions of 1a with benzonitrile. | |
 |
| | Scheme 3 Reactions of 1a with 1,1-disubstituted styrenes. | |
Oxidization of 3,4-dihydroquinolinium salts
During the chromatographic purification of dihydroquinoline, we learned that both quinoline and 3-hydroxyquinoline, resulting from the oxidation, were obtained. This observation raised our attention of the preparation of 3-hydroxyquinolines from dihydroquinoline via oxidation. Thus, further studies focused on this issue with the use of 3a as a model. Basically, we proposed that the allylic oxidation giving 6a′ followed by tautomerization should render the desired product (Scheme 4). Table 2 summarizes the results of the oxidation of 3a under various conditions.
 |
| | Scheme 4 Oxidation of dihydroquinoline 3a leading to 3-hydroxyquinoline. | |
Table 2 Optimization for oxidation of 3a into 6a and 5a

|
| Entry |
Conditions |
6a |
5 |
| Reaction conditions: a mixture of 3a (0.16 mmol) in solvent (2 mL) was treated with oxidant as indicated in each entry. |
| 1 |
H2O2 (2 eq.)/CH3CN |
— |
— |
| 2 |
H2O2 (2 eq.)/Na2CO3/CH3CN |
30% |
55% |
| 3 |
Oxone (1 eq.)/CH3CN |
10% |
58% |
| 4 |
Oxone (1 eq.)/Na2CO3 (1 eq.)/CH3CN |
30% |
43% |
| 5 |
Silica gel/NaHCO3/CH3CN |
15% |
42% |
| 6 |
I2 (1 eq.)/Na2CO3 (1 eq.)/CH3CN |
15% |
77% |
| 7 |
O2 (1 atm)/Na2CO3 (1 eq.)/CH3CN/50 °C |
14% |
30% |
| 8 |
TBHP (2 eq.)/O2 (1 atm)/CH3CN |
22% |
33% |
| 9 |
S8 (1 eq.)/N2 (1 atm)/CH3CN |
— |
76% |
| 10 |
Cu2O (1 eq.)/O2 (1 atm)/CH3CN |
26% |
37% |
| 11 |
Cu(OAc)2 (1 eq.)/O2 (1 atm)/CH3CN |
8% |
56% |
| 12 |
Na2WO4 (0.1 mmol)/H2O2 (1.2 eq.)/CH3CN/2 h |
6% |
36% |
| 13 |
TBHP(1.2 eq.)/VO(acac)2(0.1 mmol)/CH3CN/2h |
30% |
70% |
| 14 |
AIBN (1 eq.)/CH3CN/N2/reflux |
— |
93% |
| 15 |
KOH (2 eq.)/O2 (1 atm)/CH3CN |
6% |
61% |
| 16 |
Na2CO3 (1 eq.)/O2 (1 atm)/CH3CN |
11% |
51% |
| 17 |
Na2CO3 (10 eq.)/O2 (1 atm)/CH2Cl2/72 h |
73% |
25% |
| 18 |
Na2CO3 (10 eq.)/O2 (1 atm)/MeOH/72 h |
12% |
60% |
| 19 |
Na2CO3 (10 eq.)/TEMPO/O2 (1 atm)/CH2Cl2/72 h |
18% |
11% |
Quite a lot of procedures were attempted in the oxidation of 3a, but in the most cases leading the quinoline 5, to be a favoured process. Fortunately, the desired hydroxyquinoline 6a was obtained in 73% yield upon treating with O2 in the presence of excess of Na2CO3 accompanied with 25% of 5 (Table 2, entry 17). By addition of an equimolar amount of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO), the reaction was inhibited dramatically, indicating that a possible radical pathway is involved in this oxidation (Table 2, entry 19). Thus, a rationalization of formation of 5 versus 6a is summarized in Scheme 5. The abstraction of hydrogen atoms from benzylic or allylic positions would lead to the products. In particular, the reaction of O2 with intermediate 3a′′ yields the desired product 6a.
 |
| | Scheme 5 Abstraction of hydrogen from benzylic versus allylic positions. | |
Few dihydroquinolines were then transformed into the corresponding 3-hydroxyquinolines smoothly by adopting the same procedure (Scheme 6). In case of 3m, the oxidation gave 4-methyl-4-phenylquinolin-3(4H)-one (7) (eqn (2)),19 which is in agreement of the proposed pathway of this oxidation in Scheme 4.
| |
 | (2) |
 |
| | Scheme 6 Oxidation of dihydroquinlines to give 3-hydroxyquinolines (NMR yields). | |
One-pot fashion of preparation of 3-hydoxyquinolines
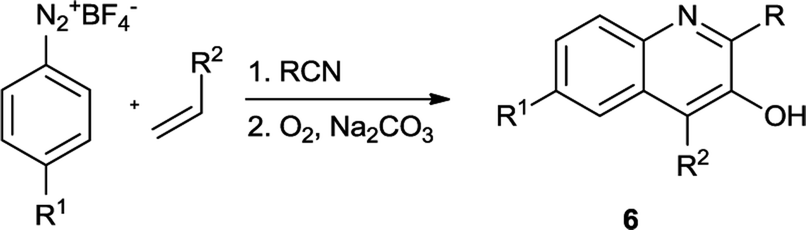
We attempted to carry out the two step reactions in a one-pot fashion (Table 3). Various aryldiazonium salts, nitriles and alkenes participated well in this oxidative transformation to afford structurally multi-substituted 3-hydroxyquinoline frameworks. Typically, phenyldiazonium tetrafluoroborate was treated with styrene in anhydrous MeCN under conditions identical to those employed previously (Table 1), followed by treatment of Na2CO3 and prolonged exposure to O2 furnished the 3-hydroxyquinoline derivative 6a in 36% isolated yield (Table 3, entry 1). Aryldiazonium salts bearing electron-donating substituents (Me-, t-Bu-) afforded the corresponding 3-hydroxyquinolines in acceptable yields (Table 3, entries 3–4). It should be noted that minor amount of 2,4-disubstituted quinoline derivatives were also isolated from the reaction mixture. Several aliphatic nitriles such as, primary, secondary, cyclic nitriles involved in the reaction and smoothly delivered the corresponding products (Table 3, entries 5–7). Unfortunately, benzylnitrile and chloroacetonitrile failed to produce the corresponding adducts. To our delight, aliphatic alkene was also a viable substrate for this oxidative transformation and provided the corresponding 2,4-dialkyl-3-hydroxyquinoline in good yield (Table 3, entry 8). As expected, benzonitrile furnished the expected adduct (6j) together with 21% of 2,4-phenylquinazoline.
Table 3 Preparation of 3-hydroxyquinolines in one-pot fashiona

|
| Entry |
Substituents in 6 |
Product (yield) |
| Reaction conditions: a mixture of arenediazonium salt (0.52 mmol), alkene (1.04 mmol) and nitrile (2 mL) in reaction tube at 80 °C for 2 h; followed by an aqueous workup with saturated aq. NaHCO3 and stirred in O2 (1 atm) with 5 g of Na2CO3 for 3 days. |
| 1 |
R = Me, R1 = H, R2 = C6H5 |
6a (36%) |
| 2 |
R = Me, R1 = H, R2 = p-(ClCH2)C6H4 |
6b (34%) |
| 3 |
R = Me, R1 = Me, R2 = C6H5, |
6d (47%) |
| 4 |
R = Me, R1 = t-Bu, R2 = C6H5 |
6e (44%) |
| 5 |
R = n-Pr, R1 = H, R2 = C6H5, |
6f (32%) |
| 6 |
R = i-Pr, R1 = H, R2 = C6H5 |
6g (39%) |
| 7 |
R = c-Pr, R1 = H, R2 = C6H5 |
6h (42%) |
| 8 |
R = Me, R1 = H, R2 = CH3(CH2)5 |
6i (56%) |
| 9 |
R = C6H5, R1 = H, R2 = C6H5 |
6j (46%) |
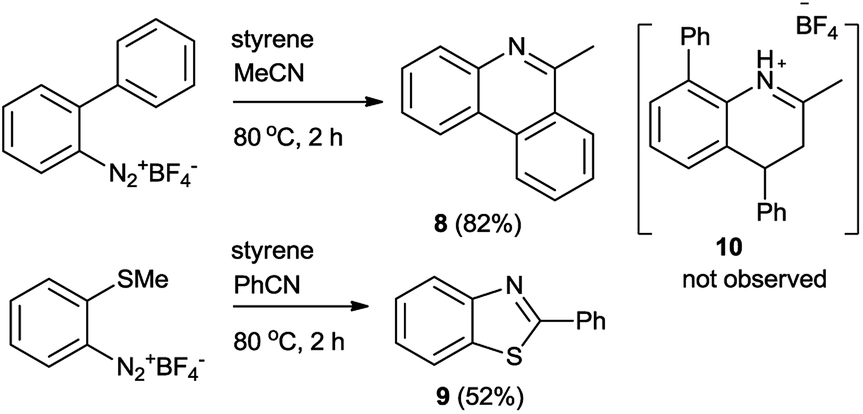
When aryldiazonium salt bearing adjacent phenyl or methylthio moiety was employed, N-arylnitrilium intermediate expeditiously underwent the intramolecular cyclization to deliver 6-methyl-phenanthridine18 (8) and 2-substituted benzothiazole (9) (Scheme 7). Presumably, the intra-molecular pathway is superior to the inter-molecular one.
 |
| | Scheme 7 Intra- versus inter-molecular reactions. | |
Besides the intermolecular reaction with styrenes, we also examined the reaction via an intramolecular fashion. Thus, o-ethenylbenzenediazonium salt 11 was used for investigation. Reaction of 11 with various nitriles gave the corresponding 2-substituted quinolines 12a–d in good yields (eqn (3)). Obviously, the nucleophilic attack of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C toward nitrilium ion followed by elimination took place immediately to deliver the final product.
C toward nitrilium ion followed by elimination took place immediately to deliver the final product.
| |
 | (3) |
Conclusions
In summary, we have demonstrated a synthetic useful approach for the preparation of 3-hydroxyquinolines from dihydroquinolines under atmospheric pressure of oxygen in the presence of sodium carbonate. This preparation can be carried out from diazonium salts in a one-pot fashion. Intramolecular trapping of in situ generated N-arylnitrilium intermediate with an alkene could generate 2-substituted quinolines in good yields. Preparation of functionalized 3-hydroxyquinoline derivatives through other known synthetic avenues generally requires a multistep synthesis.
Experimental
1H and 13C NMR were recorded in a 400 MHz Bruker spectrometer. Chemical shifts were given relative to CDCl3, CD3CN and DMSO-d6. Acetonitrile and other liquid nitriles were dried over activated molecular sieves, respectively, whereas solid nitriles were dried under vacuum. All the anilines were commercially purchased and used for diazotization without further purification. Other chemicals were used as purchased. Flash chromatography was performed using silica gel 230–400 mesh. Analytical thin layer chromatography (TLC) was performed using Merck-60 F-254 plates. In cases of known compounds, their 1H and 13C NMR values were compared with the literature values. Melting points were determined on a Fargo MP-1D instrument. All spectral data are deposited in the ESI.†
General procedure for preparation of 3,4-dihydroquinolines
In a dry 10 mL glass sealed tube, phenyldiazonium salt (100 mg, 0.52 mmol, 1.0 eq.) and styrene (108 mg, 1.04 mmol, 2.0 eq.) were suspended in 2 mL of anhydrous nitrile. The tube was sealed with a Teflon screw cap and heated in an oil bath (80 °C) for 2 h. After cooling to room temperature, reaction mixture with diethyl ether (20 mL). Corresponding 3,4-dihydroquinolinium tetrafluoroborate salt was formed as a solid which was washed with 2 × 10 mL of diethyl ether and dried under vacuum.
2-Methyl-4-phenyl-3,4-dihydroquinolinium tetrafluoroborate (3a)17
Off-white solid (130 mg, 81%); mp 193–194 °C: 1H NMR (400 MHz, CDCl3) δ 12.4 (brs, 1H), 7.52 (dd, J = 7.6, 1.6 Hz, 1H), 7.48–7.44 (m, 1H), 7.44–7.40 (m, 2H), 7.39–7.32 (m, 2H), 7.23–7.20 (m, 2H), 7.03–7.00 (m, 1H), 4.49 (t, J = 8.8 Hz, 1H), 3.48–3.32 (m, 2H), 2.66 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 189.2, 141.8, 133.2, 132.6, 131.3, 130.5, 130.3, 130.1, 129.2, 129.1, 121.8, 38.7, 38.6, 25.7. HRMS (ESI-TOF) calcd for C16H16N (M-BF4)+ m/z = 222.1277, found 222.1274. These data are in agreement with the reported data.
2,6-Dimethyl-4-phenyl-3,4-dihydroquinolinium tetrafluoro-borate (3b)
Pale yellow solid (121 mg, 72%). Mp 191–192 °C. 1H NMR (400 MHz, CD3CN): δ = 12.41 (brs, 1H), 7.41–7.37 (m, 3H), 7.35–7.31 (m, 1H), 7.27 (d, J = 8.0 Hz, 1H), 7.20 (d, J = 7.2 Hz, 2H), 6.85 (s, 1H), 4.43 (t, J = 8.8 Hz, 1H), 3.44–3.28 (m, 2H), 2.63 (s, 3H), 2.27 (s, 3H) ppm. 13C NMR (100 MHz, CD3CN): δ = 187.3, 143.3, 141.8, 130.9, 130.8, 130.6, 130.2, 129.0 (2C), 128.9, 121.4, 38.5, 38.4, 25.3, 21.5 ppm. HRMS (ESI-TOF) calcd for C17H18N (M-BF4)+ m/z = 236.1434, found 236.1443.
6-Methyl-2,4-diphenyl-3,4-dihydroquinolinium tetrafluoroborate (3c)
Bright yellow solid (98 mg, 54%). Mp 110–111 °C. 1H NMR (400 MHz, CD3CN): δ = 12.28 (brs, 1H), 8.07–8.05 (m, 2H), 7.88–7.84 (m, 1H), 7.72–7.65 (m, 3H), 7.41–7.31 (m, 4H), 7.27–7.24 (m, 2H), 4.61 (t, J = 8.4 Hz, 1H), 3.92–3.79 (m, 2H), 2.33 (s, 3H) ppm. 13C NMR (100 MHz, CD3CN): δ = 177.5, 143.8, 141.3, 137.7, 131.8, 131.5, 131.0, 130.9 (2C), 130.5, 130.4, 130.2, 128.9 (2C), 122.5, 38.8, 36.0, 21.6 ppm. HRMS (ESI-TOF) calcd for C22H20N (M-BF4)+ m/z = 298.1590, found 298.1571.
6-(tert-Butyl)-2-methyl-4-phenyl-3,4-dihydroquinolinium tetrafluoroborate (3d)
Pale yellow solid (101 mg, 68%). Mp 190–191 °C. 1H NMR (400 MHz, CD3CN): δ = 12.4 (brs, 1H), 7.51 (dd, J = 8.4, 1.6 Hz, 1H), 7.46 (d, J = 8.4 Hz, 1H), 7.40–7.37 (m, 2H), 7.34–7.30 (m, 1H), 7.20 (d, J = 7.2 Hz, 2H), 7.13 (d, J = 1.2 Hz, 1H), 4.50 (t, J = 8.4 Hz, 1H), 3.47–3.31 (m, 2H), 2.64 (s, 3H), 1.21 (s, 9H) ppm. 13C NMR (100 MHz, CD3CN): δ = 187.7, 156.0, 141.9, 130.7, 130.6, 130.2, 128.9, 128.8, 127.3, 126.8, 121.3, 38.7, 38.5, 35.8, 31.2, 25.4 ppm. HRMS (ESI-TOF) calcd for C20H24N (M-BF4)+ m/z = 278.1903, found 278.1914.
4-Phenyl-2-propyl-3,4-dihydroquinolinium tetrafluoroborate (3e)
Pale yellow solid (110 mg, 63%). Mp 132–133 °C. 1H NMR (400 MHz, CD3CN): δ = 12.40 (brs, 1H), 7.59 (d, J = 7.6 Hz, 1H), 7.51–7.44 (m, 2H), 7.40–7.30 (m, 3H), 7.17 (d, J = 7.2 Hz, 2H), 7.08 (d, J = 7.2 Hz, 1H), 4.52 (t, J = 8.0 Hz, 1H), 3.49–3.36 (m, 2H), 2.93–2.79 (m, 2H), 1.77–1.58 (m, 2H), 0.92 (t, J = 7.6 Hz, 3H) ppm. 13C NMR (100 MHz, CD3CN): δ = 191.7, 141.6, 133.3, 132.8, 131.4, 130.5, 130.4, 130.2, 129.1 (2C), 122.1, 40.9, 38.5, 37.5, 20.2, 14.0 ppm. HRMS (ESI-TOF) calcd for C18H20N (M-BF4)+ m/z = 250.1590, found 250.1593.
2-Isopropyl-4-phenyl-3,4-dihydroquinolinium tetrafluoroborate (3f)
Pale yellow solid (101 mg, 57%). Mp 148–149 °C. 1H NMR (400 MHz, CD3CN): δ = 12.20 (brs, 1H), 7.66 (dd, J = 7.2, 1.2 Hz, 1H), 7.53–7.45 (m, 2H), 7.39–7.30 (m, 3H), 7.16–7.10 (m, 3H), 4.53 (t, J = 8.0 Hz, 1H), 3.47–3.35 (m, 2H), 3.17 (sept, 6.8 Hz, 1H), 1.27 (d, J = 6.8 Hz, 3H), 1.15 (d, J = 6.8 Hz, 3H) ppm. 13C NMR (100 MHz, CD3CN): δ = 194.9, 141.4, 133.4, 132.8, 131.6, 130.4, 130.3, 130.2, 129.1, 129.0, 122.4, 38.5, 35.2, 18.6, 18.5 ppm. HRMS (ESI-TOF) calcd for C18H20N (M-BF4)+ m/z = 250.1590, found 250.1597.
2-Cyclopropyl-4-phenyl-3,4-dihydroquinolinium tetrafluoroborate (3g)
Bright yellow solid (114 mg, 66%). Mp 157–158 °C. 1H NMR (400 MHz, CD3CN): δ = 12.3 (brs, 1H), 7.52–7.45 (m, 2H), 7.41–7.31 (m, 4H), 7.17–7.14 (m, 2H), 7.03 (d, J = 7.6 Hz, 1H), 4.42 (t, J = 8.0 Hz, 1H), 2.95–2.93 (m, 2H), 2.41–2.34 (m, 1H), 1.68–1.56 (m, 2H), 1.55–1.48 (m, 1H), 1.33–1.27 (m, 1H) ppm. 13C NMR (100 MHz, CD3CN): δ = 191.3, 141.0, 133.6, 131.5, 131.2, 130.2 (2C), 129.9, 128.9 (2C), 120.8, 38.4, 32.1, 21.1, 14.6, 14.1 ppm. HRMS (ESI-TOF) calcd for C18H18N (M-BF4)+ m/z = 248.1434, found 248.1455.
2,4-Diphenyl-3,4-dihydroquinolinium tetrafluoroborate (3h)
Orange solid (129 mg, 67%). Mp 101–102 °C. 1H NMR (400 MHz, CD3CN): δ = 12.32 (brs, 1H), 8.10–8.08 (m, 2H), 7.90–7.86 (m, 1H), 7.77 (dd, J = 8.0, 1.6 Hz, 1H), 7.74–7.70 (m, 2H), 7.56–7.47 (m, 2H), 7.42–7.32 (m, 3H), 7.28–7.26 (m, 2H), 7.12 (d, J = 7.6 Hz, 1H), 4.67 (t, J = 8.4 Hz, 1H), 3.97–3.83 (m, 2H) ppm. 13C NMR (100 MHz, CD3CN): δ = 179.0, 141.1, 138.0, 133.9, 132.7, 131.7, 130.9 (2C), 130.6 (2C), 130.3, 130.0, 129.1, 129.0, 122.6, 38.8, 36.0 ppm. HRMS (ESI-TOF) calcd for C21H18N (M-BF4)+ m/z = 284.1434, found 284.1439.
4-(4-Bromophenyl)-2-methyl-3,4-dihydroquinolinium tetrafluoro-borate (3i)
Orange solid (162 mg, 80%). Mp 145–147 °C. 1H NMR (400 MHz, CD3CN): δ = 12.44 (br, 1H, N–H), 7.56–7.42 (m, 5H, Ar–H), 7.16–7.13 (m, 2H, Ar–H), 7.02 (d, J = 7.4 Hz, 1H, Ar–H), 4.50, 4.52–4.48 (t, J = 8.8 Hz, 1H, –CH), 3.47–3.29 (m, 2H, –CH2), 2.65 (s, 3H, –CH3) ppm. 13C NMR (100 MHz, CD3CN): δ = 188.7, 140.7, 133.0, 132.7, 132.3, 131.0, 130.3, 129.8(2C), 122.0, 121.4, 38.0, 37.7, 25.3 ppm. HRMS (ESI-TOF) calcd for C16H15N79Br (M-BF4): m/z = 300.0388, found 300.0379; HRMS (ESI-TOF) calcd for C16H15N81Br (M-BF4): m/z = 302.0368; found: 302.0359.
4-(4-Chlorophenyl)-2-methyl-3,4-dihydroquinolinium tetrafluoro-borate (3j)
Yellow solid (111 mg, 62%). Mp 161–162 °C. 1H NMR (400 MHz, CD3CN): δ = 7.51–7.39 (m, 5H, Ar–H), 7.22–7.19 (m, 2H, Ar–H), 7.03 (d, J = 7.2 Hz, 1H, Ar–H), 4.51 4.53–4.49 (t, J = 8.4 Hz, 1H, –CH), 3.48–3.29 (m, 2H, –CH2), 2.65 (s, 3H, –CH3) ppm. 13C NMR (100 MHz, CD3CN): δ = 188.6, 140.3, 134.0, 132.8, 132.3, 130.72, 130.4, 130.0, 129.9, 129.4, 121.4, 38.1, 37.7, 25.3 ppm. HRMS (ESI-TOF) calcd for C16H15N35Cl (M-BF4) m/z = 256.0893, found 256.0897; HRMS (ESI-TOF) calcd for C16H15N37Cl (M-BF4) m/z = 258.0864; found: 258.0964.
4-(4-(Chloromethyl)phenyl)-2-methyl-3,4-dihydroquinolinium tetrafluoroborate (3k)
Bright yellow solid (141 mg, 76%). Mp 126–127 °C. 1H NMR (400 MHz, CD3CN): δ = 12.29 (brs, 1H), 7.53–7.49 (m, 2H), 7.47–7.41 (m, 3H), 7.22 (d, J = 8.0 Hz, 2H), 7.03 (d, J = 7.6 Hz, 1H), 4.66 (s, 2H), 4.51 (t, J = 8.8 Hz, 1H), 3.48–3.31 (m, 2H), 2.65 (s, 3H) ppm. 13C NMR (100 MHz, CD3CN): δ = 189.0, 142.1, 138.9, 133.1, 132.7, 131.0, 130.8, 130.3, 130.2, 129.7, 121.8, 46.9, 38.5, 38.4, 25.7 ppm. HRMS (ESI-TOF) calcd for C17H1735ClN (M−BF4)+ m/z = 270.1044, found 270.1051; HRMS (ESI-TOF) calcd for C17H1737ClN (M-BF4)+ m/z =272.1044, found 272.1115.
4-(2,5-Dimethylphenyl)-2-isopropyl-3,4-dihydroquinolinium tetrafluoroborate (3l)
Orange solid (74 mg, 39%). Mp 158–159 °C. 1H NMR (400 MHz, CD3CN): δ = 12.04 (br, 1H, N–H), 7.64–7.62 (d, J = 7.6 Hz, 1H, Ar–H), 7.53–7.44 (m, 2H, Ar–H), 7.19–7.17 (d, J = 8.0 Hz, 1H, Ar–H), 7.05–7.00 (m, 2H, Ar–H), 6.65 (s, 1H, Ar–H), 4.74–4.70 (t, J = 8.4 Hz, 1H, –CH), 3.41–3.24 (m, 2H, –CH2), 3.21–3.11 (septet, J = 6.8 Hz, 1H, –CH(CH3) (CH3)), 2.37 (s, 3H, –CH3), 2.19 (s, 3H, –CH3), 1.29–1.27 (d, J = 6.8 Hz, 3H, –CH3), 1.18–1.16 (d, J = 6.8 Hz, 3H, –CH3) ppm. 13C NMR (100 MHz, CD3CN): δ = 194.8, 138.6, 137.3, 134.1, 133.4, 132.5, 131.9, 131.6, 129.7 (2C), 129.3, 128.5, 122.0, 38.2, 34.3, 33.7, 20.9, 19.1, 18.5, 18.0 ppm. HRMS (ESI-TOF) calcd for C20H24 (M-BF4): = 278.1909; found: 278.1906.
2,4-Dimethyl-4-phenyl-3,4-dihydroquinolinium tetrafluoroborate (3m)
White solid (60 mg, 49%). Mp 171–172 °C. 1H NMR (400 MHz, CD3CN): δ = 7.55–7.51 (m, 3H, Ar–H), 7.37–7.29 (m, 4H, Ar–H), 7.21–7.19 (m, 2H, Ar–H), 3.71–3.66 (d, J = 18.8 Hz, 1H, –CHa(Hb)), 3.23–3.19 (d, J = 18.8 Hz, 1H, –C(Ha)Hb), 2.63 (s, 3H, –CH3), 1.72 (s, 3H, –CH3) ppm. 13C NMR (100 MHz, CD3CN): δ = 188.3, 144.8, 135.8, 132.5, 132.4, 129.7, 129.6, 128.6, 128.3, 127.3, 121.7, 44.8, 40.0, 27.3, 25.4 ppm. HRMS (ESI-TOF) calcd for C17H18N (M-BF4): = 236.1439; found 236.1442.
6-Methyl-6a,7,8,9,10,10a-hexahydro-7,10-methanophenanthridinum tetrafluoroborate (3n)
Pale yellow solid (95 mg, 62%). Mp 191–192 °C. 1H NMR (400 MHz, CD3CN): δ = 12.0 (brs, 1H), 7.45 (t, J = 7.6 Hz, 1H), 7.40 (d, J = 7.2 Hz, 1H), 7.34 (t, J = 7.2 Hz, 1H), 7.29 (d, J = 7.6 Hz, 1H), 3.28 (d, J = 10.4 Hz, 1H), 3.20 (d, J = 10.4 Hz, 1H), 2.85 (s, 1H), 2.59 (s, 3H), 2.47 (s, 1H), 1.79–1.62 (m, 4H), 1.35–1.28 (m, 2H) ppm. 13C NMR (100 MHz, CD3CN): δ = 186.5, 132.5, 131.3, 131.0, 129.4, 129.1, 121.4, 50.1, 49.5, 47.1, 42.9, 36.3, 31.2, 29.3, 24.3 ppm. HRMS (ESI-TOF) calcd for C15H18N (M-BF4)+ m/z = 212.1434, found 212.1445.
2,4-Diphenylquinazoline (4)18c
White solid (26 mg, 18%). Mp 110–111 °C. 1H NMR (400 MHz, CDCl3): δ = 8.70 (d, J = 8.0 Hz, 2H), 8.15 (d, J = 8.4 Hz, 1H), 8.12 (d, J = 8.4 Hz, 1H), 7.89–7.85 (m, 3H), 7.59–7.57 (m, 3H), 7.55–7.47 (m, 4H) ppm. 13C NMR (100 MHz, CDCl3) δ 168.3, 160.2, 151.9, 138.1, 137.6, 133.5, 130.5, 130.1, 129.9, 129.1, 128.6, 128.5 (3C), 126.9, 121.6 ppm. HRMS (ESI-TOF) calcd for C20H15N2 (M + H)+ m/z = 283.1235, found 283.1231. These data are in agreement with the reported data.18c
2-Methyl-4-phenylquinoline (5)
Pale yellow solid (33 mg, 77%). 1H NMR (400 MHz, CDCl3): δ = 8.07 (d, J = 8.8 Hz, 1H), 7.82 (d, J = 8.4 Hz, 1H), 7.66–7.63 (m, 1H), 7.49–7.43 (m 5H), 7.38 (t, J = 7.2 Hz, 1H), 7.19 (s, 1H), 2.75 (s, 3H). 13C NMR (100 MHz, CDCl3): δ = 158.4, 148.4, 148.3, 138.0, 129.4, 129.2, 128.9, 128.4, 128.2, 125.6, 125.5, 125.0, 122.1, 25.2; HRMS (ESI) calcd for C16H14N (M + H)+ m/z = 220.1126 found 220.1127. These data are in agreement with the reported data.18b
General procedure for the oxidation of 3,4-dihydroquinoline with oxygen
2-Methyl-4-phenyl-3,4-dihydroquinolinium tetrafluoroborate 3a (0.3 mmol) was extracted with DCM (25 mL), washed with a saturated aqueous solution of NaHCO3 (5 mL). The organic layer was dried over MgSO4 and filtered. To this was added Na2CO3 (10 equiv) and this mixture was stirred at room temperature for 3 days. Next, the resulting mixture was filtered through a plug of Celite and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, eluent, 30% EtOAc in hexane) to give the desired product.
General procedure for one-pot preparation of 3-hydroxyquinoline
In a dry 10 mL glass sealed tube, phenyldiazonium salt (100 mg, 0.52 mmol, 1.0 eq.) and styrene (108 mg, 1.04 mmol, 2.0 eq.) were suspended in 2 mL of anhydrous nitrile. The tube was sealed with a Teflon screw cap and heated in an oil bath (80 °C) for 2 h. After cooling to room temperature, reaction mixture was diluted with DCM (20 mL) and washed with saturated aq. NaHCO3 solution (10 mL). Combined organic layer was stirred over anhydrous Na2CO3 (5 g) for 3 days. Solvents removed and purified by column chromatography (eluent: 20% to 40% EtOAc in hexane) to obtain the corresponding substituted 3-hydroxyquinolines.
2-Methyl-4-phenylquinolin-3-ol (6a)18b
Off-white solid (44 mg, 36%); mp 238–239 °C:1H NMR (400 MHz, DMSO-d6) δ 8.86 (brs, 1H), 7.87 (d, J = 8.4 Hz, 1H), 7.54 (t, J = 7.2 Hz, 2H), 7.47 (t, J = 7.2 Hz, 2H), 7.34–7.31 (m, 3H), 7.23 (d, J = 8.0 Hz, 1H), 2.62 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 152.5, 145.4, 142.1, 133.7, 130.3, 128.7, 128.4, 128.2, 127.8, 127.5, 125.8, 125.7, 124.1, 21.2. HRMS (ESI-TOF) calcd for C16H14NO (M + H)+ m/z = 236.1075, found 236.1073. These data are in agreement with the reported data.
4-(4-(Chloromethyl)phenyl)-2-methylquinolin-3-ol (6b)15
Pale yellow solid (59 mg, 69%). 1H NMR (400 MHz, DMSO-d6): δ = 8.98 (br, 1H, O–H), 7.88 (d, J = 8.4 Hz, 1H, Ar–H), 7.60 (d, J = 8.0 Hz, 2H, Ar–H), 7.50–7.46 (m, 1H, Ar–H), 7.37–7.33 (m, 3H, Ar–H), 7.23 (d, J = 8.4 Hz, 1H, Ar–H), 4.88 (s, 2H, -CH2Cl), 2.63 (s, 3H, -CH3) ppm. 13C NMR (100 MHz, DMSO-d6): δ = 152.6, 145.5, 142.1, 137.1, 133.9, 130.7, 129.0, 128.3, 128.2, 127.4, 125.8(2C), 124.0, 46.0, 21.3 ppm. HRMS (ESI-TOF) calcd for C17H1535ClNO (M + H+) m/z = 284.0842; found: 284.0840; HRMS (ESI-TOF) calcd for C17H1537ClNO (M + H+) m/z = 286.0813; found: 286.0807. These data are in agreement with the reported data.
4-(4-Chlorophenyl)-2-methylquinolin-3-ol (6c)
Pale yellow solid (41 mg, 50%). Mp 272–274 °C. 1H NMR (400 MHz, DMSO-d6): δ = 7.87 (d, J = 8.4 Hz, 1H, Ar–H), 7.59 (d, J = 8.4 Hz, 2H, Ar–H), 7.50–7.46 (m, 1H, Ar–H), 7.38–7.33 (m, 3H, Ar–H), 7.23 (d, J = 8.0 Hz, 1H, Ar–H), 2.61 (s, 3H, –CH3) ppm. 13C NMR (125 MHz, DMSO-d6): δ = 152.6, 145.7, 142.0, 132.7, 132.4, 128.6, 128.3, 127.3, 127.2, 126.0, 125.9(2C), 123.9, 21.2 ppm. HRMS (ESI-TOF) calcd for C16H1335ClNO (M + H+) m/z = 270.0680; found: 270.0695; HRMS (ESI-TOF) calcd for C16H1337ClNO (M + H+) m/z = 272.0651; found: 272.0692.
2,6-Dimethyl-4-phenylquinolin-3-ol (6d)
Pale yellow solid (56 mg, 47%). Mp 205–206 °C. 1H NMR (400 MHz, CDCl3): δ = 7.92 (d, J = 8.4 Hz, 1H), 7.57–7.47 (m, 3H), 7.37–7.32 (m, 3H), 7.06 (s, 1H), 4.32 (brs, 1H), 2.71 (s, 3H), 2.35 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3): δ = 150.3, 144.8, 141.0, 136.0, 135.8, 132.4, 130.2, 129.4, 128.8, 128.5, 127.9, 127.2, 123.3, 21.6, 20.4 ppm. HRMS (ESI-TOF) calcd for C17H16NO (M + H)+ m/z = 250.1232, found 250.1239.
6-(tert-Butyl)-2-methyl-4-phenylquinolin-3-ol (6e)
Pale yellow solid (52 mg, 44%). Mp 243–244 °C.1H NMR (400 MHz, CDCl3): δ = 7.90 (d, J = 9.2 Hz, 1H), 7.59–7.52 (m, 2H), 7.47 (t, J = 7.6 Hz, 2H), 7.38 (d, J = 7.6 Hz, 2H), 7.25–7.24 (m, 1H), 2.68 (s, 3H), 1.24 (s, 9H) ppm. 13C NMR (100 MHz, CDCl3): δ = 150.4, 148.7, 144.6, 141.2, 132.3, 130.2, 129.5, 128.9, 127.9, 127.5, 126.7, 125.1, 119.4, 34.8, 31.0, 20.5 ppm. HRMS (ESI-TOF) calcd for C20H20NO (M + H)+ m/z = 292.1701, found 292.1702.
4-Phenyl-2-propylquinolin-3-ol (6f)
Pale yellow solid (43 mg, 32%). Mp 177–178 °C.1H NMR (400 MHz, CDCl3): δ = 8.00 (d, J = 8.4 Hz, 1H), 7.57–7.54 (m, 2H), 7.50–7.46 (m, 2H), 7.38 (d, J = 7.2 Hz, 2H), 7.32–7.31 (m, 2H), 5.50 (brs, 1H), 3.02 (t, J = 8.0 Hz, 2H), 1.85 (sextet, J = 7.6 Hz, 2H), 1.05 (t, J = 7.6 Hz, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 154.7, 144.3, 142.9, 132.2, 130.3, 129.6, 129.0, 128.7, 127.5, 127.1, 126.2, 126.0, 124.3, 36.1, 21.7, 14.2 ppm. HRMS (ESI-TOF) calcd for C18H18NO (M + H)+ m/z = 264.1388, found 264.1390.
2-Isopropyl-4-phenylquinolin-3-ol (6g)
Pale yellow solid (53 mg, 39%). Mp 98–99 °C.1H NMR (400 MHz, CDCl3): δ = 8.05 (d, J = 8.4 Hz, 1H), 7.61–7.57 (m, 2H), 7.54–7.47 (m, 2H), 7.40 (d, J = 6.8 Hz, 2H), 7.32–7.31 (m, 2H), 5.25 (brs, 1H), 3.63 (sept, J = 6.8 Hz, 1H), 1.43 (d, J = 6.8 Hz, 6H) ppm. 13C NMR (100 MHz, CDCl3): δ = 158.5, 143.6, 142.9, 132.2, 130.4, 129.7, 129.1, 129.0, 127.2, 126.9, 126.0, 125.9, 124.2, 31.0, 20.8 ppm. HRMS (ESI-TOF) calcd for C18H18NO (M + H)+ m/z = 264.1388, found 264.1394.
2-Cyclopropyl-4-phenylquinolin-3-ol (6h)
Off-white solid (57 mg, 42%). Mp 127–128 °C. 1H NMR (400 MHz, CDCl3): δ = 7.97 (d, J = 8.4 Hz, 1H), 7.58 (t, J = 7.6 Hz, 2H), 7.54–7.44 (m, 2H), 7.41–7.39 (m, 2H), 7.31–7.26 (m, 2H), 5.35 (brs, 1H), 2.63 (m, 1H), 1.36–1.35 (m, 2H), 1.10–1.06 (m, 2H). 13C NMR (100 MHz, CDCl3): δ = 154.5, 144.4, 132.2, 130.3, 129.9 (2C), 129.6, 129.0, 128.4, 126.6, 126.3, 125.6, 124.3, 11.9, 9.5. HRMS (ESI-TOF) calcd for C18H16NO (M + H)+ m/z = 262.1232, found 262.1232.
4-Hexyl-2-methylquinolin-3-ol (6i)
Off-white solid (70 mg, 56%). Mp 161–162 °C. 1H NMR (400 MHz, CDCl3): δ = 8.00 (d, J = 8.0 Hz, 1H), 7.83 (dd, J = 8.0, 1.2 Hz, 1H), 7.51–7.44 (m, 2H), 3.02 (t, J = 8.0 Hz, 2H), 2.67 (s, 3H), 1.66–1.58 (m, 2H), 1.43–1.36 (m, 2H), 1.28–1.26 (m, 2H), 1.25–1.23 (m, 2H), 0.84 (t, J = 6.8 Hz, 3H) ppm. 13C NMR (100 MHz, CDCl3): δ = 150.2, 146.1, 141.5, 127.6 (2C), 126.9, 126.3, 122.9 (2C), 31.6, 29.6, 29.3, 25.2, 22.5, 20.0, 14.0 ppm. HRMS (ESI-TOF) calcd for C16H22NO (M + H)+ m/z = 244.1701, found 244.1696.
2,4-Diphenylquinolin-3-ol (6j)20
Off-white solid (68 mg, 46%). Mp 123–124 °C. 1H NMR (400 MHz, CDCl3): δ = 8.15 (d, J = 8.4 Hz, 1H), 8.01–7.93 (m, 2H), 7.62–7.58 (m, 2H), 7.58–7.53 (m, 2H), 7.52–7.48 (m, 2H), 7.48–7.42 (m, 3H), 7.42–7.37 (m, 2H), 5.45 (brs, 1H). 13C NMR (100 MHz, CDCl3): δ = 150.0, 143.9, 143.3, 137.2, 132.2, 130.3 (2C), 129.5 (2C), 129.4 (2C), 129.1, 128.4 (2C), 127.6, 126.8, 124.5. HRMS (ESI-TOF) calcd for C21H16NO (M + H)+ m/z = 298.1232, found 298.1242. These data are in agreement with the reported data.
2,4-Dimethyl-4-phenylquinolin-3(4H)-one (7)
Yellow oil (52 mg, 70%). 1H NMR (400 MHz, CDCl3): δ = 7.60 (d, J = 7.2 Hz, 1H, Ar–H), 7.46–7.42 (m, 1H, Ar–H), 7.37–7.33 (m, 1H, Ar–H), 7.29–7.23 (m, 4H, Ar–H), 7.03–7.01 (m, 2H, Ar–H), 2.28 (s, 3H, –CH3), 1.89 (s, 3H, –CH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 196.4, 160.7, 140.7, 140.6, 136.8, 129.4, 128.8, 128.7, 128.4, 127.9, 127.6, 127.2, 53.6, 23.3, 20.0 ppm. HRMS (ESI-TOF) calcd for C17H16NO (M + H+) m/z = 250.1232; found: 250.1243.
6-Methylphenanthridine (8)18b
Pale yellow oil (59 mg, 82%). 1H NMR (400 MHz, CDCl3): δ = 8.59 (d, J = 8.0 Hz, 1H), 8.50 (d, J = 8.4 Hz, 1H), 8.18 (d, J = 8.4 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.81 (t, J = 8.0 Hz, 1H), 7.70–7.64 (m, 2H), 7.59 (t, J = 7.6 Hz, 1H), 3.02 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3): δ = 158.8, 143.6, 132.5, 130.4, 129.2, 128.6, 127.2, 126.5, 126.3, 125.8, 123.7, 122.2, 121.9, 23.3 ppm. HRMS(ESI) calcd for C14H12N (M + 1)+ m/z = 194.0970, found 194.0976. These data are in agreement with the reported data.
2-Phenylbenzo[d]thiazole (9)21b
Off-white solid (45 mg, 52%). Mp 111–112 °C. 1H NMR (400 MHz, CDCl3): δ = 8.09–8.05 (m, 3H), 7.88 (dd, J = 8.0, 0.4 Hz, 1H), 7.49–7.45 (m, 4H), 7.36 (t, J = 7.2 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3) δ 168.0, 154.1, 135.0, 133.5, 130.9, 128.9, 127.5, 126.2, 125.1, 123.0, 121.5 ppm. HRMS(ESI) calcd for C13H10NS (M + 1)+ m/z = 212.0534, found 212.0539. These data are in agreement with the reported data.
General procedure for preparing 2-substituted quinolines from o-vinylaryldiazonium tetrafluoroborate
In a dry 10 mL glass sealed tube, o-vinylaryldiazonium salt (0.46 mmol) is suspended in 2 mL of anhydrous nitrile. The tube was sealed with a Teflon screw cap and heated in an oil bath (60 °C) for 12 h. After cooling to room temperature, reaction mixture was diluted with DCM (20 mL) and washed with saturated aq. NaHCO3 solution (10 mL). Organic layer was dried over anhydrous Na2SO4. Solvents removed and purified by column chromatography (eluent: 20% EtOAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane) to obtain the title compound.
:hexane) to obtain the title compound.
2-Methylquinoline (12a)21c
Brown oil (55 mg, 74%). 1H NMR (400 MHz, CDCl3): δ = 7.97 (d, J = 8.4 Hz, 1H), 7.92 (t, J = 8.0 Hz, 1H), 7.60 (d, J = 8.0 Hz, 1H), 7.61–7.57 (m, 1H), 7.40–7.36 (m, 1H), 7.18–7.15 (m, 1H), 2.66 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3); δ = 158.7, 147.6, 135.9, 129.2, 128.3, 127.2, 126.2, 125.4, 121.7, 25.1 ppm. HRMS (ESI-TOF) calcd for C10H10N (M + H)+ m/z = 144.0813, found 144.0818. These data are in agreement with the reported data.
2-Propylquinoline (12b)21d
Pale yellow oil (61 mg, 69%). 1H NMR (400 MHz, CDCl3): δ = 8.04 (dd, J = 8.4, 3.2 Hz, 2H), 7.75 (d, J = 8.4 Hz, 1H), 7.68–7.63 (m, 1H), 7.45 (t, J = 8.0 Hz, 1H), 7.27 (d, J = 8.4 Hz, 1H), 2.94 (t, J = 7.6 Hz, 2H), 1.82 (sext, J = 7.6 Hz, 2H), 1.00 (t, J = 7.6 Hz, 3H) ppm. 13C NMR (100 MHz, CDCl3): δ = 162.8, 147.7, 136.2, 129.3, 128.6, 127.4, 126.7, 125.6, 121.3, 41.1, 23.2, 13.9 ppm. HRMS (ESI-TOF) calcd for C12H14N (M + H)+ m/z = 172.1126, found 172.1126. These data are in agreement with the reported data.
2-Isopropylquinoline (12c)21e
Pale yellow oil (56 mg, 63%). 1H NMR (400 MHz, CDCl3): δ = 8.05 (d, J = 8.4 Hz, 1H), 8.00 (d, J = 8.4 Hz, 1H), 7.70 (d, J = 8.0 Hz, 1H), 7.65–7.61 (m, 1H), 7.42 (t, J = 8.4 Hz, 1H), 7.27 (d, J = 8.4 Hz, 1H), 3.24 (sept, J = 6.8 Hz, 1H), 1.37 (d, J = 6.8 Hz, 6H) ppm. 13C NMR (100 MHz, CDCl3): δ = 167.4, 147.6, 136.2, 129.0, 128.8, 127.2, 126.7, 125.4, 119.0, 37.1, 22.3. HRMS (ESI-TOF) calcd for C12H14N (M + H)+ m/z = 172.1126, found 172.1134. These data are in agreement with the reported data.
2-Phenylquinoline (12d)21c
Off-white solid (72 mg, 69%). 1H NMR (400 MHz, CDCl3): δ = 8.20–8.14 (m, 4H), 7.85 (d, J = 8.8 Hz, 1H), 7.80 (dd, J = 8.0, 0.8 Hz, 1H), 7.73–7.69 (m, 1H), 7.54–7.49 (m, 3H), 7.47–7.43 (m, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 157.3, 148.2, 139.6, 136.7, 129.7, 129.6, 129.2, 128.8, 127.5, 127.4, 127.1, 126.2, 118.9 ppm. HRMS (ESI-TOF) calcd for C15H12N (M + H)+ m/z = 206.0970, found 206.0965. These data are in agreement with the reported data.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Ministry of Science and Technology, Taiwan (Grant No. MOST106-2113-M-002-018) for the financial support. We also thank “National Taiwan University Mass Spectrometry-based Proteomics Core Facility” for the measurement of ESI mass data.
Notes and references
-
(a) S. Cretton, S. Dorsaz, A. Azzollini, Q. F. Godal, L. Marcourt, S. N. Ebrahimi, F. Voinesco, E. Michellod, D. Sanglard, M. Cuendet and P. Christen, J. Nat. Prod., 2016, 79, 300 CrossRef CAS PubMed;
(b) J. P. Michael, Nat. Prod. Rep., 2008, 25, 166 RSC;
(c) J. P. Michael, Nat. Prod. Rep., 2004, 21, 650 RSC.
-
(a) O. Afzal, S. Kumar, M. R. Haider, M. R. Ali, R. Kumar, M. Jaggi and S. Bawa, Eur. J. Med. Chem., 2015, 97, 871 CrossRef CAS PubMed;
(b) S. Kumar, S. Bawa and H. Gupta, Mini-Rev. Med. Chem., 2009, 9, 1648 CrossRef CAS PubMed;
(c) D. J. Hazuda, P. Felock and M. Witmer, Science, 2000, 287, 646 CrossRef CAS PubMed;
(d) J. Wang, M. W. Chen, Y. Ji, S. B. Hu and Y. G. Zhou, J. Am. Chem. Soc., 2016, 138, 10413 CrossRef CAS PubMed;
(e) M. Heitbaum, R. Frchlich and F. Glorius, Adv. Synth. Catal., 2010, 352, 357 CrossRef CAS , and references therein..
-
(a) L. A. Dawson, K. J. Cato, C. Scott, J. M. Watson, M. D. Wood, R. Foxton, R. De la Flor, G. A. Jones, J. N. Kew, J. E. Cluderay, E. Southam, G. S. Murkitt, J. Gartlon, D. J. Perberton, D. C. Jones, C. H. Davies and J. Hagen, Neuropsychopharmacology, 2008, 33, 1642 CrossRef CAS PubMed;
(b) P. W. Bedard, V. Clerin, N. Sushkova, B. Tchernychev, T. Antrilli, C. Resmini, J. C. Keith Jr, J. K. Hennan, N. Kaila, S. Debernardo, K. Janz, Q. Wang, D. L. Crandall, R. G. Schaub, G. D. Shaw and L. L. Carter, J. Pharmacol. Exp. Ther., 2008, 324, 497 CrossRef CAS PubMed;
(c) M. A. Massoud, S. A. El Bialy, W. A. Bayoumi and W. M. El Husseiny, Heterocycl. Commun., 2014, 20, 81 CAS.
- V. Suchaud, F. Bailly and C. Lion, Bioorg. Med. Chem. Lett., 2012, 22, 3988 CrossRef CAS PubMed.
- Y. Aoki, M. Ishiwara, A. Koda and H. Takagaki, Eur. J. Pharmacol., 2000, 409, 325 CrossRef CAS PubMed.
- A. J. Duplantier, S. L. Becker and M. J. Bohanon, J. Med. Chem., 2009, 52, 3576 CrossRef CAS PubMed.
- S. A. Jenekhe, L. Lu and M. M. Alam, Macromolecules, 2001, 34, 7315 CrossRef CAS.
- A. Heguy, P. Cai, P. Meyn, D. Houck, S. Russo, R. Michitsch, C. Pearce, B. Katz, G. Bringmann, D. Feineis, D. L. Taylor and A. S. Tyms, Antiviral Chem. Chemother., 1998, 9, 149 CrossRef CAS PubMed.
- D. L. Boger and J.-H. Chen, J. Org. Chem., 1995, 60, 7369 CrossRef CAS.
-
(a) E. J. Cragoe, M. D. Bealor, C. M. Robb, C. Ziegler and J. M. Sprague, J. Org. Chem., 1953, 18, 561 CrossRef CAS;
(b) F. J. Kreysa, P. S. Forgione and V. F. Maturi, J. Org. Chem., 1955, 20, 971 CrossRef CAS.
- W. Zhang and J. M. Ready, Angew. Chem., Int. Ed., 2014, 53, 8980 CrossRef CAS PubMed.
- Y. Kobayashi and T. Harayama, Org. Lett., 2009, 11, 1603 CrossRef CAS PubMed.
- N. H. Cromwell and R. A. Setterquist, J. Am. Chem. Soc., 1954, 76, 5752 CrossRef CAS.
- Z. He and T. F. Jamison, Angew. Chem., Int. Ed., 2014, 53, 3353 CrossRef CAS PubMed.
- P. Chauhan, M. Ravi, R. Kant and P. P. Yadav, Org. Biomol. Chem., 2017, 15, 1080 RSC.
- V. A. Mamedov, V. L. Mamedova, V. V. Sykaev, D. E. Korshin, G. Z. Khikmatova, E. V. Mironova, O. B. Bazanova, L. K. Rizvanov and S. K. Latypova, Tetrahedron, 2017, 73, 5082 CrossRef CAS.
- S. W. Youn, H. J. Yoo, E. M. Lee and S. Y. Lee, Adv. Synth. Catal., 2018, 360, 278 CrossRef CAS.
-
(a) M. Ramanathan and S.-T. Liu, Tetrahedron, 2017, 73, 4317 CrossRef CAS;
(b) M. Ramanathan and S.-T. Liu, J. Org. Chem., 2015, 80, 5329 CrossRef CAS PubMed;
(c) M. Ramanathan and S.-T. Liu, J. Org. Chem., 2017, 82, 8290 CrossRef CAS PubMed.
- M. Xie, X. Liu, Y. Zhu, X. Zhao, Y. Xia, L. Lin and X. Feng, Chem.–Eur. J., 2011, 17, 13800 CrossRef CAS PubMed.
- K. Janz and N. Kaila, J. Org. Chem., 2009, 74, 8874 CrossRef CAS PubMed.
-
(a) X. Su, C. Chen, Y. Wang, J. Chen, Z. Lou and M. Li, Chem. Commun., 2013, 49, 6752 RSC;
(b) J. I. Urzua, R. Contrera, C. O. Salasa and R. A Tapia, RSC Adv., 2016, 6, 82401 RSC;
(c) K. H. He, F. F. Tan, C. Z. Zhou, G. J. Zhou, X. L. Yang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 3080 CrossRef CAS PubMed;
(d) N. T. Patil and V. S. Raut, J. Org. Chem., 2010, 75, 6961 CrossRef CAS PubMed;
(e) Y. F. Liang, X. F. Zhou, S. Y. Tang, Y. B. Huang, Y. S. Feng and H. J. Xu, RSC Adv., 2013, 3, 7739 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectral data of all compounds and spectra. See DOI: 10.1039/c8ra07940d |
| ‡ Equal contribution. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*