
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNi(II)-based coordination polymers for efficient electrocatalytic oxygen evolution reaction
Zhi-Qiang Jiang*a,
Yu-Feng Lia,
Xue-Jun Zhua,
Jin Lua,
Lei Zhang b and
Tian Wen*b
b and
Tian Wen*b
aDeep-processing of Fine Flake Graphite Sichuan Province Key Laboratory of Colleges and Universities, Panzhihua University, Panzhihua, Sichuan 617000, P. R. China. E-mail: jiangzhiqiang@mail.pzhu.edu.cn
bSchool of Chemistry, The University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: tian.wen@unimelb.edu.au
First published on 15th November 2018
Abstract
The exploration of highly efficient, stable and cheap water oxidation electrocatalysts using earth-abundant elements is still a great challenge. Herein, alkaline-stable cationic Ni(II) coordination polymers (Ni-CPs) were successfully obtained under hydrothermal conditions, which could stabilize the incorporation of Fe(III) to form Fe-immobilized Fe@Ni-CPs. The newly developed Ni-based CPs were used for the first time as an effective electrocatalyst for the oxygen evolution reaction in strong alkaline media.
The oxygen evolution reaction (OER) plays a vital role in energy storage and conversion applications due to energy issues and the need for sustainable development.1–4 Because of the sluggish kinetics of the OER, excellent electrocatalysts are required to work in acidic or strong alkaline environments.5,6 Noble metal-based catalysts like IrO2 and RuO2 with high efficiency showed excellent OER catalytic activity.7 However, noble metal with high-cost and scarcity are impractical for scale-up applications. Currently, to substitute these precious metal-based materials, transition-metal-based (Fe, Co, Ni and so on) and metal-free (e.g., N, P and S) hybrid materials have been extensively developed.8–10 For example, metal–organic frameworks (MOFs) such as ZIF-8/67 derived metal–carbon composite materials exhibit promising electrocatalytic performance.11,12 Unfortunately, the pyrolysis process destroys the framework completely and causes agglomeration of metals, resulting in a decreased number of active sites. Therefore, to explore highly efficient and low-cost OER catalysts that can be directly used in the OER without calcination are desired, including complexes and MOFs.
Recently, much of transition bimetallic materials showed excellent electrocatalytic activity.13–15 However, a handful of examples such as Fe–Co-MOFs or Co–Ni-MOFs have been explored due to instability, poor conductivity and harsh synthesis conditions.16–20 Notably, the disadvantages of MOFs have limited their usage in the potential OER. Therefore, it is urgent to develop cheap, stable and active OER catalysts to replace the precious metals. However, this is still a great challenge. Coordination polymers (CPs) with a low-dimensional framework similar to MOFs are constructed by metal ion and organic ligands with potential active sites and functional groups, exhibiting wide applications in sensing, photoluminescence and photocatalysis.21–26 However, rare examples of CPs have been directly explored in the OER. For Fe/Ni-based bimetal electrocatalysts, a novel strategy involves doping Fe(III) into a functional Ni-based CPs, which could enhance the electrocatalytic OER activities.
Herein, we report the hydrothermal synthesis of a Ni-based CPs as a high-performance OER electrocatalyst in strong alkaline solutions (Scheme 1). The blue crystals of [Ni(bp)3·(H2O)2]·(bp)·(ClO4)2 (bp = 4,4′-biprydine) were obtained upon the reaction of bp ligands with NiClO4·6H2O under hydrothermal systems. Chemical stability tests displayed that Ni-CPs could retain their original framework in water or even a strong alkaline (pH = 14) solution after 12 hours (Fig. S1†), which is rarely reported for most transition metal CPs. The thermogravimetric analysis (TGA) showed that there is a significant change at about 110 °C due to the weight loss of the partial guest (Fig. S2†). Interestingly, the cationic Ni-CPs successfully captured the Mohr's salt (ammonium iron(II) sulfate) by taking advantage of the post-synthetic strategy. The obtained Fe@Ni-CPs exhibited a high-efficient OER activity under strong alkaline conditions.
 | ||
| Scheme 1 Illustration of the synthesis process for Fe doped Ni coordination polymers for the OER. | ||
Single-crystal X-ray diffraction analysis revealed that the Ni-CPs crystallized in the C2/c space group (Table S1†). The obtained Ni coordination polymer was the isostructural compound reported by Talham,27 but their packing modes were distinctly different (Fig. S3–S5†). It also had a similar coordination environment to the railroad-like double chains synthesized by Yaghi.28 The parallel chains were occupied by 4,4′-bpy, perchlorate and water molecules. In the Ni-CPs, most of the phenyl rings adopted the face-to-face mode. There were evident π⋯π interactions between the adjacent 4,4′-biprydine (Fig. 1a). In addition, there were strong intermolecular hydrogen bonds between 4,4′-bpy and perchlorate (strong Cl–O⋯C amongst adjacent layers) (Fig. 1a). The weak reaction increased the high density of the framework and protected the coordination bonds against external guest attacking. These chains and guests were further packed with a three-dimensional structure along the c-axis (Fig. 1b).
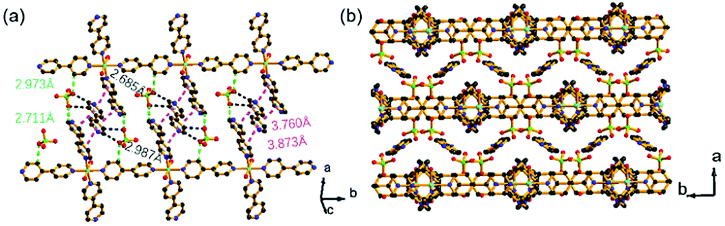 | ||
| Fig. 1 (a) The weak reaction between chains in Ni CPs, showing the Cl–O⋯C (green and black dashed line) and π⋯π interactions (pink dashed line) between the adjacent perchlorate and 4,4′-biprydine, respectively; (b) the packing view of Ni CPs along the c-axis. Cl in green, Ni in pale blue, N in blue, O in red and C in black. H atoms and partial guest molecules were omitted for clarity. | ||
The unique cationic Ni-CPs framework has the potential to immobilize some counterpart ions. To demonstrate this, Mohr's salts were investigated. Most strikingly, the color slowly changed from blue to green in an aqueous solution, given by the optical image (Fig. 2a and b), which not only indicated that Ni-CPs captured Mohr's salts via ion-exchange, but also suggested an alteration in the valence of Fe ions. It was possible that Fe2+ may have been further oxidized to Fe3+ under the O2 and water environment when we prepared the Fe@Ni-CPs (4Fe2+ + 2H2O + O2 = 4Fe3+ + 4OH−). The PXRD pattern showed that the frameworks remained unchanged after doping with Fe ions (Fig. S1†). From the transmission electron microscopy (TEM) images (Fig. 2c), after immobilization, the morphology of the Fe@Ni-CPs was still level and smooth; no ring-like patterns arose corresponding to the selected area for electron diffraction (SAED) (Fig. 2d), indicating that no bulk Fe particles formed during ion-exchange. This was further demonstrated using high resolution TEM (HRTEM) (Fig. 2e), in which there was no lattice fringe of crystallized Fe. The well distribution of C, N, O, Cl, Fe, and Ni in Fe@Ni-CPs was demonstrated by elemental mapping (Fig. 2f–l). Energy-dispersive X-ray spectroscopy (EDX) also agreed well with the above mapping data (Fig. S6†). In addition, the Fe3+ uptake was 4.1 wt%, as determined by inductively coupled plasma atomic emission spectroscopy (ICP). These results showed Fe ions to have been successfully immobilized by the Ni-CPs.
 | ||
| Fig. 2 (a–b) The optical image of Ni-CPs and Fe@Ni-CPs; (c) TEM images of Fe@ Ni-CPs; (d–e) the corresponding SAED and HRTEM pattern. (f–l) Element mapping of C, N, O, Cl, Fe, and Ni in Fe@Ni-CPs. | ||
The X-ray photoelectron spectroscopy (XPS) survey spectrum of the Fe@Ni-CPs also showed the presence of C, N, O, Cl, Fe, and Ni elements (Fig. 3a). The Fe 2p high resolution XPS spectrum exhibited peaks at 725 eV and 711 eV (Fig. 3b), further indicating the presence of the Fe3+ oxidation state. This could be explained by the transformations of Fe2+ to Fe3+ during the ion-exchange process. Similarly, in the Ni 2p spectra (Fig. 3c), two main peaks located at 855.8 eV and 873.5 eV can be ascribed to Ni2+ 2p3/2 and Ni2+ 2p1/2, respectively. These peaks are associated with two shakeup satellite peaks, indicating that Ni still remained in a divalent state. The Cl 2p and N 1s region could be corresponded to the ClO4− and biprydine, respectively (Fig. S7†). The O 1s spectrum (Fig. 3d) was divided into two peaks at 531.7 eV and 533.2 eV, which could be assigned to the OH group from filled H2O molecules and partial ClO4−, respectively.
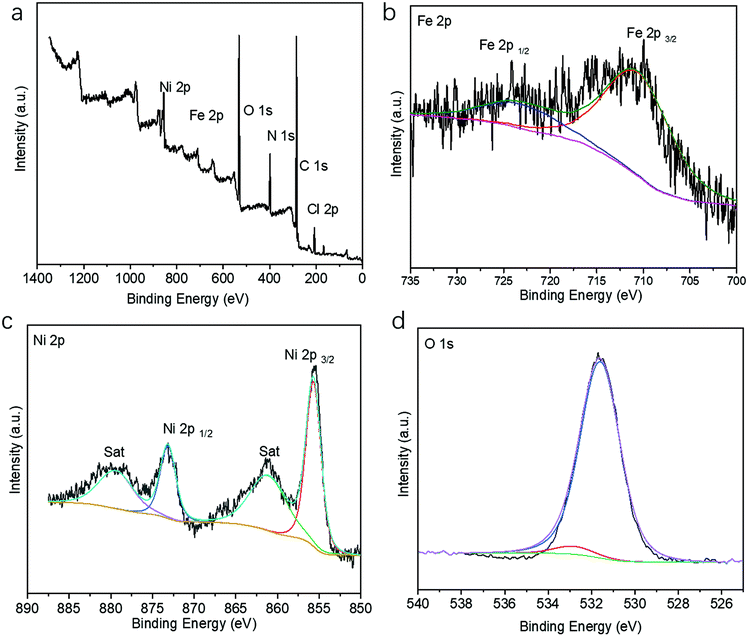 | ||
| Fig. 3 (a) XPS survey spectrum of the Ni-CPs; XPS spectra of the Ni-CPs in the (b) Fe 2p, (c) Ni 2p, and (d) O 1s regions. | ||
The above Ni-CPs with Fe doping encouraged us to investigate its electrocatalytic application in oxygen evolution reaction. To study the electrocatalytic activity of Fe@Ni-CPs for the OER, linear sweep voltammetry (LSV) was performed in a strong alkaline solution (pH = 14) for Fe@Ni-CPs@GC (fresh samples coated on glassy carbon electrode with Nafion binder). Fe@Ni-CPs@GC directly acted as working electrodes and showed good OER activity with an onset potential of 1.52 V (Fig. 4a), overpotential of 368 mV at 10 mA cm−2, and a Tafel slope of 59.3 mV dec−1 (Fig. 4b). These OER performances are close to some reported MOFs catalysts (Table S2†) and even better than commercial benchmark OER catalysts like RuO2 working at the same condition (Fig. S8†). In contrast, the electrocatalytic OER activities of the pristine Ni-CPs@GC without Fe incorporation displayed much worse activity. The onset potential, overpotential (at 10 mA cm−2), and the Tafel slope reached 1.62 V, 458 mV, and 96.8 mV dec−1, respectively (Fig. 4a and b). In addition, Fe@Ni-CPs showed a strong durability during the OER process. The chronoamperometric response of Fe@Ni-CPs displayed a slight anodic current attenuation within 12 h due to the peeling of samples during the evolution of a large amount of O2 gas (Fig. S9†). Furthermore, LSV of Fe@Ni-CPs showed negligible changes after OER tests for 12 h (Fig. S10†). These results indicate that the Fe-doped Ni-CPs with more active sites could serve as an excellent candidate for OER in strong alkaline conditions.
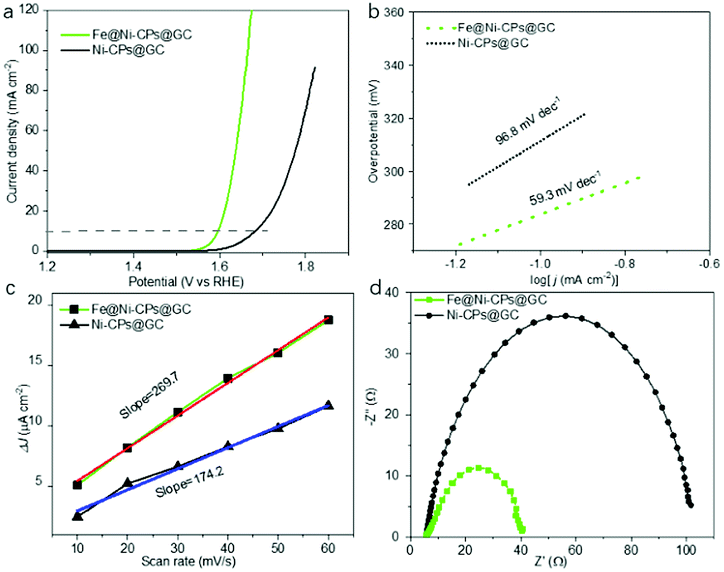 | ||
| Fig. 4 (a) OER polarization curves and (b) Tafel plots of various electrocatalysts in a 1 M KOH aqueous solution; (c) linear relationship of the current density at 1.1 V (vs. RHE) vs. scan rates for Fe@Ni-CPs@GC and Ni-CPs@GC; (d) EIS of Fe@Ni-CPs and Ni-CPs electrode. | ||
When Fe(III) was introduced, the resulting Fe@Ni-CPs catalysts greatly improved OER catalytic performance. There were dynamic collisions between Fe3+ ions and Ni-CPs, which allowed for more accessible catalytic active sites compared to the Ni-CPs. Particularly, Fe(III) doping can contribute to the adsorption and reaction of OH− groups in OER process.29 As a result, Fe@Ni-CPs enhanced charge transfer under an apt electronic environment of the mixed Fe⋯Ni systems. In addition, electrochemical impedance spectrum and double-layer capacitance (Cdl) of the Fe@Ni-CP were also studied. The Cdl of Fe@Ni-CPs was confirmed to be 269.7 μF cm−2 (Fig. 4c and S11†), which is higher than that of Ni-CPs (Cdl = 174.2 μF cm−2) (Fig. 4c and S12†). The semicircular diameter in EIS of Fe@Ni-PCP was smaller than that of Ni-CPS (Fig. 4d). These results further showed that Fe@Ni-CPs were more effective in enlarging the catalytically active surface area, conductivity and synergistic effects between Fe and Ni in comparison to Ni-CPs coated on electrodes.
In conclusion, a new alkaline-stable cationic Ni(II) coordinated polymers was synthesized under hydrothermal conditions. The Ni CPs could quickly interact with Mohr's salt. Interestingly, the Ni CPs could act as a unique oxidation matrix to realize the transformation of Fe2+ to Fe3+ during the ion-exchange process. Furthermore, the resulting Fe@Ni-CPs electrode, for the first time, showed an excellent electrocatalytic activity for OER in strong alkaline media. This study provides a new avenue to explore stable coordinated polymers by incorporating the low-cost and high-activity transition metal, Fe, which will substitute the rare noble metals used in energy-related research.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the Science and Technology Planning Project in Sichuan Province (2015RZ0029), Science and Technology Planning Project in Panzhihua City (2014CY-G-24, 2015CY-G-19, 2016CY-G-4), and the Australian Research Council (DE150100901).Notes and references
- (a) M. Gräetzel, Acc. Chem. Res., 1981, 14, 376 CrossRef; (b) A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141 CrossRef CAS.
- K. Maeda, K. Teramura, D. L. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- Y. Zhou, X. F. Guan, H. Zhou, K. Ramadoss, S. Adam, H. J. Liu, S. Lee, J. Shi, M. Tsuchiya, D. D. Fong and S. Ramanathan, Nature, 2016, 534, 231 CrossRef CAS PubMed.
- B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. Lou and X. C. Wang, Nat. Energy, 2016, 1, 15006 CrossRef CAS.
- A. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342 CrossRef PubMed.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060 RSC.
- (a) C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS PubMed; (b) C. C. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347 CrossRef CAS PubMed.
- (a) H. Zhang, Z. Ma, J. Duan, H. Liu, G. Liu, T. Wang, K. Chang, M. Li, L. Shi, X. Meng, K. Wu, J. K. Ye, M. Li, L. Shi, X. Meng, K. Wu and J. Ye, ACS Nano, 2016, 10, 684 CrossRef CAS PubMed; (b) H. Zhang, Z. Ma, G. Liu, L. Shi, J. Tang, H. Pang, K. Wu, T. Takei, J. Zhang, Y. Yamauchi and J. Ye, NPG Asia Mater., 2016, 8, 293 CrossRef.
- (a) Y. H. Qian, I. A. Khan and D. Zhao, Small, 2017, 13, 1701143 CrossRef PubMed; (b) M. Jahan, Z. L. Liu and K. P. Loh, Adv. Funct. Mater., 2013, 23, 5363 CrossRef CAS; (c) X. B. Liu, Y. C. Liu and L. Z. Fan, J. Mater. Chem. A, 2017, 5, 15310 RSC; (d) Y. J. Tang, M. R. Gao, C. H. Liu, S. L. Li, H. L. Jiang, Y. Q. Lan, M. Han and S. H. Yu, Angew. Chem., Int. Ed., 2015, 54, 12928 CrossRef CAS PubMed.
- (a) S. Peng, L. Li, X. Han, W. Sun, M. Srinivasan, S. G. Mhaisalkar, F. Cheng, Q. Yan, J. Chen and S. Ramakrishna, Angew. Chem., Int. Ed., 2014, 126, 12802 CrossRef; (b) M. Li, T. Liu, X. Bo, M. Zhou, L. Guo and S. Guo, Nano Energy, 2017, 33, 221 CrossRef CAS; (c) S. Dong, X. Chen, X. Zhang and G. Cui, Coord. Chem. Rev., 2013, 257, 1946 CrossRef CAS.
- H. B. Zhang, J. W. Nai, L. Yu and X. W. Lou, Joule, 2017, 1, 77 CrossRef CAS.
- (a) J. Yang, F. Zhang, H. Lu, X. Hong, H. Jiang, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2015, 54, 10889 CrossRef CAS PubMed; (b) J. Yang, F. Zhang, X. Wang, D. He, G. Wu, Q. Yang, X. Hong, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 12854 CrossRef CAS PubMed; (c) Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 6937 CrossRef CAS PubMed; (d) X. Wang, W. Chen, L. Zhang, T. Yao, W. Liu, Y. Lin, H. Ju, J. Dong, L. Zheng, W. Yan, X. Zheng, Z. Li, X. Wang, J. Yang, D. He, Y. Wang, Z. Deng, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 9419 CrossRef CAS PubMed.
- (a) J. Chi, H. M. Yu, B. W. Qin, L. Fu, J. Jia, B. L. Yi and Z. G. Shao, ACS Appl. Mater. Interfaces, 2017, 9, 464 CrossRef CAS PubMed; (b) Y. J. Tang, C. H. Liu, W. Huang, X. L. Wang, L. Z. Dong, S. L. Li and Y. Q. Lan, ACS Appl. Mater. Interfaces, 2017, 9, 16977 CrossRef CAS PubMed.
- F. L. Li, Q. Shao, X. Q. Huang and J. P. Lang, Angew. Chem., Int. Ed., 2018, 57, 1888 CrossRef CAS PubMed.
- (a) J. M. Lv, D. X. Bai, L. Yang, Y. Guo, H. Yan and S. L Xu, Chem. Commun., 2018, 54, 8909 RSC; (b) D. Li, H. Baydoun, C. N. Verani and S. L. Brock, J. Am. Chem. Soc., 2016, 138, 4006 CrossRef CAS PubMed.
- J. Shen, P. Liao, D. Zhou, C. He, J. Wu, W. Zhang, J. Zhang and X. Chen, J. Am. Chem. Soc., 2017, 139, 1778 CrossRef CAS PubMed.
- S. L. Zhao, Y. Wang, J. C. Dong, C. T. He, H. J. Yin, P. F. An, K. Zhao, X. F. Zhang, C. Gao, L. J. Zhang, J. W. Lv, J. X. Wang, J. Q. Zhang, A. M. Khattak, N. A. Khan, Z. X. Wei, J. Zhang, S. Q. Liu, H. J. Zhao and Z. Y. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- J. Duan, S. Chen and C. Zhao, Nat. Commun., 2017, 8, 15341 CrossRef CAS PubMed.
- (a) C. W. Kung, J. E. Mondloch, T. C. Wang, W. Bury, W. Hoffeditz, B. M. Klahr, R. C. Klet, M. J. Pellin, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2015, 7, 28223 CrossRef CAS PubMed; (b) L. Wang, Y. Z. Wu, R. Cao, L. T. Ren, M. X. Chen, X. Feng, J. W. Zhou and B. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 16736 CrossRef CAS PubMed; (c) K. Maity, K. Bhunia, D. Pradhan and K. Biradha, ACS Appl. Mater. Interfaces, 2017, 9, 37548 CrossRef CAS PubMed.
- J. Xing, K. Guo, Z. Zou, M. Cai, J. Du and C. Xu, Chem. Commun., 2018, 54, 7046 RSC.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC.
- Y. J. Cui, Y. F. Yue, G. D. Qian and B. L. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed.
- M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782 CrossRef PubMed.
- Q. L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468 RSC.
- M. Albrecht, M. Lutz, A. L. Spek and G. van Koten, Nature, 2000, 406, 970 CrossRef CAS PubMed.
- T. Wen, D. Zhang, J. Liu, R. Lin and J. Zhang, Chem. Commun., 2013, 49, 5660 RSC.
- J. D. Woodward, R. Backov, K. A. Abboud, H. Ohnuki, M. W. Meisel and D. R. Talham, Polyhedron, 2003, 22, 2821 CrossRef CAS.
- O. M. Yaghi, H. l. Li and T. L. Groy, Inorg. Chem., 1997, 36, 4292 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Chen, Y. Han and S. Z. Qiao, J. Am. Chem. Soc., 2017, 139, 3336 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC [1858717]. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra07492e |
| This journal is © The Royal Society of Chemistry 2018 |