DOI:
10.1039/C8RA07514J
(Paper)
RSC Adv., 2018,
8, 35360-35366
Vortex-assisted liquid–liquid microextraction and indirect spectrophotometric determination of chromium(VI)
Received
9th September 2018
, Accepted 8th October 2018
First published on 15th October 2018
Abstract
A novel, simple, sensitive and selective method for the indirect spectrophotometric determination of chromium(VI) was developed on the basis of vortex-assisted liquid–liquid microextraction (VALLME) of an ion association pair formed between the triiodide anion (I3−) and cationic dye Astrazone Brilliant Red 4G (ABR). The indirect spectrophotometric determination of chromium(VI) was based on the redox-reaction of Cr(VI) with iodide and formed I3−. The triiodide anion was determined spectrophotometrically at 530 nm following VALLME of an ion association pair with ABR. The calibration plot was linear in the range 0.050–4.2 μg L−1 (R2 = 0.998), and limit of detection (LOD) was 0.015 μg L−1 for Cr(VI). The suggested procedure was successfully applied for the determination Cr(VI) in water samples (RSD 3.3–5.7%, recovery 97.7–102.8% when determining ultratrace levels Cr(VI) at 0.55–5.77 μg L−1), the accuracy of the method was assessed through a comparison with reference methods and certified reference materials.
Introduction
Chromium(VI) compounds are highly toxic. They exhibit carcinogenic, mutagenic and embryotoxic effects,1–3 and they are mobile in the environment.4,5 While the natural occurrence of chromium(VI) is very rare, harmful levels of Cr(VI) in the environment occur often due to human activity. Chromium(VI) compounds are used in various industries such as metal coatings, protective paints, dyes and pigments owing to their properties in particular anti-corrosive ones. Taking into account the current number of anthropogenic sources of pollution, attention to the measurement of chromium(VI) in water samples in trace levels is particularly important.6
A variety of analytical methods have previously been used for Cr(VI) determination, including voltammetry,7–12 luminescence and chemiluminescence,13–17 flame and electrothermal AAS,18–25 chromatography,26–31 UV-vis spectrophotometry.32–41 Each of these techniques offers both advantages and disadvantages. A comparison of the methods for the determination of chromium was made in the review.42 There is no universal method for determining chromium(VI). While voltammetric methods for determining Cr(VI) are rarely used,6 luminescence and chemiluminescence have low selectivity for Cr(VI). At the same time, chromatography methods have low sensitivity to Cr(VI) and are expensive.6 For this reason they are seldom used. Meanwhile, flame and electrothermal AAS are used to determine total chromium only. These methods require the separation and concentration of chromium by liquid–liquid extraction,21–24 cloud point extraction to the surfactant-rich phase,22–27 coprecipitation,23 ion exchange34 or ion-exchange chromatography,35 organic soluble membrane filter,40 sorption or solid phase extraction.9,20,25,36,37,41,43
Spectrophotometry is the most commonly used method for determination of chromium(VI) in water samples.6 Various organic reagents have been suggested for chromium(VI) determination including 1,5-diphenylcarbazide,33–37 methyl orange in the presence of potassium bromate,38 methyl blue in the presence of sodium periodate,39 o-chlorophenylfluorone and tetradecylpyridinium chloride,40 1,4,8,11-tetraazacyclotetradecane (cyclam),41 but the number of such reagents is limited. The chemical compound known as 1,5-diphenylcarbazide (DPC), which develops an international standard for the determination of chromium(VI) in waters, is used the most.44 The sensitivity of DPC is insufficient to determine chromium in the waters on trace level, however.
The amount of chromium(VI) in the majority of samples is significantly low which makes the possibility of determining it directly very unlikely. Therefore, preliminary concentration of chromium necessitates the use of “green” technologies. Microextraction procedures are considered as green because they minimize the consumption of harmful solvents.45–47 Among the various techniques of liquid–liquid microextraction, the vortex-assisted turns out to be promising.48,49 The vortex-assisted liquid–liquid microextraction of the product of the redox reaction (I3−) of dichromate with iodides in the form of ion association pair with cationic dye was not previously used for the indirect determination of chromium(VI).
In this study, we have used the vortex-assisted liquid–liquid microextraction procedure to indirectly determine Cr(VI) by spectrophotometry. After optimizing the redox-reaction of dichromate with iodides conditions and the microextraction of the product reaction, a spectrophotometric method for determining Cr(VI) in water samples is developed.
Experimental
Instrumentations
An SPECORD® S 600 UV/Vis (Analytik Jena AG, Germany) and Lightwave II (Biochrom, UK) spectrophotometers were used to record the absorption spectra and for routine measurements with quartz ultramicrocuvettes (Starna Scientific Ltd., UK), l = 1.0 cm. The microextraction procedure was assisted by vortex mixer VM-3000MD (Medline Scientific, UK) and the separation of the phases was facilitated by a centrifuge (MCD-2000, MRC ltd. China). The pH values of solutions were measured with an InoLab pH Level 1 potentiometer (WTW, Germany) with a BlueLine 23 pH Model electrode (SI Analytics, Germany). The extraction procedure was performed in polypropylene tube with conical bottom and screw cap.
Reagents and chemicals
All chemicals and solvents used in the experiment were of analytical reagent grade. A 0.01 mol L−1 standard solution of potassium dichromate was prepared by dissolving the exact sample K2Cr2O7 (Sigma-Aldrich, Germany) in deionized water. A working solution with 0.001–0.000001 mol L−1 concentration of potassium dichromate was prepared daily by dilution of a standard stock solution with deionized water. We used a 5.0 mol L−1 solution sulfuric acid, which was obtained by dilution of concentrated sulfate acid (Sigma-Aldrich, Germany). Solution of potassium iodide was prepared by dissolving the exact sample KI (Sigma-Aldrich, Germany) in deionized water. A 0.001 mol L−1 standard solution of Astrazone Brilliant Red 4G (ABR) was prepared by dissolving the exact sample ABR (Spectrum Chemical MFG Corp., USA), in mixtures ethanol and deionized water.
Procedure of chromium(VI) determination
A 4.0 mL aliquot containing from 0.25–21.0 ng of Cr(VI) (at a higher concentration of Cr(VI), an appropriately smaller volume is added, which is adjusted to 4.0 mL with water) was placed into a polypropylene centrifuge tube, added 0.15 mL 20% aqueous solution of potassium – sodium tartrates, mixed and the tube was allowed to stand for 1 min. Then added 0.2 mL of 5 mol L−1 of H2SO4, 0.15 mL of 0.01 mol L−1 of KI, 0.5 mL of 0.001 mol L−1 of ABR and thoroughly mixed. As a result, the ion associate of triiodide anion (I3−) with ABR cation is formed. Next, 250 μL of isoamylacetate was added. The mixture was shaken on a vortex agitator at 3000 rpm for 15 s. The tube was then centrifuged at 1500 rpm for 2 min. The floated phase was withdrawn using a micropipette and transferred to an ultramicrocuvettes with l = 1.0 cm and the absorbance was measured at 530 nm against blank. The content of Cr(VI) was determined using a calibration plot constructed under similar conditions.
Results and discussion
Reaction chemistry
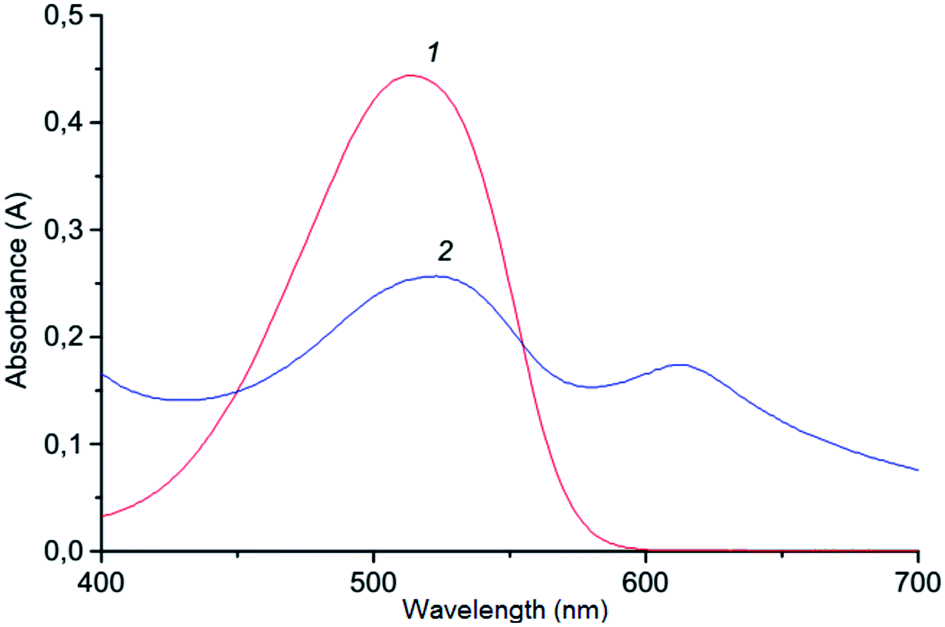
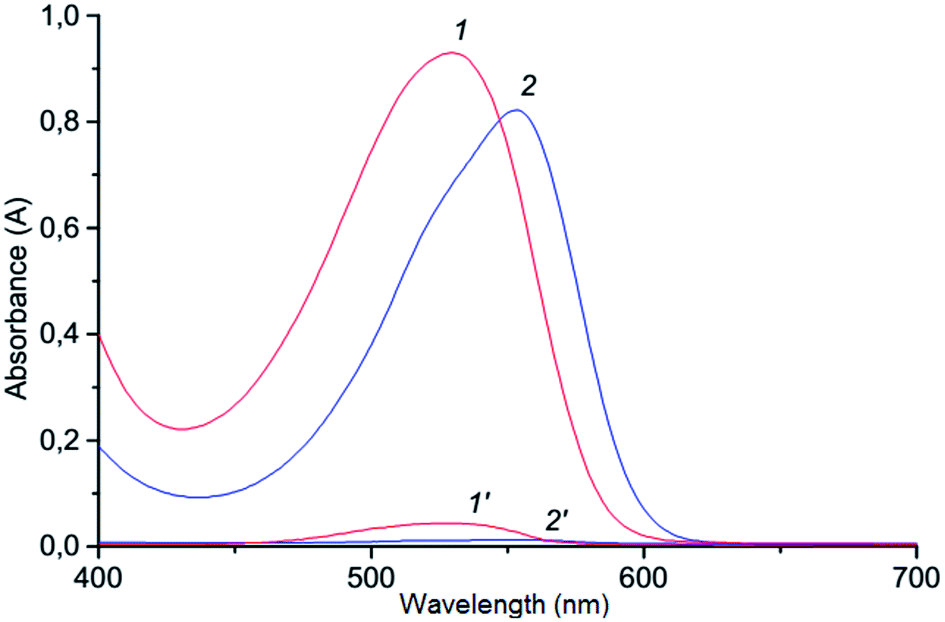
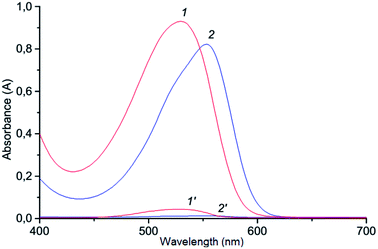
The method suggested in this paper includes indirect determination of chromium(VI). Determination of chromium(VI) based on redox-reaction of dichromate potassium with iodide and formed I3−.50 The redox-reaction of dichromate potassium with iodide is well studied,51,52 and the kinetic studies showed half order reaction as to dichromate ion and first order both for iodide ion and hydrogen ion concentrations (under optimal conditions the reaction proceeds quickly; the value of the rate constant was 8.67 mol1/2 S2 L−1/2). Generally in experiments, Cr(VI) is indirectly determined by the formation of ion association pair I3− with basic triphenylmethane dyes (LOD = 0.065 mg L−1;calibration plot was linear in the range 0–1.5 mg/25 mL with crystal violet),53 and the analytical signal is measured with the use of the resonance Rayleigh scattering but the procedure for determining chromium(VI) is rather complicated. In this study, the ion association pair, which is formed by a reaction of triiodide anion with cation Astrazone Brilliant Red 4G, is determined spectrophotometrically at 530 mn following VALLME in isoamylacetate. The formation of ion associate in aqueous solution is confirmed by bathochromic shift (Δλ = 100 nm) of the absorption of the ABR (Fig. 1). The system is not suitable for the spectrophotometric determination of chromium(VI) in the aqueous phase since the system is colloidal and unstable. It is not necessary to count on the high accuracy of measurements in the water phase, so the ion associate is extracted into the organic phase. Therefore, we can assume that the chemistry of formation and extraction of ion associate to appear is as follows:| | |
Cr2O72− + 9I− + 14H+ = 2Cr3+ + 3I3− + 7H2O
| (1) |
| | |
I3−(aq) + ABR+(aq) ↔ (I3−·ABR+)(aq) ↔ (I3−·ABR+)(org)
| (2) |
 |
| | Fig. 1 Absorption spectra of the ABR (1) and ion association pair ABR with I3− (2) in aqueous solution: 1–2.3 × 10−5 mol L−1 ABR; 0.2 mol L−1 H2SO4; 2–2.3 × 10−5 mol L−1 ABR; 1.3 × 10−5 mol L−1 Cr(VI); 3.0 × 10−4 mol L−1 KI; 0.2 mol L−1 H2SO4. | |
The concentration of Cr(VI) is determined from the corresponding calibration curve. We studied other basic dyes, such as crystal violet, brilliant green, Astra Phloxin FF, but ABR (ε510 = 1.9 × 104 L mol−1 cm−1) showed the highest extraction degree when VALLME was used.
Optimization of chemical variables for Cr(VI) determination
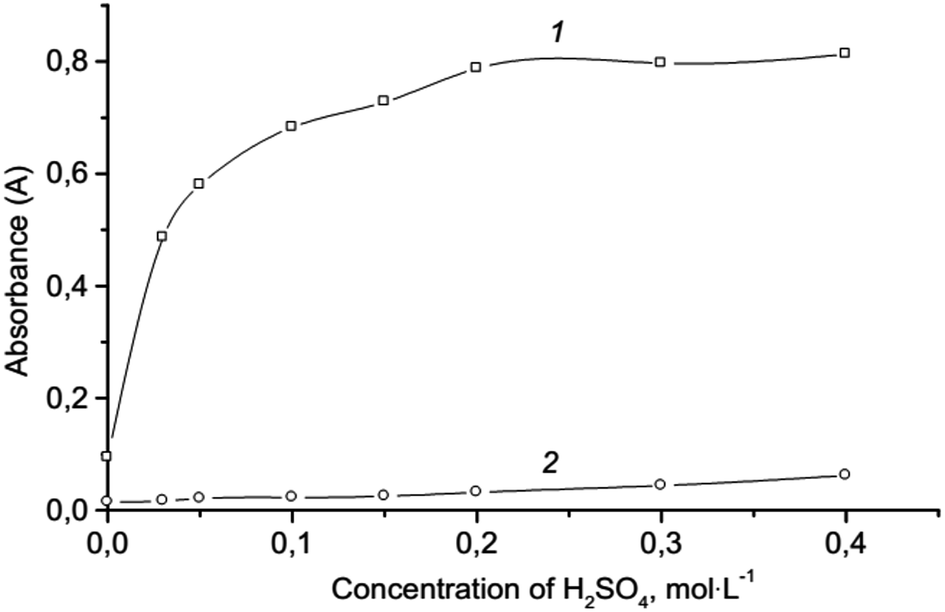
Effect of acidity. The nature and concentration of the acid are crucial parameters that influence the determination of chromium(VI) as seen in the reaction (1). Preliminary results show that it is more convenient to use sulfuric acid. The effect of sulfuric acid concentration was studied over the concentration range of 0.0–0.4 mol L−1 (Fig. 2). The absorbance of the extract increases with increasing acid concentration up to 0.2 mol L−1 with no occurrence of subsequent changes. A concentration of 0.2 mol L−1 of sulfuric acid was chosen as optimal, which provides both the necessary completeness and speed of formation of triiodide ions, and sufficient selectivity for the formation of ion association pair. It is not advisable to choose an acidic medium because the selectivity of the reaction (1) decreases, and the analytical signal of blank test increases. In a more acidic medium, some oxidants (NO3−, ClO3−) and reducing agents (C2O42−, Fe2+) hinder the determination of chromium even in equal amounts. Moreover, in a strong acidic medium, the oxidation of iodides with oxygen occurs, which increases the analytical signal of the blank test.54 In addition, in a strong acidic medium, the partial protonation of the dye ABR (pKprotonation = −0.22) occurs.
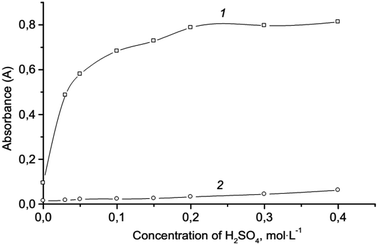 |
| | Fig. 2 Effect of the H2SO4 concentration on the microextraction: 2.0 × 10−6 mol L−1 Cr(VI); 3.0 × 10−4 mol L−1 KI; 1.6 × 10−4 mol L−1 ABR; 250 μL isoamylacetate; vortexing 15 s, 3000 rpm; centrifugation 2 min, 1500 rpm; 1 – ion associate; 2 – blank test. | |
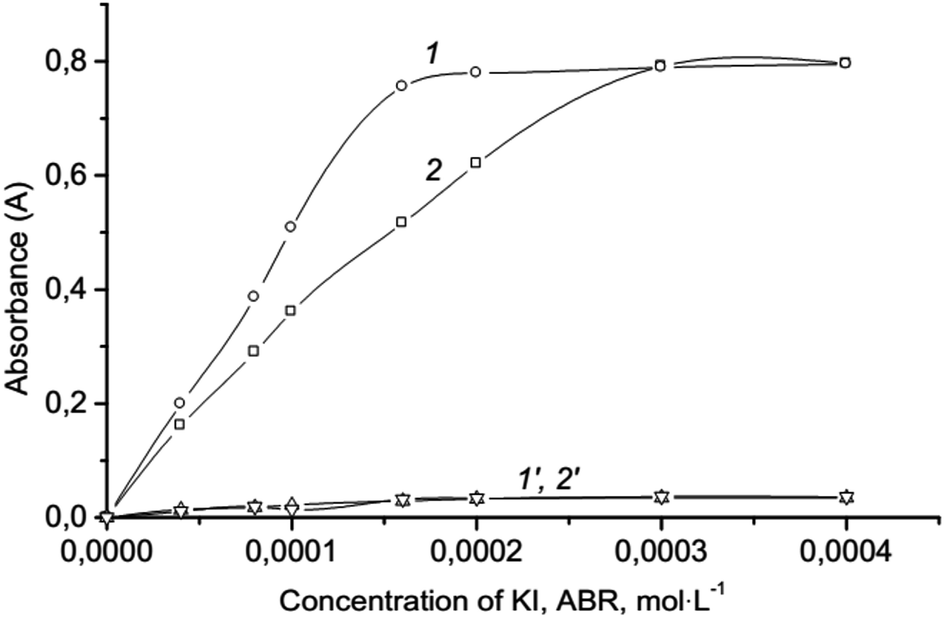
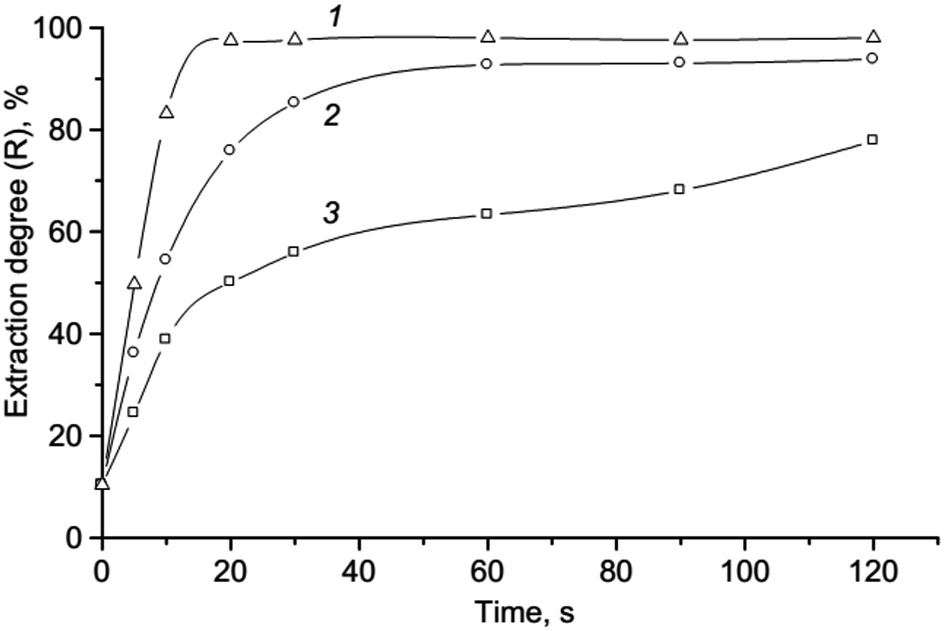
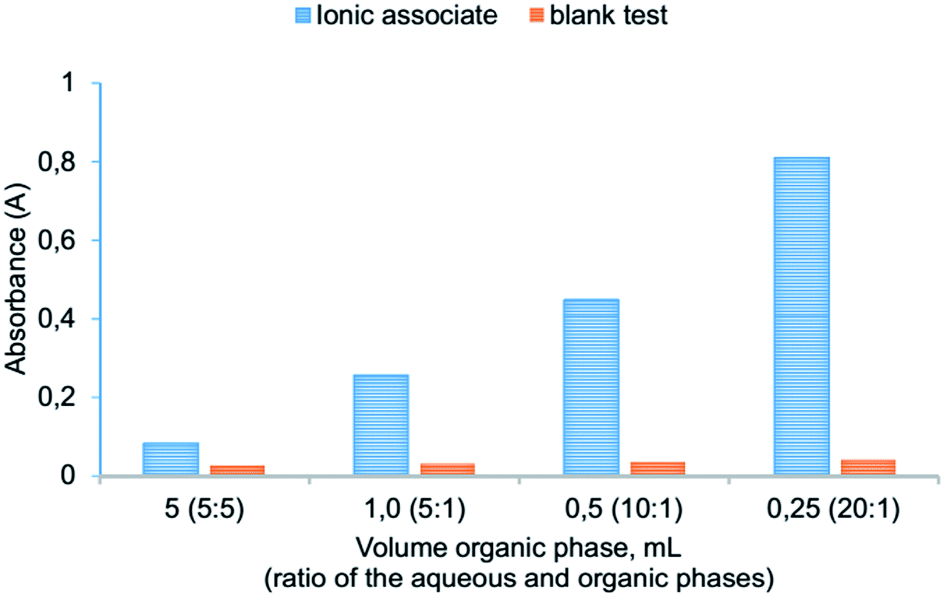
Effect of concentration reagents and extraction solvent. The effect of concentration reagents (KI, ABR) on the extraction of ion association was studied. It showed that the reaction (1) undergoes an excess of KI, as well as the formation of ion association pair with an excess of ABR cations. In this work the range of 5.0 × 10−5–4.0 × 10−4 mol L−1 was studied for optimizing of KI and ABR concentration and extraction efficiencies were determined (see Fig. 3). Results revealed that the complete extraction of ion associate occurred at concentrations higher than 3.0 × 10−4 mol L−1 KI and 1.6 × 10−4 mol L−1 ABR which was chosen as optimal.
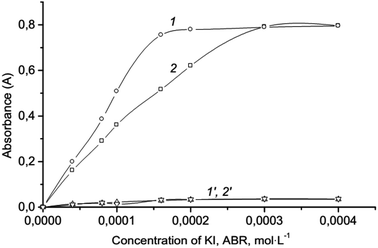 |
| | Fig. 3 Effect of the ABR (1) and KI (2) concentration on the absorbance of ionic associate extracts: 2.0 × 10−6 mol L−1 Cr(VI); 0.2 mol L−1 H2SO4; 250 μL isoamylacetate; vortexing 15 s, 3000 rpm; centrifugation 2 min, 1500 rpm; 1–3.0 × 10−4 mol L−1 KI; 2–1.6 × 10−4 mol L−1 ABR; 1′, 2′ – respectively blank tests. | |
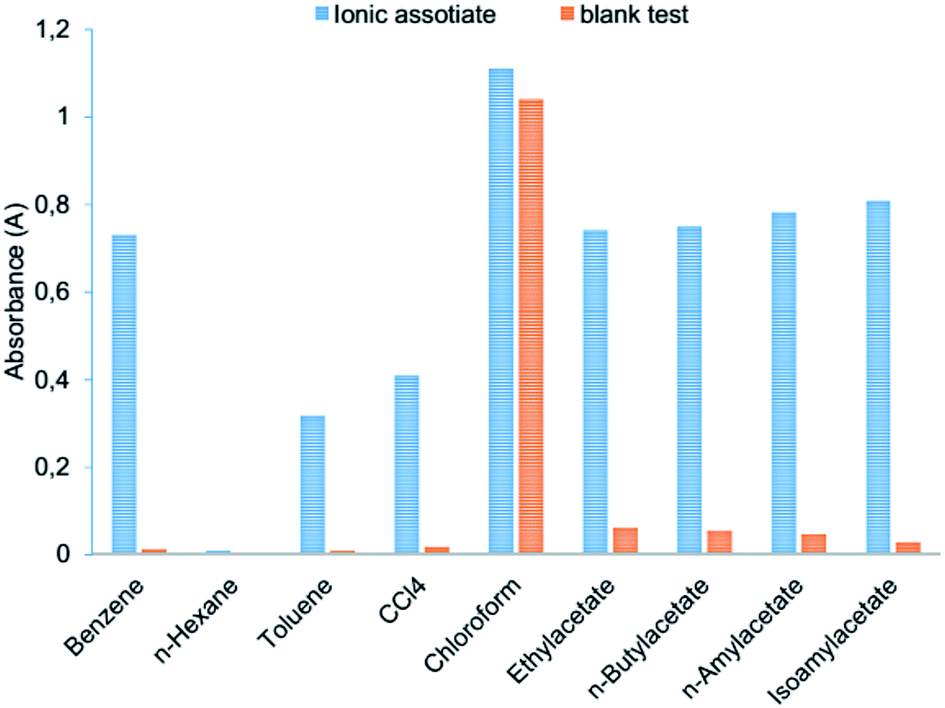
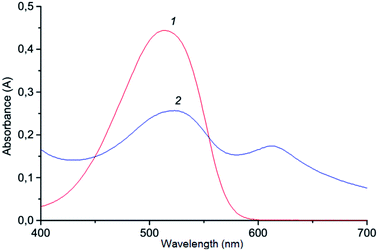
Benzene, n-hexane, toluene, carbon tetrachloride, chloroform, ethylacetate, n-butylacetate, n-amylacetate and isoamylacetate were examined as extraction solvents (Fig. 4). ABR is well soluble in chloroform which causes an increase in the absorbance of the blank test. Toluene, n-hexane, and carbon tetrachloride insufficiently extracted ion association pair (I3−·ABR+), among the esters – the best is isoamylacetate. Highly toxic benzene effectively extracted the ionic associate, but the different absorbance of the ionic associate extract and the blank test is lower than the one with isoamylacetate (Fig. 5). Therefore, the isoamylacetate was used as the extractant.
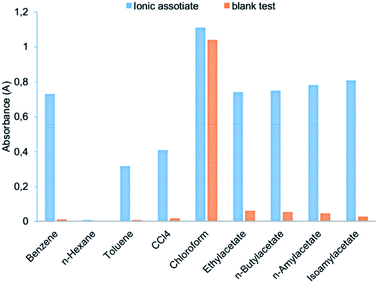 |
| | Fig. 4 Extraction efficiency using different solvents: 2.0 × 10−6 mol L−1 Cr(VI); 3.0 × 10−4 mol L−1 KI; 1.6 × 10−4 mol L−1 ABR; 0.2 mol L−1 H2SO4; 250 μL organic solvents; vortexing 15 s, 3000 rpm; centrifugation 2 min, 1500 rpm. | |
 |
| | Fig. 5 Absorption spectra of the ion association pair in organic phase: 2.2 × 10−6 mol L−1 Cr(VI); 3.0 × 10−4 mol L−1 KI; 1.6 × 10−4 mol L−1 ABR; 0.2 mol L−1 H2SO4; 250 μL organic phase; vortexing 15 s, 3000 rpm; centrifugation 2 min, 1500 rpm; 1 – isoamylacetate; 2 – benzene; 1′, 2′ – respectively blank tests. | |
Interference
Effects of various interferences possibly present in the water samples were investigated. Most cations and anions did not interfere with the determination of Cr(VI) in water samples. The tolerable amount was taken as that which resulted in an absorbance error of not more than ±5%. The results in Table 1 reveal that this method is acceptable for the determination of Cr(VI) in water samples. The determination of chromium(VI) in the presence of chromium(III) is possible. The spectrophotometric determination of chromium(VI) is already hindered by equal amounts of NO2−, BrO3−, IO3−, IO4−, ClO4−.
Table 1 Effect of interfering ions on the determination of Cr(VI)
| Interfering ions |
Tolerance limit, mg L−1 |
Tolerance ratio interfering ion/Cr(VI) |
| In the presence of a sufficient surplus of tartrates 100-fold of Cr3+, Fe3+ and Cu2+ amounts yet does not interfere to spectrophotometric determination of Cr(VI). |
| HCO3−, CH3COO−, HPO42−, SO42−, Cl−, Na+, K+, Li+, Ca2+, Sr2+, Ni2+, Co2+, Zn2+, C4H4O62− |
500 |
2000 |
| C2O42−, NO3−, ClO3−, Fe2+ |
3 |
100 |
| aCr3+ |
0.5 |
20 |
| aFe3+, Cu2+ |
0.2 |
10 |
Tartrates improve the selectivity of chromium(VI) determination. In particular, in the presence of a sufficient surplus of tartrates 100-fold of Cr3+, Fe3+ and Cu2+ the amounts do not yet interfere with the spectrophotometric determination of Cr(VI). The influence of NO2− is taken into account by its parallel determination in water samples.
Analytical figures of merit
On the basis of the obtained data, a new procedure for VALLME and indirect spectrophotometric determination of Cr(VI) was developed. The calibration curves were constructed under the optimized experimental conditions from six data points. LODs were calculated from a calibration graph based on three times the standard deviation of the blank test.
The regression equation for the calibration plot is A = (0.658 ± 0.018)C + (0.011 ± 0.004) (where A is the absorbance and C is the concentration of Cr(VI) μg L−1 in the aqueous phase) in the range of 0.050–4.2 μg L−1 chromium(VI) with a correlation coefficient of 0.9983. The limit of detection (n = 10, P = 0.95) was found to be 0.015 μg L−1 of Cr(VI).
Analytical applications
Analysis of certified sample. For the analysis, we used a certified reference material SPS-WW2 Batch 113. Matrix effects ware studied using the standard addition method. The results of the analysis of the certified sample showed satisfactory metrological characteristics: recovery – 100.7%, RSD – 2.5%. Thus, VALLME can be successfully used for the spectrophotometric determination of Cr(VI) in water samples.
Analysis of water samples. To demonstrate the practicability of the suggested approach, analyses of river and reservoir water samples were performed. The results are given in Table 2. Satisfactory recovery data were obtained and the results were in good agreement with the reference method,44 which is based on the use of 1,5-diphenylcarbazide. The proposed method is more sensitive, metrological characteristics (RSD 3.3–5.7%, recovery 97.7–102.8% when determining on ultra-trace level Cr(VI) at 0.55–5.77 μg L−1) indicate its practical use. The developed method allows determining the Cr(VI) in water samples within 0.05–4.2 μg L−1.
Table 2 Determination of Cr(VI) in water samples by the proposed and reference method (n = 6; P = 0.95)a
| Sample |
Added Cr(VI) μg L−1 |
Found this method Cr(VI) μg L−1 |
Recovery, % |
RSD, % |
Found by reference method44 Cr(VI) μg L−1 |
| ND – not detected. |
| River water, no. 1 |
0.00 |
0.55 |
— |
5.7 |
ND |
| 1.00 |
1.59 |
102.6 |
4.3 |
| 2.00 |
2.51 |
98.4 |
4.1 |
| River water, no. 2 |
0.00 |
0.73 |
— |
5.3 |
ND |
| 1.00 |
1.69 |
97.7 |
4.2 |
| 2.00 |
2.78 |
101.8 |
4.1 |
| River water, no. 3 |
0.00 |
1.21 |
— |
4.3 |
ND |
| 1.00 |
2.27 |
102.7 |
4.3 |
| 2.00 |
3.30 |
102.8 |
3.9 |
| Reservoir water |
0.00 |
3.85 |
— |
3.3 |
3.91 |
| 1.00 |
4.92 |
101.4 |
3.4 |
| 2.00 |
5.77 |
98.6 |
3.1 |
Conclusions
The first proposed indirect spectrophotometric determination of chromium(VI) was based on the VALLME, the product redox-reaction as ion association pair formed between I3− and ABR. On the basis of the findings of this study, a new, simple, rapid and sensitive spectrophotometric method for determination of Cr(VI) in water samples was suggested. Typically, toxic solvents (benzene, toluene, chloroform, carbon tetrachloride and others) are used as extractants. We used ecologically friendly organic solvent isoamylacetate the quantity of which was minimized (250 μL). Therefore, the proposed method can be considered as green. Although a high preconcentration factor was achieved, it is more sensitive than the reference and alternative spectrophotometric techniques. Reagents for the spectrophotometric determination of chromium(VI) are sufficient, with DPC (Table 3) being most commonly used. There have been attempts to improve the characteristics of the procedure for the spectrophotometric determination of chromium(VI) with DPC by modifying the procedure using ion exchange, sorption and other techniques. These procedures are difficult and lengthy whereas microextraction is a simple and fast procedure. Currently, the proposed method could play a useful role in monitoring the level of Cr(VI) in water samples.
Table 3 Spectrophotometric methods for determinations of Cr(VI) in waters samples
| Procedure/reagent |
LOD, (μg L−1) |
Linear range, (μg L−1) |
RSD, %/recovery, % |
Ref. |
| Flow injection spectrophotometry with ion exchange preconcentration/DPC |
0.044 |
0.1–50.0 |
1.3–4.1/98–103 |
34 |
| Flow injection spectrophotometry with separation by ion-exchange chromatography/DPC |
1.25 |
1–1000 |
1.21–1.66/98.0–104.0 |
35 |
| Spectrophotometry with preconcentration by modified sorbent/DPC and sodium dodecyl sulphate |
0.4 |
1.3–40.0 |
2.3/92.6–109.9 |
36 |
| Spectrophotometry with preconcentration by pyridine-functionalized mesoporous silicasorbent/DPC |
2.3 |
— |
1.8/98.5 |
37 |
| Reversed flow injection catalytic spectrophotometry/methyl orange in present's potassium bromate |
30.0 |
0–3000 |
1.2/94.5–96.7 |
38 |
| Reversed flow injection catalytic spectrophotometry/methyl blue in present's sodium periodate |
7.0 |
0–1000 |
1.5/92.9–97.5 |
39 |
| Spectrophotometry with preconcentration by organic soluble membrane filter/o-chlorophenylfluorone and tetradecylpyridinium chloride |
0.3 |
0.1–1.8 μg/5 mL |
2.4–3.2/98–105 |
40 |
| Spectrophotometry/cyclam |
1.0 |
200–20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
0.3–3.3/98.2–100.2 |
41 |
| Spectrophotometry/DPC |
— |
2–50 |
1.3–5.5/98.5–106.5 |
44 |
| Spectrophotometry with VALLME/iodide and ABR |
0.015 |
0.05–4.2 |
3.1–5.7/97.7–102.8 |
This work |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work has been supported by the Scientific Grant Agency of the Ministry of Education of the Slovak Republic and the Slovak Academy of Sciences VEGA (grant No. 1/0253/16).
Notes and references
- N. McCarroll, N. Keshava, J. Chen, G. Akerman, A. Kligerman and E. Rinde, Environ. Mol. Mutagen., 2010, 51(2), 89 CAS.
- R. Beveridge, J. Pintos, M.-É. Parent, J. Asselin and J. Siemiatycki, Am. J. Ind. Med., 2010, 53(5), 476 Search PubMed.
- A. A. Nemec, L. M. Zubritsky and A. Barchowsky, Chem. Res. Toxicol., 2010, 23(2), 396 Search PubMed.
- N. M. Dogan, C. Kantar, S. Gulcan, C. J. Dodge, B. C. Yilmaz and M. A. Mazmanci, Environ. Sci. Technol., 2011, 45(6), 2278 CrossRef CAS PubMed.
- T. Srinath, T. Verma, P. W. Ramteke and S. K. Garg, Chemosphere, 2002, 48(4), 427 CrossRef CAS PubMed.
- M. Khadem, F. Golbabaei and A. Rahmani, Int. J. Occup. Hyg., 2017, 9(3), 118 Search PubMed.
- A. Bobrowsk, A. Królicka and J. Zarębski, Electroanalysis, 2009, 21(13), 1449 CrossRef.
- W. Jin, G. Wu and A. Chen, Analyst, 2014, 139(1), 235 RSC.
- M. Korolczuk and A. Stępniowska, Talanta, 2012, 88, 427 CrossRef CAS PubMed.
- T. K. Sari, J. Jin, R. Zein and E. Munaf, Anal. Sci., 2017, 33(7), 801 CrossRef CAS PubMed.
- R. Ouyang, S. A. Bragg, J. Q. Chambers and Z.-L. Xue, Anal. Chim. Acta, 2012, 722, 1 CrossRef CAS PubMed.
- E. Punrat, C. Maksuk, S. Chuanuwatanakul, W. Wonsawat and O. Chailapakul, Talanta, 2016, 150, 198 CrossRef CAS PubMed.
- H. Zhang, Q. Liu, T. Wang, Z. Yun, G. Li, J. Liu and G. Jiang, Anal. Chim. Acta, 2013, 770, 140 CrossRef CAS PubMed.
- M. Rong, L. Lin, X. Song, Y. Wang, Y. Zhong, J. Yan, Y. Feng, X. Zeng and X. Chen, Biosens. Bioelectron., 2015, 68, 210 CrossRef CAS PubMed.
- S. Kanwal, Q. Ma, W. Dow, G. Wand and X. Su, Int. J. Environ. Anal. Chem., 2012, 92(2), 210 CrossRef CAS.
- B. B. Campos, M. Algarra, B. Alonso, C. M. Casado and J. C. G. Esteves da Silva, Talanta, 2015, 144, 862 CrossRef CAS PubMed.
- M. D. Luaces, N. C. Martínez, M. Granda, A. C. Valdés and A. M. Gutiérrez, Talanta, 2011, 85(4), 1904 CrossRef CAS PubMed.
- X. Zhao, N. Song, W. Zhou and Q. Jia, Cent. Eur. J. Chem., 2012, 10(4), 927 CAS.
- R. Rubio, A. Sahuquillo, G. Rauret and Ph. Quevauviller, Int. J. Environ. Anal. Chem., 1992, 47(2), 99 CrossRef CAS.
- C. R. T. Tarley, G. F. Lima, D. R. Nascimento, A. R. S. Assis, E. S. Ribeiro, K. M. Diniz, M. A. Bezerra and M. G. Segatelli, Talanta, 2012, 100, 71 CrossRef CAS PubMed.
- Á. Béni, R. Karosi and J. Posta, Microchem. J., 2007, 85(1), 103 CrossRef.
- K. Kiran, K. S. Kumar, B. Prasad, K. Suvardhan, R. B. Lekkala and K. Janardhanam, J. Hazard. Mater., 2008, 150(3), 582 CrossRef CAS PubMed.
- T. Minami, Y. Sohrin and J. Ueda, Anal. Sci., 2005, 21(12), 1519 CrossRef CAS PubMed.
- X. Wen, S. Yang, H. Zhang, X. Zhao and J. Guo, Int. J. Environ. Anal. Chem., 2014, 94(12), 1243 CrossRef CAS.
- X. Qi, S. Gao, G. Ding and A.-N. Tang, Talanta, 2017, 162, 345 CrossRef CAS PubMed.
- B. Markiewicz, I. Komorowicz, A. Sajnóg, M. Belter and D. Barałkiewicz, Talanta, 2015, 132, 814 CrossRef CAS PubMed.
- L.-L. Wang, J.-Q. Wang, Z.-X. Zheng and P. Xiao, J. Hazard. Mater., 2010, 177(1–3), 114 CrossRef CAS PubMed.
- R. E. Wolf, S. A. Morman, P. L. Hageman, T. M. Hoefen and G. S. Plumlee, Anal. Bioanal. Chem., 2011, 401(9), 2733 CrossRef CAS PubMed.
- B. Markiewicz, I. Komorowicz and D. Barałkiewicz, Talanta, 2016, 152, 489 CrossRef CAS PubMed.
- M. J. Powell, D. W. Boomer and D. R. Wiederin, Anal. Chem., 1995, 67(14), 2474 CrossRef CAS.
- F. Laborda, M. P. Górris, E. Bolea and J. R. Castillo, Int. J. Environ. Anal. Chem., 2007, 87(3), 227 CrossRef CAS.
- S. Ressalan, R. S. Chauhan, A. K. Goswami and D. N. Purohit, Rev. Anal. Chem., 1997, 16(2), 69 CAS.
- J. S. Kim, Y. R. Choi, Y. S. Kim, Y. J. Lee, J. H. Ko, S. Y. Kwon and S. B. Heo, Anal. Chim. Acta, 2011, 690(2), 182 CrossRef CAS PubMed.
- J. Sung-woon, L. Hyun-woo and K. Chul-ho, Bull. Korean Chem. Soc., 2011, 32(9), 3437 CrossRef.
- S. Chen, X. Zhang, L. Yu, L. Wang and H. Li, Spectrochim. Acta, Part A, 2012, 88, 49 CrossRef CAS PubMed.
- H. Sereshti, M. V. Farahani and M. Baghdadi, Talanta, 2016, 146, 662 CrossRef CAS PubMed.
- H. Ebrahimzadeh, A. A. Asgharinezhad, N. Tavassoli, O. Sadeghi, M. M. Amini and F. Kamarei, Int. J. Environ. Anal. Chem., 2012, 92(4), 509 CrossRef CAS.
- S. Ling, China Water Wastewater, 2011, 16, 96 Search PubMed.
- S. Ling and X. Hengshen, Adv. Mater. Res., 2012, 356–360, 939 Search PubMed.
- W.-J. Li, C. Tai and X.-X. Gu, Int. J. Environ. Anal. Chem., 2001, 81(2), 127 CrossRef CAS.
- M. A. Zaitoun, Int. J. Environ. Anal. Chem., 2005, 85(6), 399 CrossRef CAS.
- V. Gómez and M. P. Callao, TrAC, Trends Anal. Chem., 2006, 25(10), 1006 CrossRef.
- K. Pyrzynska, TrAC, Trends Anal. Chem., 2012, 32, 100 CrossRef CAS.
- ISO 18412:2005, Water quality. Determination of Chromium (VI). Photometric method for weakly contaminated water Search PubMed.
- A. Zgoka-Grześkowiak and T. Grześkowiak, TrAC, Trends Anal. Chem., 2011, 30(9), 1382 CrossRef.
- N. Campillo, P. Viñas, J. Šandrejová and V. Andruch, Appl. Spectrosc. Rev., 2017, 52(4), 267 CrossRef.
- P. Zhang, L. Hu, R. Lu, W. Zhou and H. Gao, Anal. Methods, 2013, 5, 5376 RSC.
- S. Zaruba, V. Bozóová, A. B. Vishnikin, Y. R. Bazeľ, J. Šandrejová, K. Gavazovc and V. Andruch, Microchem. J., 2017, 132, 59 CrossRef CAS.
- M. Rečlo, E. Yilman, Y. Bazel and M. Soylak, Int. J. Environ. Anal. Chem., 2017, 97(14–15), 1315 CrossRef.
- T. S. Riabukhina, D. Janegová and Ya. R. Bazel, Methods Objects Chem. Anal., 2018, 13(1), 44 CrossRef.
- D. C. Gaswick and J. H. Krueger, J. Am. Chem. Soc., 1969, 91(9), 2240 CrossRef CAS.
- K. E. Howlett and S. Sarsfield, J. Chem. Soc. A, 1968, 683 RSC.
- S. P. Liu, Q. Liu, Z. F. Liu, M. Li and C. Z. Huang, Anal. Chim. Acta, 1999, 379(1–2), 53 CrossRef.
- O. V. Zui, Methods Objects Chem. Anal., 2009, 4(1), 38 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a