
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvanced palladium free approach to the synthesis of substituted alkene oxindoles via aluminum-promoted Knoevenagel reaction†
Daria S. Novikova *a,
Tatyana A. Grigorevaa,
Andrey A. Zolotarevb,
Alexander V. Garabadzhiua and
Vyacheslav G. Tribulovicha
*a,
Tatyana A. Grigorevaa,
Andrey A. Zolotarevb,
Alexander V. Garabadzhiua and
Vyacheslav G. Tribulovicha
aLaboratory of Molecular Pharmacology, Saint Petersburg State Institute of Technology (Technical University), Moskovskii pr. 26, Saint Petersburg 190013, Russia. E-mail: dc.novikova@gmail.com
bDepartment of Crystallography, Saint Petersburg State University, per. Dekabristov 16, Saint Petersburg 199155, Russia
First published on 10th October 2018
Abstract
A synthetic route for the synthesis of C24, as well as for the design of focused libraries of direct AMPK activators was developed based on a convergent strategy. The proposed scheme corresponds to the current trends in C–H bond functionalization. The use of aluminum isopropoxide for the Knoevenagel condensation of oxindole with benzophenones is a noticeable point of this work.
AMP-activated protein kinase (AMPK) has been extensively studied over the last decades. A key role of AMPK in the regulation of energy balance suggests it to be a promising target for energy metabolism-related diseases.1 To date, AMPK is considered as a therapeutic target in type 2 diabetes, obesity, cardiovascular diseases, atherosclerosis, and cancer.2 A number of indirect AMPK activators are already used in clinical practice for their beneficial metabolic effects.3 However, there are no drugs that directly activate AMPK on the pharmaceutical market yet.
The demand for direct AMPK activators inspired pharmaceutical companies to start a campaign to design such molecules.4 To date, several classes of compounds that potently activate AMPK have been identified: AICAR and other AMP mimetics,5 thienopyridones,6 benzimidazoles,7 and alkene oxindole derivatives.8 The latter activate AMPK by the most interesting and least studied mechanism.9 These compounds show good target activity, and C24 (YLF-466D; (E)-3-((3-((4-chlorophenyl)(phenyl)methylene)-2-oxoindolin-1-yl)methyl) benzoic acid) is the most potent 2-oxindole derivative.8 The structure of these compounds provides ample opportunities for both the development of soluble forms and wide structural variations. Small molecules based on 2-oxindole scaffold have a broad spectrum of biological activity.10 The high druglikeness of these compounds is determined by the fact that 2-oxindole is a tryptophan metabolite and can mimic its affinity to various protein substrates. Antitumor activity, identified for 3-(benzylidene)indolin-2-ones,11 allows to design multitarget compounds that affect both the metabolic signaling cascades of a cancer cell through AMPK and the proliferative signaling pathway by reactivating p53,12 since high lipophilicity of the considered structures provides potent binding to the active cavity of the MDM2 protein.13
Despite all the advantages of 2-oxindole derivatives in studying the AMPK activation and the small molecule development, the synthetic routes for obtaining this series remain rather laborious. Here, we present a simple and effective route of C24 synthesis. This route takes into account both the possibility of synthesis of small molecule libraries as well as current trends in organic synthesis.
Linear synthesis and convergent synthesis are the main strategies in organic chemistry. The convergent strategy is much more suitable for providing the diversity of a library of complex biologically active compounds. The diversity is realized through divergent stages of the synthetic scheme. However, the divergence of the synthetic scheme at earlier stages leads to the individualization of the synthesis of each compound and, consequently, an increase in the efforts to synthesize a compound library.14
Following the linear scheme, we synthesized C24 from 3-phenylpropiolic acid by amidation with aniline, alkylation with 3-(bromomethyl)benzoic acid methyl ester, ring formation of the resulting intermediate, followed by removal of the ester group (Scheme 1).15 It is interesting that at the ring formation step we observed the formation of dehalogenated compound along with the target product. The resulting mixture of the products cannot be separated by crystallization. Single crystals of mixed composition are formed when crystallizing (Fig. 1). They can be separated only using reversed-phase silica gel, but extremely low solubility of the esters makes the purification process difficult. In addition to the above difficulties, the first stages of the synthesis are divergent, so to introduce a substituent into ring A it is necessary to obtain substituted phenylpropiolic acid, for example, by the Sonogashira coupling. It appears that the diversity is provided before the most difficult stage of building the carbon skeleton.
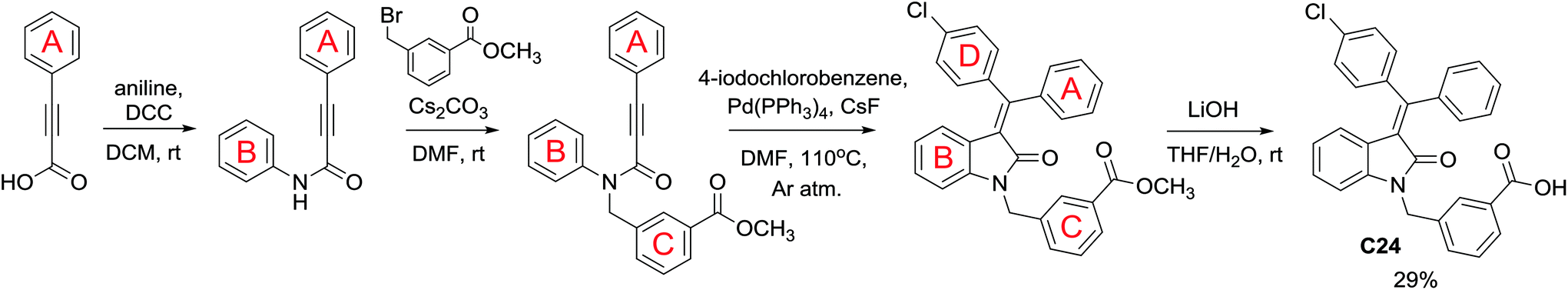 | ||
| Scheme 1 Linear scheme of C24 synthesis. | ||
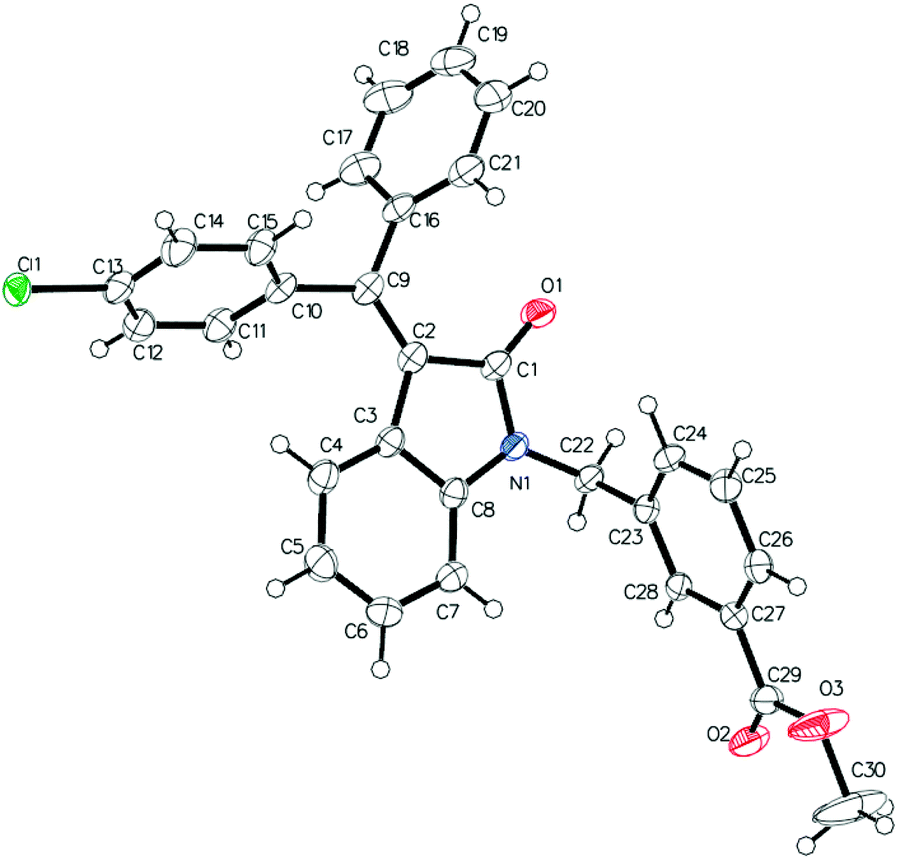 | ||
| Fig. 1 ORTEP representation of the crystal structure corresponding to C30H22.25Cl0.75NO3 crystals. | ||
Current synthetic schemes based on propiolic acid have various modifications, including one pot and domino multicomponent ones.16 However, the modern names do not relieve them of the shortcomings inherent to the linear scheme; moreover, the use of active catalysts can lead to undesirable rearrangements of the carbon skeleton of the target substrate.17
The convergent synthesis (Scheme 2) can be implemented based on the route obtained using obvious disconnections. The simplest way to build the carbon skeleton of substituted alkene oxindoles is to perform the Knoevenagel condensation of oxindole with benzophenones. The condensation of oxindole with aldehydes and alkylphenones proceeds practically with quantitative yields under common conditions: toluene, pyrrolidine, azeotropic distillation of water. Reduction of yields is observed only in the case of ketones and aldehydes substituted in position 2 of the aromatic ring. However, there is the difficulty to realize the condensation of oxindole with benzophenones,18 and under the same conditions the reaction product is formed only in trace amounts when benzophenone is taken as a carbonyl compound (Table 1).
 | ||
| Scheme 2 Convergent scheme of C24 synthesis. | ||
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Time, h | Yield (E/Z ratio) |
| a Reaction conditions: 2-oxindole (1 equiv.), corresponding ketone (1.2 equiv.), pyrrolidine (2 equiv.), toluene, reflux with the Dean–Stark trap.b E/Z ratio not determined. | ||||
| 1 | 4-Cl | H | 0.25 | 96 (63/36) |
| 2 | 2-Cl | H | 3 | 72 (98/2) |
| 3 | H | CH3 | 0.5 | 94 (96/4) |
| 4 | 4-Cl | CH3 | 1 | 91 (96/4) |
| 5 | 2-Cl | CH3 | 3 | 65 (99/1) |
| 6 | H | C2H5 | 2 | 85 (95/5) |
| 7 | H | n-C3H7 | 2 | 76 (95/5) |
| 8 | 3′,4′-Methylenedioxy | CH3 | 1 | 80 (98/2) |
| 9 | H | Ph | 24 | Traces |
| 10 | 4-Cl | Ph | 24 | Tracesb |
| 11 | 4-OCH3 | Ph | 24 | Tracesb |
Varying of solvents and bases does not lead to a significant increase in the conversion of the starting compounds. A positive result was reached using equimolar amounts of anhydrous ammonium acetate; the degree of oxindole conversion was increased, and the condensation product was obtained in 20% yield (Table 2).
|
||||
|---|---|---|---|---|
| Entry | Base/catalyst | Amount | Solvent | Yield |
| a About 20 equivalents. | ||||
| 1 | Piperidine | 2 | Ethanol | Trace |
| 2 | NH3 | Excessa | Toluene | Trace |
| 3 | NaH | 1.1 | THF | 8 |
| 4 | NaH | 1.1 | DMF | 6 |
| 5 | NaOH | 1 | Ethanol | 5 |
| 6 | NaOH | 1 | Butanol | 7 |
| 7 | NH4COOCH3 | Excessa | Toluene | 20 |
| 8 | Pyridine/Ti[OCH(CH3)2]4 | 3/2 | THF (rt) | 58 |
| 9 | Pyridine/Ti[OCH(CH3)2]4 | 3/2 | THF (60 °C) | 86 |
| 10 | Pyridine/Ti[OCH(CH3)2]4, (2 days of storage) | 3/2 | THF (60 °C) | 34 |
| 11 | Pyridine/Ti[OCH(CH3)2]4, (5 days of storage) | 3/2 | THF (60 °C) | Trace |
Among the modern methods for performing the Knoevenagel condensation, we should highlight a method with intermediate formation of titanium enolate,19 which was successfully used for the synthesis of 3-alkylidene oxindoles.20 We found that the proposed method shows good results not only for benzophenone condensation, as it was shown, but also when using diversely substituted benzophenones, for example 4-chlorobenzophenone (Table 2).
However, the reaction turned out to be very sensitive to the quality of titanium isopropoxide used. Long-term storage of the reagent resulted in a significant drop in the yield of the target compound. Accordingly, high requirements are imposed on the THF dryness (distillation over LAH).
When looking for less capricious, but no less effective condensation catalyst, we paid attention to aluminum isopropoxide. The homogeneous catalysis of the Knoevenagel reaction by aluminum alkoxides has not been described previously, although studies on heterogeneous catalysis using aluminum oxide were conducted.21
We proposed that aluminum isopropoxide is able of similar formation of enolates with carbonyl groups of oxindole observed for titanium isopropoxide. Thus, first using aluminum isopropoxide as a condensing agent in the Knoevenagel type reaction, we obtained a series of condensed oxindole derivatives with good yields under mild conditions (Table 3). Aluminum isopropoxide can be readily obtained in the laboratory, and its prolonged storage does not lead to a significant drop in the reaction yields.
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Yieldb | Isomer ratio, E/Zc |
| a Reaction conditions: 2-oxindole (1 equiv.), benzophenone (1.2 equiv.), pyridine (2 equiv.), aluminum isopropoxide (3 equiv.), THF, 40 °C, 12 h.b Total yield is given (for both E- and Z-isomer).c Determined by NMR after work up. | ||||
| 1 | H | H | 76 | — |
| 2 | 2-Cl | H | 80 | 62/38 |
| 3 | 4-Cl | H | 82 | 37/63 |
| 4 | 4-Cl | 4-Cl | 86 | — |
| 5 | 4-Cl | 2,4-Cl | 83 | 56/44 |
| 6 | 4-OH | H | 77 | 50/50 |
| 7 | 4-OCH3 | H | 73 | 38/62 |
| 8 | 3-CH3 | 4-Cl | 83 | 42/58 |
| 9 | 3-CH3 | 4-OH | 80 | 50/50 |
| 10 | 4-CH3 | 4-Cl | 79 | 46/54 |
| 11 | 4-CH3 | 4-OH | 72 | 50/50 |
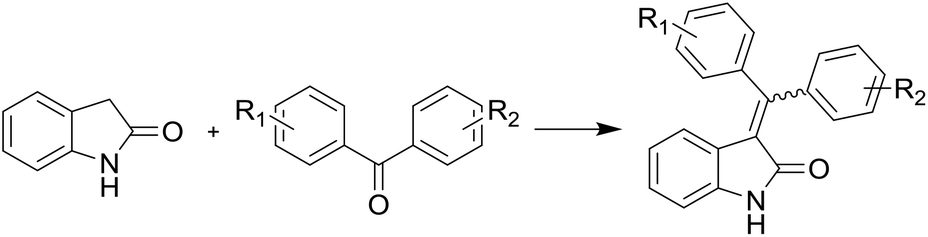
It should be noted that due to the presence of the double bond in position 3, alkene oxindoles exist as E- and Z-isomers exhibiting different biological activity.15 This requires the isolation of single isomers and separate biological evaluation.
In the case of the condensation of aldehydes and alkylphenones in the presence of pyrrolidine, E-isomers are predominantly formed. However, it was shown that the condensation of oxindole with acetophenone and propiophenone via titanium enolates leads to a prevalence of Z-isomer in the product mixture.20
Titanium- and aluminum-promoted condensation of oxindole with 4-chlorobenzophenone resulted in similar ratio of E- and Z-isomers when compared (E/Z ratio 40/60 and 37/63, respectively). This E,Z-isomer mixture of the condensation products can be successfully separated by column chromatography (Rf(E) = 0.32, Rf(Z) = 0.46, n-hexane/ethyl acetate 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The isomers can be clearly distinguished in 1H NMR spectra by the chemical shifts of protons H4 and H5 of the oxindole core (Fig. 2).
:1). The isomers can be clearly distinguished in 1H NMR spectra by the chemical shifts of protons H4 and H5 of the oxindole core (Fig. 2).
 | ||
| Fig. 2 Chemical shifts of characteristic proton signals for pair of 3((4-chlorophenyl)(phenyl)methylene)indolin-2-one isomers. | ||
Despite the differences in the E,Z-isomer ratio obtained during the condensation of oxindoles with aldehydes or unsymmetrical ketones under different synthetic conditions, from the preparative point of view, optimization of the selectivity of this stage by manipulation with the reaction mechanism is meaningless due to a relatively easy transition between the E,Z-isomers.
One isomer can be transferred to another by UV irradiation until the equilibrium is reached.22 The isomerization reaction proceeds without the by-product formation. Particularly, the Z/E equilibrium ratio for the condensation products of oxindole with 4-chlorobenzophenone is 51/49. Using a series of sequential irradiations and separations, it is possible to achieve the preparative yield of the desired isomer (E- or Z-isomer) up to 80%.
Thus, the most rational way to obtain individual isomeric forms of the compounds is to separate the mixture at the final stages of the synthetic scheme. So, methyl ester of C24 can be prepared by direct alkylation of the E,Z-isomer mixture of the condensation products with 3-(bromomethyl)benzoic acid methyl ester as an alkylating agent (Table 4) and subsequent separation of the alkylated derivatives (Rf(Ealk) = 0.21, Rf(Zalk) = 0.30, n-hexane/ethyl acetate 6:1).

Alkaline hydrolysis in a water–THF mixture is the final stage of the synthesis. It is carried out using the trivial procedure with yields close to quantitative. Scheme 2 represents a simple and effective route based on the convergent strategy, which can be used for obtaining C24 with the total yield of 58%, as well as for the synthesis of 3-(benzylidene)indolin-2-one series.
Conclusions
In summary, we developed advanced route of C24 synthesis within the convergent strategy. The used synthetic methods, despite of their simplicity, correspond to the current trends in the C–H bond functionalization without recruiting halogens or halogenoids as leaving groups.23 Thus, using the previously proposed schemes the halogen acts as a leaving group three times during the synthesis,15,16 while in the developed scheme only once at the N-alkylation step. One of the most interesting points of the work is the use of aluminum isopropoxide as a condensing agent in the Knoevenagel reaction. The given scheme and methods of synthesis provide the most comfortable opportunity for the development of focused libraries with high variability of external fragments including those with hydroxy group.Experimental
All starting compounds and reagents used, except 2-oxindole which was obtained according to the known procedure,24 are commercially available.The progress of reactions was monitored by TLC on Silica gel 60 F254 plates (Merk) using n-hexane/ethyl acetate eluent. Purification of the products was carried out using an Isolera Four flash chromatograph on SNAP KP-Sil 100 g cartridges (Biotage) with n-hexane/ethyl acetate eluent. For separation of the mixture of C24 methyl ester and its dehalogenated product, an Omnifit glass column (DIBA) packed with LiChroprep RP-18 sorbent 25–40 μm (Merck) was used.
1H and 13C NMR spectra were recorded on a Bruker Avance III 400 (400 MHz) device in DMSO-d6. To determine the spatial configuration of the compounds (E or Z isomer), previously obtained crystallographic data or NOESY/ROESY spectra were considered. The isomer ratio was evaluated only by 1H-NMR, since the isomers have different absorption at 254 nm commonly used for detection in chromatographic devices. Mass spectra were recorded on a LCMS-2020 device (Shimadzu) with a single quadrupole detector under positive mode, electrospray ionization (ESI).
General procedure for C24 synthesis using the linear scheme
Originally, the synthesis of C24 was carried out according to the known procedure15 with some modifications concerning mainly the work up of the reactions and the use of preobtained tetrakis(triphenylphosphine)palladium instead of in situ generation (total yield 29%).:1). Colorless crystals in 78% yield (5.44 g), mp 125–126 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.84 (s, 1H), 7.66 (d, J = 7.7, 4H), 7.55–7.47 (m, 3H), 7.34 (t, J = 7.8, 2H), 7.11 (t, J = 7.3, 1H); MS (ESI) m/z: 222 [M + H]+.:1), and then the title product was separated from dehalogenated product on a reversed-phase silica gel column with methanol/water eluent (3:1). Yellow crystals in 49% yield; mp 181–182 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.96 (s, 1H), 7.87 (d, J = 7.7, 1H), 7.57 (d, J = 7.8, 1H), 7.54 (d, J = 8.4, 2H), 7.47 (t, J = 7.7, 1H), 7.39–7.29 (m, 7H), 7.12 (t, J = 7.6, 1H), 6.87 (d, J = 7.8, 1H), 6.69 (t, J = 7.7, 1H), 6.33 (d, J = 7.6, 1H), 4.96 (s, 2H), 3.87 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ (ppm): 166.48, 166.06, 153.00, 142.50, 140.26, 139.90, 137.98, 134.52, 132.59, 131.17, 130.46, 130.36, 129.80, 129.71, 129.63, 129.38, 128.65, 128.45, 128.23, 124.29, 122.97, 122.85, 122.00, 109.41, 52.66, 42.62; MS (ESI) m/z: 480 [M + H]+.General procedure for Knoevenagel condensation of carbonyl compounds (aldehydes or alkylphenones) with oxindol
2-Oxindole (2 g, 0.015 mol) was suspended in 50 mL of toluene. The corresponding ketone (0.018 mol, 1.2 equiv.), 2.5 mL of pyrrolidine (0.03 mol, 2 equiv.) were added to the suspension. The reaction mixture was refluxed with the Dean–Stark trap. The reaction was monitored by TLC until the starting oxindole disappeared. Than the mixture was cooled, the solvent was evaporated under reduced pressure. The crude product was treated with a small amount of ethyl acetate and then filtered off, or recrystallized from n-hexane/ethyl acetate, or purified by flash chromatography using n-hexane/ethyl acetate eluent (if pair of isomers formed).Procedure for aluminum isopropoxide preparation
Finely flattened aluminium (2.7 g, 0.1 mol) in 100 mL isopropanol with a catalytic amount of iodine was refluxed with drying tube until aluminum was completely dissolved. The isopropanol was distilled off and the crude product was immediately purified by distillation (0.4 bar, 134–137 °C). Freshly distilled aluminum isopropoxide was obtained as high-viscosity liquid in 94% yield, solidifying within several hours.General procedure for aluminum-promoted Knoevenagel condensation of benzophenones
2-Oxindole (2 g, 0.015 mol) was dissolved in dried and freshly distilled THF, the corresponding benzophenone (0.018 mol), pyridine (2.42 mL, 0.03 mol) and aluminum isopropoxide (9.2 g, 0.045 mol) were added. The reaction mixture was heated to 40 °C and kept overnight. Then the reaction mixture was quenched with 3% Na2CO3 solution, extracted with ethyl acetate, dried over anhydrous Na2SO4 and evaporated under reduced pressure. The product (mixture of products) was isolated by flash chromatography with n-hexane/ethyl acetate eluent.General procedure for alkylation of alkene oxindole compounds with 3-(bromomethyl)benzoic acid methyl ester exemplified by the isomer mixture of 3-((4-chlorophenyl)(phenyl)methylene)indolin-2-one
Isomer mixture of 3-((4-chlorophenyl)(phenyl)methylene)indolin-2-one (2 g, 0.006 mol) and potassium carbonate (2.5 g, 0.018 mol) were dissolved in 100 mL dimethylformamide, and methyl 3-(bromomethyl)benzoate (2 g, 0.007 mol) dissolved in dimethylformamide was added gradually for an hour. The reaction mixture was stirred overnight at room temperature. The solvent was evaporated under reduced pressure, and then water was added to the residue. The precipitate was filtered off, dissolved in ethyl acetate, dried over anhydrous Na2SO4 and evaporated under reduced pressure. The product mixture was purified by flash chromatography (n-hexane/ethyl acetate 9:1).
General procedure for alkaline hydrolysis for esters of N-alkylated alkene oxindole compounds
Lithium hydroxide (1.2 g, 0.05 mol) dissolved in 50 mL water was added to the corresponding methyl ester (2.4 g, 0.005 mol) dissolved in 50 mL THF. The reaction mixture was stirred at rt and monitored by TLC. Then the mixture was acidified with 15% HCl and THF was almost completely distilled from the aqueous mixture. After THF was removed, the formed precipitate was filtered off, washed with 50 mL water and dried. The crude product was purified by recrystallization from ethyl acetate.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Russian Science Foundation (project no. 16-13-10358). XRD studies were performed at the Centre for X-ray Diffraction Methods of Research park of St. Petersburg State University.Notes and references
- (a) G. R. Steinberg and B. E. Kemp, Physiol. Rev., 2009, 89, 1025 CrossRef CAS PubMed; (b) D. G. Hardie and M. L. Ashford, Physiology, 2014, 29, 99 CrossRef CAS PubMed.
- (a) D. G. Hardie, Diabetes, 2013, 62, 2164 CrossRef CAS PubMed; (b) E. A. Day, R. J. Ford and G. R. Steinberg, Trends Endocrinol. Metab., 2017, 28, 545 CrossRef CAS PubMed.
- (a) L. G. Fryer, A. Parbu-Patel and D. Carling, J. Biol. Chem., 2002, 277, 25226 CrossRef CAS PubMed; (b) D. G. Hardie, F. A. Ross and S. A. Hawley, Chem. Biol., 2012, 19, 1222 CrossRef CAS PubMed.
- D. S. Novikova, A. V. Garabadzhiu, G. Melino, N. A. Barlev and V. G. Tribulovich, Russ. Chem. Bull., 2015, 64, 1497 CrossRef CAS.
- (a) J. E. Sullivan, K. J. Brocklehurst, A. E. Marley, F. Carey, D. Carling and R. K. Beri, FEBS Lett., 1994, 353, 33 CrossRef CAS PubMed; (b) N. Henin, M. F Vincent and G. Van den Berghe, Biochim. Biophys. Acta, 1996, 1290, 197 CrossRef PubMed; (c) C. G. Langendorf, K. R. Ngoei, J. W. Scott, N. X. Ling, S. M. Issa, M. A. Gorman, M. W. Parker, K. Sakamoto, J. S. Oakhill and B. E. Kemp, Nat. Commun., 2016, 7, 10912 CrossRef CAS PubMed.
- (a) B. Cool, B. Zinker, W. Chiou, L. Kifle, N. Cao, M. Perham, R. Dickinson, A. Adler, G. Gagne, R. Iyengar, G. Zhao, K. Marsh, P. Kym, P. Jung, H. S. Camp and E. Frevert, Cell Metab., 2006, 3, 403 CrossRef CAS PubMed; (b) G. Zhao, R. R. Iyengar, A. S. Judd, B. Cool, W. Chiou, L. Kifle, E. Frevert, H. Sham and P. R. Kym, Bioorg. Med. Chem. Lett., 2007, 17, 3254 CrossRef CAS PubMed.
- (a) J. Charton, S. Girault-Mizzi, M. A. Debreu-Fontaine, F. Foufelle, I. Hainault, J. G. Bizot-Espiard, D. H. Caignard and C. Sergheraert, Bioorg. Med. Chem., 2006, 14, 4490 CrossRef CAS PubMed; (b) B. C. Bookser, Q. Dang, T. S. Gibson, H. Jiang, D. M. Chung, J. Bao, J. Jiang, A. Kassick, A. Kekec, P. Lan, H. Lu, G. Makara, F. A. Romero, I. Sebhat, D. Wilson and D. Wodka, US Pat., 8394969B2, 2013.
- L. F. Yu, Y. Y. Li, M. B. Su, M. Zhang, W. Zhang, L. N. Zhang, T. Pang, R. T. Zhang, B. Liu, J. Y. Li, J. Li and F. J. Nan, ACS Med. Chem. Lett., 2013, 4, 475 CrossRef CAS PubMed.
- C. G. Langendorf and B. E. Kemp, Cell Res., 2015, 25, 5 CrossRef CAS PubMed.
- (a) A. Millemaggi and R. J. K. Taylor, Eur. J. Org. Chem., 2010, 24, 4527 CrossRef; (b) Z. Y. Cao, F. Zhou and J. Zhou, Acc. Chem. Res., 2018, 51, 1443 CrossRef CAS PubMed.
- (a) G. H. Zheng, J. J. Shen, Y. C. Zhan, H. Yi, S. T. Xue, Z. Wang, X. Y. Ji and Z. R. Li, Eur. J. Med. Chem., 2014, 81, 277 CrossRef CAS PubMed; (b) A. Pal, A. Ganguly, A. Ghosh, M. Yousuf, B. Rathore, R. Banerjee and S. Adhikari, ChemMedChem, 2014, 9, 727 CrossRef CAS PubMed.
- P. Li, M. Zhao, A. B. Parris, X. Feng and X. Yang, Biochem. Biophys. Res. Commun., 2015, 464, 1267 CrossRef CAS PubMed.
- (a) T. A. Grigoreva, D. S. Novikova, M. A. Gureev, A. V. Garabadzhiu and V. G. Tribulovich, Chirality, 2018, 30, 785 CrossRef CAS PubMed; (b) T. A. Grigoreva, D. S. Novikova, A. V. Petukhov, M. A. Gureev, A. V. Garabadzhiu, G. Melino, N. A. Barlev and V. G. Tribulovich, Bioorg. Med. Chem. Lett., 2017, 27, 5197 CrossRef CAS PubMed; (c) T. A. Grigoreva, A. V. Garabadzhiu and V. G. Tribulovich, Russ. J. Gen. Chem., 2016, 86, 2454 CrossRef CAS.
- J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed.
- F. Nan, L. Yu, M. Zhang, L. Chen, M. Huang, L. Feng, J. Li and T. Pang, US Pat., 8778973B2, 2014.
- (a) A. Pinto, L. Neuville, P. Retailleau and J. Zhu, Org. Lett., 2006, 8, 4927 CrossRef CAS PubMed; (b) D. Lee, S. Park, Y. Yu, K. J. Shin and J. H. Seo, Molecules, 2015, 20, 14022 CrossRef CAS PubMed; (c) S. Park, J. Lee, K. J. Shin, E. Oh and J. H. Seo, Molecules, 2017, 22, 503 CrossRef PubMed.
- (a) F. Hu, P. Nareddy, R. Lalancette, F. Jordan and M. Szostak, Org. Lett., 2017, 19, 2386 CrossRef CAS PubMed; (b) F. Hu, R. Lalancette and M. Szostak, Angew. Chem., Int. Ed., 2016, 55, 5062 CrossRef CAS PubMed.
- T. P. Du, G. G. Zhu and J. Zhou, Lett. Org. Chem., 2012, 9, 225 CrossRef CAS.
- B. A. Robichaud and K. G. Liu, Tetrahedron Lett., 2011, 52, 6935 CrossRef CAS.
- H. J. Lee, J. W. Lim, J. Yu and J. N. Kim, Tetrahedron Lett., 2014, 55, 1183 CrossRef CAS.
- F. Texier-Boullet and A. Foucaud, Tetrahedron Lett., 1982, 23, 4927 CrossRef CAS.
- L. Sun, N. Tran, F. Tang, H. App, P. Hirth, G. McMahon and C. Tang, J. Med. Chem., 1998, 41, 2588 CrossRef CAS PubMed.
- (a) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (b) F. Roudesly, J. Oble and G. Poli, J. Mol. Catal. A: Chem., 2017, 426 B, 275 CrossRef.
- C. Crestini and R. Saladino, Synth. Commun., 1994, 24, 2835 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1585543. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra07576j |
| This journal is © The Royal Society of Chemistry 2018 |