
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly(styrene)-supported N-heterocyclic carbene coordinated iron chloride as a catalyst for delayed polyurethane polymerization†
Hyeon-Jun Noh‡
 a,
T. Sadhasivam‡ab,
Do-Sung Junga,
Keundeuk Leec,
Mingu Hanc,
Ju-Young Kim*d and
Ho-Young Jung*ab
a,
T. Sadhasivam‡ab,
Do-Sung Junga,
Keundeuk Leec,
Mingu Hanc,
Ju-Young Kim*d and
Ho-Young Jung*ab
aDepartment of Environment & Energy Engineering, Chonnam National University, 77 Yongbong-ro, Buk-gu, Gwangju 61186, Republic of Korea. E-mail: jungho@jnu.ac.kr; jungho@chonnam.ac.kr
bCenter for Energy Storage System, Chonnam National University, 77 Yongbong-ro, Buk-gu, Gwangju 61186, Republic of Korea
c4th R&D Institute 2nd Directorate Agency for Defence Development, Yuseoung P. O. Box 35, Daejeon, 34186, Korea
dDepartment of Advanced Materials Engineering, Kangwon National University, Samcheok, Kangwon 25913, Republic of Korea. E-mail: juyoungk@kangwon.ac.kr
First published on 7th November 2018
Abstract
An advanced organometallic catalyst based on N-heterocyclic carbene (NHC) coordinated FeCl3 has been synthesized and used to control the reaction rate in polyurethane (PUR) polymerization. The imidazolium (Im)-based NHC was functionalized on the surface of the supporting material of bead-type chloromethyl polystyrene (PS) resin. The PS-Im–FeCl3 catalyst was synthesized through the coordination reaction between Im and FeCl3. The successful formation, functional groups, structure, and geometry of the PS-Im–FeCl3 catalysts were confirmed by Fourier transform infrared and X-ray photoelectron spectroscopy techniques. A thin layer of Im was observed to be coated uniformly on the PS bead surface and FeCl3 nanoparticles were observed to cover the coating layer homogeneously, as determined by field-emission scanning electron microscopy, transmission electron microscopy, and energy dispersive X-ray spectroscopy measurements. The PUR polymerization reaction was investigated through viscosity measurements and non-isothermal activation energy calculations by differential scanning calorimetry analysis. Based on the viscosity measurements, delayed PUR polymerization was achieved using the PS-Im–FeCl3 catalyst system. The highest viscosity (6000 cP) was achieved without any catalyst, with triphenylene bismuth, and with the PS-Im–FeCl3 catalyst after 23, 5, and 25 h of reaction time, respectively. Furthermore, the calculated activation energies (Ea) were 27.92 and 36.35 kJ mol−1 for the no-catalyst and the PS-Im–FeCl3 systems, respectively. Thus, the viscosity measurements and DSC analyses confirm that the PS-Im–FeCl3 catalyst considerably increases the PUR reaction time. The Im–FeCl3 catalyst supported by CMPS can control the reaction rate in PUR synthesis because of its high activity. Thus, the PS-Im–FeCl3 catalyst can be used as a curing retardant in the PUR industry.
Introduction
Complex catalysts specifically organometallic catalysts are used to enhance the organic chemical reactions.1 Polyurethane (PUR) is an important target of research and development because of its use in sealants, adhesives, and coating materials.2 PUR is effectively considered as a blowing agent for polymer-bonded explosives (PBX) and bonding agent for rocket propulsion.3,4 In PBX systems, retardation of PUR polymerization is required to improve the flow-ability in the Reaction Injection Molding (RIM) process for increasing the long pot lifetime. The two-component polyurethane system (2K-PUR) is the preferred and most commonly used polyurethane process.5 In 2K-PUR, one component is a reagent containing a hydroxyl group, and the other component is a reagent containing an isocyanate group.6 The curing reaction and properties of PUR can be controlled and modified using a catalyst. As PUR polymerization catalysts, organic compounds such as diazabicyclooctane (DABCO), N-alkyl morpholines, amidines (like 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU)), and dibutyltin dilaurate (DBTDL) have been considered over the past few decades.7–10 These catalysts cause fast curing because of the highly electrophilic isocyanate group, which is activated by Lewis acid. In addition, tertiary amines activate the alcohol, thus increasing the nucleophilicity of the alcohol substrate. Therefore, the polyurethane is rapidly cured at room temperature.2,11 Because of this activity, the above-mentioned catalysts cannot be used to delay the polymerization reaction.Currently, phenylmercury neodecanoate (PMND) and mercury-based catalysts are considered as suitable delayed-action PUR catalysts. However, these catalysts do not satisfy the requirements for delayed PUR polymerization because of their environmental issues and heavy metal content.12,13 Alternatives such as organic bases, N-heterocyclic carbenes (NHC), organic acids, and organic–metal complex catalysts have been studied as polyurethane catalysts.1,14–22 The organic base catalysts of ε-caprolactone and δ-valerolactone have been used as ring-opening polymerization catalysts.20,23 Alsarraf et al. investigated the polymerization performance using bicyclic penta-alkylated guanidine and a conventional tin catalyst.22 In addition, in pursuit of effective synthetic processes, organic acid catalysts have been used for polyurethane polymerization. However, the performance of acid catalysts is lower than that of commonly used metal catalysts.24 As an example, the polymerization activity of isocyanate and alcohols with the organic acid was lower than that of metal-based tin catalyst.25 Alsarraf et al. developed a new type of polyurethane delay guanidine catalyst, but it showed inferior PUR polymerization activity at room temperature and the same performance as commercial catalysts in terms of thermal activity.8 Tertiary amine blocks have also been used as polyurethane polymerization retardant. During the PUR process, the amine catalyst gradually dissociates during urethane to polyurethane polymerization with heating, which improves the flowability of the RIM system.26
NHC (also denoted as Im), which produce stable metal compounds with five-membered to seven-membered ring systems including benzimidazolium, triazolium, imidazolium, imidazolinium, pyrazolium, and pyrimidinium have been reported for various applications.27–31 Recently, polymer synthesis using NHCs has been reported and has received wide attention for its effectiveness when used to delay the PUR condensation reaction.1,8,12,15 Among the NHC catalysts, imidazolin-2-ylidene carbene has drawn significant attention because of its structure and stability.1 In addition, it has several advantages, such as nucleophilic organic catalytic properties and the ability to form complexes with transition metals. During the PUR process with NHC-based catalysts, (i) the free NHC species contributes to PUR formation, (ii) the free metal activates the isocyanate, and (iii) the NHC carbene interacts with polyol. In this process, the activated polyol reacts with isocyanate, and the polyurethane polymerization reaction is delayed. Thus, NHC-based delayed-action catalysts have emerged for industrial use because they can form polymer networks at specific temperatures.32,33 Another advantage of NHC-based delayed-action catalysts is that the PU condensation reaction can be carried out without heavy metal catalysts. Recently, the formation of NHCs with CO2 and various metal salts has been demonstrated.1,29 Naumann et al.34 reported the comparative performance of NHC-based catalysts and commercial (DBTDL and PMND) catalysts for polyurethane production at room temperature. Notably, the performance of the NHC-based metal adduct showed excellent activity during PUR polymerization. In addition, a new class of NHC–Mg, NHC–Al, NHC–Zn, and NHC–Sn catalysts have been reported.1,8,12,30,35–37 However, reports of NHC–metal adducts for PUR polymerization are still scarce. Considering these factors, we have developed an NHC–FeCl3 complex catalyst supported on chloromethyl polystyrene (CMPS) beads for delayed PUR synthesis. The catalyst is prepared via a facile synthetic process. The structural and microstructural properties of the catalyst have been analyzed using spectroscopic and microscopic techniques. In addition, the catalytic activity, degree of polymerization, and process have been analyzed using viscosity measurements and differential scanning calorimetry analyses.
Materials and methods
Reagents and analysis
Dimethylformamide (DMF, 99%), tetrahydrofuran (THF, 99%), dichloromethane (DCM, 99.8%), and 1-methylimidazolium (99%) were purchased form Duksan Pure Chemicals Co., Ltd. Methanol (MeOH, 99.9%, Daejung Co., Ltd), chloromethyl polystyrene (CMPS, BeadTech), potassium tert-butoxide (98%, Acros Organics), iron(III) chloride (FeCl3, 98%, Samchun), isophorone diisocyanate (IPDI, 98%, Sigma-aldrich) and hydroxyl-terminated polybutadiene (HTPB, Samyang fine chemical Co.,Ltd.) were purchased and used without further purification.Preparation of the catalyst
1-Methylimidazole (0.4926 g, 6 mmol) and CMPS beads (10 g, 2.1 mmol Cl/g) were added to 200 mL of DMF. The solution was stirred at 80 °C for 24 h. After complete the reaction, the as-prepared polystyrene (PS)-imidazole (Im) solution was poured into the 150 mesh standard for removing the aggregated PS and/or PS-Im. The filtrated PS-Im solution was further treated with an excess amount of DMF to remove the unreacted methylimidazole. For removing the unreacted CMPS, the PS-Im suspension was filtered through the 250 mesh standard. The unreacted CMPS was passed through the pores of 250 mesh standard due to the smaller size of CMPS. The PS-Im was collected from the 250 mesh standard screen and dried at 80 °C for 24. For the synthesis of PS-Im–FeCl3, the as-prepared PS-Im beads were placed in 100 mL of THF/DMF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with continuous stirring in a 100 mL three-neck round-bottomed flask. After complete dispersion of the PS-Im beads, a desired amount of iron(III) chloride (0.0146 g, 0.9 mmol) and potassium tert-butoxide (0.011 g, 1 mmol) were added to the bead suspension, and the suspension was stirred at 80 °C for 8 h under nitrogen atmosphere. After the reaction had completed, the resultant material was filtered through a 150-mesh standard screen with DMF, MeOH, and salt water (30%). The obtained solid products were dried in a vacuum oven. This synthetic method for PS-Im–FeCl3 was followed and inspired by Kim et al.,37 and a schematic of the preparation of PS-Im–FeCl3 is shown in Fig. 1.
:1) with continuous stirring in a 100 mL three-neck round-bottomed flask. After complete dispersion of the PS-Im beads, a desired amount of iron(III) chloride (0.0146 g, 0.9 mmol) and potassium tert-butoxide (0.011 g, 1 mmol) were added to the bead suspension, and the suspension was stirred at 80 °C for 8 h under nitrogen atmosphere. After the reaction had completed, the resultant material was filtered through a 150-mesh standard screen with DMF, MeOH, and salt water (30%). The obtained solid products were dried in a vacuum oven. This synthetic method for PS-Im–FeCl3 was followed and inspired by Kim et al.,37 and a schematic of the preparation of PS-Im–FeCl3 is shown in Fig. 1.
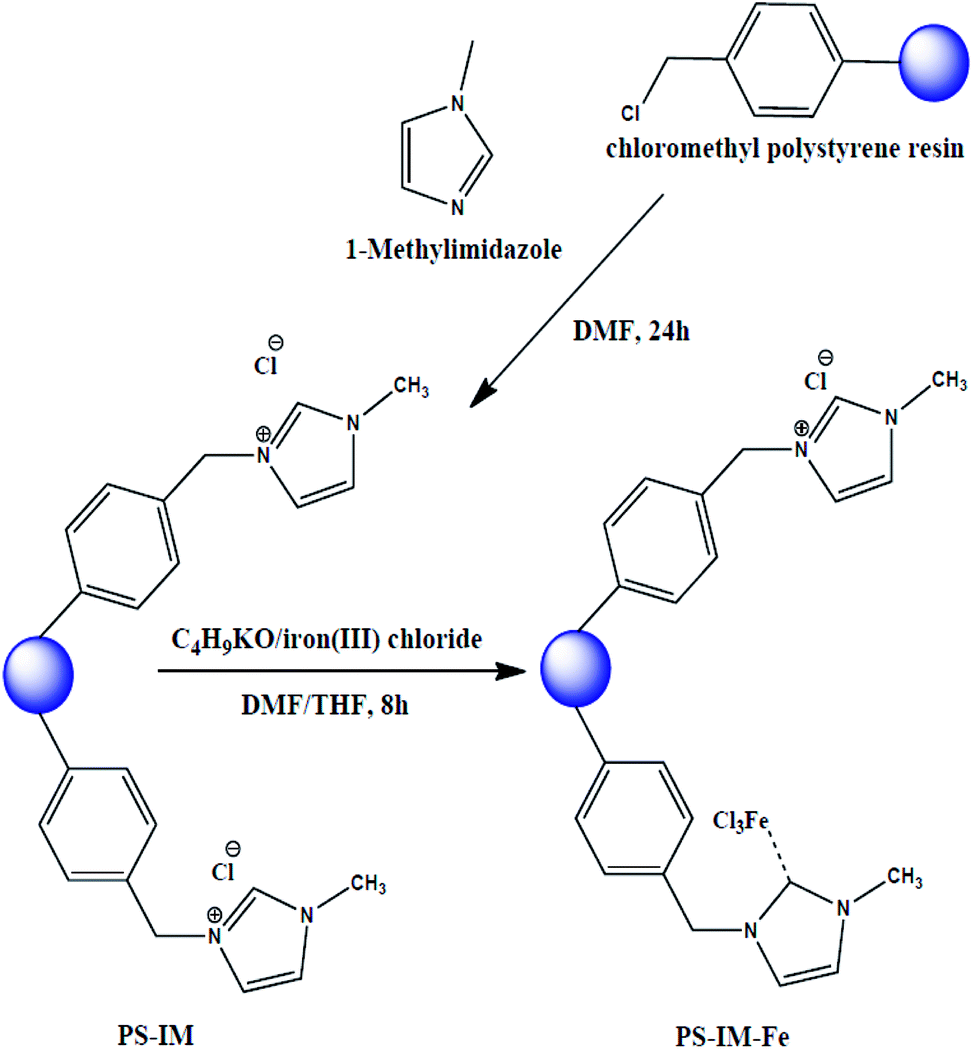 | ||
| Fig. 1 Schematic diagram of synthesis of PS-Im and PS-Im–FeCl3 catalyst. | ||
Structural and microstructural characterization
The functional groups of the as-received and as-synthesized materials were confirmed using Fourier transform infrared (FTIR) spectrometry in attenuated total reflectance (ATR) mode (Thermo Nicolet iS5). The elemental compositions and binding energies of the materials were analyzed by X-ray photoelectron spectroscopy (XPS, VG Multilab 2000). The surface morphology was analyzed using a field emission scanning electron microscope (FE-SEM) with attached energy dispersive X-ray (EDX) spectrometer (Jeol/Oxford, JSM-7500F+). The surface properties, particle size, and presence of elements in the as-prepared materials were determined using a transmission electron microscope (TEM) (Jeol JEM-2100F).Polyurethane polymerization process
To investigate the effect of the catalyst on the PUR polymerization process, hydroxyl-terminated polybutadiene (HTPB, 14 g), isophorone diisocyanate (IPDI, 1.11 g), and as-synthesized PS-Im–FeCl3 catalyst (0.14 g) were mixed in a batch-type reactor. To obtain PUR, the polymerization reaction was carried out with continuous stirring at 60 °C. A similar synthetic process was carried out without the addition of the PS-Im–FeCl3 catalyst (denoted the no-catalyst system). In addition, a commercial triphenylene bismuth (TPB) catalyst was used for comparison with PS-Im–FeCl3. To determine the PUR polymerization degree, a Brookfield viscometer was used. This was used to monitor the polymerization process based on the viscosity of the reactant solution at 60 °C.PUR polymerization degree measurements
The degree of polymerization was measured by two different methods. First, the polymerization degree was determined based on the change in viscosity of the reactants, measured using a rotary viscometer (Model 98936, Cole-Parmer). An electric furnace was used to control the curing temperature. The viscosities of the samples were measured at specific time intervals. Secondly, the degree of polymerization degree was determined using differential scanning calorimetry (DSC 131-EVO) through activation energy calculations. To assess the polymerization reaction by DSC quantitatively, a desired amount of sample (ca. 8–10 mg) was weighed and loaded into an aluminum DSC pan. Thermal (non-isothermal) analysis was conducted by increasing the temperature constantly from room temperature to 400 °C. The DSC measurements were carried out at different heating rates 5, 10, and 20 °C min−1. The activation energy (Ea) of PUR was calculated using the Kissinger equation.38,39Results and discussion
The functional groups on CMPS and the as-prepared Im-functionalized PS (PS-Im and PS-Im–FeCl3) were examined via FT-IR analysis. Fig. 2 shows the FTIR data of the CMPS, PS-Im, and PS-Im–FeCl3 catalyst materials. In the spectra of all materials, peaks at 1452 and 1492 cm−1 correspond to the benzene overtones and the benzene C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations in CMPS, respectively. The peaks observed at 696 and 759 cm−1 related to C–H out-of-plane bending and C–H bending vibrations, respectively. Furthermore, the presence of methylene and methylene groups in CMPS was confirmed by the presence of peaks between 2824 and 2975 cm−1.40,41 In the spectra of PS-Im and PS-Im–FeCl3, new peaks appeared around 1151, 1253, 1384, and 1362 cm−1, which correspond to the quaternary ammonium, amide CN stretching, imidazole ring skeleton stretching vibration, and CN stretching of imidazolium, respectively.42 Thus, the functionalization of PS by the NHC in PS-Im was confirmed. Furthermore, a small peak shift was observed in the imidazolium group peaks in the spectrum of PS-Im–FeCl3 compared to those in the spectrum of PS-Im; this is caused by the coordination of imidazolium to FeCl3.
C stretching vibrations in CMPS, respectively. The peaks observed at 696 and 759 cm−1 related to C–H out-of-plane bending and C–H bending vibrations, respectively. Furthermore, the presence of methylene and methylene groups in CMPS was confirmed by the presence of peaks between 2824 and 2975 cm−1.40,41 In the spectra of PS-Im and PS-Im–FeCl3, new peaks appeared around 1151, 1253, 1384, and 1362 cm−1, which correspond to the quaternary ammonium, amide CN stretching, imidazole ring skeleton stretching vibration, and CN stretching of imidazolium, respectively.42 Thus, the functionalization of PS by the NHC in PS-Im was confirmed. Furthermore, a small peak shift was observed in the imidazolium group peaks in the spectrum of PS-Im–FeCl3 compared to those in the spectrum of PS-Im; this is caused by the coordination of imidazolium to FeCl3.
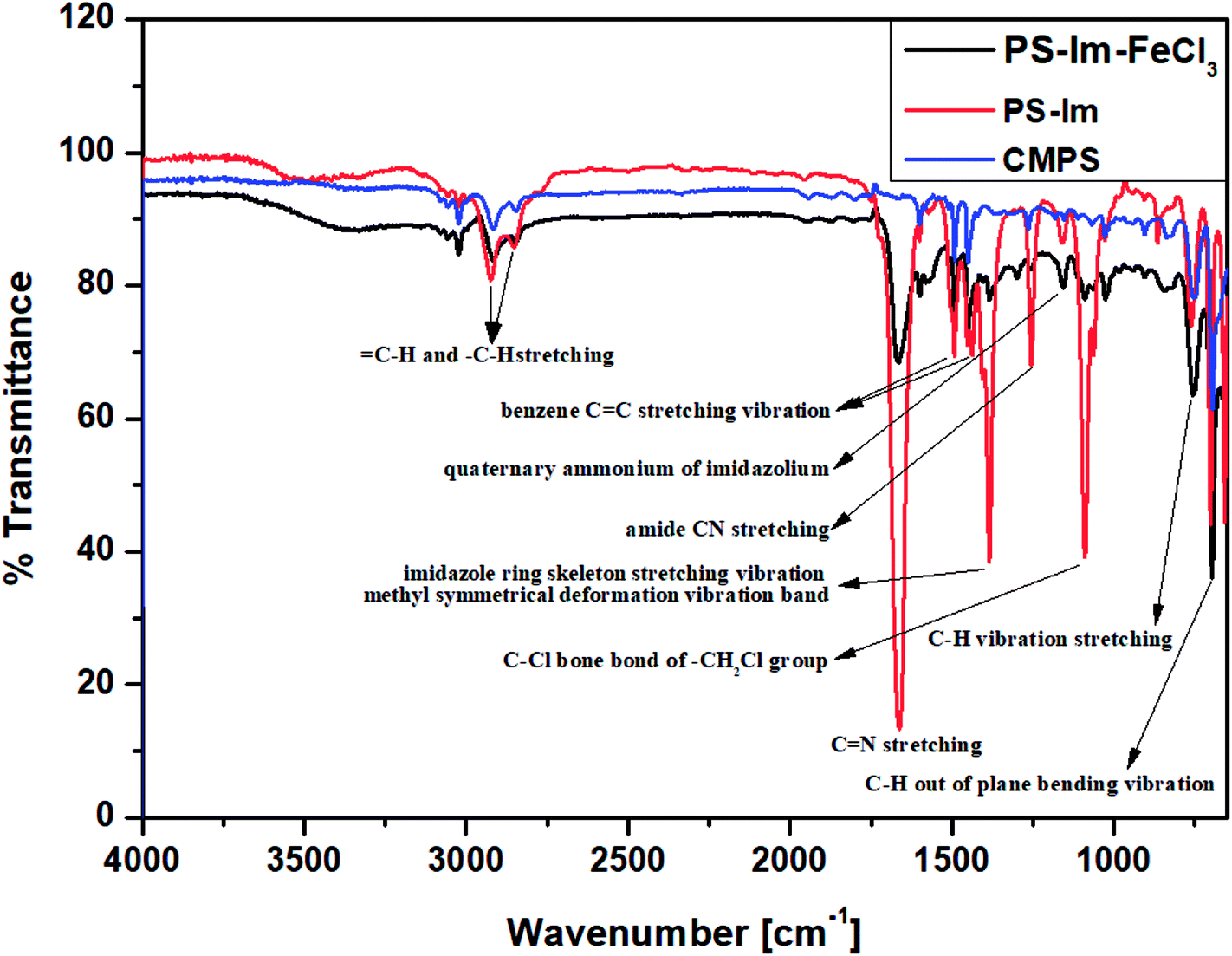 | ||
| Fig. 2 FTIR spectra of CMPS, PS-Im, and PS-Im–FeCl3. | ||
To investigate the Im–FeCl3 complex on PS, the as-prepared materials were analyzed by XPS. Fig. 3 shows the XPS results of (a) CMPS, (b) PS-Im, and (c) PS-Im–FeCl3. As shown in Fig. 3(a), the peaks at 200.8, 285.08, and 532.08 eV correspond to Cl, C, and O, respectively, which is consistent with the elemental content of CMPS.43 In the spectrum of PS-Im, an additional N peak was observed at 401.8 eV,44 as shown in Fig. 3(b). Thus, we confirmed that the PS-Im structure had formed. The coexistence of Fe in PS-Im was also confirmed by XPS measurement of PS-Im–FeCl3, as shown in Fig. 3(c). This is mainly attributed to the coordination of PS-Im and FeCl3. Comparing Fig. 3(b) and (c), a shift in the N1s peak can be seen. The XPS deconvoluted curves of N1s and Cl2p are shown in the ESI (Fig. S1(a–d)†). The N1s peaks at 401.8 and 400.08 eV in the spectra of PS-Im and PS-Im–FeCl3 confirm the presence of quaternary ammonium and trialkylamine groups, respectively. To confirm the coordination bond between FeCl3 and PS-Im, the XPS spectra were magnified and peak fitting was carried out for Fe, as shown in Fig. 3(d). Two major peaks for Fe were observed at 711.08 and 726.08 eV, which correspond to Fe2p3/2 and Fe2p1/2, respectively. From these curves, four fitting peaks were obtained at 710.54, 713.45, 723.44, and 725.75 eV. The peaks at 713.45 and 725.75 eV correspond to trivalent Fe,45 whereas the peaks at 710.54 and 723.44 eV correspond to the divalent state,45 which is attributed to the coordination of the metal in PS-Im–FeCl3. Thus, the coordination reaction of FeCl3 with Im occurred,37 and the presence of a coordinate bond between FeCl3 and PS-Im has been confirmed.
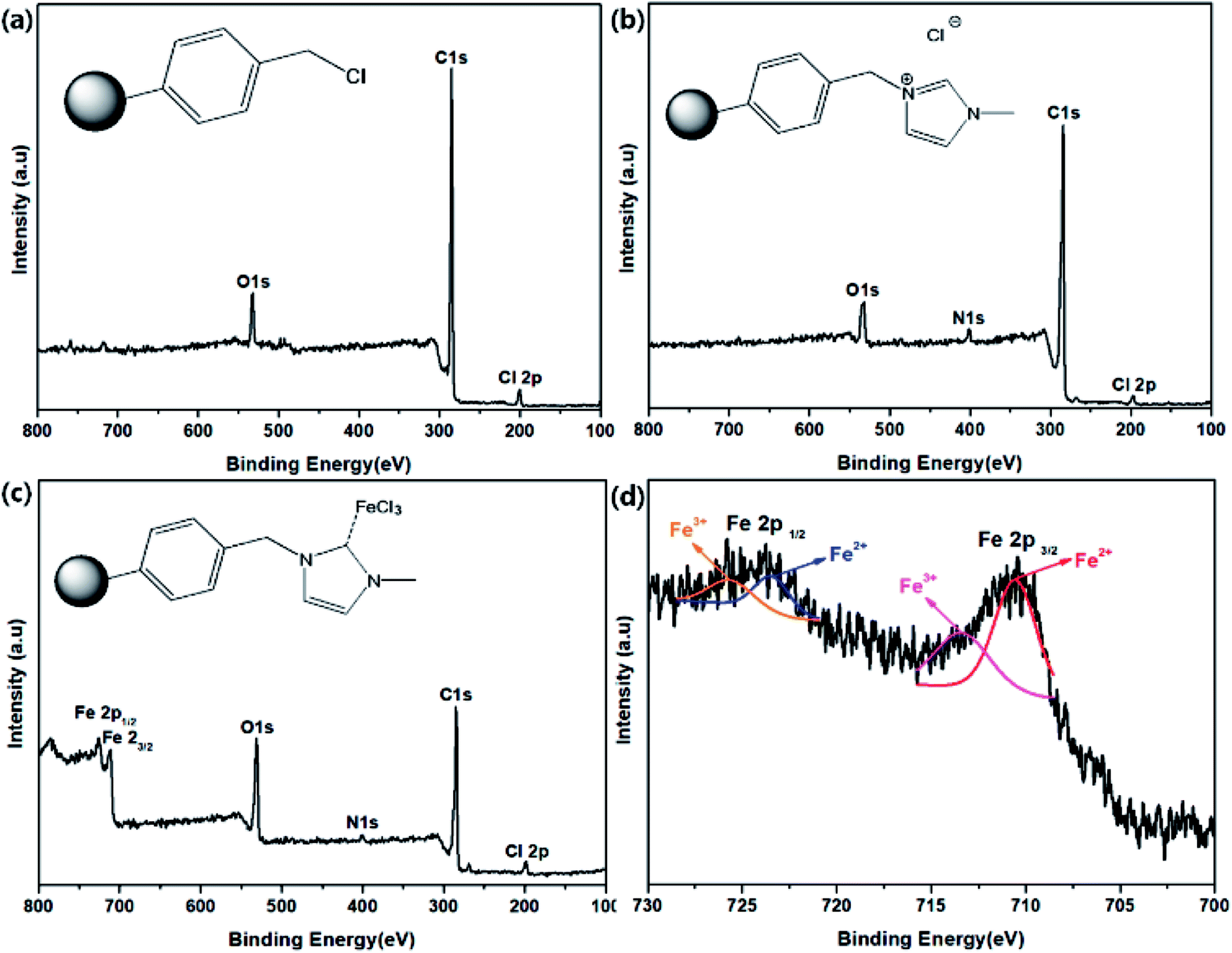 | ||
| Fig. 3 XPS spectra of (a) CMPS, (b) PS-Im, (c) PS-Im–FeCl3, and (d) deconvoluted Fe spectrum of PS-Im–FeCl3. | ||
Surface analysis was carried out via SEM characterization to observe the structural and morphological properties of the as-prepared materials. The surface morphologies of CMPS, PS-Im, and PS-Im–FeCl3 are shown in Fig. 4. As shown in Fig. 4(a), as-received CMPS has a spherical structure with a smooth surface. However, the surface morphology has been modified after reaction with imidazolium, as shown in Fig. 4(b). In the SEM image of the modified CMPS beads, the imidazolium has formed a layer on the surface of the spherical CMPS beads. The changed surface morphology is mainly due to the coupling reaction of imidazolium on the CMPS beads. The SEM image of PS-Im–FeCl3 in Fig. 4(c) shows that the small-sized particles on the spherical surface of PS-Im. In addition, the FeCl3 particles are homogeneously dispersed on the surface. Thus, the SEM micrographs confirm the successful formation of PS-Im–FeCl3, which results from the cohesion of FeCl3 because of the formation of a coordination bond between FeCl3 and Im.
 | ||
| Fig. 4 SEM micrograph images of (a) CMPS, (b) PS-Im, and (c) PS-Im–FeCl3. | ||
TEM and EDX analyses were used to confirm the formation of PS-Im–FeCl3. Fig. 5 shows the TEM image and EDX pattern of the as-synthesized PS-Im–FeCl3 catalyst. As shown in the TEM image (Fig. 5(a)), a thin layer is coated uniformly on the PS bead surface, which confirms the formation of the Im on PS. Furthermore, FeCl3 nanoparticles are attached over the surface. The particles are coordinated to the outermost layer of the Im because Im exists in multiple amorphous layers on the PS beads. Additionally, consistent with the SEM observations, the particles are uniformly distributed on the PS-Im. To investigate the uniform dispersion of the Im–FeCl3 catalyst on the PS beads, EDX analysis was carried out. The EDX spectra (Fig. 5(b)) reveal that C, N, O, Cl, and Fe were present in PS-Im–FeCl3. The percentage weight of C, N, O, Cl, and Fe are 98.58, 0.33, 0.87, 0.06, and 0.16 wt%, respectively. Based on the presence of N, we concluded that the Im had been successfully loaded on the PS bead surface. Furthermore, the presence of Fe and Cl confirms the presence of FeCl3 in the PS-Im–FeCl3 catalyst, indicating the homogeneous formation of coordination bonds between PS-Im and FeCl3.
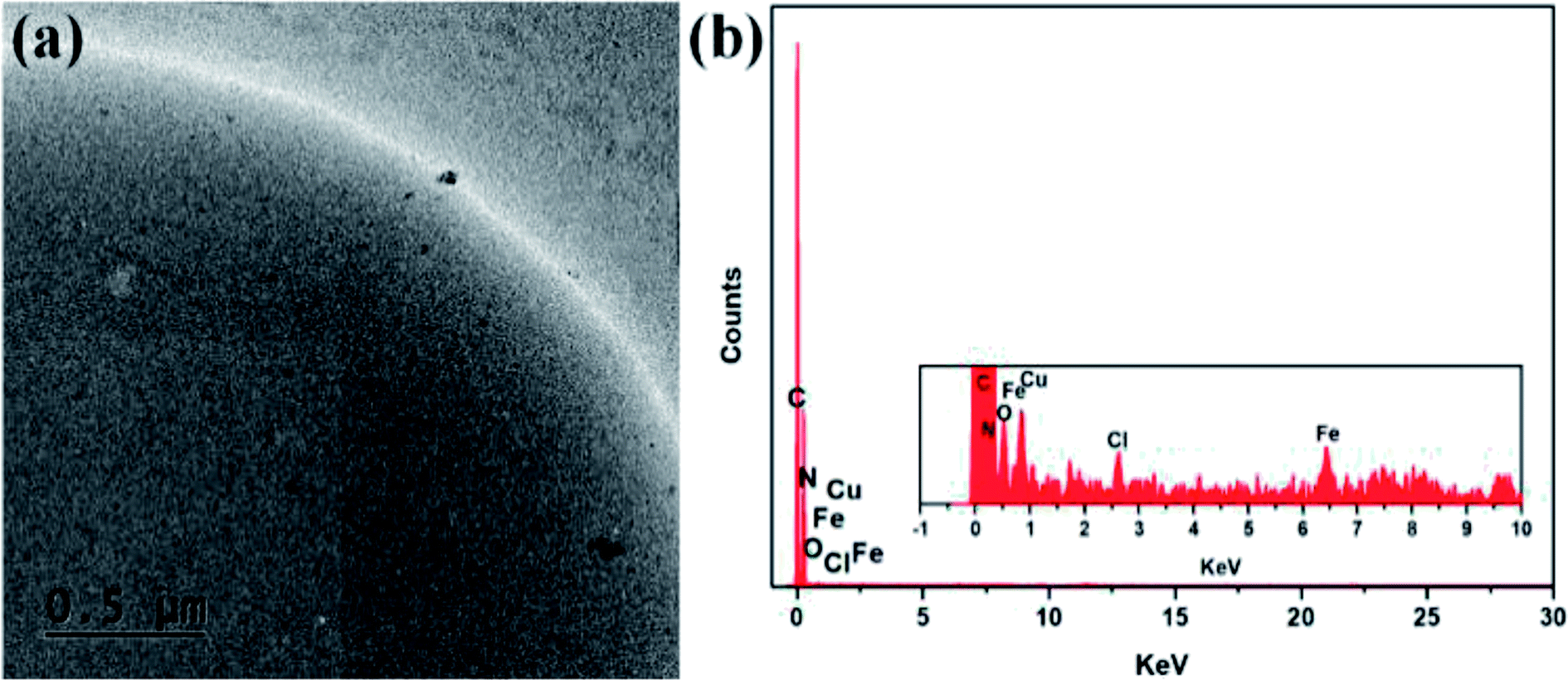 | ||
| Fig. 5 (a) TEM image and (b) EDX pattern of the PS-Im–FeCl3 catalyst. | ||
After successful preparation of the PS-Im–FeCl3 catalyst, it was used for PUR polymerization. A schematic of the catalyst mechanism for PUR polymerization is shown in Fig. 6. A possible reaction of PS-Im–FeCl3 during thermal treatment is represented in Fig. 6(a). At elevated temperatures, the catalyst1,12,34 can be activated and initiated to separate the PS-Im and FeCl3 from the PS-Im–FeCl3 catalyst system. Fig. 6(b) shows a probable reaction mechanism for the polymerization of PUR via the PS-Im–FeCl3 catalyst. The required quantities and operating parameters for PUR polymerization are given in polyurethane polymerization process section. As shown in Fig. 6(b), the initial step of the reaction involves the breakage of the coordination bond between PS-Im and FeCl3 as a function of temperature. Afterwards, the Im interacts with the polyol and, simultaneously, the metal chloride reacts with isocyanate. Here, the Im activates the polyol by acting as a Lewis acid, and the isocyanate is activated by the electrophilic reaction with FeCl3. During this process, polyurethane polymerization occurs by reaction between the polyol and isocyanate.1,12,24,25,34
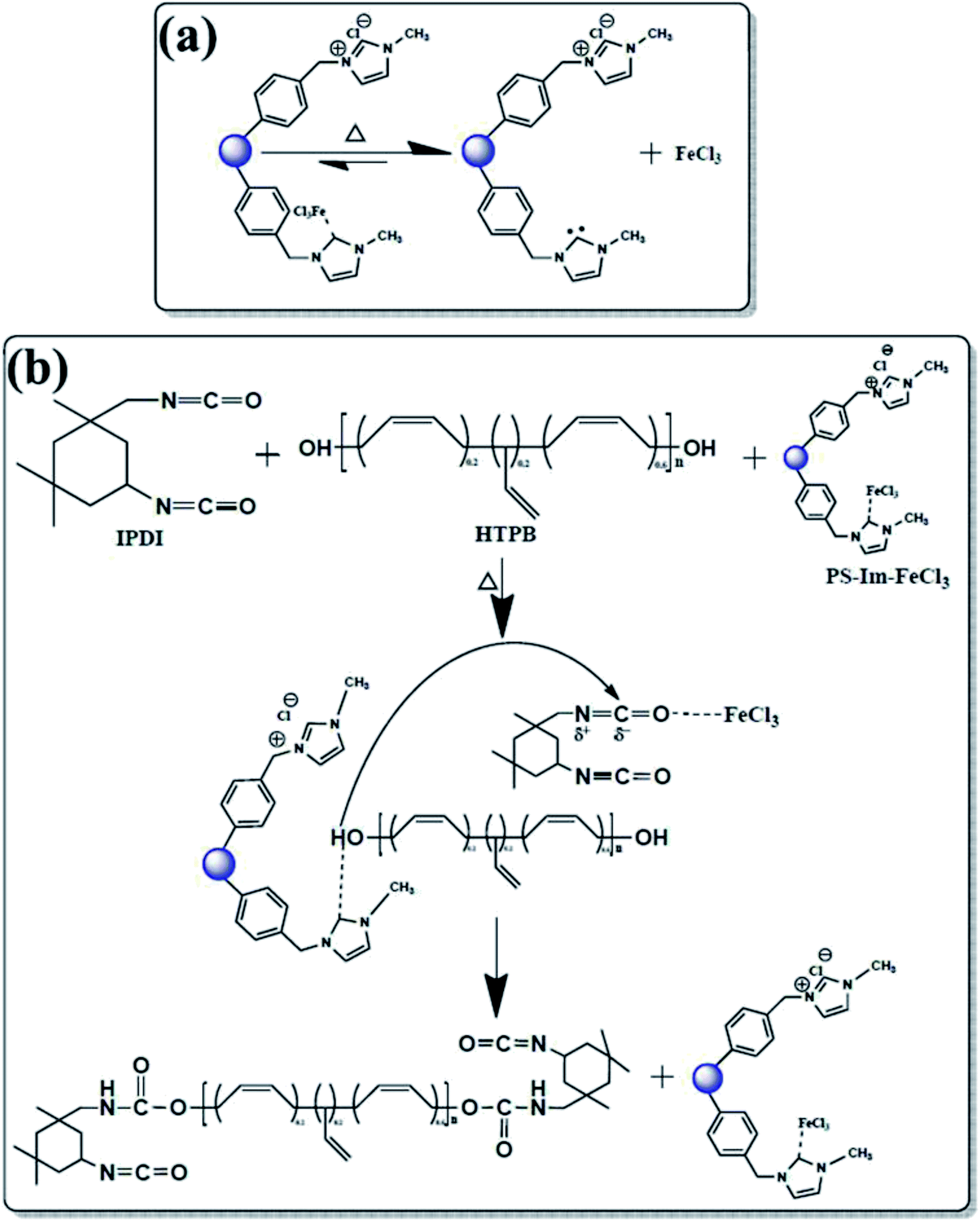 | ||
| Fig. 6 (a) Reaction process of the PS-Im–FeCl3 catalyst under applied heat and (b) catalytic activity mechanism. Dual activation of both polyol and isocyanate functional groups by the dissociated PS-Im–FeCl3 catalyst. | ||
Viscosity measurements are often used to optimize the polymerization reactions.46,47 To measure the rate of time delay for the curing of polyurethane (PU), viscosity analysis was carried out at 60 °C. The viscosity measurement data for PUR polymerization with TPB, PS-Im–FeCl3, and no catalyst are shown in Fig. 7(a). Fig. 7(b) displays the digital photographs of PUR after the polymerization reaction without catalyst and with TPB and PS-Im–FeCl3. Compared to the conventional TPB catalyst, delayed polyurethane curing occurred when no catalyst and as-prepared PS-Im–FeCl3 catalyst was used as shown in Fig. 7(a). For the TPB catalysts system, the viscosity rapidly increased to 6000 cP after 3 h of reaction; thus, the commercial TPB catalyst cannot be used to retardant PUR polymerization. In the no-catalyst system, a stable viscosity was obtained up to 8 h of polymerization reaction. The viscosity in the no-catalyst system gradually increased to 1025 cP over 14 h. However, the polymerization rate increased after 14 h, where the viscosity reached 6000 cP within 23 h. In comparison with the no-catalyst system, the delayed PUR polymerization effect was improved when using PS-Im–FeCl3, as shown in Fig. 7(a) and Table S1.† Similar to the no-catalyst system, the polymerization reaction did not occur before 8 h of reaction. Afterward, the viscosity slightly increased with increasing reaction time, up to 15 h. The viscosity of the system with PS-Im–FeCl3 was 418 cP at 15 h, which is considerably lower than that of the system without catalyst (2052 cP at 15 h) under identical conditions. Furthermore, the highest viscosity point of 6000 cP was obtained for the PS-Im–FeCl3 system after 25 h of reaction time, which is longer than those of the TPB catalyst (5 h) and no-catalyst systems (23 h). Therefore, the PS-Im–FeCl3 catalyst is effective in delaying PU polymerization. Based on these viscosity measurements, the PS-Im–FeCl3 catalyst can be considered as a delayed-action catalyst for commercial polyurethane curing. Furthermore, because TPB was not suitable as a delayed PUR polymerization catalyst (based on the rapid PUR curing39,48), we did not use it further analyses.
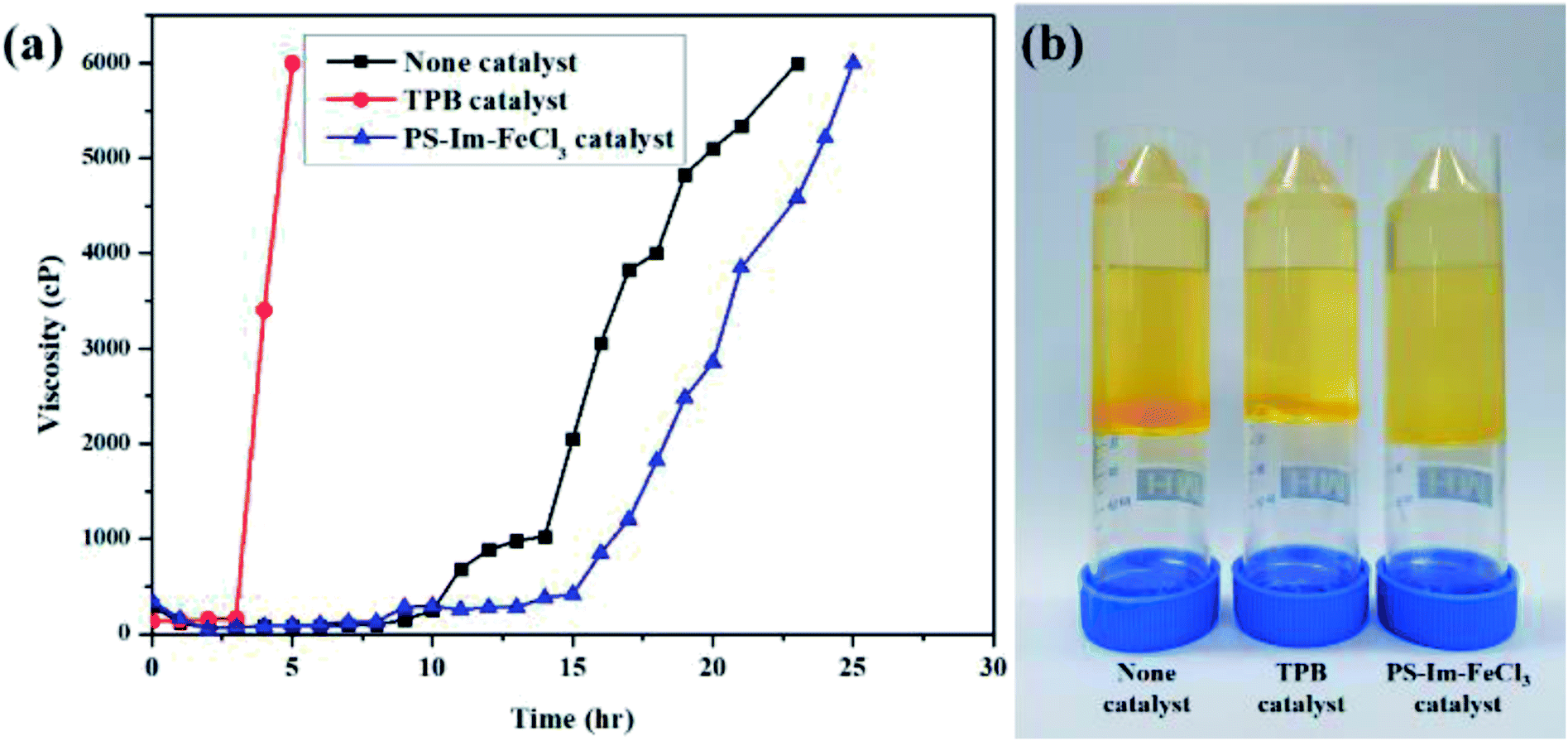 | ||
| Fig. 7 (a) Viscosity data for polyurethane curing at 60 °C using TPB, no catalyst, and PS-Im–FeCl3. (b) Digital images of PU after polymerization. | ||
To investigate PUR polymerization, DSC analysis was performed to identify the curing temperature.39,49 A comparative DSC analysis of (a) no-catalyst and (b) PS-Im–FeCl3 systems at 5 °C is shown in Fig. S2.† Soft curing occurred during the initial PUR polymerization process, and the obtained curing peaks at 149 and 170 °C correspond to the no-catalyst and PS-Im–FeCl3 systems, respectively. The kinetics of the PUR polymerization reaction is considerably lower in the PS-Im–FeCl3 system than that of the no-catalyst system. The catalyst materials are activated during the thermal treatment, which is the reason for the change in PUR polymerization temperature and activation kinetics. To calculate the activation energy for PUR polymerization, DSC measurements were carried out at various heating rates. Fig. 8 shows the DSC curves of (a) no-catalyst and (b) PS-Im–FeCl3 catalyst systems at different applied heating rates (5, 10, and 20 °C). To determine the reaction kinetics and activation energy, the initial soft curing peak was considered, as shown in Fig. 8. By increasing the heating rate, the curing peak position shifted to higher temperatures. The soft curing peak positions of the no-catalyst and PS-Im–FeCl3 catalyst systems are tabulated in Table 1. The activation energy of the PUR polymerization was calculated using the Kissinger equation.38,39
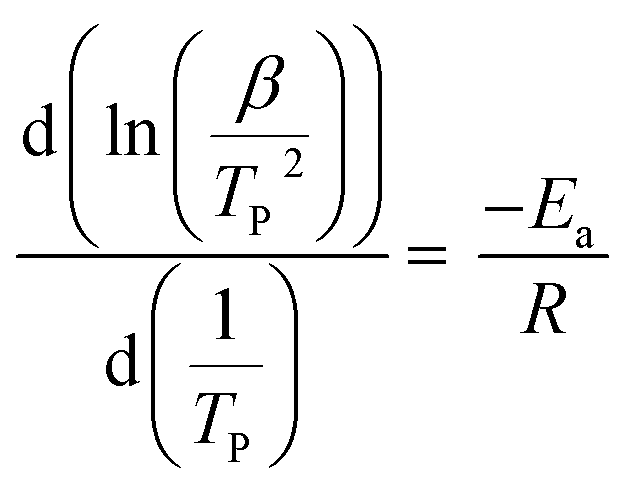
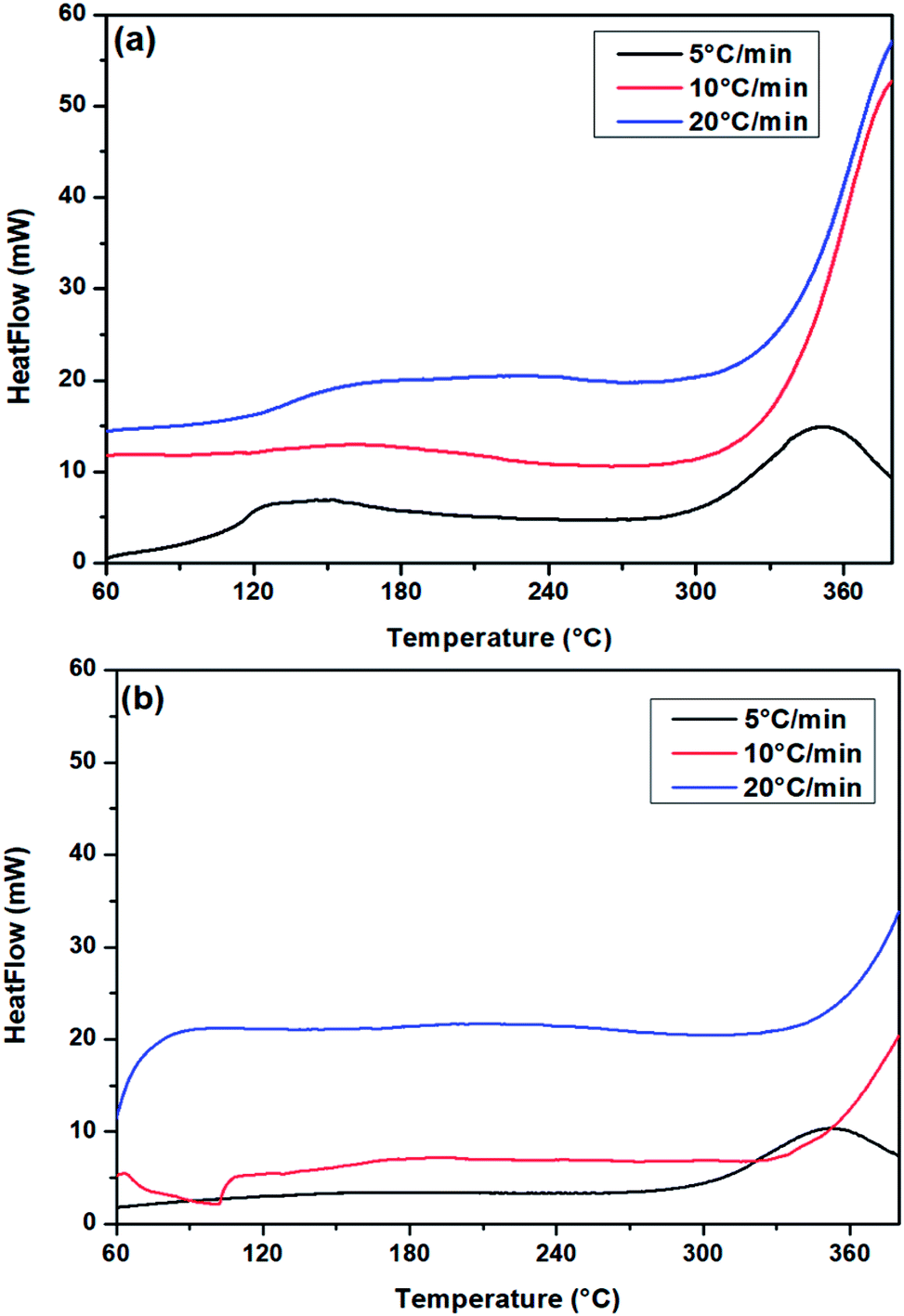 | ||
| Fig. 8 DSC curves of PUR (HTPB/IPDI) with (a) no catalyst and (b) PS-Im–FeCl3 at different heating rate. | ||
| Heating rate (°C min−1) | Peak temperature (°C) | |
|---|---|---|
| No catalyst | PS-Im–FeCl3 catalyst | |
| 5 | 149 | 170 |
| 10 | 161 | 191 |
| 20 | 209 | 227 |
Here, β is the heating rate, TP is the absolute temperature of the first curing peak (K), R is the gas constant (8.314 J mol−1 K−1) and Ea is the activation energy. The activation energy was determined from the slope of the linear plot as shown in Fig. S3(a)† for the no-catalyst system and Fig. S3(b)† for the PS-Im–FeCl3 system. The calculated activation energies for the no-catalyst and PS-Im–FeCl3 systems are 27.92 and 36.35 kJ mol−1, respectively. Compared to the no-catalyst system, the activation energy when using the PS-Im–FeCl3 catalyst for PUR polymerization is higher. Thus, we have confirmed that the PS-Im–FeCl3 catalyst can considerably reduce the reaction kinetics and increase the reaction time for PUR polymerization. In addition, the experimental results confirm that the PS-Im–FeCl3 catalyst is an effective catalyst for delaying PUR polymerization.
Conclusions
The PS-Im–FeCl3 catalyst was successfully synthesized, and its structural/microstructural properties were investigated through sophisticated characterization techniques. In addition, the catalyst was tested for its ability to delay the PUR polymerization reaction. The formation of PS-Im–FeCl3 was confirmed by FTIR and XPS analyses. Based on TEM measurements, we found that a thin layer of Im was coated on the surface of the PS bead support, and FeCl3 nanoparticles were homogeneously anchored on the Im via coordination bonds between Im and FeCl3. The as-prepared PS-Im–FeCl3 catalyst was introduced to the PUR polymerization system, and polymerization was carried out. Viscosity measurements with respect to time, as well as DSC analysis were carried out during the reaction. Furthermore, the catalytic effect and PUR performances of the PS-Im–FeCl3 catalyst were compared to those of the no-catalyst and commercial TPB catalyst systems. Time-delayed PUR polymerization was achieved for the PS-Im–FeCl3 (25 h) catalyst system compared to the commercial TPB catalyst (5 h) and no catalyst (23 h). Furthermore, the resulting activation energies of the no-catalyst and PS-Im–FeCl3 catalyst systems were found to be 27.92 and 36.35 kJ mol−1, respectively. A higher viscosity with time and higher activation energy for PUR polymerization were obtained with the PS-Im–FeCl3 catalyst compared to that of the other tested systems. Therefore, the PS-Im–FeCl3 catalyst is suitable for delayed PUR curing, as well as other potential applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was funded by the 2016 Defense Acquisition Program Administration/Agency for Defense Development (No. 220160003), Republic of Korea. We gratefully acknowledge the financial support from the DAPA/ADD of Korea and Converged Energy Materials Research Center in Yonsei University, Republic of Korea. This research was supported by Gwangju Green Environment Center (Research No. 15-05-04-13-09), Republic of Korea.Notes and references
- B. Bantu, G. Manohar Pawar, K. Wurst, U. Decker, A. M. Schmidt and M. R. Buchmeiser, Eur. J. Inorg. Chem., 2009, 2009, 1970–1976 CrossRef.
- M. Patri, S. Rath and U. Suryavansi, J. Appl. Polym. Sci., 2006, 99, 884–890 CrossRef CAS.
- A. G. Ajaz, Rubber Chem. Technol., 1995, 68, 481–506 CrossRef CAS.
- Q. An, S. V. Zybin, W. A. Goddard III, A. J. Botero, M. Blanco and S.-N. Luo, Phys. Rev. B, 2011, 22, 220101 CrossRef.
- M. Sonntage, Surf. Coat. Int., 1999, 9, 456–459 CrossRef.
- D. K. Chattopadhyay and K. Raju, Prog. Polym. Sci., 2007, 32, 352–418 CrossRef CAS.
- A. L. Silva and J. C. Bordado, Catal. Rev., 2004, 46, 31–51 CrossRef.
- J. Alsarraf, F. Robert, H. Cramail and Y. Landais, Polym. Chem., 2013, 4, 904–907 RSC.
- S. Niyogi, S. Sarkar and B. Adhikari, Indian J. Chem. Technol., 2002, 9, 330 CAS.
- H. Ni, H. A. Nash, J. G. Worden and M. D. Soucek, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1677–1688 CrossRef CAS.
- O. Coulembier, A. P. Dove, R. C. Pratt, A. C. Sentman, D. A. Culkin, L. Mespouille, P. Dubois, R. M. Waymouth and J. L. Hedrick, Angew. Chem., Int. Ed., 2005, 44, 4964–4968 CrossRef CAS PubMed.
- B. Bantu, G. M. Pawar, U. Decker, K. Wurst, A. M. Schmidt and M. R. Buchmeiser, Chem. - Eur. J., 2009, 15, 3103–3109 CrossRef CAS PubMed.
- L. Paches Samblas, PhD thesis, University of Bath, 2010.
- R. C. Pratt, B. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS PubMed.
- M. Fèvre, J. Pinaud, A. Leteneur, Y. Gnanou, J. Vignolle, D. Taton, K. Miqueu and J.-M. Sotiropoulos, J. Am. Chem. Soc., 2012, 134, 6776–6784 CrossRef PubMed.
- D. J. Coady, H. W. Horn, G. O. Jones, H. Sardon, A. C. Engler, R. M. Waymouth, J. E. Rice, Y. Y. Yang and J. L. Hedrick, ACS Macro Lett., 2013, 2, 306–312 CrossRef CAS.
- K. Makiguchi, T. Satoh and T. Kakuchi, Macromolecules, 2011, 44, 1999–2005 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef CAS.
- J. Raynaud, C. Absalon, Y. Gnanou and D. Taton, J. Am. Chem. Soc., 2009, 131, 3201–3209 CrossRef CAS PubMed.
- J. Alsarraf, Y. A. Ammar, F. r. Robert, E. Cloutet, H. Cramail and Y. Landais, Macromolecules, 2012, 45, 2249–2256 CrossRef CAS.
- N. Susperregui, D. Delcroix, B. Martin-Vaca, D. Bourissou and L. Maron, J. Org. Chem., 2010, 75, 6581–6587 CrossRef CAS PubMed.
- R. P. Subrayan, S. Zhang, F. N. Jones, V. Swarup and A. I. Yezrielev, J. Appl. Polym. Sci., 2000, 77, 2212–2228 CrossRef CAS.
- H. Sardon, A. C. Engler, J. M. Chan, J. M. García, D. J. Coady, A. Pascual, D. Mecerreyes, G. O. Jones, J. E. Rice and H. W. Horn, J. Am. Chem. Soc., 2013, 135, 16235–16241 CrossRef CAS PubMed.
- L. J. Lee, Rubber Chem. Technol., 1980, 53, 542–599 CrossRef CAS.
- I. J. Lin and C. S. Vasam, Coord. Chem. Rev., 2007, 251, 642–670 CrossRef CAS.
- S. Naumann and A. P. Dove, Polym. Chem., 2015, 6, 3185–3200 RSC.
- O. Coutelier, M. El Ezzi, M. Destarac, F. Bonnette, T. Kato, A. Baceiredo, G. Sivasankarapillai, Y. Gnanou and D. Taton, Polym. Chem., 2012, 3, 605–608 RSC.
- D. Bezier, J. B. Sortais and C. Darcel, Adv. Synth. Catal., 2013, 355, 19–33 CrossRef CAS.
- R. Zhong, A. C. Lindhorst, F. J. Groche and F. E. Kühn, Chem. Rev., 2017, 117, 1970–2058 CrossRef CAS PubMed.
- S. Naumann, M. Speiser, R. Schowner, E. Giebel and M. R. Buchmeiser, Macromolecules, 2014, 14, 4548–4556 CrossRef.
- H. Kalita, Shape Memory Polymers: Theory and Application, Walter de Gruyter GmbH & Co KG, 2018 Search PubMed.
- S. Naumann and M. R. Buchmeiser, Catal. Sci. Technol., 2014, 4, 2466–2479 RSC.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed.
- K. Hara, K. Iwahashi, S. Takakusagi, K. Uosaki and M. Sawamura, Surf. Sci., 2007, 601, 5127–5132 CrossRef CAS.
- Y. H. Kim, S. Shin, H. J. Yoon, J. W. Kim, J. K. Cho and Y.-S. Lee, Catal. Commun., 2013, 40, 18–22 CrossRef CAS.
- H. E. Kissinger, Anal. Chem., 1957, 29, 1702–1706 CrossRef CAS.
- S. Lee, J. H. Choi, I.-K. Hong and J. W. Lee, J. Ind. Eng. Chem., 2015, 21, 980–985 CrossRef CAS.
- M. H. Omidi, M. Alibeygi, F. Piri and M. Masoudifarid, Mater. Sci.-Pol., 2017, 35, 105–110 CAS.
- K. D. Safa, H. A. Eram and M. H. Nasirtabrizi, Iran. Polym. J., 2006, 15, 249–257 CAS.
- P. H. Wang, T. L. Wang, W. C. Lin, H. Y. Lin, M. H Lee and C. H. Yang, Nanomaterials, 2018, 4, 225 CrossRef PubMed.
- J. Zhang, Y. Chen, W. Zhao and Y. Li, R. Soc. Open Sci., 2018, 9, 181013 CrossRef.
- S. Eyley and W. Thielemans, Chem. Commun., 2011, 14, 4177–4179 RSC.
- B. Wang, M. Anpo, J. Lin, C. Yang, Y. Zhang and X. Wang, Catal. Today, 2018 DOI:10.1016/j.cattod.2018.07.001.
- T. Chai, Y. C. Liu, H. Ma, Y. Yu, J. M. Yuan, J. H. Wang and J. H. Guo, in IOP Conference Series: Materials Science and Engineering, IOP Publishing, 2016, vol. 137, p. 012069 Search PubMed.
- R. Vesna and M. Petric, Sci.-Tech. Rev., 2005, 1, 46–49 Search PubMed.
- Y. Ou, Q. Jiao, S. Yan and Y. Zhu, Cent. Eur. J. Energ. Mater., 2018, 1, 131–149 CrossRef.
- K. Catherine, K. Krishnan and K. Ninan, J. Therm. Anal. Calorim., 2000, 59, 93–100 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07677d |
| ‡ Authors Hyeon-Jun Noh and T. Sadhasivam contributed equally to this work and should be considered as co-first authors. |
| This journal is © The Royal Society of Chemistry 2018 |