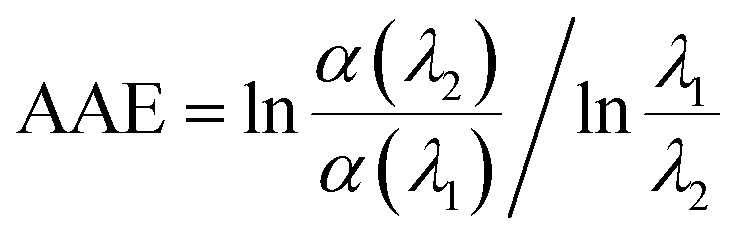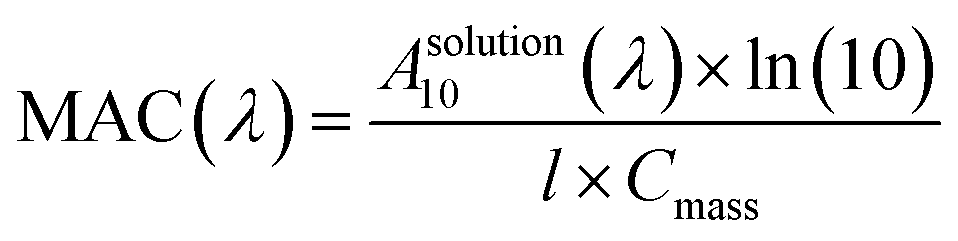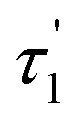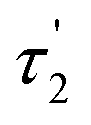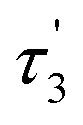DOI:
10.1039/C8RA06913A
(Paper)
RSC Adv., 2018,
8, 38566-38573
Formation and photochemical properties of aqueous brown carbon through glyoxal reactions with glycine†
Received
18th August 2018
, Accepted 29th October 2018
First published on 15th November 2018
Abstract
In recent years, brown carbon aerosols, as important contributors to light absorption and climate forcing by aerosols, have been forefront in the field of atmospheric research. Aqueous brown carbon can be formed through the aqueous reaction of glyoxal (GX) with glycine (Gly). GX–Gly mixtures exhibit changes in their optical properties in the ultraviolet and near visible regions, which can be monitored with ultraviolet/visible and fluorescence spectroscopy. In this study, we quantified the absorption and excitation–emission matrix spectra during the formation of aqueous brown carbon, which was generated from GX–Gly mixtures. The formation of brown carbon was further evidenced using several optical parameters, including absorption coefficient, absorption Angstrom exponents, mass absorption coefficient, effective quantum yields and fluorescence lifetime values. The results of hydrogen peroxide oxidation photolysis revealed the probable removal processes of the atmospheric aqueous brown carbon. The fluorescence lifetime values of the brown carbon samples were less than 10 ns. Liquid chromatography combined with mass spectrometry analysis was used to investigate the probable chemical composition of the brown carbon samples from GX–Gly mixtures.
Introduction
Atmospheric aerosols play important roles in determining light absorption and climate forcing, which follow direct or indirect mechanisms.1–3 Aerosol particles can both scatter and absorb UV-Visible sunlight. Atmospheric “brown carbon” is the collective term for the organic compounds in the atmosphere, which are responsible for light absorption and climate forcing, but are poorly characterized. In recent years, brown carbon aerosols have been forefront in the field of atmospheric research.4–10 Due to the contribution to radiative forcing, regional air quality variation, and global climate change, brown carbon aerosols have drawn much attention considering atmospheric aerosols.11 As a category of condensed-phase organic carbonaceous compounds, brown carbon can efficiently absorb solar radiation in the ultraviolet (UV) and near visible (Vis) regions with strong wavelength dependency.12,13 The light absorption capacity of brown carbon in the ultraviolet band increases with a decrease in wavelength. Therefore, it has a significant impact on the global climate.14–17 Brown carbon substantially influences and even dominates the total aerosol absorption at specific wavelengths in certain geographic areas.18–21 Atmospheric brown carbon particles have a high degree of complexity in terms of their chemical compositions and sources.22–27 Their sources can be generally classified into two major categories: (i) primary emissions from biomass burning and fossil fuel combustion and (ii) secondary anthropogenic emissions from a variety of precursors through solid- or aqueous-phase reactions (e.g., oligomerization).1,14
The formation of brown carbon through aqueous reactions of small carbonyl molecules has been widely reported in the literature. Powelson et al.1 studied the formation of brown carbon through the reactions of aqueous phase carbonyl compounds with amines and ammonium sulfate (AS). They concluded that amines were much more effective than AS on a per mole basis by comparing the absorption intensity of the brown carbon in the UV/Vis region. Tang et al.4 reported the optical and physicochemical properties of brown carbon aerosols formed through the reactions of methylglyoxal (MG) with AS. As a class of secondary organic aerosols, they were useful references for atmospheric chemistry and climate studies. Aiona et al.7 examined the molecular composition, mass absorption coefficients, fluorescence spectra, and photolytic efficiency of brown carbon samples, which were produced through the evaporation of aqueous MG–AS mixtures. They found that the MG–AS brown carbon system may affect the atmospheric photochemistry in aerosol particles, but with limited impact on the direct radiative forcing of climate. Hawkins et al.8 provided evidence for the pyrazine-based chromophores in cloud-water mimics containing MG and AS, which had implications in the further clarification of the reactivity and degradation pathways of brown carbon. According to the optical properties and chemical composition of individual brown carbon components produced through the reactions of MG and AS, Lin et al.21 found that the reduced-nitrogen organic compounds formed in the reactions between atmospheric carbonyls and ammonia/amines are important brown carbon chromophores.
Although numerous studies have been reported1,4,5,8,21 on brown carbon aerosols, knowledge on the optical and chemical properties of that formed through the aqueous reaction of carbonyl compounds with nitrogen-containing compounds is scarce. In our previous study we demonstrated that Maillard (browning) reactions of carbonyl compound hydroxyacetone (HA) with Gly lead to products with significantly enhanced absorption coefficients.10 In the current study we also observed that the formation of secondary organic material brown carbon through aqueous reactions between GX and Gly was a complex chemical reaction process. In our studies, the fluorescence intensities of GX–Gly mixtures were found to increase with reaction time, reaching a maximum value after approximately 7 days and manifesting very obvious changes at 13 days and 19 days. The reason for choosing 19 days is that it is comparable to (or longer than) the average lifetime of aerosol particles in the troposphere.4 Specifically, the reaction time (1–19 days) was optimized. The formation of aqueous brown carbon aerosols from GX–Gly mixtures studied herein was further elucidated using the absorption Angstrom exponent (AAE). The AAE was calculated to be approximately 5 for the visible wavelength range of 400–480 nm, which is closer to the range of 2 to 7 measured for brown carbon in field studies.5,6 Besides, some other fundamental optical parameters were also obtained, such as the wavelength-dependent absorption coefficient (α), mass absorption coefficient (MAC), effective quantum yield (QY) and fluorescence lifetime values. Furthermore, the photochemical catalytic properties of the as-obtained brown carbon in aqueous phase were studied in terms of hydrogen peroxide (H2O2) oxidation. Liquid chromatography (LC) combined with mass spectrometry (MS) analysis was used to investigate the probable chemical composition of the brown carbon samples from GX–Gly mixtures. This study aimed to characterize the optical properties and chemical composition of aqueous brown carbon samples formed from GX–Gly mixtures by using UV/Vis spectroscopy, fluorescence spectroscopy and LC/MS analysis, providing fundamental understanding of the important effects of aqueous brown carbon for atmospheric chemistry and climate protection.
Experimental section
Aqueous brown carbon stock solutions were prepared in ultrapure water with ammonium sulfate (AS, >99.0%, Beijing Chemical Works), glycine (Gly, >99.0%, Tianjin Guangfu Fine Chemical Research Institute) and glyoxal (GX, >40%, Tianjin FuChen Chemical Reagents Factory). Brown carbon stock solutions from GX–Gly were prepared by mixing 15 mL Gly (0.75 M), 15 mL GX (0.75 M) and 15 mL ultrapure water. The final concentrations of the stock solutions of GX–Gly were ∼0.25 M for Gly and ∼0.25 M for GX. The stock solutions were stored in 100 mL transparent glass beakers covered with plastic wrap, and kept in dark at room temperature.28
A U-3900 UV/Vis spectrophotometer (Hitachi, Japan) was employed to record the absorbance of the samples in the wavelength range of 200–700 nm in a 1 cm-quartz cell. A reference absorption spectrum of ultrapure water was used as the background. An F-97 fluorescence spectrometer (Pgeneral, Shanghai, China) was used in EEM mode to map the development of fluorescence over the entire range of excitation (λex) and emission (λem) bands. In brief, for the EEM spectra, the excitation wavelength was varied over 250–480 nm at a step of 10 nm, while the emitted fluorescence strength was recorded over 300–600 nm at a step of 1 nm. In the H2O2 oxidation experiment, 2.98 mL 0.025 M brown carbon sample (containing 0.025 M GX) and 0.02 mL 0.75 M hydrogen peroxide (30%, Wuxi City Yasheng Chemical CO., LTD) were mixed thoroughly. The mixed solutions were then placed in a 1 cm quartz cuvette for 254 nm illumination (Ultraviolet Analyzer, Hangzhou Qi Wei Instrument CO., LTD) and absorption measurements. The final concentration of H2O2 was ∼5 mM in the brown carbon solutions. Besides, the fluorescence lifetimes of the aqueous brown carbon samples were measured using a time-correlated single-photon counting fluorescence spectrometer (FLS920, Edinburgh Instruments Ltd.) with a TCSPC Laser. The fixed excitation wavelength of the TCSPC Laser in the instrument was 404 nm. Thus, the measured parameters of fluorescence lifetime were λex = 404 nm and λem = 600 nm.
The brown carbon solutions were analysed using an Agilent 6520 Accurate-Mass Q-TOF LC/MS. The column used for HPLC analysis was an Imtakt Scherzo SM-C18 (130 Å, 3 μm, 150 × 3.0 mm), part number SM035. The SM-C18 column was operated at a flow rate of 0.3 mL min−1 with the following elution protocol: hold for 5 min at 3% of CH3CN, 1 min for 10% CH3CN, 4 min hold at this level, 43 min linear gradient to 90% CH3CN, 7 min hold at this level, 1 min return to 10% CH3CN, and another hold until the total scan time reached 85 min. In the experiment, the column temperature was maintained at ambient temperature and the sample injection volume was 2 μL. In the scan mode, positive ions entering the MS were analyzed over the m/z range of 100 to 400 nm. The acquisition rate was 1 spectra per s with the acquisition time of 1000 ms per spectrum.
Results and discussion
UV/Vis light absorption
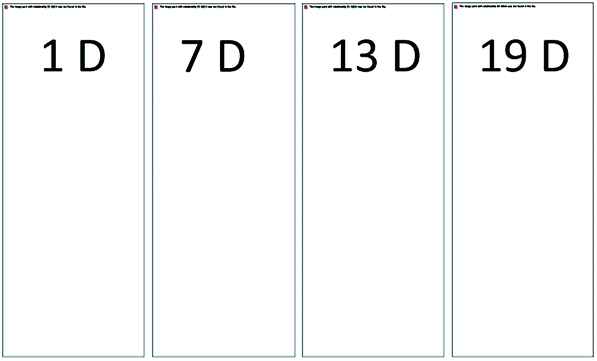
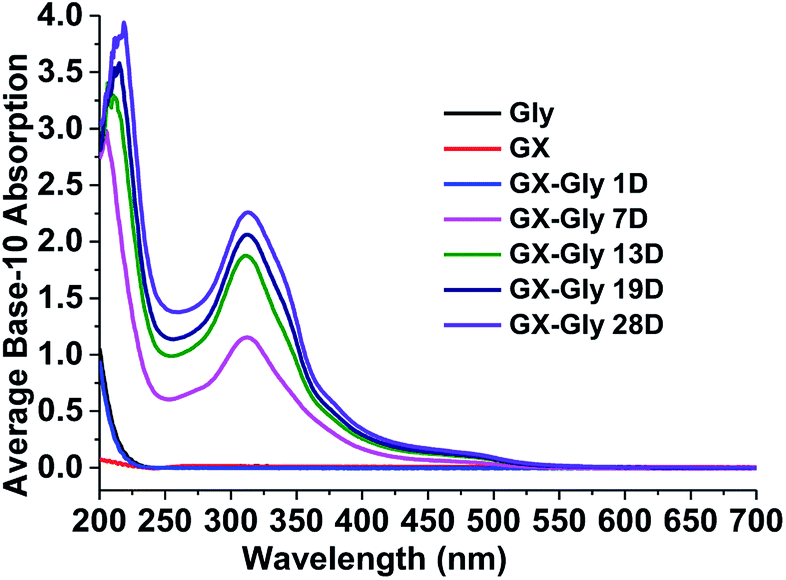
For the stock GX–Gly solutions, visual inspection revealed that their brown coloration increased with an increase in reaction time, as shown in Fig. 1. After reaction for a certain number of days (D), 1.00 mL GX–Gly stock solutions were diluted with 9.00 mL ultrapure water, yielding a GX concentration of 0.025 M. Fig. 2 shows UV/Vis the absorption spectra of the diluted GX–Gly mixtures after reaction for different times. The base-10 absorption coefficient, α, of the GX–Gly mixtures was calculated using the formula αSOA(λ) = A(λ)/l,29 where, A(λ) stands for the base-10 absorption strength and l stands for the quartz cuvette path length. The background was determined by measuring the absorption of ultrapure water.
 |
| | Fig. 1 Photographs of the 0.25 M GX–Gly stock solutions (containing 0.25 M GX) with a change in color over time. | |
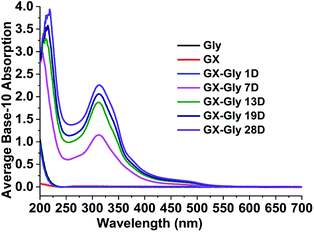 |
| | Fig. 2 UV/Vis absorption spectra of the GX–Gly mixtures after reaction for different times over 250–480 nm. | |
As shown in Fig. 2, compared with 0.025 M Gly and 0.025 M GX, the base-10 absorption coefficient, α, of the GX–Gly mixtures significantly increased with an increase in reaction time in the ultraviolet (250–400 nm) and near visible (400–480 nm) regions. It should be noted that their absorption at longer wavelengths (>480 nm) also increased with reaction time (19 days); however, the absolute changes are very small, which can be omitted. The spectral data also show that the GX–Gly mixtures displayed a broad absorption band, with a peak at about 310 nm. For the GX–Gly mixtures, a new peak at 314 nm was observed in the production of light-absorbing species compared with the un-reacted solutions. Also, 314 nm was close to that obtained in the previous measurements by Powelson et al.1 (320 nm). The differences were likely due to the effects of pH and temperature.
The absorption Angstrom exponent (AAE) of the brown carbon samples was given by the following equation,
| |
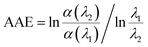 | (1) |
where, AAE differentiates the black and brown carbons and model the effects of light-absorbing aerosol on atmospheric radiative transfer.
1 Generally, AAE = 1 is for BC, while atmospheric brown carbon has an AAE in the range of 2–7,
30 which is as high as 11.4 for pine smoke extracts.
31 The AAE of the GX–Gly mixtures in this study is about 5 (
Table 1) over the near visible wavelength range of 400–480 nm, which overlaps with that of atmospheric brown carbon. Therefore, the GX–Gly mixtures can be regarded as brown carbon with a moderate absorption.
Table 1 Measured base-10 absorption coefficients (α) corresponding to 5.8 × 10−7 M QS solutions and 0.025 M GX–Gly mixtures. The 362–600 nm integrated emission intensities correspond to 350 nm excitation. The effective quantum yield (QY) values were calculated with eqn (2). The last two columns contain the fluorescence index (FI) and the index of recent autochthonous contribution (BIX)
| Solution |
α (350 nm) cm−1 |
Iarea |
QY% |
AAE 400–480 nm |
FI |
BIX |
| QS (5.8 × 10−7 M) |
0.00603 |
12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 269.4 269.4 |
51 |
— |
— |
— |
| GX–Gly (7 D) |
0.598 |
14119.0 |
0.59 |
6.73 |
1.57 |
0.59 |
| GX–Gly (13 D) |
0.930 |
12752.1 |
0.34 |
5.32 |
1.67 |
0.58 |
| GX–Gly (19 D) |
1.107 |
10789.3 |
0.24 |
5.35 |
1.69 |
0.59 |
| GX–Gly (28 D), before photolysis |
1.298 |
10241.6 |
0.20 |
5.39 |
1.71 |
0.57 |
| GX–Gly (28 D), after photolysis |
1.036 |
15030.6 |
0.36 |
7.39 |
1.80 |
0.57 |
Excitation–emission spectra and effective quantum yield
Excitation–emission spectra (EEM plot). The emission intensity is usually displayed in a contour plot as a function of the excitation and emission wavelengths in EEM plot.32,33 Fig. 3 shows EEM plots of the GX–Gly mixtures after reaction for different times. Similar fluorescence contour patterns were observed at 7 days, 13 days and 19 days; however, the relative emission intensities and the peak shape exhibited no significant change. There were more wide fluorescence absorption peaks in the excitation range of 350–430 nm. The brown carbon samples of the GX–Gly mixtures after reaction for 7 days, 13 days and 19 days exhibited maximum emission intensities when they were excited at wavelengths of 350 nm and 400 nm. The average Stokes shift, which is defined as the average difference between λmaxem and λex, was about 92, 94 and 97 nm, respectively.
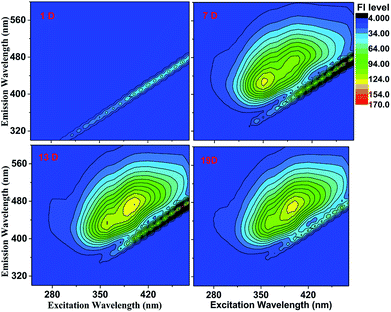 |
| | Fig. 3 EEM plots of the GX–Gly mixtures after reaction for different times. The fluorescence intensity (FI level, color coded as shown on the right) increased slightly with an increase in reaction time. | |
The EEM intensities at different excitation and emission wavelengths can be used to indicate the type and source of the dissolved organic matters.29,34 According to Birdwell et al.,35 the index of recent autochthonous (i.e., produced by the original source of dissolved organic matter as opposed to externally produced29) contribution (BIX) was calculated using the ratio of the emission intensities at 380 and 430 nm to that at 310 nm,35 while the fluorescence index (FI) was calculated using the ratio of the emission intensities at 450 and 500 nm to that at 370 nm.35 In general, FI ≈ 1.4 is observed for terrestrially-derived fulvic acids, while FI ≈ 1.9 corresponds to fulvic acids of microbial origin.35 The degree of aromaticity in fulvic acids (as assessed by the fraction of sp2 carbon) decreased from 30% to 10%, and FI increased from 1.4 to 1.9.35
If the observed FI values fall in range of 1.5 ± 0.4, the intermediate fluorescent properties are relative to the terrestrial and biologic dissolved organic matters.29 Table 1 lists the two parameters calculated for the GX–Gly mixtures in this study. Here, the observed FI values of the GX–Gly mixtures increased from 1.57 to 1.69, which are quite close to 1.9. Therefore, the GX–Gly mixtures probably correspond to fulvic acids of microbial origin, where the degree of aromaticity in the fulvic acids probably decreased from 30% to 10%. According to Huguet et al.,36 BIX ∼ 0.6 corresponds to DOM with a low content of autochthonous component, while BIX > 1 is observed for dissolved organic matters of biological origin. The BIX values for the GX–Gly mixtures had an average level of 0.6 (Table 1), which is consistent with that of a low content of autochthonous component. In summary, during the reaction of the GX–Gly mixtures, there may have been fulvic acids of microbial origin, but the product concentration is very low.
Effective quantum yields (QY). The effective quantum yields (QY) of the samples were measured, as described by Lee et al.29 using quinine sulfate (QS). QS is a convenient standard with QYQS = 51%.37,38 In our previous studies, we calculated the absorption coefficient (0.00603 cm−1) of QS at the peak absorption (350 nm) and the corresponding integral area (12269.4) between 362 and 600 nm using 5.8 × 10−7 M QS solutions.10 The QY values of the brown carbon samples were calculated using the following formula:| |
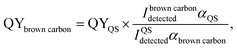 | (2) |
where, I is the measured fluorescence emission intensities integrated over all the emission wavelengths and α represents the absorption coefficients. The resulting QY values of the GX–Gly mixtures are listed in Table 1. Fig. 2 demonstrates that α increased significantly during the reaction periods. However, the QY values decreased slightly from 0.59% to 0.24% and they had the same order of magnitude (∼0.01), as shown in Table 1.
H2O2 oxidation photolysis of the brown carbon solutions
Mass absorption coefficients of the brown carbon solutions. To understand the atmospheric removal processes of the aerosol brown carbon, the photochemical behavior of the brown carbon in terms of H2O2 oxidation in the aqueous phase was studied. The extent of browning of the GX–Gly mixtures was quantified by using effective mass absorption coefficient (MAC). The wavelength-dependent MAC (cm2 g−1) can be directly calculated busing the following formula:| |
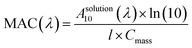 | (3) |
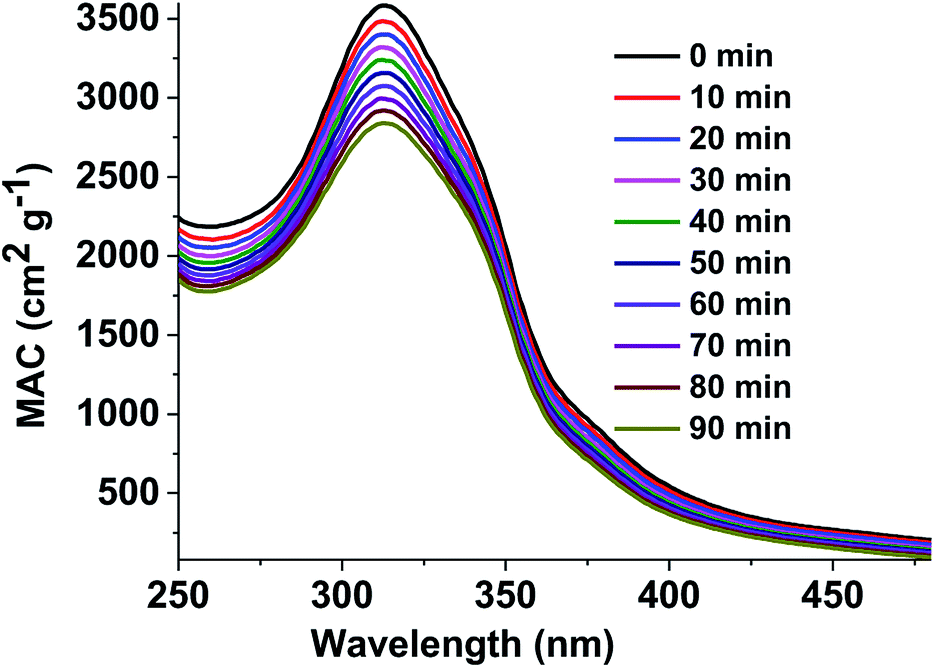
where, A10 represents the base-10 absorbance, Cmass (g cm−3) represents mass concentration, and l (cm) represents path length. The MAC spectra of the GX–Gly mixtures before and after H2O2 oxidation at different photolysis times are shown in Fig. 4. The absorption coefficient is maximum in the UV range of 250–400 nm and there is a tail in the visible range of 400–480 nm. The shapes of the spectra1 agree with the characteristics of typical atmospheric brown carbon material. Although omitted from Fig. 4, the MAC continues to decline at approximately the same rate upon further photolysis, which is consistent with the previous measurements by Lee et al.27
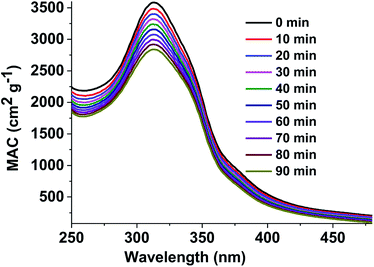 |
| | Fig. 4 UV/Vis absorption spectra recorded during the photolysis of the GX–Gly mixtures. The vertical axis corresponds to the mass absorption coefficient (MAC) calculated with eqn (3). | |
Effective rate of photolysis of the aqueous brown carbon samples. Fig. 4 shows UV/Vis absorption spectra of the GX–Gly mixtures recorded during photolysis. The exposure of the aqueous GX–Gly mixtures to an ultraviolet analyzer moderately reduced the MAC, probably due to the photodegradation of the chromophores of the GX–Gly mixtures. Although they are not included in Fig. 4, the MAC data continued to decline at the same rate with an increase in photolysis time. Due to the very large number of individual chromophores in the GX–Gly mixtures, quantitative interpretation of these measurements is rather complicated.The effective rate constant, κ, considering the first-order kinetic process, is commonly used to approximate the MAC decay,27
| | |
MACt = MAC0 × exp(−κt).
| (4) |
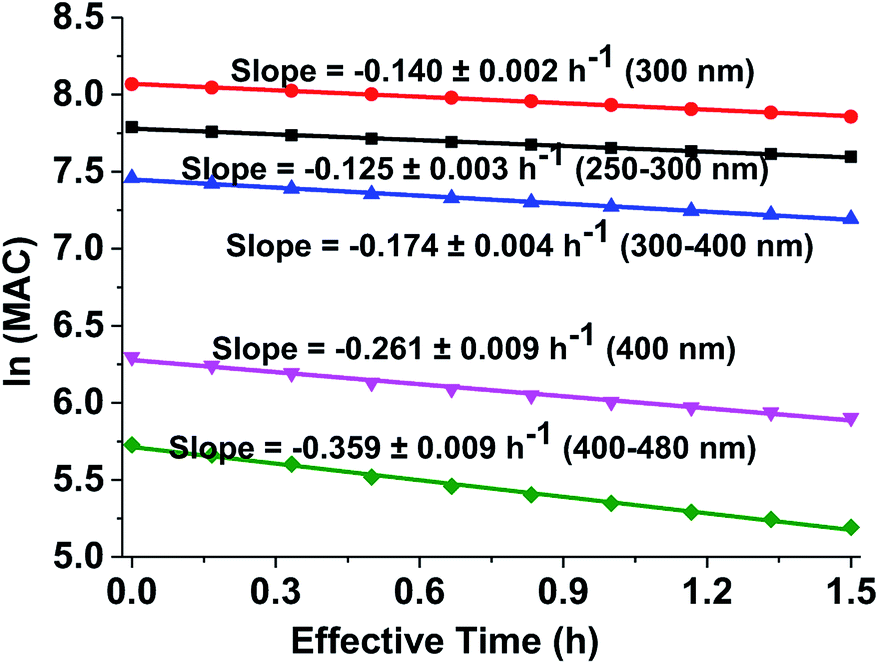
Fig. 5 shows the dependence of the average MAC of the brown carbon samples on the effective photolysis time of H2O2 oxidation. The effective rate constant κ followed the order in the visible range of (κ = 0.359 h−1) > 400 nm (κ = 0.261 h−1) > the near-UV range (κ = 0.174 h−1) > 300 nm (κ = 0.140 h−1) > short-UV range (κ = 0.125 h−1).
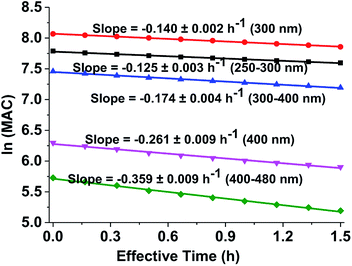 |
| | Fig. 5 Decay in absorbance of the GX–Gly aqueous mixtures during irradiation. The average ln(MAC) was plotted against the effective photolysis time in the short-UV region (250–300 nm, black square) at 300 nm (red circle), in the near-UV region (300–400 nm, blue regular triangle) at 400 nm (pink inverted triangle), and in the visible region (400–480 nm, green rhombus). | |
κ values can be used to derive the empirical half-lives, τ = ln(2)/κ,27 which are listed in Table 2. The data on photodegradation of the GX–Gly mixtures obtained according to the oxidation of H2O2 is also included. The distinctive peak at about 312 nm was significantly weakened after photolysis for 1.5 h and the brown color of the GX–Gly mixtures became lighter during the H2O2 oxidation photolysis process.
Table 2 Effective rate constant, κ, (h−1) and effective half-life, τ, (h) for the disappearance of absorbance at different wavelengths in the GX–Gly mixtures
| Wavelength (nm) |
Short-UV |
300 |
Near-UV |
400 |
Visible |
| κ |
0.125 |
0.140 |
0.174 |
0.261 |
0.359 |
| τ |
5.55 |
4.95 |
3.98 |
2.66 |
1.93 |
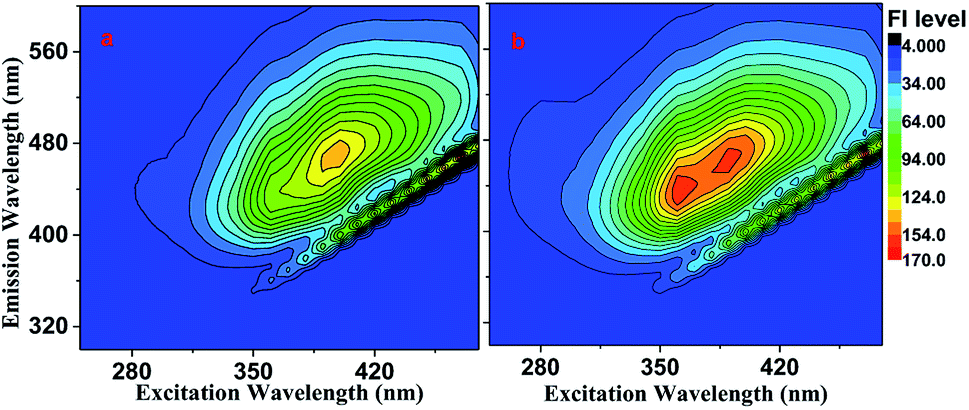
Fluorescence of the brown carbon samples. Fig. 6 shows the EEM spectra of the GX–Gly mixtures, which were moderately fluorescent both before and after the H2O2 oxidation photolysis. The QY values were ∼0.20% and ∼0.36% before and after the H2O2 oxidation photolysis, respectively, as shown in Table 1. The overall absorption coefficient of the GX–Gly mixtures was reduced due to the H2O2 oxidation photolysis (Fig. 4). However, their fluorescence intensity slightly increased, as shown in Fig. 6b. One possible reason for the increased fluorescence intensity is the loss of nitroaromatic compounds during the photolysis.27
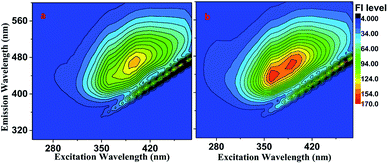 |
| | Fig. 6 EEM spectra of the GX–Gly mixtures recorded (a) before and (b) after H2O2 oxidation photolysis. | |
Measurement of fluorescence lifetime. Fluorescence lifetime, as an intrinsic property of a fluorophore, can be considered a state function because it does not depend on the initial perturbation conditions, such as excitation wavelength and method of measurement.39,40 In addition, fluorescence lifetime is a parameter largely independent of the fluorescence intensity and fluorophore concentration.41 As shown in Fig. 3, the maximum emission was observed at the wavelength of 600 nm. The fluorescence decay curve of the brown carbon samples of the GX–Gly mixtures that were reacted for 36 days is shown in Fig. 7. Due to the variability of the products in the GX–Gly mixtures, the calculated fluorescence lifetime values are not unique, which are 0.44, 2.37, 8.03, respectively, as shown in Table 3.
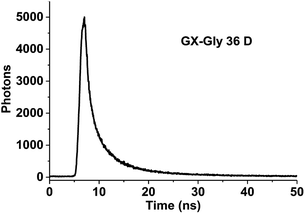 |
| | Fig. 7 Fluorescence decay curve of the GX–Gly mixtures (λex = 404 nm and λem = 600 nm). | |
Table 3 The fluorescence lifetimes of the brown carbon samples
| Solution |
(ns) |
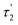
(ns) |
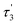
(ns) |
χ2 |
| GX–Gly |
0.44 |
2.37 |
8.03 |
1.001 |
Liquid chromatography/mass spectrometry analysis
Although we investigated the optical properties of the GX–Gly mixtures by UV/Vis and fluorescence spectra, their detailed chemical composition should be well understood. Therefore, Fourier transform infrared (FTIR) and Raman spectroscopy were employed to further chemically characterize the brown carbon samples, as shown in Fig. S1 and S2.† However, the FTIR and Raman spectral properties of the brown carbon samples were very similar to the relevant Gly solutions, which means that these two techniques are unsuitable for detecting the organic components of this type of brown carbon mixture. Lin et al.21 noted that liquid chromatography (LC) coupled with mass spectrometry (MS) to investigate the chemical composition of brown carbon samples from methylglyoxal and ammonium sulfate mixtures resulted in improved separation, which will significantly advance understanding of brown carbon chemistry. In their studies, the SM-C18 column showed better capacity for resolving the brown carbon chromophores than five other columns.21 Thus, we also used LC/MS analysis to investigate the probable chemical composition of the brown carbon samples from GX–Gly mixtures.
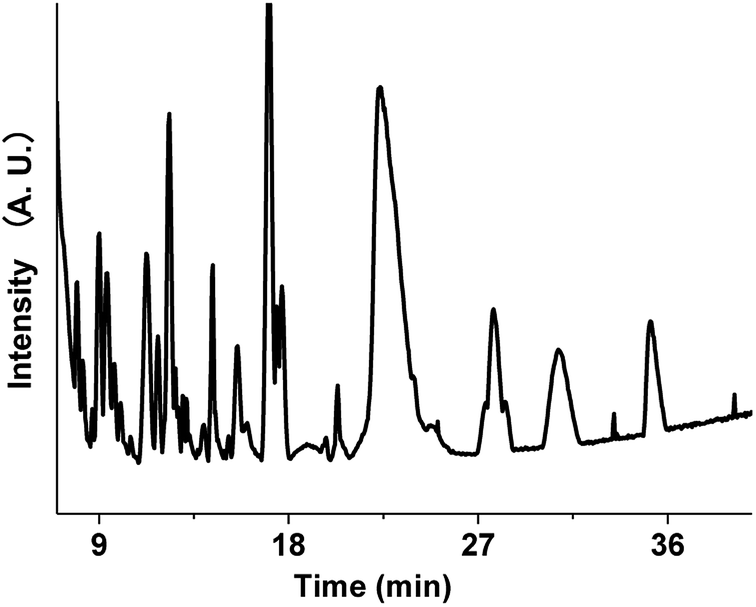
The aqueous brown carbon samples were stored in the dark and incubated at room temperature for ∼32 days prior to their analysis. The brown carbon materials produced by the reactions between GX and Gly are complicated, and their detailed chemical composition has not been fully elucidated. We have examined the base peak chromatograms (BPCs) of the brown carbon samples, as shown in Fig. 8. Fig. 8 clearly shows the presence of abundant organic products from the GX–Gly mixtures, which are not volatile and thus retained in the particles. Determination of the chemical structures of the products was out the scope of this paper. Nevertheless, the LC/MS experimental results are consistent with the UV/Vis light absorption results.
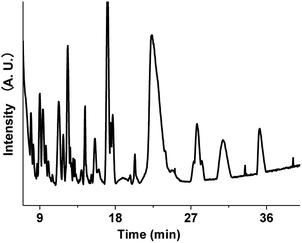 |
| | Fig. 8 Base peak chromatograms (BPCs) of the brown carbon mixtures separated using the SM-C18 column. | |
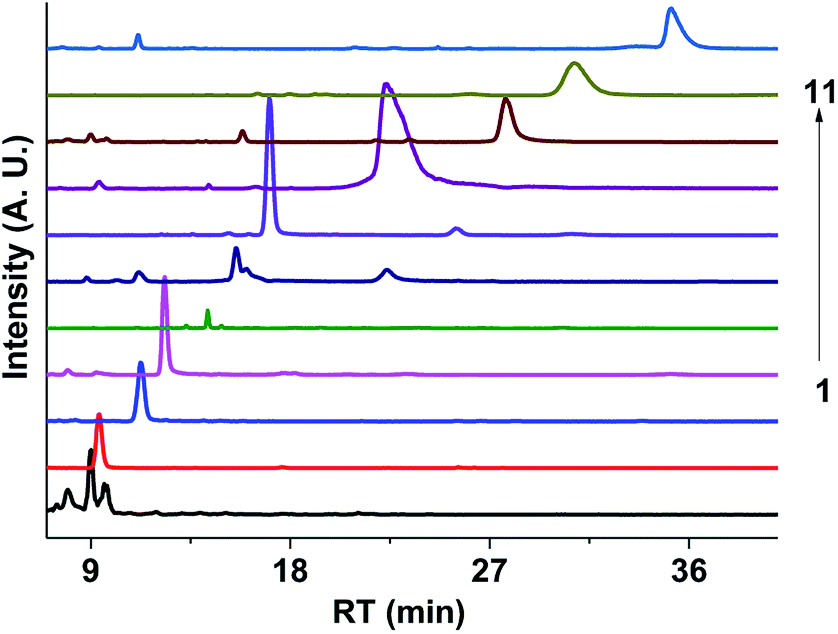
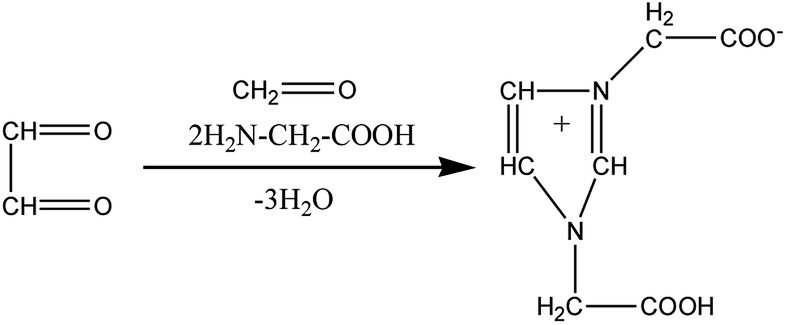
Fig. 9 shows the extracted ion chromatograms (EICs) of 11 selected compounds. The EICs indicated that the separation of the GX–Gly mixtures was the best when using the SM-C18 column. This was evident from the widest spread of the observed retention times for the neutral formulas. The double-bond equivalent (DBE) values for the neutral formulas in Table 4 were calculated with the equation: DBE = c − h/2 + n/2 + 1, where c, h, and n are the number of carbon, hydrogen and nitrogen atoms, respectively. Oligomerization of the carbonyl compounds catalyzed by Gly and C–N bond formation were suggested as the pathways for the formation of brown carbon.42,43 The formulas listed in Table 4 contain 3 nitrogen atoms, 1–9 oxygen atoms and 10–15 carbon atoms, suggesting the presence of both pathways. These results are essentially consistent with the results reported by Lin et al.21 Velisek et al.44 studied the reaction products of GX with Gly in detail. As shown in Fig. 10, 3-carboxymethyl-1-imidazoliumethanoate was formed in the reaction between GX and Gly. Powelson et al.1 also reported that 1,3-diglycine-imidazole was produced in the GX–Gly mixtures. Thus, we speculated that the selected 11 compounds were probably imidazole derivatives. Combined with previous reports,45,46 the major brown carbon chromophores in the GX–Gly mixtures probably correspond to reduced-nitrogen compounds formed from reactions between carbonyls and amines.
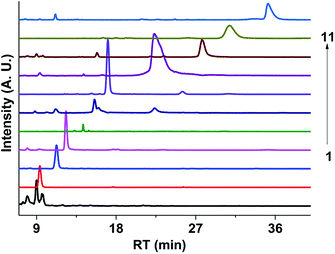 |
| | Fig. 9 Extracted ion chromatograms (EICs) of the 11 selected compounds in the GX–Gly mixture separated using the SM-C18 column. | |
Table 4 Neutral formulas tentatively confirmed by assuming that the positive mode mass spectral features were either the protonated molecule: a [M + H]+, b [M + Na]+, c [M + K]+ or d [M + NH4]+
| Serial number |
Neutral formula |
Measured m/z |
Theoretical mass |
DBE |
| 1 |
C11H14N2 |
213.0800 |
c 213.0789 |
6 |
| 2 |
C11H9N3O4 |
248.0677 |
a 248.0666 |
9 |
| 3 |
C13H14N2O2 |
269.0682 |
c 269.0687 |
8 |
| 4 |
C13H15NO9 |
330.0836 |
a 330.0820 |
7 |
| 5 |
C14H19NO9 |
368.0956 |
b 368.0952 |
6 |
| 6 |
C10H9NO |
198.0335 |
c 198.0316 |
7 |
| 7 |
C15H17N3O3 |
326.0907 |
c 326.0901 |
9 |
| 8 |
C13H10N2O2 |
249.0647 |
b 249.0634 |
10 |
| 9 |
C10H9NO2 |
214.0293 |
c 214.0265 |
7 |
| 10 |
C15H15N3O3 |
324.0748 |
c 324.0745 |
10 |
| 11 |
C12H12N2O2 |
255.0553 |
c 255.0530 |
8 |
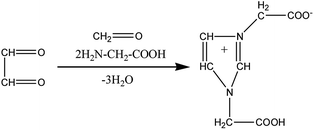 |
| | Fig. 10 Formation of 3-carboxymethyl-1-imidazoliumethanoate.46 | |
Conclusions
The reacted solutions showed a pronounced brownish decoloration behaviour, which increased with an increase in reaction time. The UV/Vis absorption of the GX–Gly mixtures was significantly enhanced in the range of 250–480 nm due to the formation of organic chromophores during the reactions. The aqueous brown carbon solutions from GX–Gly mixtures underwent rapid photobleaching due to the H2O2 oxidation photolysis. Various experimental data confirmed that the aqueous brown carbon contained complex organic components, with at least five-member heterocyclic compounds.21 This is consistent with the previous conclusion that the reaction may have been triggered by the dissolved organic matter of biological origin with a low content of autochthonous component. From the LC/MS analysis, it can be concluded that the abundant organic products formed from the GX–Gly mixtures are not volatile, thus they remain in the particles.
Conflicts of interest
We declare we have no competing financial interests.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 91544223, 21473009 and 21373026) and the Program of College Students Innovation of Laboratory Construction (2016ckjh116).
Notes and references
- M. H. Powelson, B. M. Espelien, L. N. Hawkins, M. M. Galloway and D. O. De Haan, Environ. Sci. Technol., 2014, 48, 985–993 CrossRef CAS PubMed.
- V. Ramanathan and G. Carmichael, Nat. Geosci., 2008, 1, 221–227 CrossRef CAS.
- S. Q. D. Solomon, M. Manning, Z. Chen, M. Marquis, K. Averyt, M. Tignor and H. L. Miller, Contribution of Working Gruop 1 to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, New York, 2007 Search PubMed.
- M. Tang, J. M. Alexander, D. Kwon, A. D. Estillore, O. Laskina, M. A. Young, P. D. Kleiber and V. H. Grassian, J. Phys. Chem. A, 2016, 120, 4155–4166 CrossRef CAS PubMed.
- U. Poschl, Anal. Bioanal. Chem., 2003, 375, 30–32 CrossRef PubMed.
- R. W. Bergstrom, P. Pilewskie, P. B. Russell, J. Redemann, T. C. Bond, P. K. Quinn and B. Sierau, Atmospheric Chemistry and Physics Discussions, 2007, 7, 10669–10686 CrossRef.
- P. K. Aiona, H. J. Lee, R. Leslie, P. Lin, A. Laskin, J. Laskin and S. A. Nizkorodov, ACS Earth Space Chem., 2017, 1, 522–532 CrossRef CAS.
- L. N. Hawkins, H. G. Welsh and M. V. Alexander, Atmospheric Chemistry and Physics Discussions, 2018, 1–29 CrossRef.
- R. K. Chakrabarty, H. Moosmuller, L. W. A. Chen, K. Lewis, W. P. Arnott, C. Mazzoleni, M. K. Dubey, C. E. Wold, W. M. Hao and S. M. Kreidenweis, Atmos. Chem. Phys., 2010, 10, 6363–6370 CrossRef CAS.
- Y. Gao and Y. Zhang, RSC Adv., 2018, 8, 20719–20725 RSC.
- C. Q. Yan, M. Zheng and Y. H. Zhang, Environmental Science, 2014, 35, 4404–4414 Search PubMed.
- J. Cai, G. Zhi, Y. Chen, F. Meng, Z. Xue, J. Li and Y. Fang, Res. Environ. Sci., 2014, 27, 455–461 CAS.
- M. Yang, S. G. Howell, J. Zhuang and B. J. Huebert, Atmos. Chem. Phys., 2009, 9, 2035–2050 CrossRef CAS.
- E. R. Graber and Y. Rudich, Atmos. Chem. Phys., 2006, 6, 729–753 CrossRef CAS.
- K. M. Updyke, T. B. Nguyen and S. A. Nizkorodov, Atmos. Environ., 2012, 63, 22–31 CrossRef CAS.
- T. B. Nguyen, J. Laskin, A. Laskin and S. A. Nizkorodov, Environ. Sci. Technol., 2011, 45, 6908–6918 CrossRef CAS PubMed.
- D. T. L. Alexander, P. A. Crozier and J. R. Anderson, Science, 2008, 321, 833–836 CrossRef CAS PubMed.
- R. Bahadur, P. S. Praveen, Y. Xu and V. Ramanathan, PNAS, 2012, 109, 17366–17371 CrossRef CAS PubMed.
- C. E. Chung, V. Ramanathan and D. Decremer, PNAS, 2012, 109, 11624–11629 CrossRef CAS PubMed.
- Y. Feng, V. Ramanathan and V. R. Kotamarthi, Atmos. Chem. Phys., 2013, 13, 8607–8621 CrossRef.
- P. Lin, J. Laskin, S. A. Nizkorodov and A. Laskin, Environ. Sci. Technol., 2015, 49, 14257–14266 CrossRef CAS PubMed.
- D. A. Lack, J. M. Langridge, R. Bahreini, C. D. Cappa, A. M. Middlebrook and J. P. Schwarz, PNAS, 2012, 109, 14802–14807 CrossRef CAS PubMed.
- A. Hoffer, A. Gelencser, P. Guyon, G. Kiss, O. Schmid, G. P. Frank, P. Artaxo and M. O. Andreae, Atmos. Chem. Phys., 2006, 6, 3563–3570 CrossRef CAS.
- S. M. Phillips and G. D. Smith, J. Phys. Chem., 2015, 119, 4545–4551 CrossRef CAS PubMed.
- J. M. Flores, D. F. Zhao, L. Segev, P. Schlag, A. Kiendker-Scharr, H. Fuchs, A. K. Watne, N. Bluvshtein, Th. F. Mentel, M. Hallquist and Y. Rudich, Atmospheric Chemistry and Physics Discussions, 2014, 14, 4149–4187 CrossRef.
- Y. Lei, Z. Shen, Q. Wang, T. Zhang, J. Cao, J. Sun, Q. Zhang, L. Wang, H. Xu, J. Tian and J. Wu,, Atmos. Res., 2018, 213, 27–33 CrossRef CAS.
- H. J. Lee, P. K. Aiona, A. Laskin, J. Laskin and S. A. Nizkorodov, Environ. Sci. Technol., 2014, 48, 10217–10226 CrossRef CAS PubMed.
- N. Sareen, A. N. Schwier, E. L. Shapiro, D. Mitroo and V. F. McNeill, Atmos. Chem. Phys., 2010, 10, 997–1016 CrossRef CAS.
- H. J. Lee, A. Laskin, J. Laskin and S. A. Nizkorodov, Environ. Sci. Technol., 2013, 47, 5763–5770 CrossRef CAS PubMed.
- M. O. Andreae and A. Gelencser, Atmos. Chem. Phys., 2006, 6, 3131–3148 CrossRef CAS.
- Y. Chen and T. C. Bond, Atmos. Chem. Phys., 2010, 10, 1773–1787 CrossRef CAS.
- N. Hudson, A. Baker and D. Reynolds, River. Res. Appl., 2007, 23, 631–649 CrossRef.
- R. M. B. O. Duarte, C. A. Pio and A. C. Duarte, J. Atmos. Chem., 2004, 48, 157–171 CrossRef CAS.
- D. M. McKnight, E. W. Boyer, P. K. Westerhoff, P. T. Doran, T. Kulbe and D. T. Andersen, Limnol. Oceanogr., 2001, 46, 38–48 CrossRef CAS.
- J. E. Birdwell and K. T. Valsaraj, Atmos. Environ., 2010, 44, 3246–3253 CrossRef CAS.
- A. Huguet, L. Vacher, S. Relexans, S. Saubusse, J. M. Froidefond and E. Parlanti, Org. Geochem., 2009, 40, 706–719 CrossRef CAS.
- R. G. Zepp, W. M. Sheldon and M. A. Moran, Mar. Chem., 2004, 89, 15–36 CrossRef CAS.
- R. G. Zepp, G. L. Baughman and P. F. Schlotzhauer, Chemosphere, 1981, 10, 119–126 CrossRef CAS.
- M. Y. Berezin and S. Achilefu, Chem. Rev., 2010, 110, 2641–2684 CrossRef CAS PubMed.
- E. M. Merzlyak, J. Goedhart, D. Shcherbo, M. E. Bulina, A. S. Shcheglov, A. F. Fradkov, A. Gaintzeva, K. A. Lukyanov, S. Lukyanov, T. W. J. Gadella and D. M. Chudakov, Nat. Methods, 2007, 4, 555–557 CrossRef CAS PubMed.
- W. Becker, A. Bergmann, M. A. Hink, K. Konig, K. Benndorf and C. Biskup, Microsc. Res. Tech., 2004, 63, 58–66 CrossRef CAS PubMed.
- A. Laskin, J. Laskin and S. A. Nizkorodov, Chem. Rev., 2015, 115, 4335–4382 CrossRef CAS PubMed.
- T. Moise, J. M. Flores and Y. Rudich, Chem. Rev., 2015, 115, 4400–4439 CrossRef CAS PubMed.
- J. Velisek, T. Davidek and J. Davidek, Flavour Chemistry, 2005, pp. 204–208 Search PubMed.
- A. K. Y. Lee, R. Zhao, R. Li, J. Liggio, S. M. Li and J. P. D Abbatt, Environ. Sci. Technol., 2013, 47, 12819–12826 CrossRef CAS PubMed.
- M. M. Galloway, M. H. Powelson, N. Sedehi, S. E. Wood, K. D. Millage, J. A. Kononenko, A. D. Rynaski and D. O. De Haan, Environ. Sci. Technol., 2014, 48, 14417–14425 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06913a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab and
Yunhong Zhang*a
ab and
Yunhong Zhang*a