
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA promising visible light-driven photocatalytic activity of conjugated polymer nanocrystals†
Chanon Pornrungroj‡
 ,
Mamiko Ozawa,
Tsunenobu Onodera and
Hidetoshi Oikawa*
,
Mamiko Ozawa,
Tsunenobu Onodera and
Hidetoshi Oikawa*
Institute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai 980-8577, Japan. E-mail: hidetoshi.oikawa.e8@tohoku.ac.jp
First published on 19th November 2018
Abstract
Conjugated polymers have recently emerged as a new class of photocatalysts. However, many conjugated polymer photocatalysts are not as effective as inorganic materials due to the limited electronic properties of their LUMO and HOMO and low long-term stability caused by the degradation of the conjugated backbone. In the present study, we have demonstrated, for the first time, the superior Visible (Vis) light-driven photocatalytic activity of conjugated polymer nanocrystals (NCs) of polydiacetylene (PDA) derivatives, namely, p-DCHD NCs for the photodegradation of Rhodamine B (RhB) compared with that of the state-of-the-art P25 TiO2 nanoparticles (NPs). In addition, we have revealed the morphological effects of p-DCHD NCs, long-term stability, and the photocatalytic degradation mechanisms. These results will open a new pathway for the further development and understanding of conjugated polymer photocatalysts toward various potential applications.
1. Introduction
Photocatalysts are important materials for various industries, including water treatment and hydrogen production.1,2 Currently, most photocatalysts are oxide-based inorganic materials, among which titanium oxide (TiO2) is the most superior due to its low-cost, high efficiency, and good long-term stability.3 However, the main drawback of TiO2 is the inability to utilize Visible (Vis) light, which accounts for 43% of the solar energy available on the earth surface,4,5 due to its wide bandgap.6 For over three decades, much efforts have been made toward extending the photocatalytic activity of these inorganic materials from the ultraviolet (UV) region toward the Vis region through various techniques, including energy band modulation by elemental doping (for example, N, C, and S)7,8 and dye sensitization as well as density functional theory (DFT) computer simulation to design new semiconductor materials with suitable electronic band structure.9–11 Moreover, conjugated polymers have recently emerged as a promising class of photocatalysts because of their narrow band gap, low-cost synthesis, and flexible mechanical properties.12,13 Despite great efforts toward the potential use of conjugated polymers as photocatalysts, many early reports indicated poor electronic properties and high rates of electron and hole (e–h) recombination, which provided significant challenges for the development of organic photocatalysts.12,14,15 Although the photocatalytic activity of non-crystalline organic nanostructures including amorphous or 2D and 3D porous network structures have been reported,16–20 to the best of our knowledge, the photocatalytic activity of “conjugated polymer crystal” nanostructures has not been explored so far.In the present study, we have presented a conjugated polymer crystal nanostructure, termed as “conjugated polymer nanocrystals (NCs)”, as a novel design approach toward the realization of high-performance conjugated polymer-based photocatalysts. More specifically, we have provided the first experimental evidence of poly[1,6-di(N-carbazolyl)-2,4-hexadiyne] (p-DCHD) NCs, which are typical one-dimensional conjugated polymer NCs of polydiacetylene (PDA) derivatives,21 as highly effective photocatalysts even when compared with the state-of-the-art P25 TiO2. PDA derivatives synthesized by solid-state polymerization in a crystal state are basically single crystals22 and are well known as one of the most promising nonlinear optical materials.23 We will discuss Vis-light-driven photocatalytic activity and the long-term stability for p-DCHD NCs as highly potential photocatalysts as well as their photodegradation mechanism compared with that of P25 TiO2 nanoparticles (NPs). This demonstration of a conjugated polymer NCs system is an essential first step toward the development of high-performance conjugated polymer-based photocatalysts.
2. Experimental section
2.1 Materials
1,6-Di(N-carbazolyl)-2,4-hexadiyne (DCHD), dioctyl sulfosuccinate sodium (AOT), and Rhodamine B (RhB) were purchased from Wako Pure Chemical Industries, Ltd. TiO2 NPs (P25 TiO2: avg. diameter = 30 nm, specific surface area = 50 m2 g−1) was supplied by Degussa Co. All the chemicals were of analytical reagent grade and used without further purification, except for DCHD, which was recrystallized for purification. Deionized and distilled water used in this study were further purified up to 18.2 MΩ cm using a water purification system (Sartorius Atrium 611 UV).2.2 Preparation of p-DCHD NCs
p-DCHD NCs were prepared by the conventional reprecipitation method and solid-state polymerization with minor modified conditions, as illustrated in Fig. S1:†24 4 mL of DCHD (5.0 mM, 24.5 mM)-THF solution containing 7.5 mM of AOT was injected into vigorously stirred water (200 mL) as a poor solvent at a given temperature using a pulseless pump (ISCO, 260D Syringe Pump). After 30 min of retention time, DCHD NCs were solid-state polymerized by UV irradiation (λ = 254 nm) for ca. 90 min. The resulting aqueous dispersion liquid of p-DCHD NCs was then filtered through a hydrophilic poly(tetrafluoroethylene) membrane (pore size: 0.05 μm) and washed several times with distilled water to ensure the removal of added surfactants. Finally, p-DCHD NCs were stably re-dispersed in an aqueous medium. The resulting samples were assigned as samples 1–6 (Table S1†).2.3 Characterizations
The size and morphology of the p-DCHD NCs were evaluated via scanning electron microscopy (SEM) using JEOL JSM-6700F. The samples were sputtered with platinum by JEOL JFC-1200 prior to SEM observation. Vis absorption spectrum and IR spectrum were measured using a UV-Vis spectrometer (JASCO, V-550) and an ATR-FT-IR spectrometer (AVATAR, 360 FT-IR), respectively. Crystal structure of the p-DCHD NCs was evaluated using a powder X-ray diffractometer (XRD) (Bruker, D8-advance X-ray diffractometer).2.4 Photocatalytic evaluation
The initial dispersion concentration of the p-DCHD NCs was fixed at 0.04 mg mL−1 (0.1 mM). 3.0 mL of p-DCHD NCs dispersion was mixed with 12 μL of RhB aqueous solution (5.12 mM) in a quartz cell with an optical path length of 10 mm, resulting in 0.010 mg mL−1 (2.1 × 10−5 M) of the initial concentration of RhB. The mixed sample was stirred for 30 min under dark condition to ensure an equilibrium state, and then irradiated for a given time at 25 °C by UV, Vis, and UV-Vis light using a Xenon lamp (USHIO SX-UI 501XQ, 500 W) equipped with the following suitable optical filters: UV: λ = 300–400 nm (filters: HA50 + U330); Vis: λ = 420–700 nm (filters: HA50 + L42); UV-Vis: λ = 300–700 nm (filters: HA50). The photocatalytic degradation of RhB was monitored with the changes in absorbance at a fixed wavelength λ = 554 nm in the Vis absorption spectra. Similarly, photocatalytic degradation was performed using TiO2 NPs. When an inert and non-oxidizing condition was required, N2 gas (>99.99%) was purged through the mixed dispersion liquid for 2 h prior to the light irradiation. The following scavengers were utilized to investigate the photocatalytic degradation mechanism of RhB by p-DCHD NCs: Cu2+ (CuSO4·5H2O) as the electron scavenger,25 MeOH as the hydroxyl radical scavenger,26 and benzoquinone as the superoxide radical scavenger.27 In addition, the dependence of the photocatalytic activity for the p-DCHD NCs on wavelength in the Vis region was investigated using an ASAHI SPECTRA MAW-302 equipped with a suitable band-pass filter (450 nm, 500 nm, 550 nm, 600 nm, 650 nm, and 700 nm). The intensity of incident light was almost the same (37.4 mW cm−2) at any wavelength in the Vis region.3. Results and discussion
3.1 Characterization of p-DCHD NCs
Fig. 1 shows the SEM images of the as-prepared p-DCHD NCs, corresponding to samples 1–6 with various morphologies having different diameter (d) and length (L) by adding surfactant AOT and/or by changing the temperature of the poor (water) medium.28 As summarized in Fig. S2,† the value of d was in the range of ca. 40–60 nm, while L changed from 70 nm to several μm for samples 1–6. In general, PDA can be formed through a solid-state polymerization process in both nano and bulk crystal states. p-DCHD is a typical PDA derivative produced by a peculiar monomer single crystal-to-polymer single crystal phase transition.22,29 All of the solid-state polymerized p-DCHD NCs samples 1–6 were not amorphous but crystalline with the same crystal structure as the bulk crystal, as confirmed by powder XRD measurements (Fig. S3†). | ||
| Fig. 1 SEM images of p-DCHD NCs (samples 1–6) with different length (L) and diameter (d). | ||
The color of dispersions of all the samples 1–6 is deep blue, which originated from the excitonic absorption peak at around λ = 650 nm and the phonon sideband at around λ = 600 nm in the extended π-conjugated backbone of PDA, as shown in Fig. 2.30,31 The excitonic absorption peak intensity clearly increased relative to the phonon sideband from sample 1 to sample 6.28 The red-shift of the excitonic absorption peak was also observed as the value of L became larger, as listed in the second column of Table S2.†32 These phenomena are mainly due to the extension of the effective conjugated length of PDA, which is closely related to the morphology (size and shape) of p-DCHD NCs, resulting in the delocalization of the excitons along the conjugated backbone, as discussed in detail elsewhere.32,33
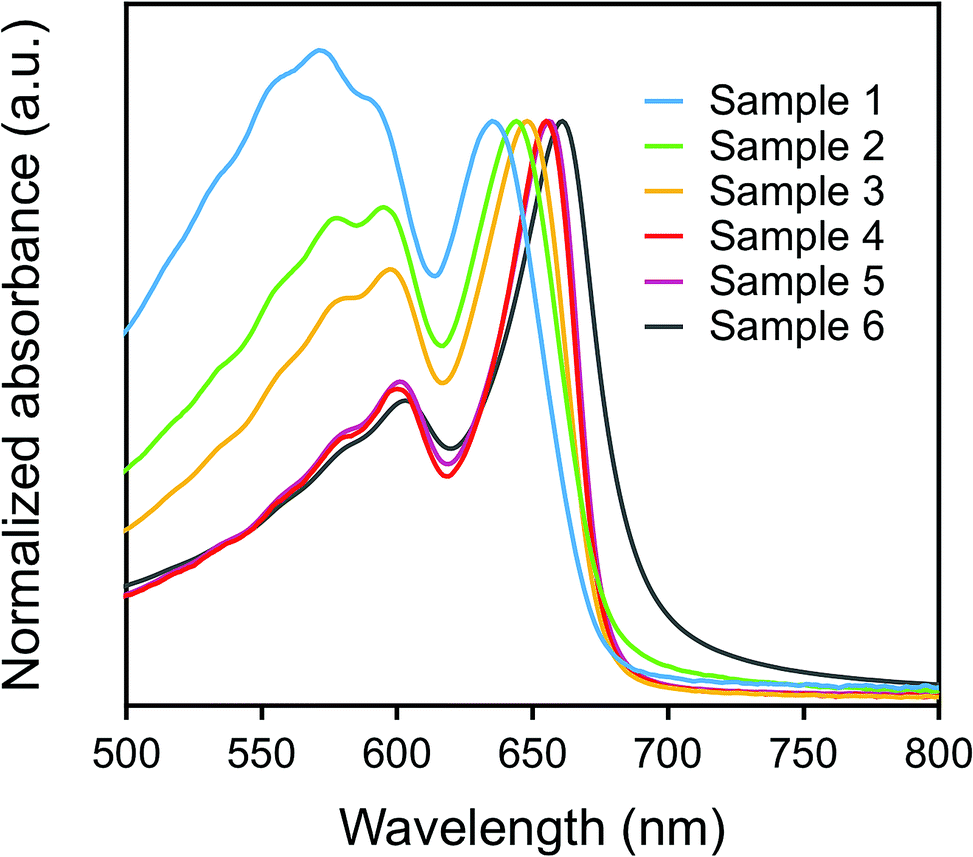 | ||
| Fig. 2 Vis absorption spectra of p-DCHD NCs (samples 1–6). | ||
3.2 Photocatalytic activity of p-DCHD NCs
Fig. 3(a) shows the typical Vis absorption spectral changes of RhB during the photodegradation reactions in presence of p-DCHD NCs (sample 3) under UV-Vis irradiation condition. Throughout the photocatalytic degradation process, the absorption peak positions were blue-shifted, and the absorbance was reduced gradually. As described in the literature,34–36 there are two competitive processes for RhB photodegradation: one is the stepwise de-ethylation of the RhB molecules, and the other is the direct degradation of the fused aromatic ring structure. These two reaction processes are considered to be the main routes for the photocatalytic degradation of RhB even in the presence of p-DCHD NCs. Herein, the photodegradation of RhB was monitored by measuring the absorbance at λ = 554 nm as a function of irradiation time and then, the apparent rate constant (k) of photocatalytic degradation for RhB was experimentally determined by assuming a pseudo-first order reaction model.31 Thus, the value of k was evaluated from the slope of the plot of ln(C/C0) as a function of irradiation time, where C was the molar concentration of RhB at a given time and C0 was the initial concentration. The calculated values of k are listed in the last column of Table S2.†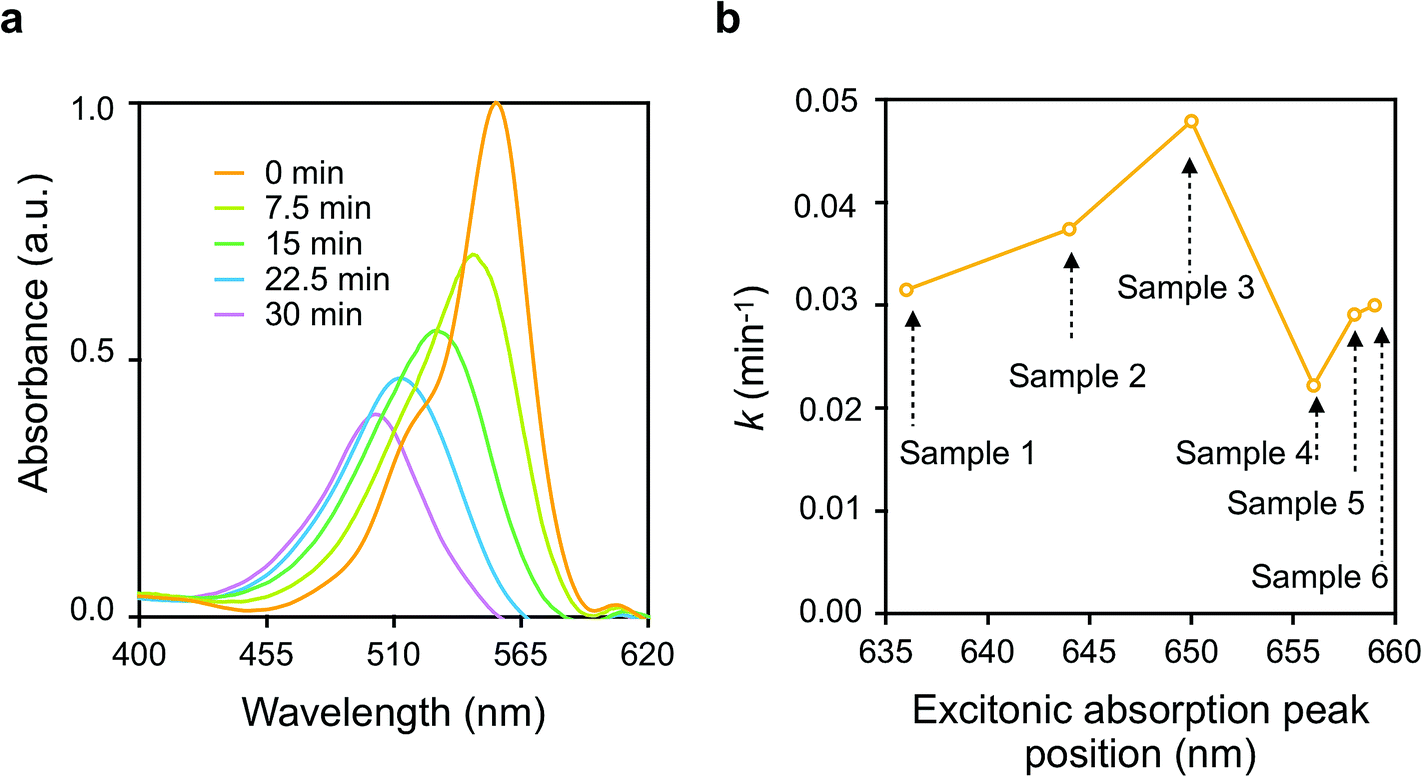 | ||
| Fig. 3 (a) Vis absorption spectral changes during the photocatalytic degradation of RhB (2.1 × 10−5 M) dispersed in aqueous p-DCHD NCs (sample 3: 0.04 mg mL−1) dispersion liquids as a function of UV-Vis irradiation time, and (b) the plots of apparent rate constants of the photocatalytic degradation (k) for RhB vs. excitonic absorption peak positions of p-DCHD NCs (samples 1–6). | ||
Fig. 3(b) displays the correlation of k values and the morphology of the as-prepared p-DCHD NCs (samples 1–6) in the case of UV-Vis irradiation. The horizontal axis in Fig. 3(b) indicates the excitonic absorption peak positions of p-DCHD NCs (samples 1–6) (Table S2†). Interestingly, the value of k is dependent not simply on the excitonic absorption peak positions but on the morphology of p-DCHD NCs. As a result, sample 3 provided the highest and maximum k value. In fact, the value of k is an apparent parameter and affected by many factors such as crystallinity, specific surface area, crystal defects, delocalization of the exciton, and charge-separated electron (e)–hole (h) pairs from the excitons.37–39
The increase in k values for samples 1–3 is due to the improved efficient absorption of the extended conjugated backbone. However, we can see a significant decrease in the k value for samples 4–6. This reduction in photocatalytic activity, despite the stronger Vis absorption, is speculated to be due to the following two reasons: one is the decrease in specific surface area and reaction site,40 and the other is the higher recombination probability of charge-separated e–h pairs in the photo-excited state.41 As discussed in detail later, the charge separation process from the photoexcited e–h pairs (the exciton) is a key step in the photocatalytic degradation activity of p-DCHD NCs.36 In any case, the most important point is to suitably control the morphology, that is, to optimize the conjugation length for p-DCHD NCs in the reprecipitation method so as to realize the highly effective photocatalytic activity. Hereafter, we will only focus on “sample 3” of the p-DCHD NCs photocatalysts.
Fig. 4 exhibits the photocatalytic activity of the p-DCHD NCs (sample 3) under (a) Vis, (b) UV, and (c) UV-Vis light irradiation conditions compared with that of the commercially available P25 TiO2 NPs. First, we can see that RhB did not photodegrade under any light irradiation conditions in the absence of photocatalysts.
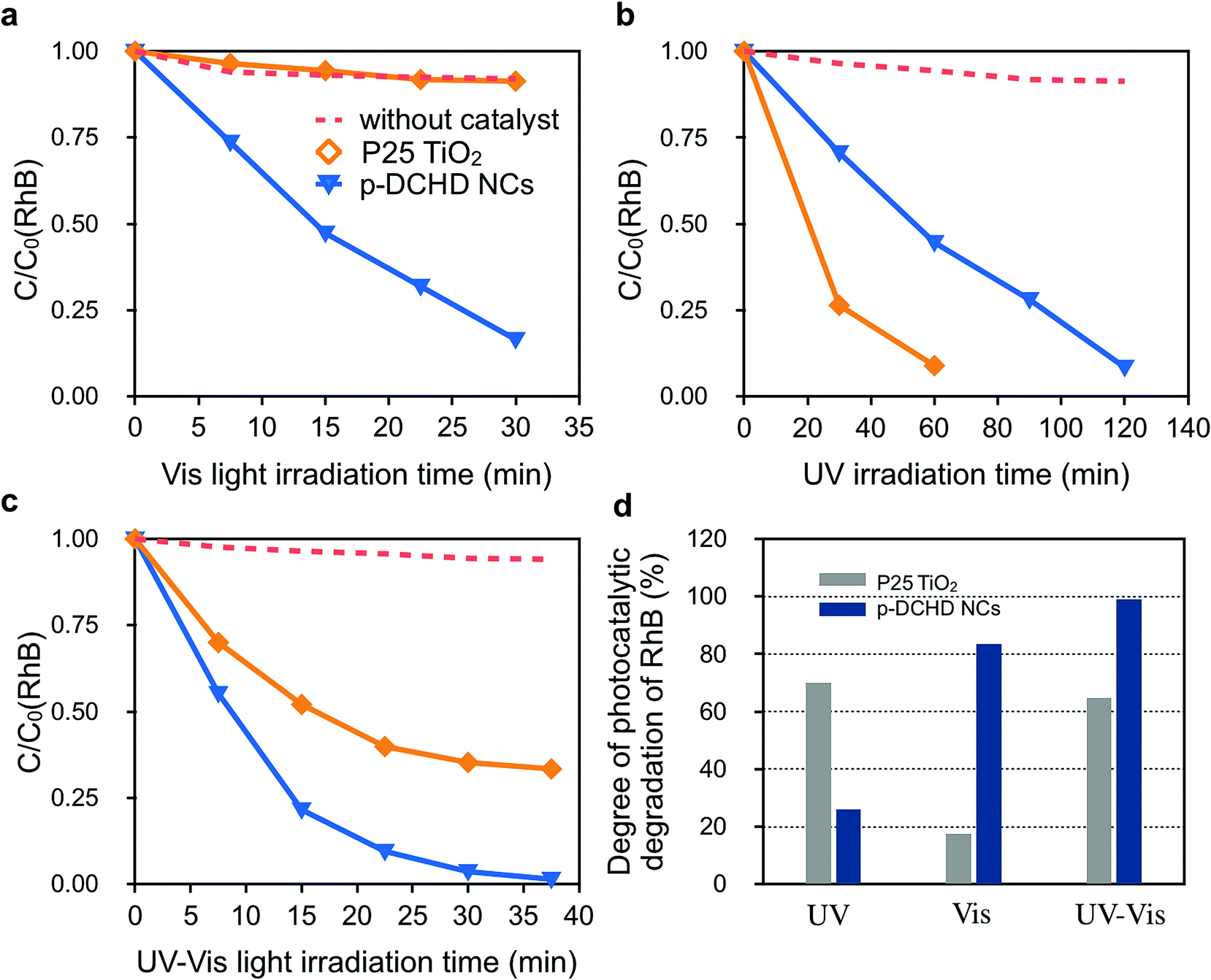 | ||
| Fig. 4 Relative concentration changes (C/C0) of residual RhB photodegraded by p-DCHD NCs and/or P25 TiO2 NPs as photocatalyst under (a) Vis light (420–700 nm), (b) UV light (300–400 nm), and (c) UV-Vis (300–700 nm) irradiation conditions. (d) Degree of photocatalytic degradation of RhB after 30 min of irradiation by UV, Vis, and UV-Vis light sources. The concentrations of p-DCHD NCs and P25 TiO2 NPs dispersed in an aqueous medium were 0.04 mg mL−1. The initial concentrations of RhB (C0) were 2.1 × 10−5 M. The legend in (a) are the same as symbols in both (b) and (c). | ||
Under Vis light irradiation conditions (Fig. 4(a)), RhB was not photodegraded by P25 TiO2 NPs due to the wide bandgap energy of TiO2 (3.2 eV).42 Interestingly, the apparent rate constant of the photocatalytic degradation for RhB by p-DCHD NCs (kp-DCHD = 2.57 × 10−2 min−1) was significantly higher than that by P25 TiO2 NPs (kP25 = 0.13 × 10−2 min−1) and about 89% of RhB was photodegraded after 30 min of Vis light irradiation. In contrast, as expected, the photocatalytic degradation activity of P25 TiO2 NPs (kP25 = 1.53 × 10−2 min−1) was superior under UV light irradiation condition (Fig. 4(b)), and p-DCHD NCs (kp-DCHD = 0.85 × 10−2 min−1) also demonstrated photocatalytic activity to some extent under UV light irradiation condition. Consequently, about 91% of RhB was photodegraded by P25 TiO2 NPs after 60 min, while about 51% of RhB was photodegraded by p-DCHD NCs after 120 min. In addition, the p-DCHD NPs (kp-DCHD = 4.79 × 10−2 min−1) showed photocatalytic activity of about three times higher than that of P25 TiO2 NPs (kTiO2 = 1.75 × 10−2 min−1) under UV-Vis light irradiation condition (Fig. 4(c)).
Fig. 4(d) displays the degree of photocatalytic degradation of RhB under UV, Vis, and UV-Vis light irradiation conditions for 30 min. These facts clearly demonstrated the superior performance of p-DCHD NCs under both UV-Vis and Vis light irradiation compared with that of P25 TiO2 NPs. This suggests that p-DCHD NCs could be regarded as a new class of Vis-light-driven photocatalysts.
The overall superior photocatalytic activity of p-DCHD NCs is likely due to two main factors: the intrinsic electronic property of the conjugated p-DCHD backbone and the high degree of crystallinity. Previous studies on conjugated polymer photocatalysts have elucidated that a more linear and longer conjugated chain would result in the enhancement of photocatalytic activity of such polymer systems by reducing charge-trap sites and increasing excitonic absorption.43,44 On the contrary, p-DCHD NCs possess unique physicochemical properties, namely, a narrow bandgap (2.33 eV),45 high excitonic absorbance, good charge mobility along the conjugated backbone (6 × 103 cm2 V−1 s−1),45 and a high degree of crystallinity that emerges from single crystal-to-polymer single crystal phase transition, as described in Fig. 1.22,29 These excellent properties could be responsible for the high photocatalytic activity of p-DCHD NCs.
In other words, the crystal lattice of p-DCHD NCs is thermally softened and relatively strain-free during solid-state polymerization,28,46 and the conversion usually reaches ca. 100%.22,29 p-DCHD NCs with a high degree crystallinity are almost defect-free. In fact, p-DCHD bulk crystals provided much low photocatalytic activity due to their low specific surface area, crystal lattice defects, and polycrystalline domains47 (Fig. S4†).
3.3 Long-term stability and post-catalysis characterization
The photocatalytic activity of p-DCHD NCs could be successfully maintained over 10 successive recycling experiments (Fig. S5†). There were no changes in the Vis absorption spectra, SEM images, FT-IR spectra, and powder XRD patterns within experimental errors before and after the recycling experiments (Fig. S6†), in contrast to the previously reported low long-term recovery of PPP14 due to the degradation of the conjugated backbone by reactive oxygen species (ROS). Such long-term stability and good physicochemical resistance of the p-DCHD NCs are mainly due to the high degree of crystallinity and close-packed (or defect-free) rigid structure in the crystal lattices of the p-DCHD NCs, which would protect and prevent the reaction of ROS with the polymer backbone.48 Moreover, such a polymer structure also contributes to the high thermal stability of p-DCHD NCs even above 250 °C (Fig. S7†).493.4 Proposed photocatalytic degradation mechanism of Rhodamine B by p-DCHD NCs
In general, the photocatalytic degradation of RhB involves the charge separation of e–h pairs and the subsequent formation of ROS such as hydroxyl radicals (OH˙) and superoxide anion radicals (O2˙−),26 which are formed by the reaction of O2 with the photoexcited electrons at the conduction band (CB). Finally, organic dyes such as RhB are decomposed and degraded by these ROS.34,50 Selected scavengers were employed to qualitatively discuss the degradation mechanism of RhB under the presence of p-DCHD NCs, although electron spin resonance (ESR) measurements could be more useful in directly detecting ROS.51Fig. 5(a) shows a significant decrease in photocatalytic activity of p-DCHD NCs under the co-existence of Cu2+. After 30 min of UV-Vis light irradiation, the degree of photocatalytic degradation of RhB remained ca. 55% with the addition of Cu2+, a typical electron scavenger.16 Probably, the electron charge separated from the exciton was quenched with Cu2+ to yield Cu+. In addition, Fig. 5(b) indicates the photocatalytic activity for the aqueous dispersion sample containing p-DCHD NCs and RhB, purged by nitrogen gas before UV-Vis light irradiation. The degree of photocatalytic degradation of RhB became distinctly lower, which suggested that the reduction in the ROS concentration was due to the removal of oxygen molecules dissolved in an aqueous medium.
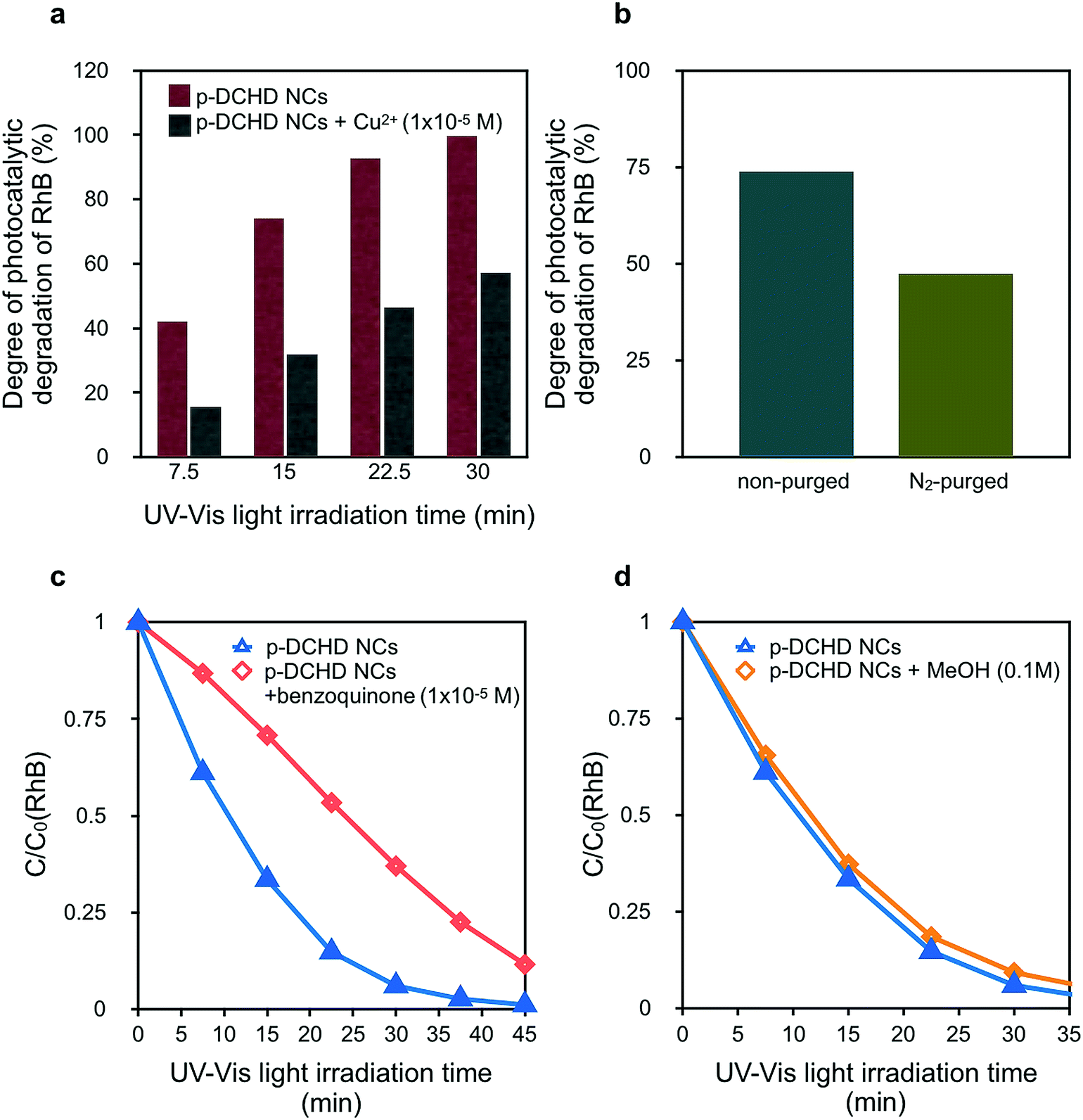 | ||
| Fig. 5 (a) Degree of the photocatalytic degradation of RhB in the presence of p-DCHD NCs with and without Cu2+ (1 × 10−5 M) under UV-Vis light irradiation, (b) comparison of the degree of photocatalytic degradation of the RhB with and without N2 purging treatment before UV-Vis light irradiation for 15 min, (c) and (d) the C/C0 (RhB) decay curves under UV-Vis light irradiation by adding benzoquinone (1 × 10−5 M) and methanol (0.1 M), respectively. | ||
Contrary to the case of TiO2, OH˙ could not be generated directly from the hole reacting with H2O45 in the case of the p-DCHD NCs system. Since the redox potential of p-DCHD (VB: +1.27 eV vs. SHE)45 is more negative than that of OH˙/OH− (+1.99 eV),30 OH˙ cannot be formed via p-DCHD NCs in the same manner as generated via TiO2 NPs. Thus, OH˙ might not be the main ROS for the photocatalytic degradation of RhB. Hence, the photocatalytic activity was evaluated to reveal the abovementioned hypothesis using benzoquinone as a known O2˙− scavenger (Fig. 5(c))26 and MeOH as a known OH˙ scavenger (Fig. 5(d)).27 It became apparent that benzoquinone considerably affected the decay curve of C/C0 (RhB), but MeOH scarcely had an influence. These results suggest that the O2˙− species was mainly responsible for the photodegradation of RhB in the present case rather than OH˙ as in the case of the TiO2 catalyst.52
We further considered the dependence of photocatalytic activity on wavelength in the Vis region. Fig. 6 displays the plots of k values for RhB degradation vs. wavelength. Sample 3 was the most efficient among other samples, as already discussed in Fig. 3(b), but all the samples did exhibit photocatalytic activity at a maximum wavelength of around 450 nm, a region which almost corresponded to the increase in photocurrent in the action spectrum of p-DCHD.48 Evidently, it was found that the charge separation of e–h pairs generated in the photoexcited p-DCHD NCs was an initial process.
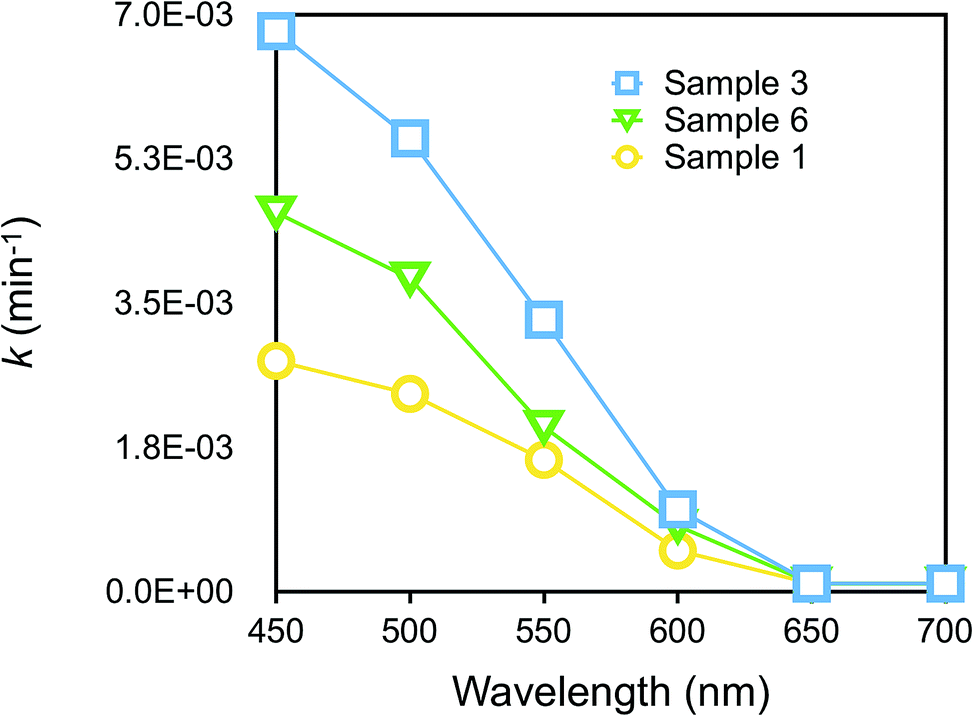 | ||
| Fig. 6 Plots of k values for RhB degradation vs. wavelength in the Vis region. | ||
4. Conclusions
We have successfully demonstrated promising photocatalytic activity of conjugated polymer nanocrystals, namely, p-DCHD NCs. In particular, the p-DCHD NCs have provided superior Vis-light-driven photocatalytic activity when compared with that of P25 TiO2 NPs. Interestingly, the photocatalytic activity was strongly affected by the morphology (size and shape) of p-DCHD NCs due to high crystallinity and the corresponding electron state, for example, the charge separation of e–h pairs (exciton) generated in photo-excited p-DCHD NCs. The excellent photocatalytic activity and the good long-term stability of p-DCHD NCs could lead to new advances in conjugated polymer-based photocatalysts for various applications, including water purification, CO2 reduction, and water-splitting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by JSPS KAKENHI Grant Number JS15K05618 and JS18K05227, and the Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT).Note and references
- M. N. Chong, B. Jin, C. W. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- K. Ujiiye-Ishii, E. Kwon, H. Kasai, H. Nakanishi and H. Oikawa, Cryst. Growth Des., 2008, 8, 369–371 CrossRef CAS.
- M. Ni, M. K. Leung, D. Y. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- S. Maity, M. Zhu, R. S. Shinabery and N. Zheng, Angew. Chem., Int. Ed., 2012, 51, 222–226 CrossRef CAS PubMed.
- T. Ohno, T. Mitsui and M. Matsumura, Chem. Lett., 2003, 32, 364–365 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- S. In, A. Orlov, R. Berg, F. García, S. Pedrosa-Jimenez, M. S. Tikhov, D. S. Wright and R. M. Lambert, J. Am. Chem. Soc., 2007, 129, 13790–13791 CrossRef CAS PubMed.
- N.-G. Park, J. Van de Lagemaat and A. Frank, J. Phys. Chem. B, 2000, 104, 8989–8994 CrossRef CAS.
- I. E. Castelli, T. Olsen, S. Datta, D. D. Landis, S. Dahl, K. S. Thygesen and K. W. Jacobsen, Energy Environ. Sci., 2012, 5, 5814–5819 RSC.
- Y. Wu, P. Lazic, G. Hautier, K. Persson and G. Ceder, Energy Environ. Sci., 2013, 6, 157–168 RSC.
- T. Shibata, A. Kabumoto, T. Shiragami, O. Ishitani, C. Pac and S. Yanagida, J. Phys. Chem., 1990, 94, 2068–2076 CrossRef CAS.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS PubMed.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 474–475 RSC.
- J. Park, J. Ind. Eng. Chem., 2017, 51, 27–43 CrossRef CAS.
- S. Ghosh, N. A. Kouamé, L. Ramos, S. Remita, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P.-H. Aubert and H. Remita, Nat. Mater., 2015, 14, 505–511 CrossRef CAS PubMed.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed.
- L. Li, Z. Cai, Q. Wu, W.-Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef CAS PubMed.
- B. C. Ma, S. Ghasimi, K. Landfester, F. Vilela and K. A. Zhang, J. Mater. Chem. A, 2015, 3, 16064–16071 RSC.
- Y. Li, Q. Duan, H. Wang, B. Gao, N. Qiu and Y. Li, J. Photochem. Photobiol., A, 2018, 356, 370–378 CrossRef CAS.
- H. Katagi, H. Kasai, S. Okada, H. Oikawa, H. Matsuda and H. Nakanishi, J. Macromol. Sci., Part A: Pure Appl. Chem., 1997, 34, 2013–2024 CrossRef.
- K. Baba, H. Kasai, S. Okada, H. Nakanishi and H. Oikawa, Mol. Cryst. Liq. Cryst., 2006, 445, 161/[451]–166/[456] Search PubMed.
- H. Matsuda, S. Yamada, E. R. Van Keuren, H. Katagi, H. Kasai, S. Okada, H. Oikawa, H. Nakanishi, E. C. Smith, A. K. Kar and B. S. Kar, Proc. SPIE, 1997, 2998, 241–248 CrossRef CAS.
- H. Kasai, H. S. Nalwa, H. Oikawa, S. Okada, H. Matsuda, N. Minami, A. Kakuta, K. Ono, A. Mukoh and H. Nakanishi, Jpn. J. Appl. Phys., 1992, 31, L1132 CrossRef CAS.
- S. Ghosh, N. A. Kouame, S. Remita, L. Ramos, F. Goubard, P.-H. Aubert, A. Dazzi, A. Deniset-Besseau and H. Remita, Sci. Rep., 2015, 5, srep18002 CrossRef PubMed.
- Y. Chen, S. Yang, K. Wang and L. Lou, J. Photochem. Photobiol., A, 2005, 172, 47–54 CrossRef CAS.
- Y. Zhang, N. Zhang, Z.-R. Tang and Y.-J. Xu, Chem. Sci., 2013, 4, 1820–1824 RSC.
- T. Onodera, T. Oshikiri, H. Katagi, H. Kasai, S. Okada, H. Oikawa, M. Terauchi, M. Tanaka and H. Nakanishi, J. Cryst. Growth, 2001, 229, 586–590 CrossRef CAS.
- V. Enkelmann, R. Leyrer, G. Schleier and G. Wegner, J. Mater. Sci., 1980, 15, 168–176 CrossRef CAS.
- D. Batchelder and D. Bloor, J. Phys. C: Solid State Phys., 1982, 15, 3005 CrossRef CAS.
- G. Weiser, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 14076 CrossRef CAS.
- H. Masuhara, H. Nakanishi and K. Sasaki, Single Organic Nanoparticles, Springer Science & Business Media, 2003 Search PubMed.
- V. V. Volkov, T. Asahi, H. Masuhara, A. Masuhara, H. Kasai, H. Oikawa and H. Nakanishi, J. Phys. Chem. B, 2004, 108, 7674–7680 CrossRef CAS.
- T. Wu, G. Liu, J. Zhao, H. Hidaka and N. Serpone, J. Phys. Chem. B, 1998, 102, 5845–5851 CrossRef CAS.
- H. Fu, C. Pan, W. Yao and Y. Zhu, J. Phys. Chem. B, 2005, 109, 22432–22439 CrossRef CAS PubMed.
- Y. Li, S. Sun, M. Ma, Y. Ouyang and W. Yan, Chem. Eng. J., 2008, 142, 147–155 CrossRef CAS.
- A. Naldoni, M. Allieta, S. Santangelo, M. Marelli, F. Fabbri, S. Cappelli, C. L. Bianchi, R. Psaro and V. Dal Santo, J. Am. Chem. Soc., 2012, 134, 7600–7603 CrossRef CAS PubMed.
- T. Hirakawa and P. V. Kamat, J. Am. Chem. Soc., 2005, 127, 3928–3934 CrossRef CAS PubMed.
- K. M. Lee, C. W. Lai, K. S. Ngai and J. C. Juan, Water Res., 2016, 88, 428–448 CrossRef CAS PubMed.
- H. Lin, C. Huang, W. Li, C. Ni, S. I. Shah and Y.-H. Tseng, Appl. Catal., B, 2006, 68, 1–11 CrossRef CAS.
- H. Harada and T. Ueda, Chem. Phys. Lett., 1984, 106, 229–231 CrossRef CAS.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. Dunlop, J. W. Hamilton, J. A. Byrne and K. O'shea, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS.
- S. Matsuoka, T. Kohzuki, Y. Kuwana, A. Nakamura and S. Yanagida, J. Chem. Soc., Perkin Trans. 2, 1992, 679–685 RSC.
- M. S. Vezie, S. Few, I. Meager, G. Pieridou, B. Dörling, R. S. Ashraf, A. R. Goñi, H. Bronstein, I. McCulloch and S. C. Hayes, Nat. Mater., 2016, 15, 746 CrossRef CAS PubMed.
- W. Spannring and H. Bässler, Chem. Phys. Lett., 1981, 84, 54–58 CrossRef CAS.
- S. Takahashi, H. Miura, H. Kasai, S. Okada, H. Oikawa and H. Nakanishi, J. Am. Chem. Soc., 2002, 124, 10944–10945 CrossRef CAS PubMed.
- P. M. Wilson and D. C. Martin, J. Mater. Res., 1992, 7, 3150–3158 CrossRef CAS.
- D. Batchelder, N. Poole and D. Bloor, Chem. Phys. Lett., 1981, 81, 560–564 CrossRef CAS.
- F. Bradner and J. Shapiro, Polym. Int., 1991, 26, 195–202 CrossRef CAS.
- I. K. Konstantinou and T. A. Albanis, Appl. Catal., B, 2004, 49, 1–14 CrossRef CAS.
- J. Zhao, C. Chen and W. Ma, Top. Catal., 2005, 35, 269–278 CrossRef CAS.
- C. Chen, W. Ma and J. Zhao, Chem. Soc. Rev., 2010, 39, 4206–4219 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07837h |
| ‡ Current address: Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK, E-mail: E-mail: cp651@cam.ac.uk |
| This journal is © The Royal Society of Chemistry 2018 |