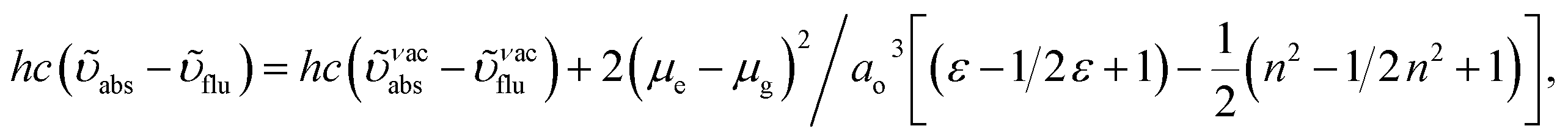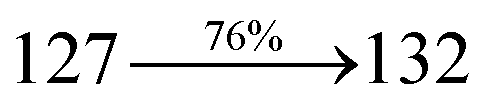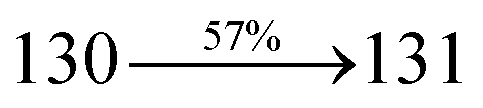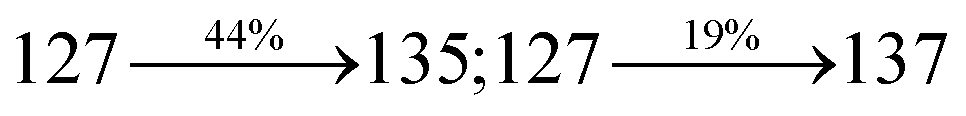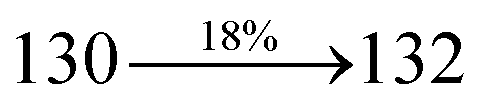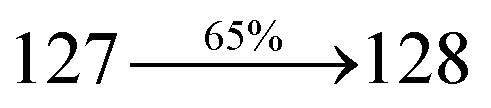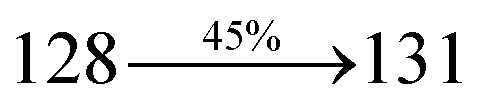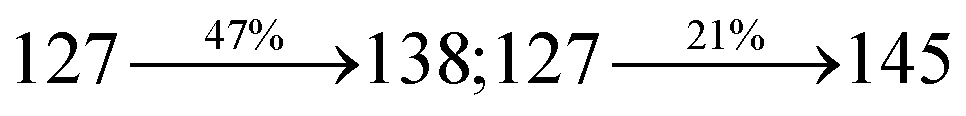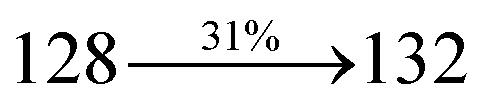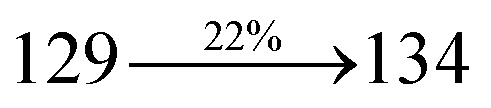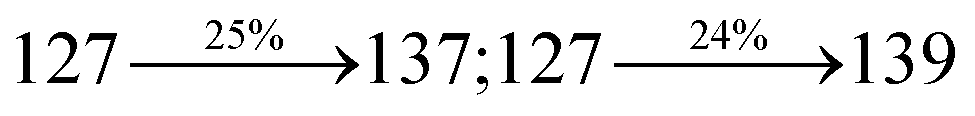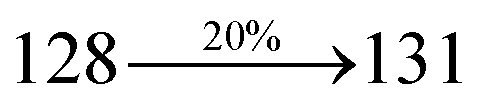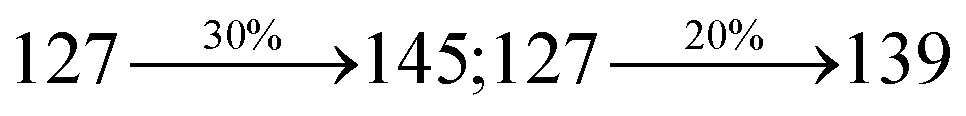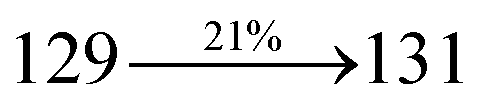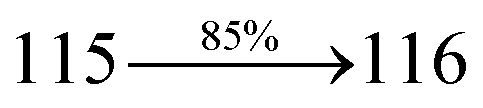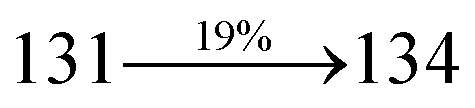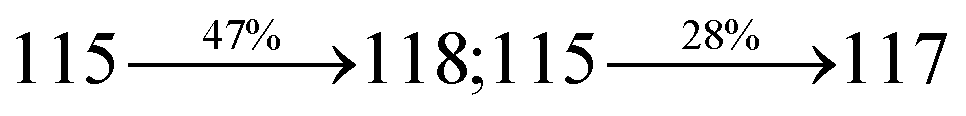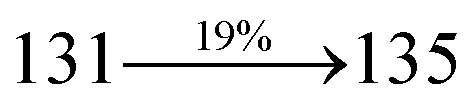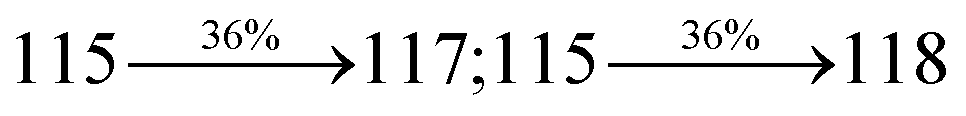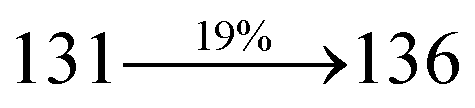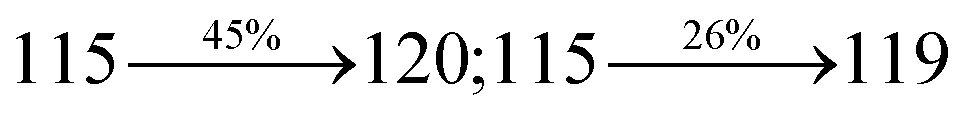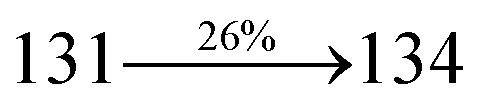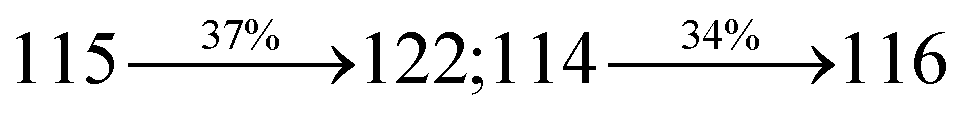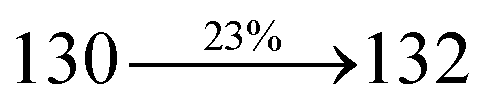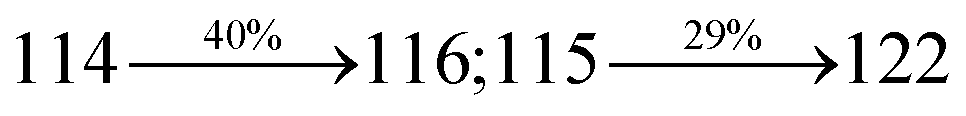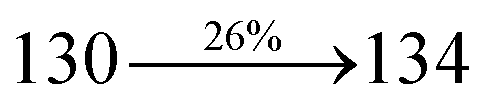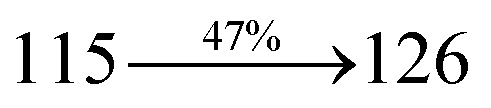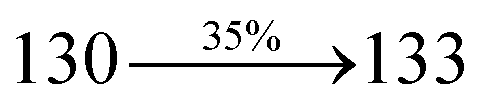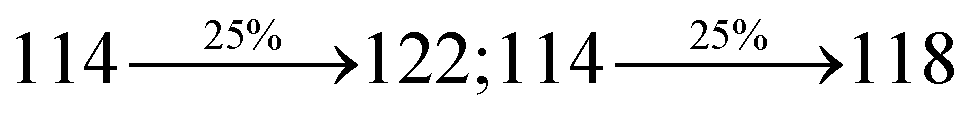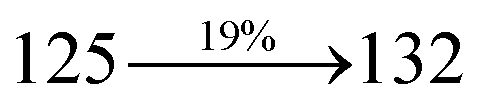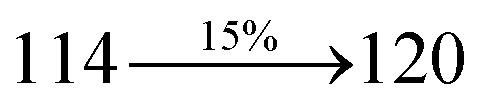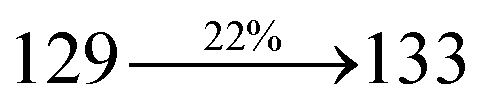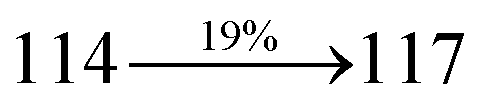DOI:
10.1039/C8RA07891B
(Paper)
RSC Adv., 2018,
8, 37324-37338
Hot exciton transition for organic light-emitting diodes: tailoring excited-state properties and electroluminescence performances of donor–spacer–acceptor molecules†
Received
22nd September 2018
, Accepted 23rd October 2018
First published on 6th November 2018
Abstract
The photophysical, electrochemical and electroluminescent properties of newly synthesized blue emitters with donor–π–acceptor geometry, namely, 4′-(1-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)-N,N-diphenyl-(2-[1,1′-biphenyl]vinyl)-4-amine (NSPI-TPA), 4′-(1-(2-methylnaphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)-N,N-diphenyl-(2-[1,1′-biphenyl]vinyl)-4-amine (MNSPI-TPA), 4-(2-(4′-(diphenylamino)-(2-[1,1′-biphenyl]vinyl)-4-yl)-1H-phenanthro[9,10-d]imidazol-1-yl)-1-naphthalene-1-carbonitrile (SPNCN-TPA) and 4-(2-(4-(9H-carbazol-9-yl)styryl)-1H-phenanthro[9,10-d]imidazol-1-yl)naphthalene-1-carbonitrile (SPNCN-Cz) were analyzed. The conjugation length in the emitters is not conducive to pure emission and hence, a molecular twisting strategy was adopted in NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz to enhance pure emission. The emissive state (HLCT) of twisted D–π–A molecules containing both LE and CT (HLCT) states was tuned for high PL (ηPL) (LE) and high exciton utilization (ηs) (CT) efficiencies by replacing triphenylamine (strong donor) with carbazole (weak donor). Among strong donor compounds, namely, NSPI-TPA, MNSPI-TPA and SPNCN-TPA, the SPNCN-TPA-based device exhibited blue emission (451 nm) with CIE coordinates (0.15, 0.08), maximum current efficiency (ηc) of 2.32 cd A−1, power efficiency (ηp) of 2.01 lm W−1 and external quantum efficiency (ηex) of 3.02%. The device with SPNCN-Cz emitter exhibited higher electroluminescence efficiencies than the SPNCN-TPA-based device, with pure blue emission (443 nm, CIE: 0.15,0.07), ηex of 3.15%, ηc of 2.56 cd A−1 and ηp of 2.45 lm W−1.
1 Introduction
Organic light-emitting diodes (OLEDs) have been widely investigated and tested commercially in recent decades and utilized in flat-panel displays.1 However, their commercialization is still restricted because of the scarcity of blue emitters.2–4 Efficient green and red emissive materials are highly exploited. However, fabrication of blue OLEDs is a major problem due to wide band gap and unbalanced carrier injection.5–10 Therefore, the design of efficient pure blue emitters with narrow full width at half maximum (FWHM) is still an important task.11,12 The aggregated arrangement of the emitter-induced molecular interaction results in bathochromic shift with low quantum efficiency.13 However, non-doped blue emitters with restricted intermolecular interaction exhibit expected quantum yields.14 Their potential carrier injection and transporting abilities provide balanced charge recombination, which results in enhanced efficiency.15–17 For TV displays, blue OLEDs with CIE (0.14, 0.08) and (0.15, 0.06) are required by the National Television System Committee (NTSC) and high-definition television (HDTV), respectively. Pyrene18 and anthracene19 derivative-based blue OLEDs exhibit high efficiency with poor color purity. Therefore, to achieve pure blue emission, emitters having D–π–A geometry with twisted configuration are employed as they reduce the π-conjugation and consequently exhibit blue emission.20–26 Furthermore, the emissive state of twisted D–π–A molecules27–30 possesses both LE and CT states (hybridized local and charge transfer state/HLCT) and shows high PL efficiency (LE) and high exciton utilization (ηs) (CT), which are attributed to hot exciton mechanism.31–33 The LE-dominated (low lying) HLCT state provided high radiative rate (kr), resulting in high photoluminescence efficiency (ηPL) of the film, whereas CT-dominated HLCT state is responsible for high ηS through RISC via the hot exciton principle.34 The larger energy gap between T2 and T1 states greatly reduces the internal conversion (IC)  , resulting in hot RISC
, resulting in hot RISC 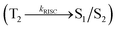 rather than cold RISC (T1 → S1) TADF mechanism.35,36 Hot exciton process with HLCT increases external quantum efficiency (ηex) because of high ηPL and high ηS. ηex can be calculated as follows: ηex = ηIQE × ηout = ηrec × ηPL × ηS × ηout, where ηIQE is the internal quantum efficiency, ηout is the light out coupling efficiency (20%), ηrec is the efficiency for electron–hole recombination (100%), ηPL is the photoluminescence efficiency of the film and ηS is the exciton utilization efficiency [ηS = ηrec × ηPL × ηout ÷ ηEL].37 The ambipolar phenanthrimidazole derivatives have been shown as potential blue emitters. The increase in conjugation in phenanthrimidazoles influenced the blue emission,38–42 and conjugation length was restricted by incorporating a bulky fragment in the core molecule, which resulted in twisted conformation.43–46 In line with this discussion and our own research interest, we report donor–spacer–acceptor derivatives, namely, 4′-(1-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)-N,N-diphenyl-(2-[1,1′-biphenyl]vinyl)-4-amine (NSPI-TPA), 4′-(1-(2-methylnaphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)-N,N-diphenyl-(2-[1,1′-biphenyl]vinyl)-4-amine (MNSPI-TPA), 4-(2-(4′-(diphenylamino)-(2-[1,1′-biphenyl]vinyl)-4-yl)-1H-phenanthro[9,10-d]imidazol-1-yl)-1-naphthalene-1-carbonitrile (SPNCN-TPA) and 4-(2-(4-(9H-carbazol-9-yl)styryl)-1H-phenanthro[9,10-d]imidazol-1-yl)naphthalene-1-carbonitrile (SPNCN-Cz) using triphenylamine as a strong donor and carbazole as weak donor (Scheme 1). The H–H repulsion of bulky styryl fragment with the phenyl moiety of TPA and the carbazole moieties leads to twisted configuration, which enhances the twist angle, thus shortening the conjugation length. The solvatochromic effect of NPSI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz was examined to understand the excited state characteristics and interstate coupling strength of LE and CT components. Combining theoretical (TD-DFT) and experimental data, the LE and CT compositions were discussed using natural transition orbital (NTO), centroids of charges and transition density matrix (TDM) analysis. Hybridization of LE and CT energy states was used for molecular design, and their composition in HLCT was tuned, resulting in high EL efficiency. Triphenylamine (TPA) of SPNCN-TPA was replaced by carbazole (Cz) in SPNCN-Cz, both of which differ in electron donating ability. As a result, the CT composition decreases with an increase in LE composition in the S1 HLCT state. Thus, the SPNCN-Cz-based device exhibited maximum electroluminescent efficiency with ηex of 3.15%, ηc of 2.56 cd A−1 and ηp of 2.45 lm W−1 and CIE of (0.15, 0.07), which were higher than those of the SPNCN-TPA-based device. Higher ηPL of SPNCN-Cz film was observed compared with that of SPNCN-TPA film; thus, high ηPL and high ηS enhanced the ηex of the SPNCN-Cz-based device. These results can be used to design low cost fluorescent materials via subtle molecular modifications using the HLCT emissive state principle.
rather than cold RISC (T1 → S1) TADF mechanism.35,36 Hot exciton process with HLCT increases external quantum efficiency (ηex) because of high ηPL and high ηS. ηex can be calculated as follows: ηex = ηIQE × ηout = ηrec × ηPL × ηS × ηout, where ηIQE is the internal quantum efficiency, ηout is the light out coupling efficiency (20%), ηrec is the efficiency for electron–hole recombination (100%), ηPL is the photoluminescence efficiency of the film and ηS is the exciton utilization efficiency [ηS = ηrec × ηPL × ηout ÷ ηEL].37 The ambipolar phenanthrimidazole derivatives have been shown as potential blue emitters. The increase in conjugation in phenanthrimidazoles influenced the blue emission,38–42 and conjugation length was restricted by incorporating a bulky fragment in the core molecule, which resulted in twisted conformation.43–46 In line with this discussion and our own research interest, we report donor–spacer–acceptor derivatives, namely, 4′-(1-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)-N,N-diphenyl-(2-[1,1′-biphenyl]vinyl)-4-amine (NSPI-TPA), 4′-(1-(2-methylnaphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)-N,N-diphenyl-(2-[1,1′-biphenyl]vinyl)-4-amine (MNSPI-TPA), 4-(2-(4′-(diphenylamino)-(2-[1,1′-biphenyl]vinyl)-4-yl)-1H-phenanthro[9,10-d]imidazol-1-yl)-1-naphthalene-1-carbonitrile (SPNCN-TPA) and 4-(2-(4-(9H-carbazol-9-yl)styryl)-1H-phenanthro[9,10-d]imidazol-1-yl)naphthalene-1-carbonitrile (SPNCN-Cz) using triphenylamine as a strong donor and carbazole as weak donor (Scheme 1). The H–H repulsion of bulky styryl fragment with the phenyl moiety of TPA and the carbazole moieties leads to twisted configuration, which enhances the twist angle, thus shortening the conjugation length. The solvatochromic effect of NPSI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz was examined to understand the excited state characteristics and interstate coupling strength of LE and CT components. Combining theoretical (TD-DFT) and experimental data, the LE and CT compositions were discussed using natural transition orbital (NTO), centroids of charges and transition density matrix (TDM) analysis. Hybridization of LE and CT energy states was used for molecular design, and their composition in HLCT was tuned, resulting in high EL efficiency. Triphenylamine (TPA) of SPNCN-TPA was replaced by carbazole (Cz) in SPNCN-Cz, both of which differ in electron donating ability. As a result, the CT composition decreases with an increase in LE composition in the S1 HLCT state. Thus, the SPNCN-Cz-based device exhibited maximum electroluminescent efficiency with ηex of 3.15%, ηc of 2.56 cd A−1 and ηp of 2.45 lm W−1 and CIE of (0.15, 0.07), which were higher than those of the SPNCN-TPA-based device. Higher ηPL of SPNCN-Cz film was observed compared with that of SPNCN-TPA film; thus, high ηPL and high ηS enhanced the ηex of the SPNCN-Cz-based device. These results can be used to design low cost fluorescent materials via subtle molecular modifications using the HLCT emissive state principle.
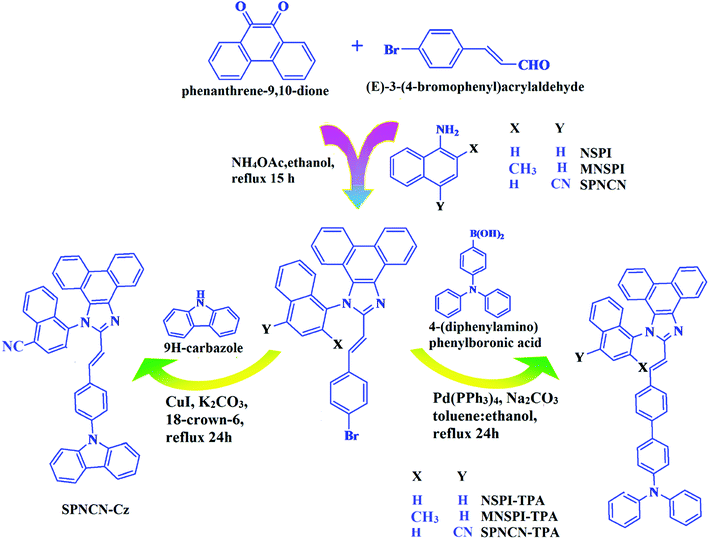 |
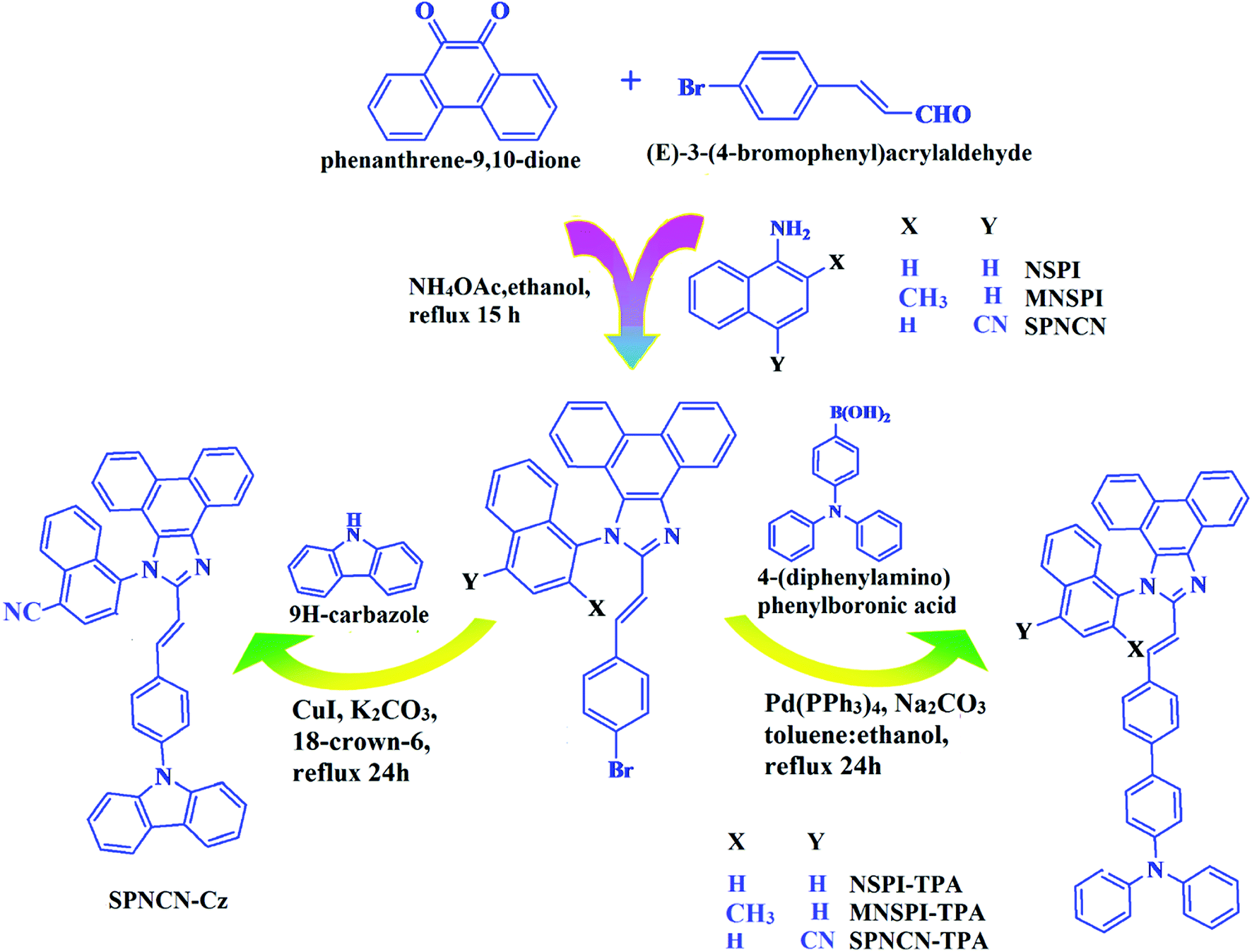
| | Scheme 1 Synthesis routes of NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz. | |
2 Experimental
2.1 Synthesis of p-bromostyrylphenanthroimidazoles (NSPI, MNSPI & SPNCN)
The synthesis routes of p-bromostyrylphenanthroimidazoles, namely, 2-(4-bromostyryl)-1-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazole (NSPI), 2-(4-bromostyryl)-1-(2-methylnaphthalen-1-yl)-1H-phenanthro[9,10-d]imidazole (MNSPI) and 4-(2-(4-bromostyryl)-1H-phenanthro[9,10-d]imidazol-1-yl)naphthalene-1-carbonitrile (SPNCN), along with donor–spacer–acceptor derivatives, namely, NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz, are displayed in Scheme 1. Phenanthrene-9,10-dione (1 mmol), 4-bromo-1-cinnamaldehyde (1 mmol), 1-naphthylamine (NSPI)/2-methylnaphthalen-1-amine (MNSPI)/4-aminonaphthalene-1-carbonitrile (SPNCN) (1 mmol) and ammonium acetate (1 mmol) were refluxed in 25 mL acetic acid. The crude product was column chromatographed (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethylacetate) and the pure sample was used for synthesizing the blue emissive materials.
:ethylacetate) and the pure sample was used for synthesizing the blue emissive materials.
2.1.1 4′-(1-(Naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)-N,N-diphenyl-(2-[1,1′-biphenyl]vinyl)-4-amine (NSPI-TPA). Mixture of 2-(4-bromostyryl)-1-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazole (NSPI) (4.5 mmol), 4-(diphenylamino)phenylboronic acid (7.5 mmol), Pd(PPh3)4 (0.25 mmol) and aqueous Na2CO3 (15 mL) in toluene:ethanol (20:15 mL) was refluxed (N2 atmosphere) for 18 h. The reaction mixture was extracted with dichloromethane and then, the solvent was distilled. The purified NSPI-TPA was used for further analysis. Yield 68%. Anal. calcd: C51H35N3: C, 88.79; H, 5.11; N, 6.09. Found: C, 88.72; H, 5.06; N, 6.01. 400 MHz 1H NMR (CDCl3): δ 6.58–6.61 (m, 6H), 6.64 (d, J = 8.2 Hz, 2H), 6.98 (d, J = 16.4 Hz, 2H), 7.14 (t, 4H), 7.31–7.38 (m, 10H), 7.7 (d, J = 8.4 Hz, 3H), 7.83–7.89 (m, 4H), 8.12 (d, J = 8.3 Hz, 2H), 8.93 (d, J = 8.6 Hz, 2H) (Fig. S1†). 400 MHz 13C NMR (CDCl3): δ 112.7, 122.32, 122.61, 124.21, 126.52, 126.84, 127.75, 127.94, 129.64, 130.68, 130.80, 131.47, 132.35, 133.58, 134.26, 134.68, 135.85, 139.56, 141.11, 141.62 (Fig. S2†). MALDI TOF MS: m/z. 689.21 [M+] (Fig. S9†). Calcd: 689.28.
2.1.2 4′-(1-(2-Methylnaphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)-N,N-diphenyl-(2-[1,1′-biphenyl]vinyl)-4-amine (MNSPI-TPA). A mixture of 2-(4-bromostyryl)-1-(2-methylnaphthalen-1-yl)-1H-phenanthro[9,10-d]imidazole (MNSPI) (4.5 mmol), 4-(diphenylamino)phenylboronic acid (7.5 mmol), Pd(PPh3)4 (0.25 mmol) and aqueous Na2CO3 (15 mL) in toluene:ethanol (20:15 mL) was refluxed for 24 h and extracted with dichloromethane. Yield 58%. Anal. calcd: C52H37N3: C, 88.73; H, 5.30; N, 5.97. Found: C, 88.68; H, 5.25; N, 5.91. 400 MHz 1H NMR (CDCl3): δ 2.45 (s, 3H), 6.46–6.54 (m, 6H), 6.66 (d, J = 8.1 Hz, 2H), 6.91 (d, J = 16.3 Hz, 2H), 7.02 (t, 4H), 7.12–7.45 (m, 9H), 7.51–7.62 (m, 3H), 7.81–7.85 (m, 4H), 8.11 (d, J = 8.4 Hz, 2H), 8.91 (d, J = 8.2 Hz, 2H) (Fig. S3†). 400 MHz 13C NMR (CDCl3): δ 15.51, 112.70, 122.31, 123.15, 125.25, 126.36, 126.69, 126.81, 127.54, 127.72, 127.84, 128.41, 129.86, 131.44, 133.57, 134.82, 135.66, 139.80, 141.15, 147.75 (Fig. S4†). MALDI TOF MS: m/z. 703.26 [M+] (Fig. S9†). Calcd: 703.30.
2.1.3 4-(2-(4′-(Diphenylamino)-(2-[1,1′-biphenyl]vinyl)-4-yl)-1H-phenanthro[9,10-d]imidazol-1-yl)-1-naphthonitrile (SPNCN-TPA). A mixture of 4-(2-(4-bromostyryl)-1H-phenanthro[9,10-d]imidazol-1-yl)naphthalene-1-carbonitrile (SPNCN) (4.5 mmol), 4-(diphenylamino)phenylboronic acid (7.5 mmol), Pd(PPh3)4 (0.25 mmol) and aqueous Na2CO3 (15 mL) in toluene:ethanol (20:15 mL) was refluxed under N2 stream. Yield 60%. Anal. calcd: C52H34N4: C, 87.37; H, 4.79; N, 7.84. Found: C, 87.31; H, 4.72; N, 7.78. 400 MHz 1H NMR (CDCl3): δ 6.59–6.61 (m, 6H), 6.99 (d, J = 16 Hz, 2H), 7.15 (t, 4H), 7.25 (d, J = 8.2 Hz, 2H), 7.36–7.45 (m, 7H), 7.56–7.88 (m, 9H), 8.12 (d, J = 8.4 Hz, 2H), 8.93 (d, J = 8.5 Hz, 2H) (Fig. S5†). 400 MHz 13C NMR (CDCl3): δ 109.40, 110.61, 115.88, 121.65, 122.51, 123.49, 126.64, 126.81, 128.52, 128.64, 129.74, 131.87, 132.85, 133.58, 134.56, 135.18, 139.55 (Fig. S6†). MALDI TOF MS: m/z. 714.21 [M+] (Fig. S9†). Calcd: 714.28.
2.1.4 4-(2-(4-(9H-Carbazol-9-yl)styryl)-1H-phenanthro[9,10-d]imidazol-1-yl)naphthalene-1-carbonitrile (SPNCN-Cz). A mixture of 4-(2-styryl-1H-phenanthro[9,10-d]imidazol-1-yl)naphthalene-1-carbonitrile (SPNCN) (4.5 mmol), 9H-carbazole (7.5 mmol), (0.45 g, 1.0 mmol), CuI (10.0 mg, 0.05 mmol), 18-crown-6 (13.2 mg, 0.05 mmol) and K2CO3 (0.83 g, 6.0 mmol) in tetrahydro-1,3-dimethylpyrimidin-2(1H)-one (2.0 mL) was refluxed in ethanol (20 mL) under nitrogen atmosphere. Yield 61%. Anal. calcd: C46H28N4: C, 86.77; H, 4.43; N, 8.80. Found: C, 86.72; H, 4.38; N, 8.73. 400 MHz 1H NMR (CDCl3): δ 6.99 (d, J = 16.1 Hz, 2H), 7.0–7.08 (m, 4H), 7.22–7.31 (m, 4H), 7.45–7.66 (m, 7H), 7.80–7.89 (m, 6H), 8.12 (d, J = 8.4 Hz, 3H), 8.93 (d, J = 8.0 Hz, 2H) (Fig. S7†). 400 MHz 13C NMR (CDCl3): δ 109.50, 111.61, 112.81, 115.87, 119.12, 120.13, 121.01, 121.59, 121.74, 122.24, 122.48, 123.81, 126.54, 126.67, 127.56, 128.61, 131.51, 132.30, 132.75, 133.46, 136.55, 139.74, 14.35, 141.56 (Fig. S8†). MALDI TOF MS: m/z. 636.15 [M+] (Fig. S9†). Calcd: 636.23.
2.2 Measurements and general methods
Reagents and solvents were purchased from commercial sources. 1H and 13C NMR spectra were recorded on a Bruker (400 MHz) spectrometer and an Agilent instrument (LCMS VL SD) was employed to record the mass spectra. UV-vis absorption was measured using a Perkin-Elmer Lambda 35 (solution) and Lambda 35 spectrophotometer with an integrated sphere (RSA-PE-20) (film). Photoluminescence (PL) spectra were recorded on a PerkinElmer LS55 fluorescence spectrometer. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed using a PerkinElmer thermal analysis system and NETZSCH-DSC-204, respectively. Decay analysis was conducted using a nanosecond time correlated single photon counting (TCSPC) spectrometer (Horiba Fluorocube-01-NL lifetime system) with nanoLED excitation source and TBX-PS detector; DAS6 software was used to fit the decay curve and χ2 values lie between 0.8 and 1.2. The absolute photoluminescence quantum yield (PLQY) was determined using a fluorescence spectrometer (Model-F7100). Cyclic voltammetry was performed using Potentiostat CHI 630A electrochemical analyzer (platinum electrode and platinum wire as the working electrode and counter electrode, respectively; Ag/Ag+ as the reference electrode, with 100 mV s−1 scan). Ferrocene was used as the internal standard, with the highest occupied molecular orbital energy of −4.80 eV, and 0.1 M tetrabutylammoniumperchlorate in CH2Cl2 was used as the supporting electrolyte. The HOMO energies (EHOMO) were calculated using oxidation potential [EHOMO = −(Eox + 4.8 eV)] and the LUMO energies (ELUMO) were calculated by subtracting the optical band gap from the HOMO energy [ELUMO = EHOMO − 1239/λonset]. For theoretical calculations, ground state (DFT)/excited state (TD-DFT) geometrical properties were optimized by employing Gaussian 09 program.47 Multifunctional wavefunction analyzer (Multiwfn)29 was used to determine the nature of electronic transitions of excited states and natural transition orbitals (NTOs).
2.3 Fabrication of devices
Devices with structure of ITO/NPB (70 nm)/NSPI-TPA or MNSPI-TPA or SPNCN-TPA or SPNCN-Cz (100 nm)/TPBI (20 nm)/LiF (0.5 nm)/Al (120 nm) were fabricated on pre-cleaned ITO-coated glass substrates with 20 Ω sq−1 sheet resistance. Current density–voltage characteristics were recorded on a Keithley 2400 power source. EL spectra were recorded on a USB-650-VIS-NIR spectrometer (Ocean Optics, Inc, USA).
3 Results and discussion
3.1 Synthesis, thermal and photophysical properties
The synthesis routes of p-bromostyrylphenanthrimidazoles (NSPI, MNSPI and SPNCN) and donor–spacer–acceptor derivatives (NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz) are shown in Scheme 1. The blue emitters were synthesized via Suzuki coupling reactions48 of p-bromostyrylphenanthrimidazoles with 4-diphenylaminophenylboronic acid (NSPI-TPA, MNSPI-TPA and SPNCN-TPA)/9H-carbazole (SPNCN-Cz) with yields of 68, 58, 60 and 61% of NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz, respectively. All these as-synthesized emitters were confirmed via 1H and 13C NMR spectroscopy, elemental analysis and high-resolution mass spectrometry. The two new efficient cyano-substituted blue emissive materials, SPNCN-TPA and SPNCN-Cz, consist of triphenylamine-styryl-naphthalenecarbonitrilephenathrimidazole (TPA-strong donor) and carbazole-styryl-naphthalenecarbonitrilephenathrimidazole (Cz-weak donor) backbones and the orthogonal naphthonitrile acceptor. In NSPI-TPA, MNSPI-TPA and SPNCN-TPA, the styryl ring is inserted between triphenylamine and naphthonitrilephenanthrimidazole to extend the π-conjugation (increase the LE component), which results in the enhancement of PL efficiency (ηPL) while simultaneously maintaining the strength of both donor (LE component) and acceptor (CT component). However, carbazole in SPNCN-Cz induces higher LE magnitude than SPNCN-TPA. To improve photoluminescence efficiency (ηPL), the strong donor TPA moiety in SPNCN-TPA is replaced with weaker donor carbazole to decrease the CT component and increase the LE component in the emissive state (S1 HLCT) of SPNCN-Cz.
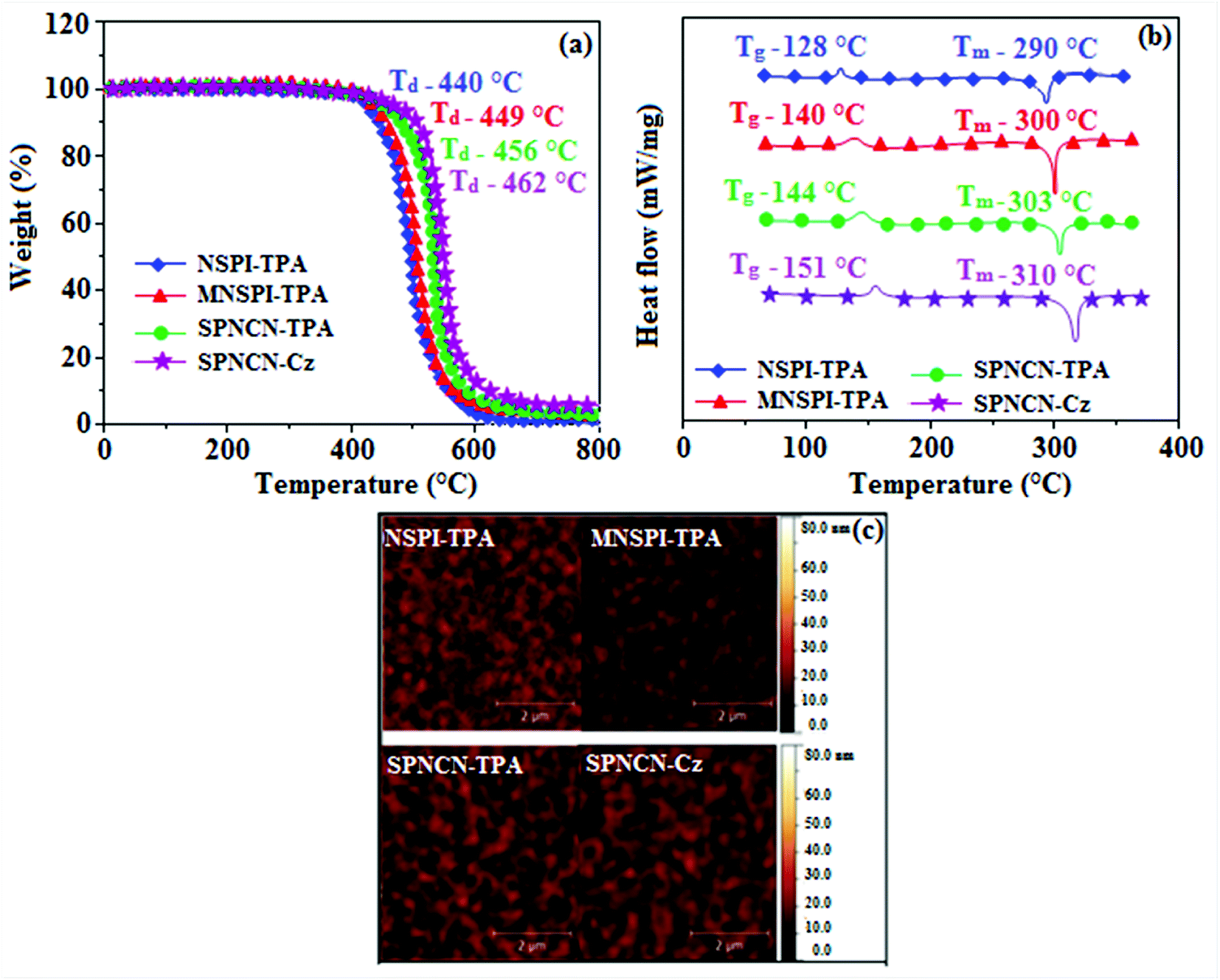
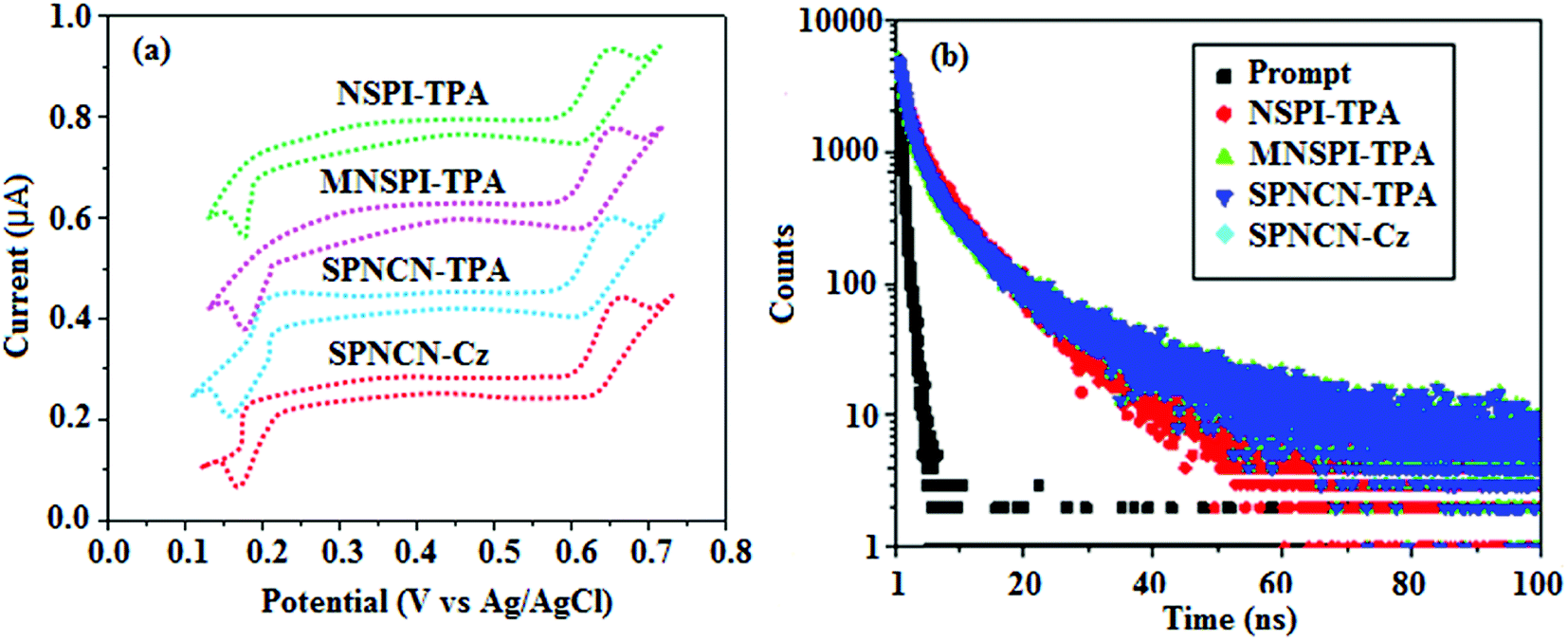
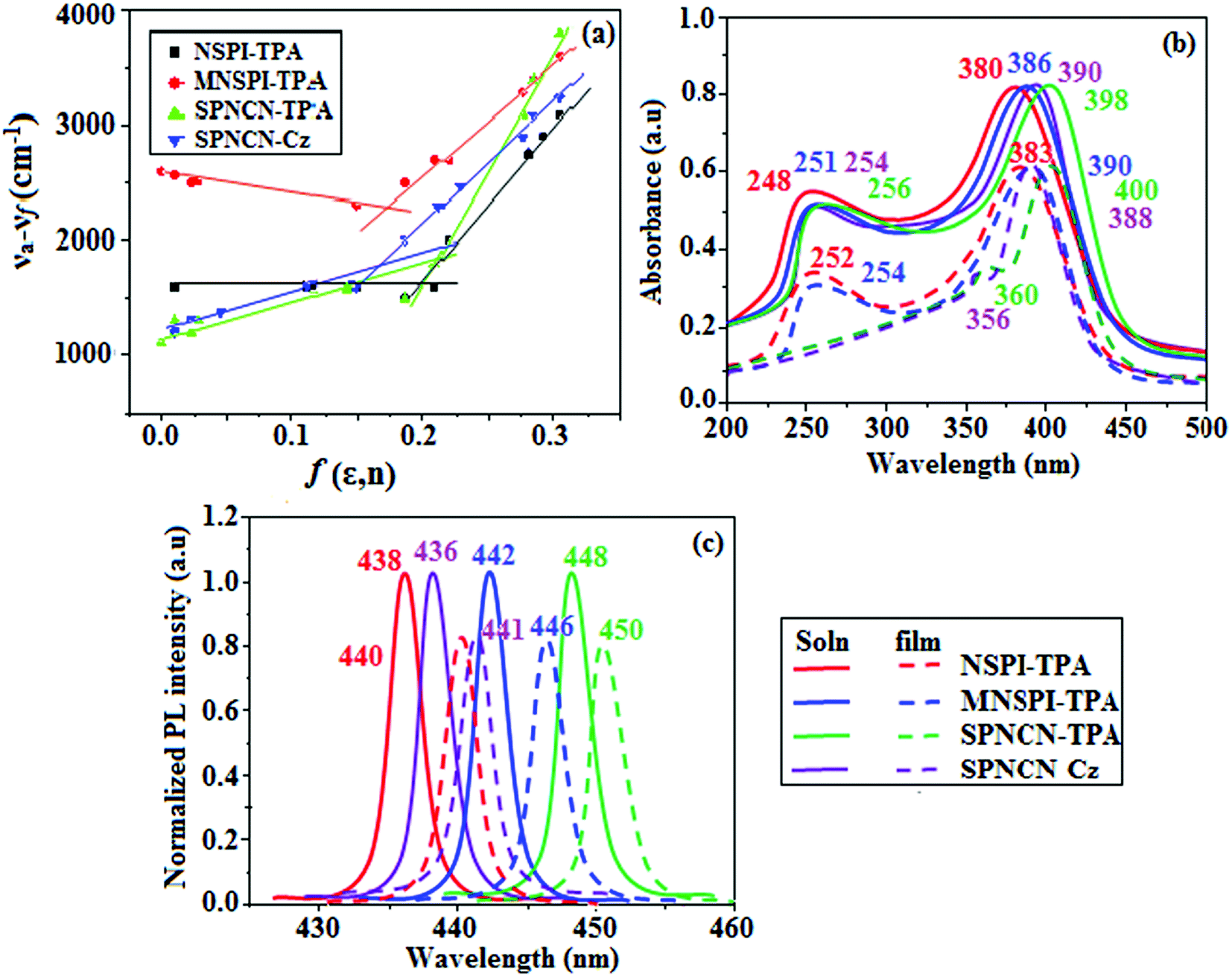
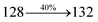
The decomposition temperatures (Td5) of NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz were measured to be 440, 449, 456 and 462 °C, respectively. The harvested high glass-transition temperature (Tg) of 128, 140, 144 and 151 °C (Fig. 1) along with high melting temperatures (Tm) of 290, 300, 303 and 310 °C are attributed to the non-coplanar geometry shown by bulky naphthonitrilephenanthrimidazole block along with triphenylamine/carbazole and styryl linker of rigid D–π–A molecules (Table 1). The thermal morphological stabilities of NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz thin films were examined by atomic force microscopy (AFM) at room temperature and at 100 °C for 16 h. The root-mean-square roughness (RMS) of NSPI-TPA (0.25 nm), MNSPI-TPA (0.29 nm), SPNCN-TPA (0.35 nm) and SPNCN-Cz (0.39 nm) thin-film surfaces show that there are no substantial changes before and after annealing (100 °C, Fig. 1). The glass transition (Tg) and decomposition temperatures (Td5) and the morphologies of NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz thin films support the suitability of these materials for fabrication of OLEDs and hence, it is expected that the as-synthesized D–π–A materials will lower the turn on voltage in the device performances.49,50 The small HOMO energies (EHOMO) of −5.26 eV (NSPI-TPA), −5.23 eV (MNSPI-TPA), −5.20 eV (SPNCN-TPA) and −5.21 eV (SPNCN-Cz) were estimated from their respective oxidation onset potentials [0.46 V (NSPI-TPA), 0.43 V (MNSPI-TPA), 0.40 V (SPNCN-TPA) and 0.41 V (SPNCN-Cz)], supporting the hole-injection property of D–π–A materials (Fig. 2, Table 1). Since the LUMO energies (ELUMO) of −2.04 eV (NSPI-TPA), −2.06 eV (MNSPI-TPA), −2.02 eV (SPNCN-TPA) and 2.15 eV (SPNCN-Cz) are close to that of 1,3,5-tris(N-phenylimidazol-2-yl)benzene (−2.40 eV), these materials also possess electron-injection abilities.51 Furthermore, the frontier molecular orbital (HOMO–LUMO) analysis confirms the carrier injection abilities and hence, these materials can be employed as potential emitters in OLEDs.52,53 The calculated high fluorescence quantum yields, which are 0.58/0.46 (NPS-TPA), 0.69/0.56 (MNPS-TPA), 0.48/0.42 (SPNCN-TPA) and 0.52/0.49 (SPNCN-Cz), in both solution and film support the co-emission phenomenon from LE and CT of emissive materials. The radiative (kr) and non-radiative (knr) transition rates of SPNCN-TPA and SPNCN-Cz were calculated from lifetime (τ) and quantum yield (ϕ) data. Compared with SPNCN-TPA, the increase in radiative rate (kr) and decrease in non-radiative rate (knr) of SPNCN-Cz are also in agreement with the aim of our molecular design. Compared with parent compounds, the blue emitting NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz materials show very strong absorption [solution (εmax)/film] at 380 (εmax = 26315 cm−1 M−1)/383 nm, 386 (εmax = 25906 cm−1 M−1)/390 nm, 398 (εmax = 25125 cm−1 M−1)/400 nm and 390 (εmax = 25641 cm−1 M−1)/388 nm (Fig. 3). The intramolecular charge transfer (ICT) from donor triphenylamine/carbazole to acceptor (naphthonitrilephenanthrimidazole) is likely to be the cause for strong absorption, and the absorption at around 248 nm is attributed to π–π* transition.54 The intramolecular charge transfer (ICT) was also confirmed by the MEP diagram (Fig. 4). Compared with solution of the materials, negligible absorption shifts in their corresponding films reveal that suppressed π–π* stacking exists in the solid state.55 The observed larger red shift further supports the charge-transfer (CT) in the twisted geometry of NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz. Compared with SPNCN-TPA, SPNCN-Cz exhibits higher blue shift for both absorption and emission, which is attributed to the poor electron donor ability of Cz relative to TPA. The increase in LE composition with decrease in CT in the S1-emissive HLCT state is likely to be the reason for this blue shift. The full width at half maximum in the absorption spectrum of SPNCN-Cz (40 nm) is narrowed relative to that of SPNCN-TPA (58 nm, Fig. 3). This observation indicated that the decrease in CT component of SPNCN-Cz in the S1 state is in good agreement with the NTO description for S0 → S1 transition. However, the absorption peaks of both SPNCN-TPA and SPNCN-Cz are narrower than those of the cyano-free parent compounds, indicating more LE character. The emission peaks of SPNCN-TPA and SPNCN-Cz show blue shift relative to those of their parent compounds, which is in contrast to the general observation, i.e., extension of π-conjugation leads to a red-shifted emission.56 The enhanced LE component is equivalent to the suppressed CT component in the emissive states of SPNCN-TPA and SPNCN-Cz, resulting in blue shift. The increase in LE composition is expected to result in a red-shifted PL spectrum, whereas the suppressed CT results in a blue-shifted PL spectrum. From the experimental observation, it is known that the latter factor is more dominant than the former. In addition, there is an overlap between UV and PL spectra of both SPNCN-TPA and SPNCN-Cz because of the enhanced LE character in SPNCN-TPA and SPNCN-Cz than that in their respective parent compounds. SPNCN-Cz exhibits solvatochromic red shift (45 nm), which is smaller than that of SPNCN-TPA (75 nm) (Fig. S10, Table S1 and S2†). Similarly, small absorption shifts of 22 nm and 32 nm were observed for SPNCN-Cz and SPNCN-TPA, respectively (Fig. S11, Tables S1 and S2†). These solvatochromic shifts confirmed that the low-lying S1-excited states of SPNCN-Cz and SPNCN-TPA must possess CT character.57–59 The % CT character of the S1 state of SPNCN-Cz is lower than that of the S1 state of SPNCN-TPA, whereas the % LE character of the S1 state of SPNCN-Cz is higher than that of the S1 state of SPNCN-TPA (Table 2). In hexane, both SPNCN-Cz and SPNCN-TPA show LE-like character because of vibronic PL spectrum. Compared with the broad and smooth PL spectrum of cyano-free compounds, there is higher % LE in the S1 state of both SPNCN-Cz and SPNCN-TPA. The full width at half maximum (FWHM) of SPNCN-Cz (40 nm) and SPNCN-TPA (58 nm) indicates that these compounds possess higher PL efficiency due to higher % LE in the S1 emissive state. The solvatochromic effect using Lippert–Mataga plot is displayed in Fig. 3 (Tables S1 and S2†). When the solvent polarity increased, the blue emitters exhibited larger red shift, which supports charge transfer (CT) in these molecules.57 From the plot of Stokes shift against solvent polarity function, ground state dipole moment (μg) can be calculated:
where
μg and
μe are the ground state and excited state dipole moments,
![[small upsilon, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e131.gif) abs
abs and
νacabs are the solvent-equilibrated absorption maximum and that extrapolated to gas phase, respectively,
flu and
νacflu are the solvent equilibrated fluorescence maximum and that extrapolated to gas-phase, respectively,
ao is the Onsager cavity and
ε and
n are the solvent dielectric constant and refractive index, respectively. The non-linear correlation of Stokes shift
vs. solvent polarity function reveals that there is a transformation of the fitted line between ethyl ether and methylene chloride. This non-linear correlation supports the presence of both locally exited state (LE) and charge transfer exited state (CT). Ground state dipole (
μg) of blue-emitting materials NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz could be estimated from density functional theory (DFT) calculations as 2.98, 3.39, 5.94 and 7.02
D, respectively, and the corresponding calculated
μe values in high polarity solvents are 22.8, 23.6, 25.8 and 30.8
D. The small
μg estimated from the slope is 2.98, 3.39, 5.94 and 7.02
D for NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz, respectively, which is attributed to local exciton (LE) transition.
57 The large calculated
μe values in high polarity solvents (22.8, 23.6, 25.8 and 30.8
D)
are close to the
μe of the charge-transfer molecule 4-(
N,
N-dimethylamino)-benzonitrile (23.0
D).
59 All these results show that CT dominates in high polarity medium, whereas LE dominates in low polarity medium and there is mixed contribution of LE and CT in medium polarity solvents (ethyl ether and methylene chloride).
31 The oscillator strengths of NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz are displayed in
Tables 3 and
4. The oscillator strength of S
1 state of SPNCN-Cz (0.2554,
Table 4) is higher than that of SPNCN-TPA (0.1261,
Table 4), which results in the higher PL efficiency (
ηPL) of SPNCN-Cz. Molecular modification from TPA to Cz caused an increase in % LE in the S
1 emissive state and enhanced the
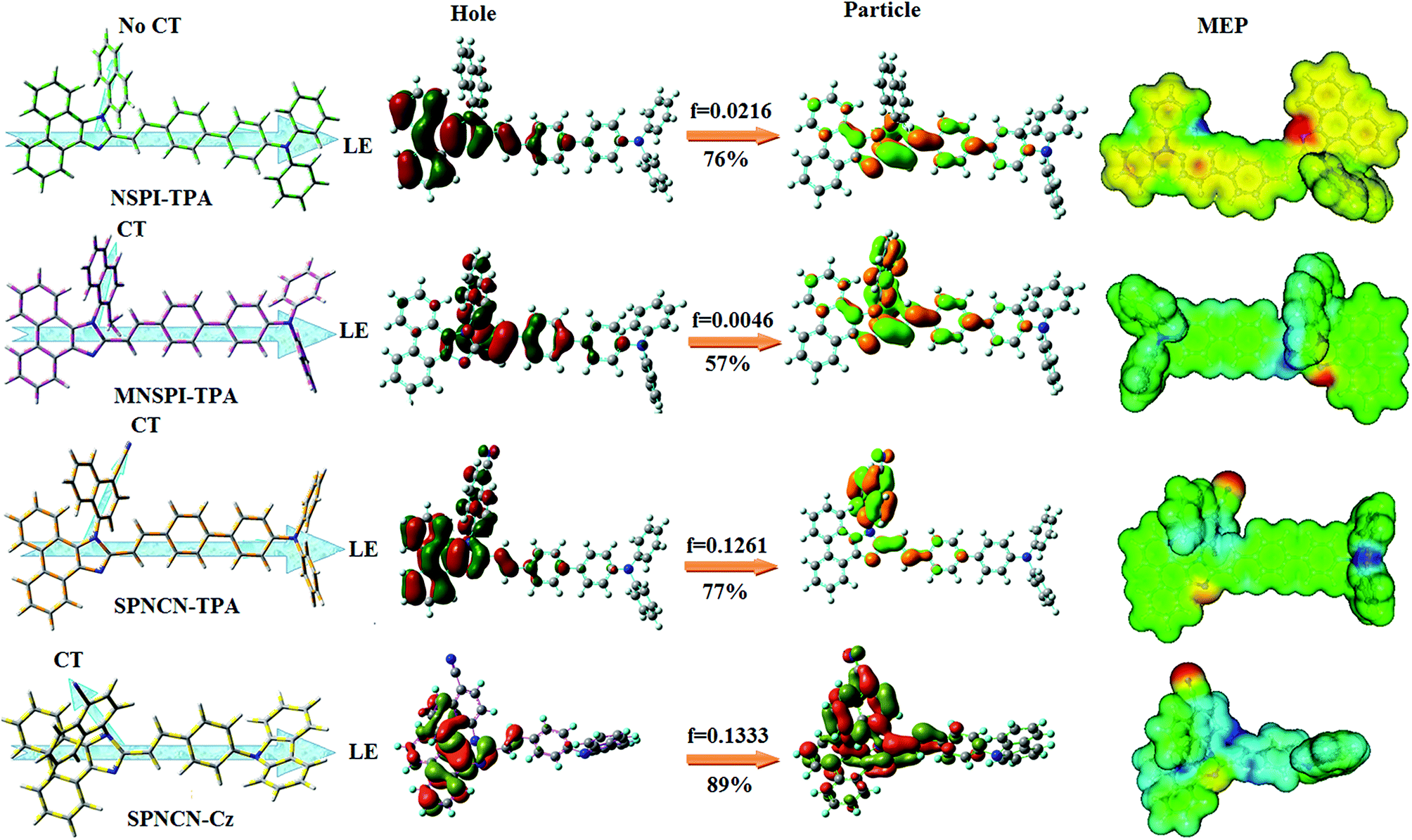
ηPL of SPNCN-Cz. In order to supplement experimental results, theoretical calculations (natural transition orbital analysis) were performed to describe the excited state characteristics of SPNCN-TPA and SPNCN-Cz materials (
Fig. 4). In SPNCN-TPA and SPNCN-Cz, holes are located over the horizontal backbone, whereas particles are located on vertical naphthonitrile in the S
1 state. CT transition is maintained from the horizontal backbone to vertical naphthonitrile in the S
1 states of SPNCN-TPA and SPNCN-Cz. The overlap density between a hole and a particle is expanded due to spacer styryl moiety, indicating an enhanced % LE in the S
1 state (Fig. S12 and S13
†). The NTOs of S
1 and S
2 excited states of SPNCN-TPA and SPNCN-Cz exhibit a hybrid splitting state character that derives from the interstate coupling of LE and CT levels (
Tables 3,
4, Fig. S14 and S15
†),
i.e., formation of HLCT state. The wave function symmetry of a particle on naphthonitrile is opposite between S
1 and S
2 states, indicating interstate hybridization coupling
ΨS1/S2 =
cLEΨLE ±
cCTΨCT. Similar hole–electron wave functions between S
1 and S
2 are observed in both SPNCN-TPA and SPNCN-Cz, indicating a quasi-equivalent hybridization between LE and CT states as a result of the almost isoenergies of initial LE and CT states (
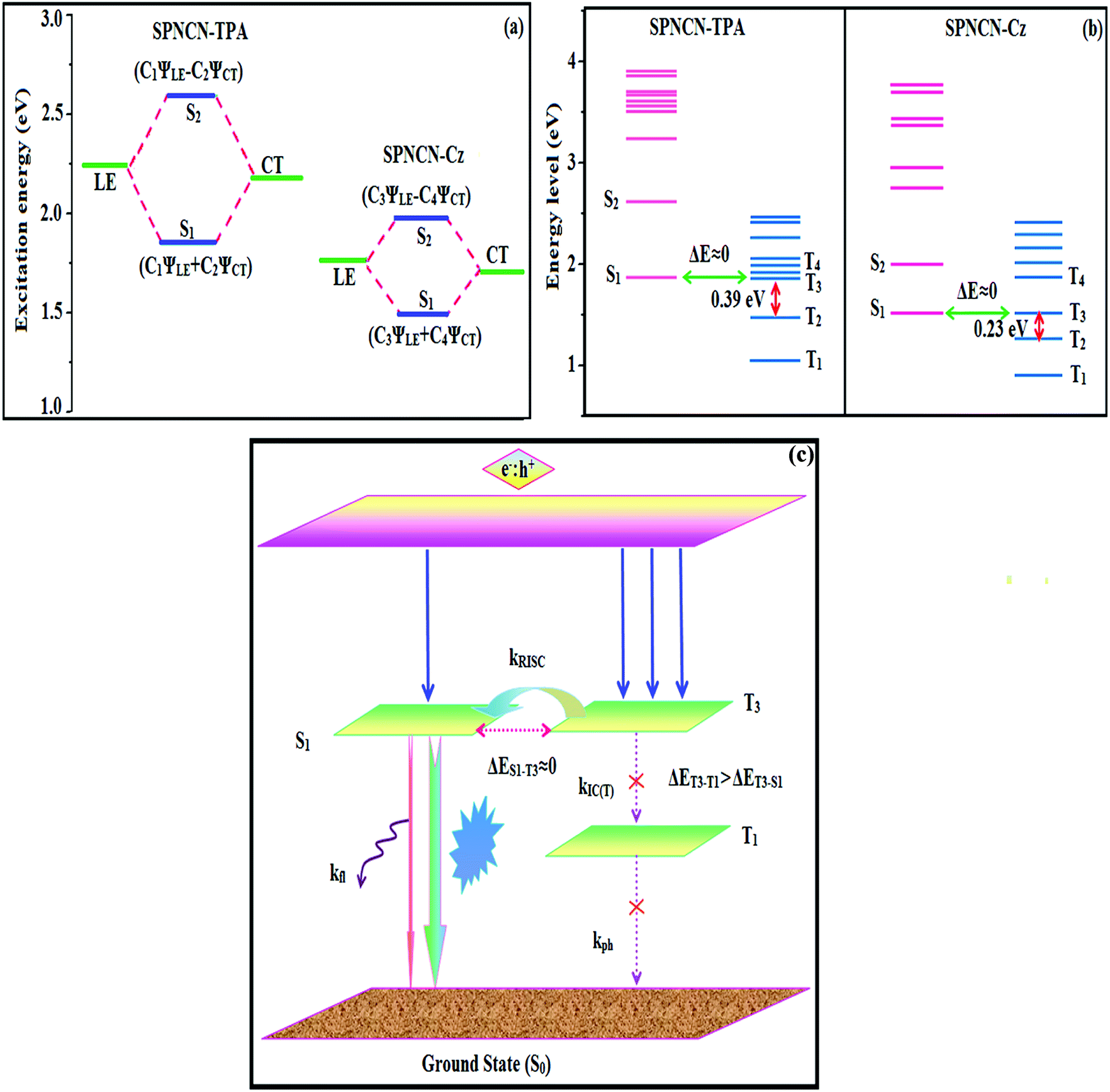
Fig. 5). Therefore, the degree of hybridization between LE and CT states is dependent not only on the initial
ELE–
ECT energy gap but also on their interstate coupling strength.
60 Compared with non-equivalent hybridization, quasi-equivalent hybridization is expected to achieve the combination of high
ηPL and high
ηs to maximize the EL efficiency of fluorescent OLED materials due to the more balanced LE and CT components in HLCT states of SPNCN-TPA and SPNCN-Cz. The formation of the HLCT state can be analysed through the excitation energies of LE and CT states (
Tables 3 and
4). In SPNCN-Cz and SPNCN-TPA, the LE state is stabilized as compared with the CT state and energy gap (
ES2–
ES1) is small when compared with their cyano-free parent compounds, resulting in quasi-hybridization. In the case of SPNCN-Cz, the energy gap (
ES2–
ES1) is reduced, resulting in effective hybridization and improved OLED efficiency. The overlap between the hole and the particle is also displayed in Fig. S12
† (SPNCN-TPA) and Fig. S13
† (SPNCN-Cz). Composition of HLCT can be determined from the wave function of electron–hole pair transition density matrix (TDM) plotted in a two-dimensional color-filled map (Fig. S16 and S17
†). The axes represent the atom in a molecule, which is proportional to the probability of finding an electron and a hole in an atomic orbital located on a non-hydrogen atom. The diagonal represents the LE component localized on the main backbone, while the off–diagonal region shows the CT component. The qualitatively calculated percentages of LE and CT in S
1–S
10 and T
1–T
10 states are displayed in
Table 2. This finding also supports that HLCT state contributes to hybridization, in addition to the LE and CT states. Upon excitation, an electron is transferred from a donor and localized on an acceptor. Depending upon intramolecular geometrical and electronic coupling, the transferred electron is delocalized from the region of nearby the donor molecule to the vicinity of the acceptor. This effect can be qualitatively studied by analyzing the electron density distribution at the ground and excited states. Computed electron–hole properties, distance between hole and electron, transition density,
H and
t indexes and RMSD of electrons and holes of SPNCN-Cz and SPNCN-TPA are displayed in Table S3–S8.
† The integral value of a hole and an electron of SPICN-Cz is less than that in SPICN-TPA with transition density. The integral overlap of hole–electron distribution (
S) is a measure of spatial separation of holes and electrons. The integral overlap (
S) of holes and electrons and the distance (
D) between centroids of holes and electrons evidence the existence of LE and CT states (Tables S5 and S7
†). When compared with parent compounds, SPNCN-TPA and SPNCN-Cz have small
D and high
S values. However, small
D and high
S of SPNCN-TPA indicate that charge transfer (CT) is higher in percentage for the SPNCN-TPA isomer. The variation in dipole moment with respect to S
0 state was also outputted, which was directly evaluated based on the position of centroids of holes and electrons. RMSD of holes or electrons characterizes their distribution breadth (Tables S6–S8
†). RMSDs of both electron and hole in SPNCN-Cz are higher in the
X direction, indicating that electron and hole distributions are much broader in the
X direction, whereas RMSD of the electron of SPNCN-TPA is smaller and that of the hole is higher than those of SPNCN-Cz. The
H index (half sum of the axis of anisotropic density variation distribution) measures the spread of positive and negative regions related to CT. The CT index,
i.e.,
t index difference between
DCT and
H index, is another measure of the separation of hole–electron (eqn (S15) and (S16)
†). For both SPNCN-Cz and SPNCN-TPA, the non-zero
t is negative in all directions. The overlap of the hole and electron is very severe (Table S9
†) and the eigenvalue is greater than 0.96, supporting the hybridization and described in terms of dominant excitation pair in terms of 94% of transition. This is further evidenced by Δ
r index (Tables S3 and S4
†). The Δ
r index (eqn (S1)
†) is the average hole (h
+)–electron (e
−) distance (
dh+–e−) upon excitation, which reveals the nature of the excitation,
viz., LE or CT. Valence excitation (LE) is related to short distances (
dh+–e−), while larger distances (
dh+–e−) are related to CT excitation. The triplet exciton is transformed to the singlet excitons in SPNCN-TPA and SPNCN-Cz
via RISC process with high energy excited state (hot CT channel),
61,62 which is beneficial for triplet exciton conversion in electroluminescence processes without any delayed fluorescence. CT excitons are formed with weak binding energy (
Eb) in higher excited states.
63 As a result, the exciton utilization can be harvested in SPNCN-TPA and SPNCN-Cz, similar to that observed in phosphorescent materials. The quasi-equivalent hybridized materials SPNCN-TPA and SPNCN-Cz exhibit excellent device performances due to fine modulation in the excited states. The enhanced LE component and hybridization between LE and CT components results in high
ηPL and high
ηs. The coexisting LE/CT composition in SPNCN-TPA and SPNCN-Cz harvested high
ηPL and high
ηs and enhanced the OLEDs' performances (
Table 1). Ground (S
0) and excited (S
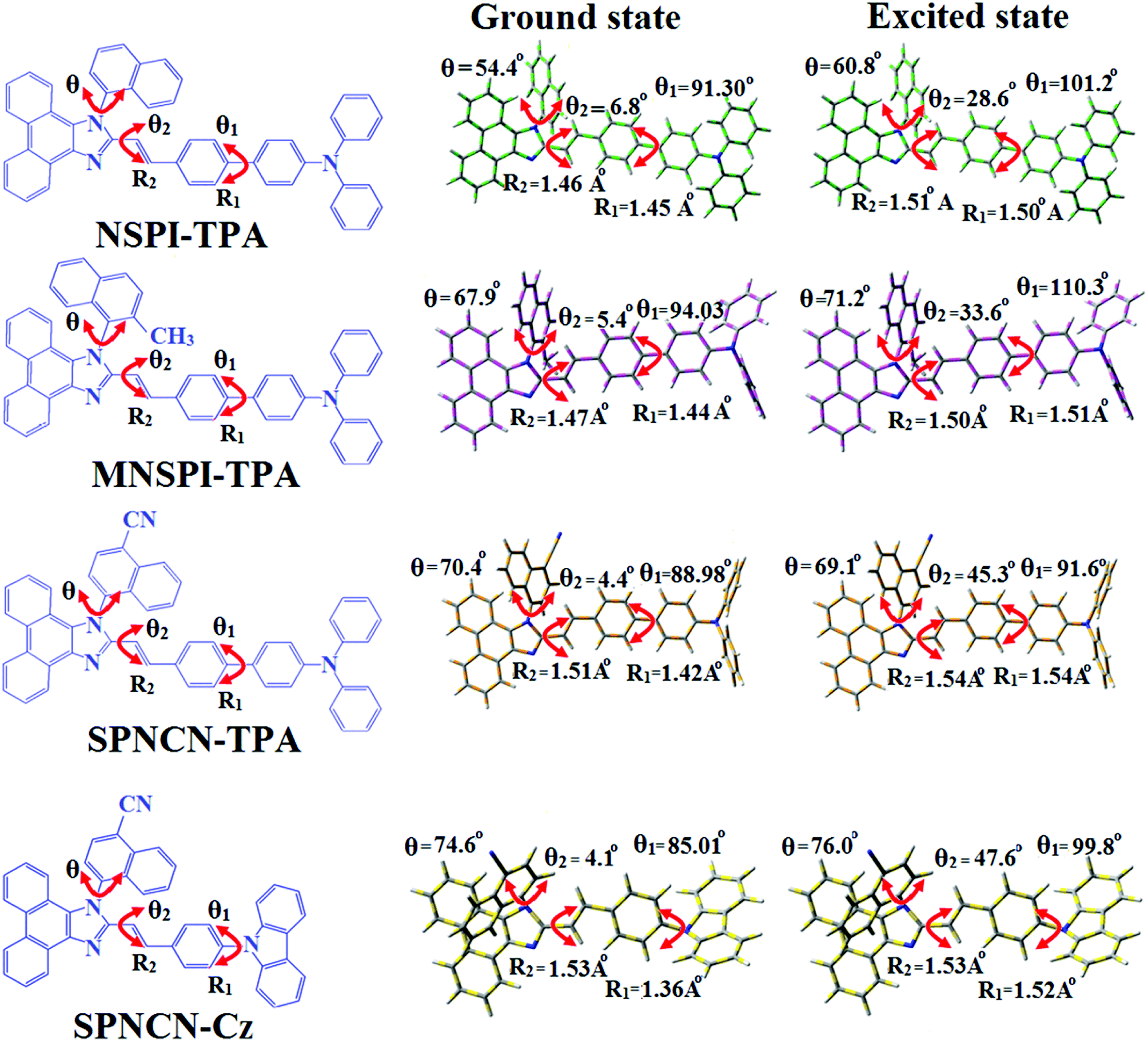
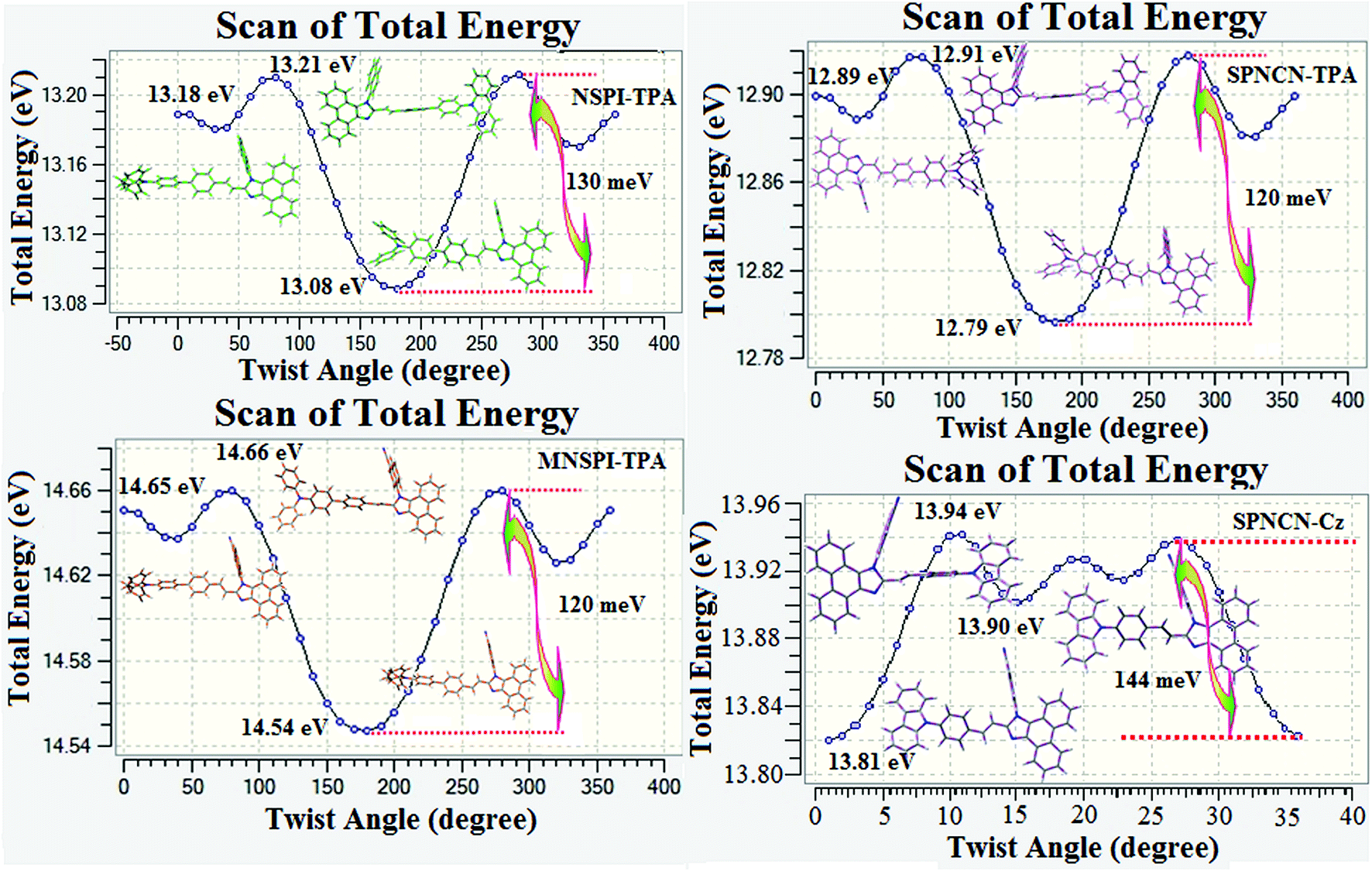
1) geometries of NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz were optimized by DFT/B3LYP/6-31G (d,p) and TD-DFT/B3LYP/6-31G (d,p). The optimized geometry shows styryl in NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz and the naphthonitrilephenanthrimidazole ring adopts a coplanar configuration with dihedral angles (
θ2) of 6.8, 5.4, 4.4 and 4.1°, respectively, leading to an
extended molecule. However, the dihedral angles (
θ1) between the phenyl moiety of TPA and styryl ring are 91.30, 94.03, 88.98 and 85.01°. The highly twisted dihedral angles are beneficial to adjusting the π-conjugation length between acceptor and donor groups. Naphthonitrile is twisted with respect to the phenanthrimidazole ring with dihedral angles (
θ) of 54.4 (NSPI-TPA), 67.9 (MNSPI-TPA), 70.4 (MNPS-TPA) and 74.6° (SPNCN-Cz). The donor TPA and acceptor phenanthrimidazole lead to electrostatic interactions
52 in these molecules. The dihedral angle (
θ3) between phenyl of TPA and the styryl moiety is a key parameter: larger angle suppresses π–π stacking in film, resulting in the prevention of the self-quenching of fluorescence. The twist angle (
θ2) of the emitters in excited state is increased to 28.6, 33.6, 45.3 and 47.6° compared with ground state twist angle
θ2 and bond length (
R1) is elongated S
1 → S
0 by 0.05, 0.07, 0.14 and 0.16 Å (
Fig. 6). The smaller change in geometry (S
0 to S
1) decreases the non-radiative emission (
knr), which results in enhanced photoluminescence efficiency (
ηPL). The twisted naphthonitrilephenanthrimidazoles NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz can effectively suppress molecular aggregation and the almost orthogonal dihedral angles (∼89.0°) between styryl and phenyl of TPA/Cz core can effectively minimize the intermolecular packing (
Fig. 7) and can be used as hole-trapping sites, whereas the peripheral phenanthrimidazole core blocks electron-trapping sites. Thus, both carrier injection and transport ability are expected from these reported emitters. Furthermore, the relative carrier transport of the title materials was investigated by fabricating hole-only devices as well as electron-only devices (
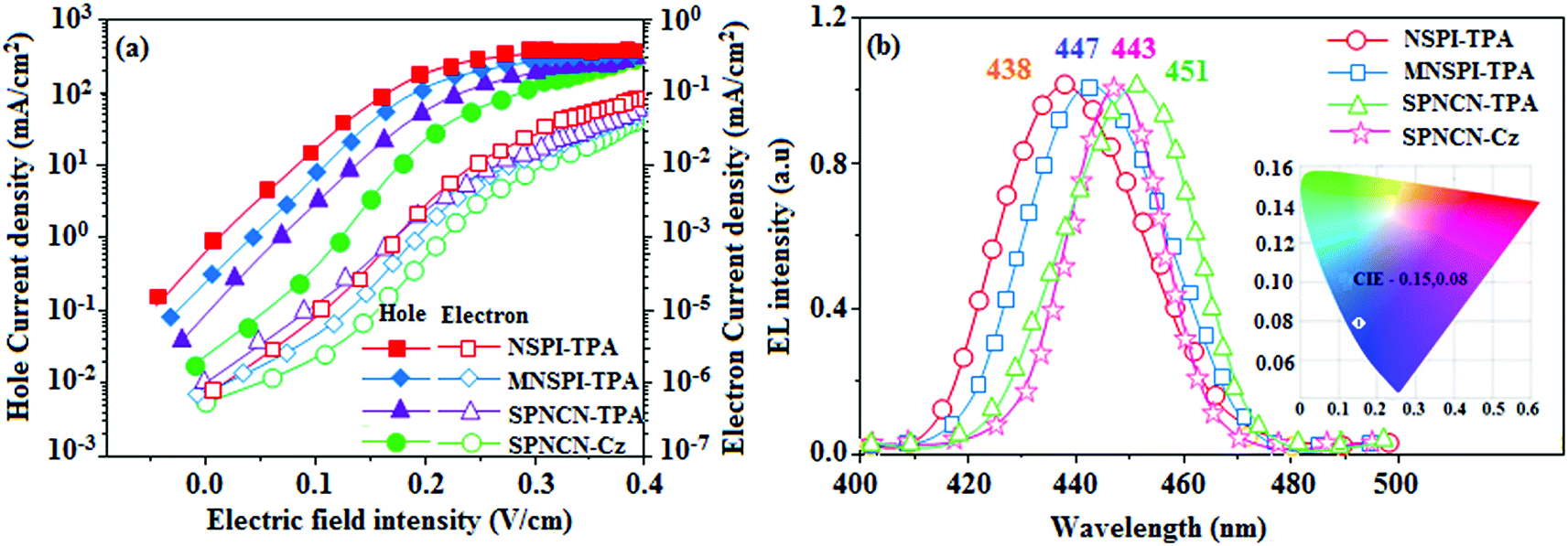
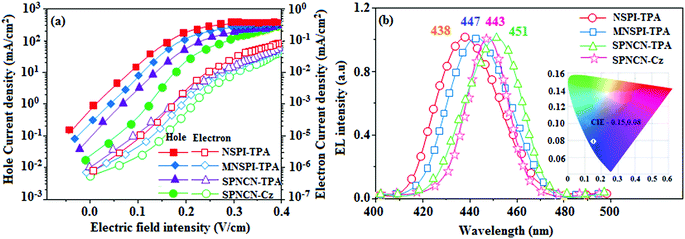
Fig. 8). The hole-only and electron-only devices with configurations of ITO/HATCN (10 nm)/NPB (70 nm)/NSPI-TPA or MNSPI-TPA or SPNCN-TPA or SPNCN-Cz (100 nm)/NPB (70 nm)/Al (120 nm) (hole-only device IV) and ITO/TPBi (10 nm)/NSPI-TPA or MNSPI-TPA or SPNCN-TPA or SPNCN-Cz (100 nm)/TPBi (10 nm)/LiF (1 nm)/Al (120 nm) (electron-only device V) were fabricated. For the hole-only device, NPB adjacent to Al was deposited (to block electrons), whereas for the electron-only device, TPBi was deposited close to the anode (to avoid hole injection).
64 The title materials show bipolar transporting abilities: the carrier mobility of compounds SPNCN-Cz and SPNCN-TPA was more effective than that of NPS-TPA and MNPS-TPA because of the naphthonitrile fragment. Hence, the devices based on SPNCN-TPA and SPNCN-Cz achieved relatively lower turn-on voltages with higher device efficiencies. Generally, TADF materials exhibit flat decay curves due to the time-consuming TADF process for exciton conversion from triplet to singlet. SPNCN-TPA and SPNCN-Cz decay sharply (
Fig. 2). Hence, radiative excitons in SPNCN-TPA and SPNCN-Cz are short-lived without TADF contribution. Exciton utilization efficiency (
ηs) in SPNCN-TPA and SPNCN-Cz follows neither TTA nor TADF mechanism.
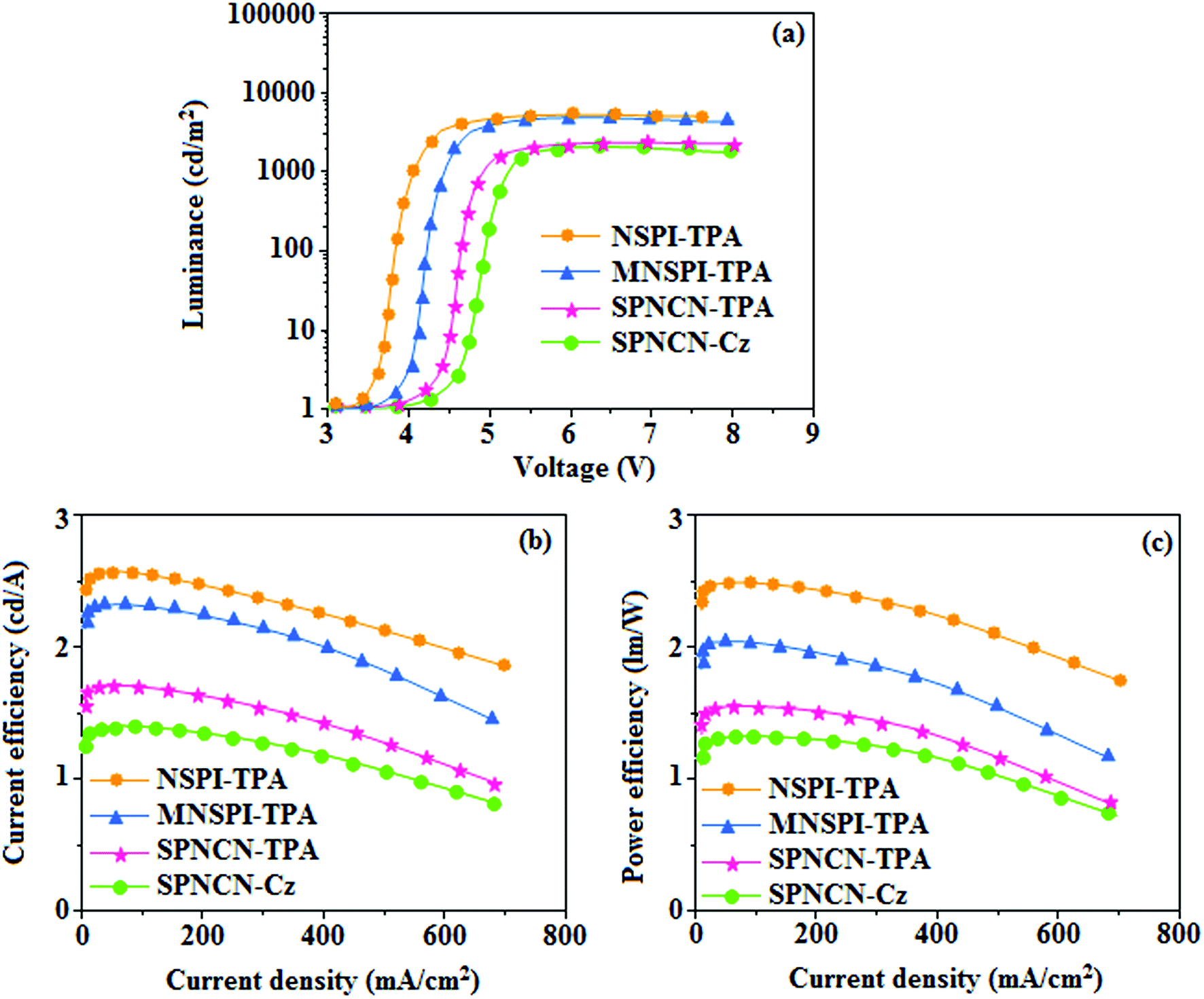
65 The non-doped EL devices were fabricated to investigate the relationship between excited state properties and EL performances of NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz (
Fig. 9,
Table 1). The device with configuration of ITO/NPB (70 nm)/NSPI-TPA (100 nm) or MNSPI-TPA (100 nm) or SPNCN-TPA (100 nm) or SPNCN-Cz (100 nm)/TPBi (20 nm)/LiF (1 nm)/Al (120 nm) were fabricated. The devices based on SPNCN-TPA and SPNCN-Cz exhibit blue EL emission with CIE of (0.15, 0.08) for SPNCN-TPA and (0.15, 0.07) for SPNCN-Cz. Among strong donor compounds, the SPNCN-TPA-based device exhibited blue emission at 451 nm, maximum current efficiency of 2.32 cd A
−1, power efficiency of 2.01 lm W
−1 and external quantum efficiency of 3.02%. The non-doped device with SPNCN-Cz exhibited better electroluminescent performance than the SPNCN-TPA-based device: high external quantum efficiency of 3.15%, current efficiency of 2.56 cd A
−1 and power efficiency of 2.45 lm W
−1. The
ηs of SPNCN-Cz and SPNCN-TPA was calculated to be 32.14 and 30.8%, respectively, which was superior to the 25% spin statistics limit. The increased
ηs and
ηIQE (15.75 for SPNCN-Cz and 15.10 for SPNCN-TPA) was due to the CT state contributed from the cyano group. The twisted geometry of emissive materials through introduction of a sterically hindered group enhanced the color purity. The fabricated devices also show excellent efficiency with no roll-off external quantum efficiency. Efficiencies of the as-fabricated current devices were compared with literature reported efficiencies
66–79 and displayed in Table S10,
† which shows that newly synthesized twisted D–π–A blue emitters are the best in view of efficiency (
Table 5).
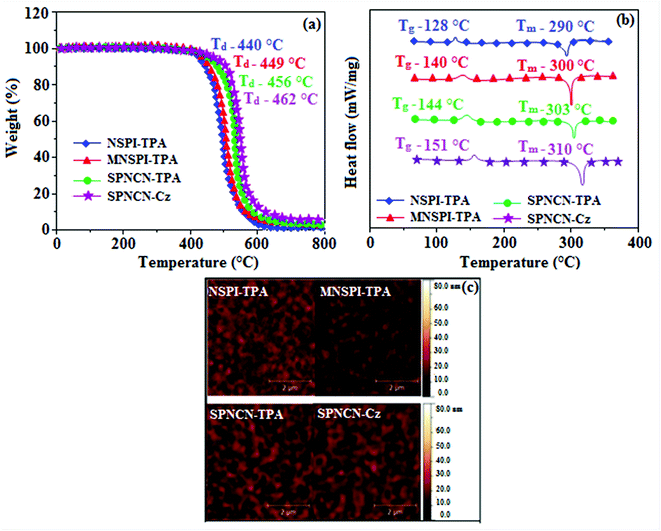 |
| | Fig. 1 (a) TGA, (b) DSC graphs, and (c) AFM images of NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz. | |
Table 1 Photophysical and thermal properties and device efficiencies of NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz
| Parameters |
NSPI-TPA |
MNSPI-TPA |
SPNCN-TPA |
SPNCN-Cz |
| Normalized absorption (λab) and emission (λem) spectra of NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz in CH2Cl2 (10–5 M)/film. Tg/Td-glass transition temperature/thermal decomposition temperature at a weight percentage of 95%. ϕ (soln/film) – PL quantum yield was calculated in dichloromethane/solid state quantum yield was measured on the quartz plate using an integrating sphere. HOMO/LUMO – EHOMO = (Eox + 4.8 eV)/ELUMO = EHOMO − 1239/λonset. Eg – energy gap (HOMO–LUMO). ηex – external quantum efficiency; maximum internal quantum efficiency. ηIQE = ηex/ηout, ηout light out coupling efficiency (−20%); excitation utilization efficiency. ηs = ηIQE/ηPL. |
| Photophysical & thermal properties |
| aλab (nm) (sol/film) |
248, 380/252, 383 |
251, 386/254, 390 |
256, 398/360, 400 |
254, 390/356, 388 |
| aλem (nm) (sol/film) |
438/440 |
442/446 |
448, 450 |
439/441 |
| FWHM |
75 |
69 |
58 |
40 |
| bTm/Tg/Td5 (°C) |
290/128/440 |
300/140/449 |
303/144/456 |
310/151/462 |
| cϕ (sol/film) |
0.58/0.46 |
0.69/0.56 |
0.48/0.42 |
0.52/0.49 |
| τ (ns) |
2.5 |
2.1 |
1.7 |
1.8 |
| dHOMO/LUMO (eV) |
−5.26/−2.04 |
−5.23/−2.06 |
−5.20/−2.02 |
−5.21/−2.15 |
| kr × 108 (s−1) |
2.3 |
3.2 |
2.8 |
2.9 |
| knr × 108 (s−1) |
1.7 |
1.5 |
3.0 |
2.7 |
| eEg (eV) |
3.22 |
3.17 |
3.18 |
2.86 |
|
| Device efficiency |
| gηIQE (%) |
10.1 |
11.8 |
15.1 |
15.75 |
| hηs (%) |
20.6 |
21.1 |
30.8 |
32.14 |
| Von (V) |
4.5 |
4.3 |
3.8 |
3.5 |
| L (cd m−2) |
2013 |
2158 |
4362 |
4826 |
| fηex (%) |
2.01 |
2.36 |
3.02 |
3.15 |
| ηc (cd A−1) |
1.38 |
1.68 |
2.32 |
2.56 |
| ηp (lm W−1) |
1.29 |
1.51 |
2.01 |
2.45 |
| EL (nm) |
438 |
447 |
451 |
443 |
| CIE (x, y) |
(0.15, 0.12) |
(0.15, 0.10) |
(0.15, 0.08) |
(0.15, 0.07) |
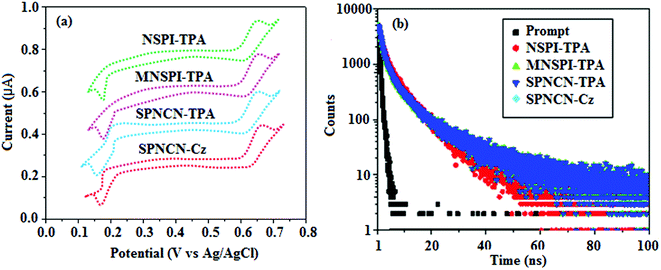 |
| | Fig. 2 (a) Cyclic voltammogram and (b) lifetime spectra of NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz. | |
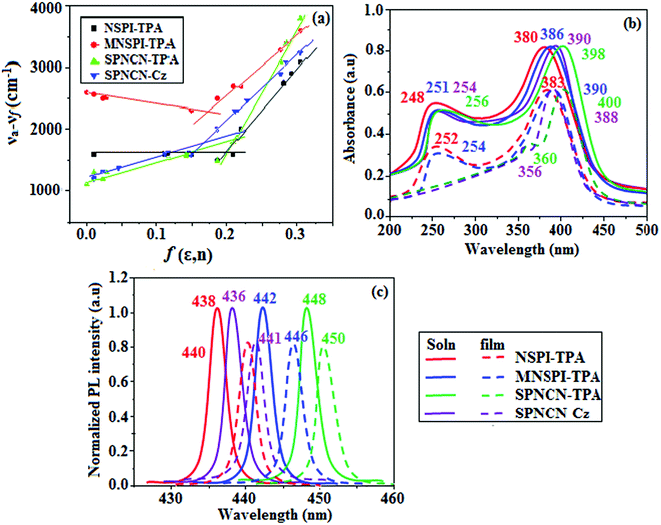 |
| | Fig. 3 (a) Lippert–Mataga plots, (b) normalized absorption spectra, and (c) emission spectra of NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz. | |
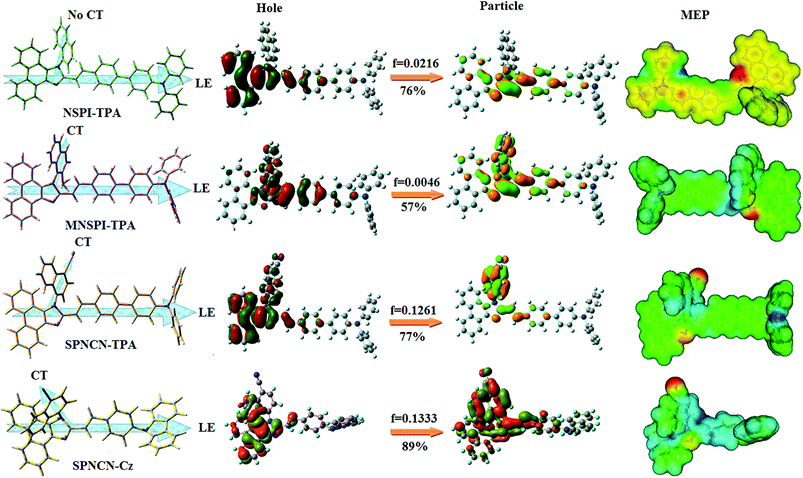 |
| | Fig. 4 HOMO, LUMO contour maps and molecular electrostatic potential (ESP) surface along with CT/LE direction in NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz. | |
Table 2 Percentage of LE and CT in SPNCN-TPA and SPNCN-Cz
| Percentage of transition |
SPNCN-TPA |
SPNCN-Cz |
| Singlet S1–S10 |
Triplet T1–T10 |
Singlet S1–S10 |
Triplet T1–T10 |
| % LE |
10 |
60 |
30 |
40 |
| % CT |
90 |
40 |
70 |
60 |
Table 3 Calculated energies (E, eV) and oscillator strengths (f) of S0–S10 transitions from NTO of NSPI-TPA and MNSPI-TPA
| Transitions |
NSPI-TPA |
MNSPI-TPA |
| E |
f |
NTO |
E |
f |
NTO |
| S0 → S1 |
1.06 |
0.0216 |
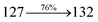 |
1.93 |
0.0187 |
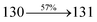 |
| S0 → S2 |
1.43 |
0.0215 |
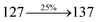 |
2.76 |
0.0850 |
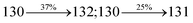 |
| S0 → S3 |
1.65 |
0.0056 |
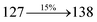 |
3.17 |
0.5536 |
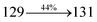 |
| S0 → S4 |
1.81 |
0.0011 |
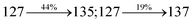 |
3.24 |
0.0370 |
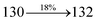 |
| S0 → S5 |
2.90 |
0.0024 |
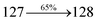 |
3.37 |
0.1357 |
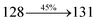 |
| S0 → S6 |
3.05 |
0.1315 |
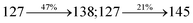 |
3.56 |
0.1629 |
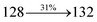 |
| S0 → S7 |
3.24 |
0.5378 |
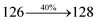 |
3.61 |
0.0669 |
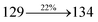 |
| S0 → S8 |
3.27 |
0.0334 |
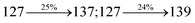 |
3.66 |
0.2855 |
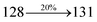 |
| S0 → S9 |
3.32 |
0.0369 |
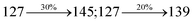 |
3.87 |
0.0723 |
 |
| S0 → S10 |
3.40 |
0.5391 |
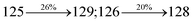 |
3.91 |
0.2707 |
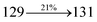 |
Table 4 Calculated energies (E, eV) and oscillator strengths (f) of S0–S10 transitions from NTO of SPNCN-TPA and SPNCN-Cz
| Transitions |
SPNCN-TPA |
SPNCN-Cz |
| E |
f |
NTO |
E |
f |
NTO |
| S0 → S1 |
2.57 |
0.1261 |
 |
2.08 |
0.2554 |
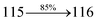 |
| S0 → S2 |
3.23 |
0.2437 |
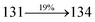 |
2.72 |
0.2762 |
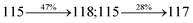 |
| S0 → S3 |
3.23 |
0.5362 |
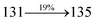 |
2.74 |
0.0865 |
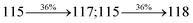 |
| S0 → S4 |
3.51 |
0.0665 |
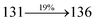 |
2.94 |
0.0637 |
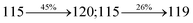 |
| S0 → S5 |
3.58 |
0.1319 |
 |
3.34 |
0.1165 |
 |
| S0 → S6 |
3.66 |
0.0972 |
 |
3.42 |
0.1741 |
 |
| S0 → S7 |
3.86 |
0.2489 |
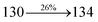 |
3.68 |
0.0986 |
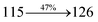 |
| S0 → S8 |
3.87 |
0.4764 |
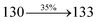 |
3.75 |
0.1179 |
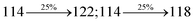 |
| S0 → S9 |
3.98 |
0.1723 |
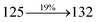 |
3.86 |
0.2818 |
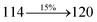 |
| S0 → S10 |
4.05 |
0.2212 |
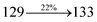 |
3.91 |
0.0054 |
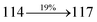 |
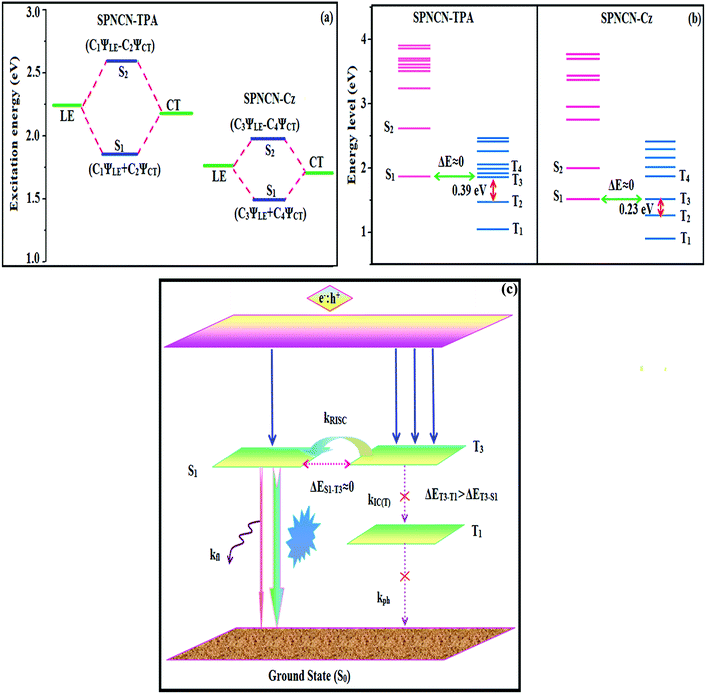 |
| | Fig. 5 (a) Schematic of hybridization processes of LE and CT states of SPNCN-TPA and SPNCN-Cz; (b) energy levels of singlet (S) and triplet (T) states of SPNCN-TPA and SPNCN-Cz; (c) scheme of exciton decay process after hole and electron recombination in OLEDs of D–π–A molecules. | |
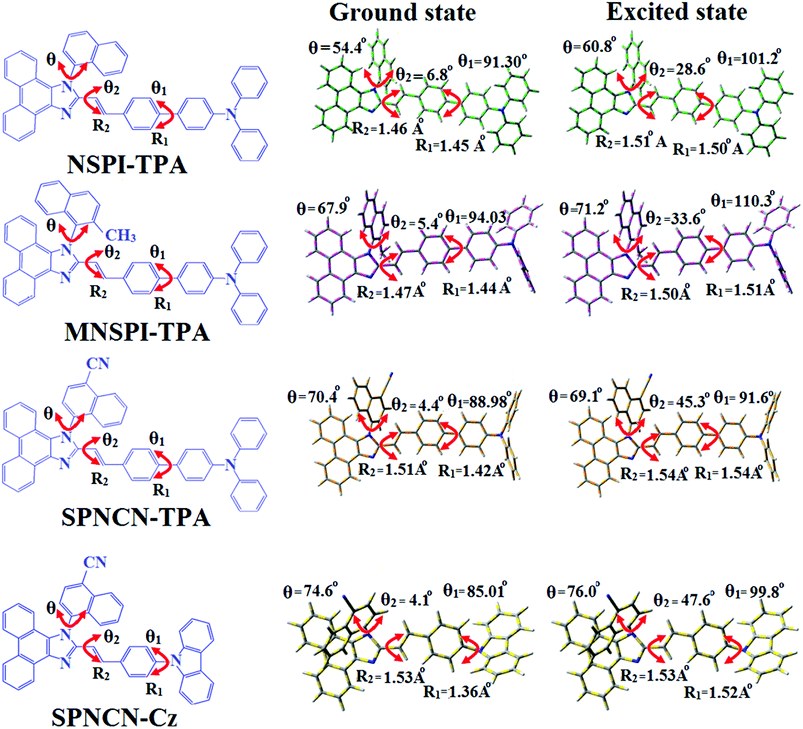 |
| | Fig. 6 Ground state and excited state geometries with dihedral angles and bond lengths of NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz. | |
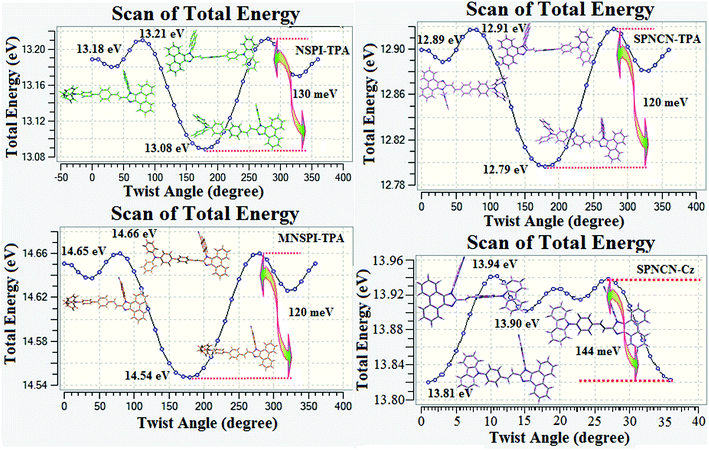 |
| | Fig. 7 Potential energy curves of NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz. | |
 |
| | Fig. 8 Hole-only and electron-only devices based on NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz. | |
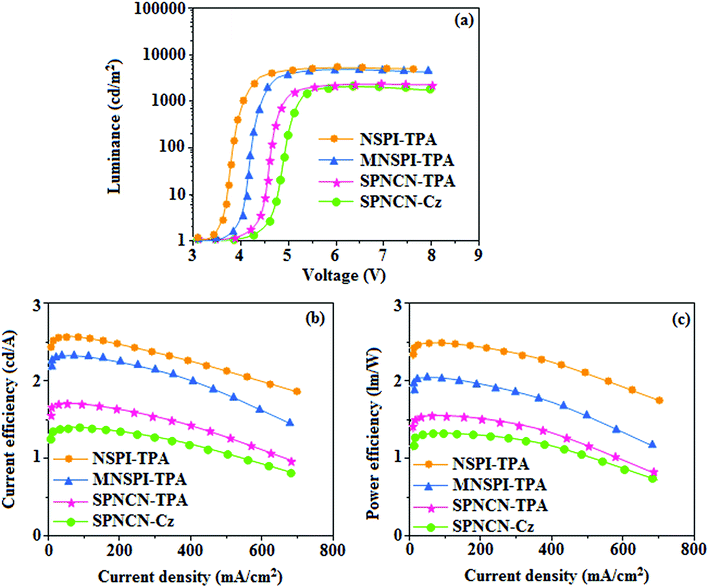 |
| | Fig. 9 Electroluminescence performances of non-doped EL devices with NSPI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz as emitters: (a) luminance versus voltage, (b) current efficiency versus current density and (c) power efficiency versus current density. | |
Table 5 Excitation energy (E, eV), excitation co-efficient (ε), overlap integral (Δr, Å) and nature of transition of S1 → S10 states of NSPI-TPA and MNSPI-TPA
| Excited states |
NSPI-TPA |
MNSPI-TPA |
| E |
ε |
Δr |
E |
ε |
Δr |
| S1 |
1.0619 |
0.4317 |
1.4304 |
1.9290 |
0.4138 |
1.0838 |
| S2 |
1.4258 |
0.4552 |
3.3486 |
2.7584 |
0.3950 |
1.6500 |
| S3 |
1.6516 |
0.4627 |
3.6408 |
3.1700 |
0.3886 |
4.0225 |
| S4 |
1.8057 |
0.4813 |
2.0202 |
3.2444 |
0.3897 |
3.0161 |
| S5 |
2.8968 |
0.4279 |
5.3083 |
3.3693 |
0.3787 |
3.1412 |
| S6 |
3.0497 |
0.4038 |
3.1612 |
3.5633 |
0.3749 |
3.6268 |
| S7 |
3.2426 |
0.3771 |
3.0296 |
3.0096 |
0.4094 |
3.8344 |
| S8 |
3.2672 |
0.4361 |
2.1519 |
3.6624 |
0.3527 |
2.9801 |
| S9 |
3.3180 |
0.3911 |
2.7920 |
3.8681 |
0.3589 |
2.9531 |
| S10 |
3.3992 |
0.3976 |
2.8895 |
3.9141 |
0.3501 |
3.8249 |
4 Conclusions
We have reported new deep-blue emitters using twisted donor–π–acceptor molecular design strategy. The photophysical and thermal stabilities and electrochemical properties of cyano-substituted blue fluorescent materials SPNCN-TPA and SPNCN-Cz can be modulated by the chemical modification of TPA moiety by a Cz fragment, which results in an HLCT emissive state with the increase in LE, decrease in CT and an increase in quantum efficiency. A fine modulation of the emissive state was performed between LE and CT composition to form a quasi-equivalent hybridized HLCT state in SPNCN-Cz and SPNCN-TPA, in which the LE component contributes high ηPL, whereas the CT component generates high ηs. SPNCN-Cz-based device shows external quantum efficiency of 3.28%, current efficiency of 2.90 cd A−1 and power efficiency of 2.26 lm W−1. The molecular design of the twisted conformation can be used to fabricate low cost fluorescent OLED materials using HLCT state principle.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
Dr J. Jayabharathi thank DST (Department of Science and Technology – EMR/2014/000094, F. No. SR/S1/1C-73/2010, F. No. SR/S1/1C-07/2007), DRDO (Defence Research and Development Organization – 213/MAT/10-11), CSIR (Council of Scientific and Industrial Research – No. 01/ (2707)/13EMR-II), UGC (University Grant Commission – 36-21/2008, F. No. 30-71/2004(SR)) and DST-Nano Mission (SR/NM/NS-1001/2016) for financial support. Authors would like to thank Dr P. Justin Jesuraj, Korea University, South Korea for scientific discussions.
References
-
(a) C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS
 ;
(b) J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Born and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS .
;
(b) J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Born and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS . - H. Kaji, H. Suzuki, T. Fukushima, K. Shizu, K. Suzuki, S. Kubo, T. Komino, H. Oiwa, F. Suzuki, A. Wakamiya, Y. Murata and C. Adachi, Nat. Commun., 2015, 6, 84761–84768 Search PubMed .
- J. Zhao, B. Liu, Z. Wang, Q. X. Tong, X. Du, C. J. Zheng, H. Lin, S. L. Tao and X. H. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 9629–9637 CrossRef CAS PubMed .
- Z. ZHU, S. Ni, W. Chen, M. Chen, J. Zhu, Y. Yuan, Q. Tong, F. WONG and C. Lee, J. Mater. Chem. C, 2018, 6, 3584–3592 RSC .
- A. Islam, Q. Wang, L. Zhang, T. Lei, L. Hong, R. Yang, Z. Liu, R. Peng, L. S. Liao and Z. Ge, Dyes Pigm., 2017, 142, 499–506 CrossRef CAS .
- Y. Park, J. H. Lee, D. H. Jung, S. H. Liu, Y. H. Lin and L. Y. Chen, et al., J. Mater. Chem., 2010, 20, 5930–5936 RSC .
- S. J. Su, C. Cai and J. J. Kido, Chem. Mater., 2011, 23, 274–284 CrossRef CAS .
- N. Matsumoto, T. Miyazaki, M. Nishiyama and C. Adachi, J. Phys. Chem. C, 2009, 113, 6261–6266 CrossRef CAS .
- M. A. Baldo, O. D. F. Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS .
- K. Mullen and U. Scherf, Cent. Eur. J. Chem., 2006, 9, 1126–1132 Search PubMed .
- C. Xiang, W. Koo and F. So, Light: Sci. Appl., 2013, 2, 74–77 CrossRef .
-
(a) G. M. Farinola and R. Ragni, Chem. Soc. Rev., 2011, 40, 3467–3482 RSC ;
(b) M. C. Gather, A. Kohnen and K. Meerholz, Adv. Mater., 2011, 23, 233–248 CrossRef CAS PubMed ;
(c) D. M. Kang, J. W. Kang, J. W. Park, S. O. Jung, S. H. Lee, H. D. Park, Y. H. Kim, S. C. Shun, J. J. Kim and S. K. Kwon, Adv. Mater., 2008, 20, 2003–2007 CrossRef CAS ;
(d) T. J. Park, W. S. Jeon, J. J. Park, S. Y. Kim, Y. K. Lee, J. Jang, J. H. Kwon and R. Pode, Appl. Phys. Lett., 2008, 92, 113308–113313 CrossRef .
-
(a) P. I. Shih, C. Y. Chuang, C. H. Chien, E. W. G. Diau and C. F. Shu, Adv. Funct. Mater., 2007, 17, 3141–3146 CrossRef CAS ;
(b) S. J. Lee, J. S. Park, K. J. Yoon, Y. I. Kim, S. H. Jin, S. K. Kang, Y. S. Gal, S. Kang, J. Y. Lee, J. W. Kang, S. H. Lee, H. D. Park and J. J. Kim, Adv. Funct. Mater., 2008, 18, 3922–3930 CrossRef CAS .
- B. Wei, J. Z. Liu, Y. Zhang, J. H. Zhang, H. N. Peng, H. L. Fan, Y. B. He and X. C. Gao, Adv. Funct. Mater., 2010, 20, 2448–2458 CrossRef CAS .
- J. W. G. Hunt, Z. Q. Jiang, Z. Y. Liu, C. L. Yang, C. Zhong, J. G. Qin, G. Yu and Y. Q. Liu, Adv. Funct. Mater., 2009, 19, 3987–3995 CrossRef .
- Z. Q. Gao, Z. H. Li, P. F. Xia, M. S. Wong, K. W. Cheah and C. H. Chen, Adv. Funct. Mater., 2007, 17, 3194–3199 CrossRef CAS .
- Y. H. Kim, H. C. Jeong, S. H. Kim, K. Yang and S. K. Kwon, Adv. Funct. Mater., 2005, 15, 1799–1805 CrossRef CAS .
- S. Tao, Z. Peng, X. Zhang, P. Wang, C. S. Lee and S. T. Lee, Adv. Funct. Mater., 2005, 15, 1716–1721 CrossRef CAS .
- C. H. Chien, C. K. Chen, F. M. Hsu, C. F. Shu, P. T. Chou and C. H. Lai, Adv. Funct. Mater., 2009, 19, 560–566 CrossRef CAS .
-
(a) M. Y. Lai, C. H. Chen, W. S. Huang, J. T. Lin, T. H. Ke, L. Y. Chen, M. H. Tsai and C. Wu, Angew. Chem., Int. Ed., 2008, 4, 591–595 CrossRef ;
(b) J. Ye, Z. Chen, M. K. Fung, C. Zheng, X. Ou and X. H. Zhang, Chem. Mater., 2013, 25, 2630–2637 CrossRef CAS .
- W. Qin, Z. Yang, Y. Jiang, J. W. Y. Lam, G. Liang, H. S. Kwok and B. Z. Tang, Chem. Mater., 2015, 27, 3892–3901 CrossRef CAS .
-
(a) B. Liu, J. W. Zhao, C. Y. Luo, F. Lu, S. L. Tao and Q. X. Tong, J. Mater. Chem. C, 2016, 4, 2003–2010 RSC ;
(b) G. Li, J. W. Zhao, D. Zhang, Z. C. Shi, Z. L. Zhu, H. Q. Song, J. J. Zhu, S. L. Tao, F. Lu and Q. X. Tong, J. Mater. Chem. C, 2016, 4, 8787–8794 RSC .
- W. C. Chen, Y. Yuan, G. F. Wu, H. X. Wei, L. Tang, Q. X. Tong, F. L. Wong and C. S. Lee, Adv. Opt. Mater., 2014, 2, 626–631 CrossRef CAS .
- W. C. Chen, G. F. Wu, Y. Yuan, H. X. Wei, F. L. Wong, Q. X. Tong and C. S. Lee, RSC Adv., 2015, 5, 18067–18074 RSC .
- C. L. Li, J. Wei, J. Han, Z. Li, X. Song, Z. Zhang, J. Zhang and Y. Wang, J. Mater. Chem. C, 2016, 4, 10120–10129 RSC .
-
(a) Y. Qian, Q. Wei, G. D. Pozo, M. M. Mróz, L. Lüer, S. Casado, J. C. Gonzalez, Q. Zhang, L. Xie, R. Xia and W. Huang, Adv. Mater., 2014, 26, 2937–2942 CrossRef CAS PubMed ;
(b) D. He, Y. Yuan, B. Liu, D. Y. Huang, C. Y. Luo, F. Lu, Q. X. Tong and C. S. Lee, Dyes Pigm., 2017, 136, 347–353 CrossRef CAS .
- M. Zhu and C. Yang, Chem. Soc. Rev., 2013, 42, 4963–4976 RSC .
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS .
- T. Le Bahers, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2011, 7, 2498–2506 CrossRef CAS PubMed .
- S. Tretiak and S. Mukamel, Chem. Rev., 2002, 102, 3171–3212 CrossRef CAS PubMed .
- W. J. Li, D. D. Liu, F. Z. Shen, D. G. Ma, Z. M. Wang, T. Fei, B. Yang and Y. G. Ma, Adv. Funct. Mater., 2012, 22, 2797–2803 CrossRef CAS .
- W. J. Li, Y. Y. Pan, R. Xiao, Q. M. Peng, S. T. Zhang, D. G. Ma, F. Li, F. Z. Shen, Y. H. W. B. Yang and Y. G. Ma, Adv. Funct. Mater., 2014, 24, 1609–1614 CrossRef CAS .
- S. Tang, W. J. Li, F. Z. Shen, D. D. Liu, B. Yang and Y. G. Ma, J. Mater. Chem., 2012, 22, 4401–4408 RSC .
- D. Chaudhuri, E. Sigmund, A. Meyer, L. Rcck, P. Klemm, S. Lautenschlager, A. Schmid, S. R. Yost, T. Van, S. Bange, S. Hcger and J. M. Lupton, Angew. Chem., 2013, 52, 13449–13452 CrossRef CAS PubMed .
- M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 60, 14422–14428 CAS .
- T. Forster, Discuss. Faraday Soc., 1959, 27, 7–17 RSC .
- J. R. Sheats, H. Antoniadis, M. Hueschen, W. Leonard, J. Miller, R. Moon, D. Roitman and A. Stocking, science, 1996, 273, 884–888 CrossRef CAS PubMed .
- X. Ouyang, X. Li, L. Ai, D. Mi, Z. Ge and S. J. Su, ACS Appl. Mater. Interfaces, 2015, 7, 7869–7877 CrossRef CAS PubMed .
- K. Wang, S. Wang, J. Wei, Y. Miao, Y. Liu and Y. Wang, Org. Electron., 2014, 15, 3211–3220 CrossRef CAS .
- N. Su, F. Meng, J. Chen, Y. Wang, H. Tan, S. Su and W. Zhu, Dyes Pigm., 2016, 128, 68–74 CrossRef CAS .
- H. Liu, P. Chen, D. Hu, X. Tang, Y. Pan, H. Zhang, W. Zhang, X. Han, Q. Bai, P. Lu and Y. Ma, Chem.–Eur. J., 2014, 20, 2149–2153 CrossRef CAS PubMed .
-
(a) Y. Yuan, J. X. Chen, F. Lu, Q. X. Tong, Q. D. Yang, H. W. Mo, T. W. Ng, F. L. Wong, Z. Q. Guo, J. Ye, Z. Chen, X. H. Zhang and C. S. Lee, Chem. Mater., 2013, 25, 4957–4965 CrossRef CAS ;
(b) Z. L. Zhu, W. C. Chen, L. D. Zhang, X. L. Liu, Q. X. Tong, F. L. Wong, F. Lu and C. S. Lee, J. Mater. Chem. C, 2016, 4, 6249–6257 RSC ;
(c) B. Liu, Y. Yuan, D. He, D. Y. Huang, C. Y. Luo, Z. L. Zhu, F. Lu, Q. X. Tong and C. S. Lee, Chem.–Eur. J., 2016, 22, 12130–12137 CrossRef CAS PubMed .
- Z. L. Zhu, M. Chen, W. C. Chen, S. F. Ni, Y. Y. Peng, C. Zhang, Q. X. Tong, F. Lu and C. S. Lee, Org. Electron., 2016, 38, 323–329 CrossRef CAS .
-
(a) R. Kim, S. Lee, K. H. Kim, Y. J. Lee, S. K. Kwon, J. J. Kim and Y. H. Kim, Chem. Commun., 2013, 49, 4664–4666 RSC ;
(b) C. J. Zheng, W. M. Zhao, Z. Q. Wang, D. Huang, J. Ye, X. M. Ou, X. H. Zhang, C. S. Lee and S. T. Lee, J. Mater. Chem., 2010, 20, 1560–1566 RSC ;
(c) J. N. Moorthy, P. Venkatakrishnan, P. Natarajan, D. F. Huang and T. J. Chow, J. Am. Chem. Soc., 2008, 130, 17320–17333 CrossRef CAS PubMed .
-
(a) B. Kim, Y. Park, J. Lee, D. Yokoyama, J. H. Lee, J. Kido and J. Park, J. Mater. Chem. C, 2013, 1, 432–440 RSC ;
(b) S. Lee, B. Kim, H. Jung, H. Shin, H. Lee, J. Lee and J. Park, Dyes Pigm., 2017, 136, 255–261 CrossRef CAS .
- H. Huang, Y. Wang, S. Zhuang, X. Yang, L. Wang and C. Yang, J. Phys. Chem. C, 2012, 116, 19458–19466 CrossRef CAS .
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, (Revision A.02), Gaussian, Inc., Wallingford., CT. 2009 Search PubMed .
-
(a) Y. Zhang, S. L. Lai, Q. X. Tong, M. Y. Chan, T. W. Ng, Z. C. Wen, G. Q. Zhang, S. T. Lee, F. L. Wong and C. S. Lee, J. Mater. Chem., 2011, 21, 8206–8214 RSC ;
(b) M. Liu, Y. Seino, D. Chen, S. Inomata, S. J. Su, H. Sasabe and J. Kido, Chem. Commun., 2015, 51, 16353–16356 RSC .
- Z. Lu, N. Liu, S. J. Lord, S. D. Bunge, W. E. Moerner and R. J. Twieg, Chem. Mater., 2009, 21, 797–810 CrossRef CAS PubMed .
- J. H. Huang, J. H. Su, X. Li, M. K. Lam, K. M. Fung, H. H. Fan, K. W. Cheah, C. H. Chen and H. J. Tian, Mater. Chem., 2011, 21, 2957–2964 RSC .
- K. C. Wu, P. J. Ku and C. S. Lin, et al., Adv. Funct. Mater., 2008, 18, 67–75 CrossRef CAS .
- J. N. Moorthy, P. Natarajan, P. Venkatakrishnan, D. F. Huang and T. J. Chow, Org. Lett., 2007, 9, 5215–5218 CrossRef CAS PubMed .
- S. Tang, M. R. Liu, P. Lu, H. Xia, M. Li, Z. Q. Xie, F. Z. Shen, C. Gu, H. Wang, B. Yang and Y. G. Ma, Adv. Funct. Mater., 2007, 17, 2869–2877 CrossRef CAS .
- Y. N. Yan, W. L. Pan and H. C. Song, Dyes Pigm., 2010, 86, 249–258 CrossRef CAS .
- Z. Ma, E. Wang, M. E. Jarvid, P. Henriksson, O. Inganas, F. Zhang and M. R. Andersson, J. Mater. Chem., 2012, 22, 2306–2314 RSC .
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed .
- C. Liu, Q. Fu, Y. Zou, C. Yang, D. Ma and J. Qin, Chem. Mater., 2014, 26, 3074–3083 CrossRef CAS .
- E. Lippert, W. Lüder and H. Boos, Advances in molecular spectroscopy, ed. A. Mangini, Pergamon Press, Oxford, 1962 Search PubMed .
-
(a) S. P. Jagtap, S. Mukhopadhyay, V. Coropceanu, G. L. Brizius, J. Bré das and D. M. Collard, J. Am. Chem. Soc., 2012, 134, 7176–7185 CrossRef CAS PubMed ;
(b) S. Shirai, S. Iwata, T. Tani and S. Inagaki, J. Phys. Chem. A, 2011, 11(5), 7687–7699 CrossRef PubMed .
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed .
- S. T. Zhang, W. J. Li, L. Yao, Y. Y. Pan, B. Yang and Y. G. Ma, Chem. Commun., 2013, 49, 11302–11304 RSC .
-
(a) M. Segal, M. Singh, K. Rivoir, S. Difley, T. V. Voorhis and M. A. Baldo, Nat. Mater., 2007, 6, 374–378 CrossRef CAS PubMed ;
(b) W. Barford, Phys. Rev. B, 2004, 70, 205204–205208 CrossRef .
- W. Jiang, L. Duan and J. Qiao, et al., Org. Lett., 2011, 13, 3146–3149 CrossRef CAS PubMed .
-
(a) S. Zhang, L. Yao, Q. Peng, W. Li, Y. Pan, R. Xiao, Y. Gao, C. Gu, Z. Wang, P. Lu, F. Li, S. Su, B. Yang and Y. Ma, Adv. Funct. Mater., 2015, 25, 1755–1762 CrossRef CAS ;
(b) J. Jayabharathi, P. Jeeva, V. Thanikachalam and S. Panimozhi, J. Photochem. Photobiol., A, 2017, 17, 30559–30562 Search PubMed .
- H. Liu, Q. Bai, L. Yao, H. Zhang, H. Xu, S. Zhang, W. Li, Y. Gao, J. Li, P. Lu, H. Wang, B. Yang and Y. Ma, Chem. Sci., 2015, 6, 3797–3804 RSC .
- Z. Wang, Y. Feng, H. Li, Z. Gao, X. Zhang, P. Lu, P. Chen, Y. Ma and S. Liu, Phys. Chem. Chem. Phys., 2014, 16, 10837–10843 RSC .
- W. Li, L. Yao, H. Liu, Z. Wang, S. Zhang, R. Xiao, H. Zhang, P. Lu, B. Yang and Y. Ma, J. Mater. Chem. C, 2014, 2, 4733–4736 RSC .
- M. F. Lin, L. Wang, W. K. Wong, K. W. Cheah, H. L. Tam, M. T. Lee and C. H. Chen, Appl. Phys. Lett., 2006, 89, 121913–121916 CrossRef .
- Z. Gao, Y. Liu, Z. Wang, F. Shen, H. Liu, G. Sun, L. Yao, Y. Lv, P. Lu and Y. Ma, Chem.–Eur. J., 2013, 19, 2602–2605 CrossRef CAS PubMed .
- X. L. Li, X. Ouyang, D. Chen, X. Cai, M. Liu, Z. Ge, Y. Cao and S. J. Su, Nanotechnology, 2016, 27, 124001–124011 CrossRef PubMed .
- Y. Zhang, T. W. Ng, F. Lu, Q. X. Tong, S. L. Lai, M. Y. Chan, H. L. Kwong and C. S. Lee, Dyes Pigm., 2013, 98, 190–194 CrossRef CAS .
- Y. Zhang, S. L. Lai, Q. X. Tong, M. F. Lo, T. W. Ng, M. Y. Chan, Z. C. Wen, J. He, K. S. Jeff, X. L. Tang, W. M. Liu, C. C. Ko, P. F. Wang and C. S. Lee, Chem. Mater., 2012, 24, 61–70 CrossRef .
- F. I. Wu, P. I. Shih, M. C. Yuan, A. K. Dixit, C. F. Shu, Z. M. Chung and E. W. G. Diau, J. Mater. Chem., 2005, 15, 4753–4760 RSC .
- Y. L. Liao, C. Y. Lin, K. T. Wong, T. H. Hou and W. Y. Hung, Org. Lett., 2007, 9, 4511–4514 CrossRef CAS PubMed .
- D. G. Yu, F. C. Zhao, Z. Zhang, C. M. Han, H. Xu and J. Li, et al., Chem. Commun., 2012, 48, 6157–6159 RSC .
- M. T. Lee, H. H. Chen, C. H. Liao, C. H. Tsai and C. H. Chen, Appl. Phys. Lett., 2004, 85, 3301–3303 CrossRef CAS .
- J. Y. Park, S. Y. Jung, J. Y. Lee and Y. G. Baek, Thin Solid Films, 2008, 516, 2917–2921 CrossRef CAS .
- C. Tang, F. Liu, Y. J. Xia, J. Lin, L. H. Xie and G. Y. Zhong, et al., Org. Electron., 2006, 7, 155–162 CrossRef CAS .
- V. Thanikachalam, P. Jeeva and J. Jayabharathi, J. Phys. Org. Chem., 2017, 3695, 1–13 Search PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07891b |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Sekar Panimozhi and
Venugopal Thanikachalam
*,
Sekar Panimozhi and
Venugopal Thanikachalam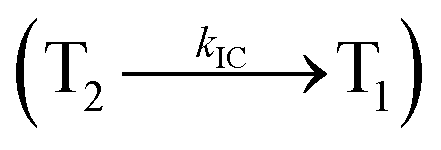 , resulting in hot RISC
, resulting in hot RISC 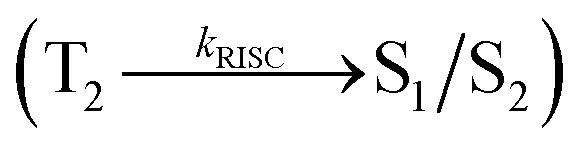 rather than cold RISC (T1 → S1) TADF mechanism.35,36 Hot exciton process with HLCT increases external quantum efficiency (ηex) because of high ηPL and high ηS. ηex can be calculated as follows: ηex = ηIQE × ηout = ηrec × ηPL × ηS × ηout, where ηIQE is the internal quantum efficiency, ηout is the light out coupling efficiency (20%), ηrec is the efficiency for electron–hole recombination (100%), ηPL is the photoluminescence efficiency of the film and ηS is the exciton utilization efficiency [ηS = ηrec × ηPL × ηout ÷ ηEL].37 The ambipolar phenanthrimidazole derivatives have been shown as potential blue emitters. The increase in conjugation in phenanthrimidazoles influenced the blue emission,38–42 and conjugation length was restricted by incorporating a bulky fragment in the core molecule, which resulted in twisted conformation.43–46 In line with this discussion and our own research interest, we report donor–spacer–acceptor derivatives, namely, 4′-(1-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)-N,N-diphenyl-(2-[1,1′-biphenyl]vinyl)-4-amine (NSPI-TPA), 4′-(1-(2-methylnaphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)-N,N-diphenyl-(2-[1,1′-biphenyl]vinyl)-4-amine (MNSPI-TPA), 4-(2-(4′-(diphenylamino)-(2-[1,1′-biphenyl]vinyl)-4-yl)-1H-phenanthro[9,10-d]imidazol-1-yl)-1-naphthalene-1-carbonitrile (SPNCN-TPA) and 4-(2-(4-(9H-carbazol-9-yl)styryl)-1H-phenanthro[9,10-d]imidazol-1-yl)naphthalene-1-carbonitrile (SPNCN-Cz) using triphenylamine as a strong donor and carbazole as weak donor (Scheme 1). The H–H repulsion of bulky styryl fragment with the phenyl moiety of TPA and the carbazole moieties leads to twisted configuration, which enhances the twist angle, thus shortening the conjugation length. The solvatochromic effect of NPSI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz was examined to understand the excited state characteristics and interstate coupling strength of LE and CT components. Combining theoretical (TD-DFT) and experimental data, the LE and CT compositions were discussed using natural transition orbital (NTO), centroids of charges and transition density matrix (TDM) analysis. Hybridization of LE and CT energy states was used for molecular design, and their composition in HLCT was tuned, resulting in high EL efficiency. Triphenylamine (TPA) of SPNCN-TPA was replaced by carbazole (Cz) in SPNCN-Cz, both of which differ in electron donating ability. As a result, the CT composition decreases with an increase in LE composition in the S1 HLCT state. Thus, the SPNCN-Cz-based device exhibited maximum electroluminescent efficiency with ηex of 3.15%, ηc of 2.56 cd A−1 and ηp of 2.45 lm W−1 and CIE of (0.15, 0.07), which were higher than those of the SPNCN-TPA-based device. Higher ηPL of SPNCN-Cz film was observed compared with that of SPNCN-TPA film; thus, high ηPL and high ηS enhanced the ηex of the SPNCN-Cz-based device. These results can be used to design low cost fluorescent materials via subtle molecular modifications using the HLCT emissive state principle.
rather than cold RISC (T1 → S1) TADF mechanism.35,36 Hot exciton process with HLCT increases external quantum efficiency (ηex) because of high ηPL and high ηS. ηex can be calculated as follows: ηex = ηIQE × ηout = ηrec × ηPL × ηS × ηout, where ηIQE is the internal quantum efficiency, ηout is the light out coupling efficiency (20%), ηrec is the efficiency for electron–hole recombination (100%), ηPL is the photoluminescence efficiency of the film and ηS is the exciton utilization efficiency [ηS = ηrec × ηPL × ηout ÷ ηEL].37 The ambipolar phenanthrimidazole derivatives have been shown as potential blue emitters. The increase in conjugation in phenanthrimidazoles influenced the blue emission,38–42 and conjugation length was restricted by incorporating a bulky fragment in the core molecule, which resulted in twisted conformation.43–46 In line with this discussion and our own research interest, we report donor–spacer–acceptor derivatives, namely, 4′-(1-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)-N,N-diphenyl-(2-[1,1′-biphenyl]vinyl)-4-amine (NSPI-TPA), 4′-(1-(2-methylnaphthalen-1-yl)-1H-phenanthro[9,10-d]imidazol-2-yl)-N,N-diphenyl-(2-[1,1′-biphenyl]vinyl)-4-amine (MNSPI-TPA), 4-(2-(4′-(diphenylamino)-(2-[1,1′-biphenyl]vinyl)-4-yl)-1H-phenanthro[9,10-d]imidazol-1-yl)-1-naphthalene-1-carbonitrile (SPNCN-TPA) and 4-(2-(4-(9H-carbazol-9-yl)styryl)-1H-phenanthro[9,10-d]imidazol-1-yl)naphthalene-1-carbonitrile (SPNCN-Cz) using triphenylamine as a strong donor and carbazole as weak donor (Scheme 1). The H–H repulsion of bulky styryl fragment with the phenyl moiety of TPA and the carbazole moieties leads to twisted configuration, which enhances the twist angle, thus shortening the conjugation length. The solvatochromic effect of NPSI-TPA, MNSPI-TPA, SPNCN-TPA and SPNCN-Cz was examined to understand the excited state characteristics and interstate coupling strength of LE and CT components. Combining theoretical (TD-DFT) and experimental data, the LE and CT compositions were discussed using natural transition orbital (NTO), centroids of charges and transition density matrix (TDM) analysis. Hybridization of LE and CT energy states was used for molecular design, and their composition in HLCT was tuned, resulting in high EL efficiency. Triphenylamine (TPA) of SPNCN-TPA was replaced by carbazole (Cz) in SPNCN-Cz, both of which differ in electron donating ability. As a result, the CT composition decreases with an increase in LE composition in the S1 HLCT state. Thus, the SPNCN-Cz-based device exhibited maximum electroluminescent efficiency with ηex of 3.15%, ηc of 2.56 cd A−1 and ηp of 2.45 lm W−1 and CIE of (0.15, 0.07), which were higher than those of the SPNCN-TPA-based device. Higher ηPL of SPNCN-Cz film was observed compared with that of SPNCN-TPA film; thus, high ηPL and high ηS enhanced the ηex of the SPNCN-Cz-based device. These results can be used to design low cost fluorescent materials via subtle molecular modifications using the HLCT emissive state principle.