
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExperimental and computational preference for phosphine regioselectivity and stereoselective tripodal rotation in HOs3(CO)8(PPh3)2(μ-1,2-N,C-η1,κ1-C7H4NS)†
Shahin A. Beguma,
Md. Arshad H. Chowdhurya,
Shishir Ghosh a,
Derek A. Tocherb,
Michael G. Richmondc,
Li Yangc,
K. I. Hardcastled,
Edward Rosenberge and
Shariff E. Kabir*a
a,
Derek A. Tocherb,
Michael G. Richmondc,
Li Yangc,
K. I. Hardcastled,
Edward Rosenberge and
Shariff E. Kabir*a
aDepartment of Chemistry, Jahangirnagar University, Savar, Dhaka-1342, Bangladesh. E-mail: skabir_ju@yahoo.com; Tel: +880-27791099
bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H OAJ, UK
cDepartment of Chemistry, University of North Texas, Denton, TX 76209, USA
dDepartment of Chemistry, Emory University, Atlanta, GA 30322, USA
eDepartment of Chemistry, University of Montana, Missoula, MT 59812, USA
First published on 21st September 2018
Abstract
The site preference for ligand substitution in the benzothiazolate-bridged cluster HOs3(CO)10(μ-1,2-N,C-η1,κ1-C7H4NS) (1) has been investigated using PPh3. 1 reacts with PPh3 in the presence of Me3NO to afford the mono- and bisphosphine substituted clusters HOs3(CO)9(PPh3)(μ-1,2-N,C-η1,κ1-C7H4NS) (2) and HOs3(CO)8(PPh3)2(μ-1,2-N,C-η1,κ1-C7H4NS) (3), respectively. 2 exists as a pair of non-interconverting isomers where the PPh3 ligand is situated at one of the equatorial sites syn to the edge-bridging hydride that shares a common Os–Os bond with the metalated heterocycle. The solid-state structure of the major isomer establishes the PPh3 regiochemistry at the N-substituted osmium center. DFT calculations confirm the thermodynamic preference for this particular isomer relative to the minor isomer whose phosphine ligand is located at the adjacent C-metalated osmium center. 2 also reacts with PPh3 to give 3. The locus of the second substitution occurs at one of the two equatorial sites at the Os(CO)4 moiety in 2 and gives rise to a pair of fluxional stereoisomers where the new phosphine ligand is scrambled between the two equatorial sites at the Os(CO)3P moiety. The molecular structure of the major isomer has been determined by X-ray diffraction analysis and found to represent the lowest energy structure of the different stereoisomers computed for HOs3(CO)8(PPh3)2(μ-1,2-N,C-η1,κ1-C7H4NS). The fluxional behavior displayed by 3 has been examined by VT NMR spectroscopy, and DFT calculations provide evidence for stereoselective tripodal rotation at the Os(CO)3P moiety that serves to equilibrate the second phosphine between the two available equatorial sites.
Introduction
The activation and value-added functionalization of benzoannulated heterocycles containing a pyridyl nitrogen by low-valent trimetallic clusters, M3(CO)12−n(NCMe)n (M = Os, Ru; n = 0, 1, 2), have been extensively studied over the past few decades.1–11 While the early impetus involved industrial considerations with respect to hydrotreatment of nitrogen-contaminated fuel stocks,1,3,12,13 current interests remain focused on the ability to use the cluster as a templating agent to help guide the selective functionalization of the bound benzoheterocycle.6,7 Recently, we have also reported our data on the biological applications of such complexes and their ability to serve as potential bioorganometallic conjugates in enzyme and telomerase inhibition studies, as well as tagging agents for use in protein crystallography.2,9The ligation of benzoheterocycles at trimetallic clusters is readily achieved via the direct reaction of a suitably activated cluster with the corresponding benzoheterocycle. For example, the labile cluster 1,2-Os3(CO)10(NCMe)2 reacts with quinoline (C9H7N) to afford isomeric products with the formula HOs3(CO)10(μ-η1,κ1-C9H6N) which differ in the coordination mode adopted by the activated heterocycle.6a Here the quinolate residue bridges one of the three metal–metal edges via the pyridyl nitrogen and either a metalated aryl C-8 or C-2 carbon atom (Scheme 1). The 1,8 isomer has been extensively examined by us and it undergoes CO loss during controlled thermolysis or photolysis to give the face-capped cluster HOs3(CO)9(μ3-η1,κ1-C9H6N). The nonacarbonyl product reacts with soft pnictogen nucleophiles at an osmium center to yield HOs3(CO)9P(μ-η1,κ1-C9H6N) (where P = phosphine, phosphite) and with hard nucleophiles (RLi and RMgX) at the heterocycle to give HOs3(CO)10(μ-η1,κ1-RC9H7N).4a,6 The latter product, which may be isolated after protonation, provides the basis for many of our reported heterocyclic functionalizations (Scheme 2).6c In comparison to numerous studies involving the 1,8 isomer, the corresponding 1,2 isomer has received relatively little attention.14
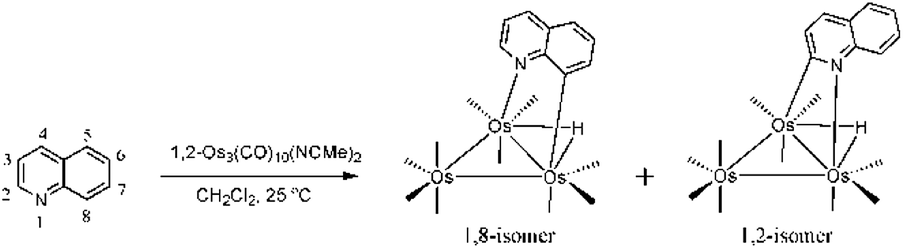 | ||
| Scheme 1 Reaction of quinoline with 1,2-Os3(CO)10(MeCN)2 to give the 1,8- and 1,2-metalated isomers of HOs3(CO)10(μ-η1,κ1-C9H6N). | ||
 | ||
| Scheme 2 Decarbonylation in HOs3(CO)10(μ-1,8-C9H6N) and the different reaction pathways exhibited by HOs3(CO)9(μ3-η1,κ1-C9H6N). | ||
Metal cluster activation of heterocycles such as benzothiazole is accompanied by added complexity in the form of potential sulfur versus nitrogen coordination. We have demonstrated that benzothiazole reacts with 1,2-Os3(CO)10(NCMe)2 to furnish the metalated products HOs3(CO)10(μ-1,7-N,C-η1,κ1-C7H4NS) and HOs3(CO)10(μ-1,2-N,C-η1,κ1-C7H4NS) where the metalation occurs with coordination of the nitrogen and carbon atoms (Scheme 3).6c,15 The benzothiazolate-substituted clusters are formed in comparable yields and may be isolated by column chromatography, albeit with material loss in the case of the 1,2 derivative. This complication in the isolation of the 1,2 isomer, coupled with its limited thermal stability vis-à-vis the 1,7 isomer, has hampered meaningful reactivity studies designed to exploit this cluster-activated heterocycle. Systematic reactivity studies are important if we are to gain a better understanding of such systems, especially in light of our earlier observations that have demonstrated that the outcome of such functionalizations is known to be sensitive to the presence of ring substituents and the ligand environment at the metal cluster.
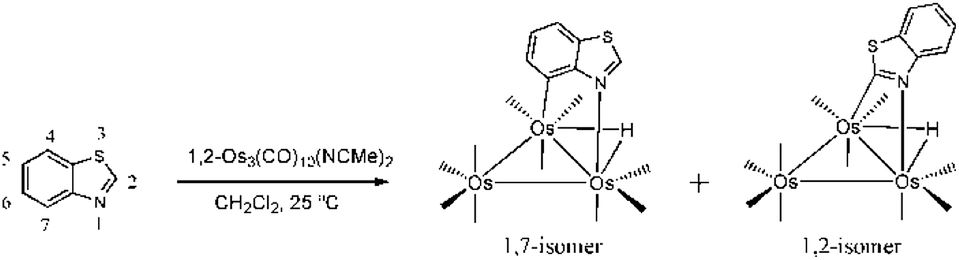 | ||
| Scheme 3 Reaction of benzothiazole with 1,2-Os3(CO)10(MeCN)2 to give the 1,7- and 1,2-N,C metalated isomers of HOs3(CO)10(μ-η1,κ1-C7H4NS). | ||
Given the significant influence the heterocyclic auxiliary has on the reactivity in this genre of metal clusters, we have initiated reactivity studies on HOs3(CO)10(μ-1,2-N,C-η1,κ1-C7H4NS) that will allow us to better understand the observed chemistry as a function of the metalated heterocycle and its control on ligand substitution and value-added functionalizations. The cluster HOs3(CO)10(μ-1,7-N,C-η1,κ1-C7H4NS), whose benzothiazolate bridging ligand has a larger backbone, has received greater attention than its smaller ring orthometalated analogue. Accordingly, we wished to study the reactivity of HOs3(CO)10(μ-1,2-N,C-η1,κ1-C7H4NS) with added ligand in order to establish the regiochemistry and thermodynamic stability of the resulting products. The effect of the 1,7-N,C ring in HOs3(CO)10(μ-1,7-N,C-η1,κ1-C7H4NS) in ligand addition and alkyne insertion reactions has recently been reported by us.5c,e,16 To our knowledge, only one reactivity report exists for the 1,2-N,C isomer to date and this involves the cyclometalation of thiourea at cluster 1.17 Herein we present our data on the regiochemical preference for PPh3 substitution in cluster 1, along with the results of electronic structure calculations that help place the observed outcome in each substitution step in context with the NMR properties and the solid-state structures determined for 2 and 3.
Results and discussion
Metalation selectivity in the reaction of benzothiazole with 1,2-Os3(CO)10(MeCN)2
Wishing to reconcile the thermodynamic stability of the 1,2- and 1,7-N,C metalated isomers from the reaction of benzothiazole with 1,2-Os3(CO)10(MeCN)2 relative to their reaction yields, along with the relative energies of the corresponding S,C-bridging isomers, we have performed DFT calculations. The input data for 1,7-N,C isomer was taken from our earlier study,5e while the 1,2 isomer was modeled using the structure of the thiazolide derivative HOs3(CO)10(1,2-N,C-C3H2NS-3-Me) after the coordinated heterocycle was converted to a benzothiazolate ligand.18 The unknown S,C isomers were modeled by assuming a bridging motif similar to the N,C-ligated clusters. Fig. 1 shows the optimized structures C1–C4 for the isomeric N,C- and S,C-metalated clusters and the energy ordering of these products (plus the release of two MeCN) relative to 1,2-Os3(CO)10(MeCN)2 (A) and benzothiazole (B). The N,C-metalated isomers C1 and C2 are thermodynamically favored compared to the S,C derivatives C3 and C4. The 1,2-N,C isomer C1 is the most stable of the four isomers and it lies 17.3 kcal mol−1 below the reactants. The 1,7-N,C isomer C2 is 16.1 kcal mol−1 less stable than C1 and its formation is exergonic by 1.2 kcal mol−1. The experimental observation and isolation of these two isomers in comparable yields suggest that these products are produced in a kinetically controlled sequence and their formation is not subject to thermodynamic control. The optimized structures of C1 and C2 exhibit good agreement with related crystallographically determined structures. Compared to the N,C isomers, both S,C isomers are less stable and lie 9.0 kcal mol−1 (C3) and 24.2 kcal mol−1 (C4) higher in energy than the reactants. Whereas the 1,2-N,C isomer is more stable than the 1,7-N,C species, the reverse trend is found for the S,C isomers. The least stable of these S,C isomers is the 1,2-S,C species (C4). Both C3 and C4 display a highly pyramidalized sulfur atom that results from sulfur coordination at the osmium center, and this perturbation induces a significant loss in the overall aromaticity in the cluster-bound heterocycle. Similar sulfur pyramidalization has been observed in organic alkylation reactions involving sulfur-containing heterocycles19 and serves as a sufficient driving force that prevents the formation of the two S,C isomers in the reaction of 1,2-Os3(CO)10(MeCN)2 with benzothiazole. Fig. S1† shows the ChemDraw pictures of C1–C4.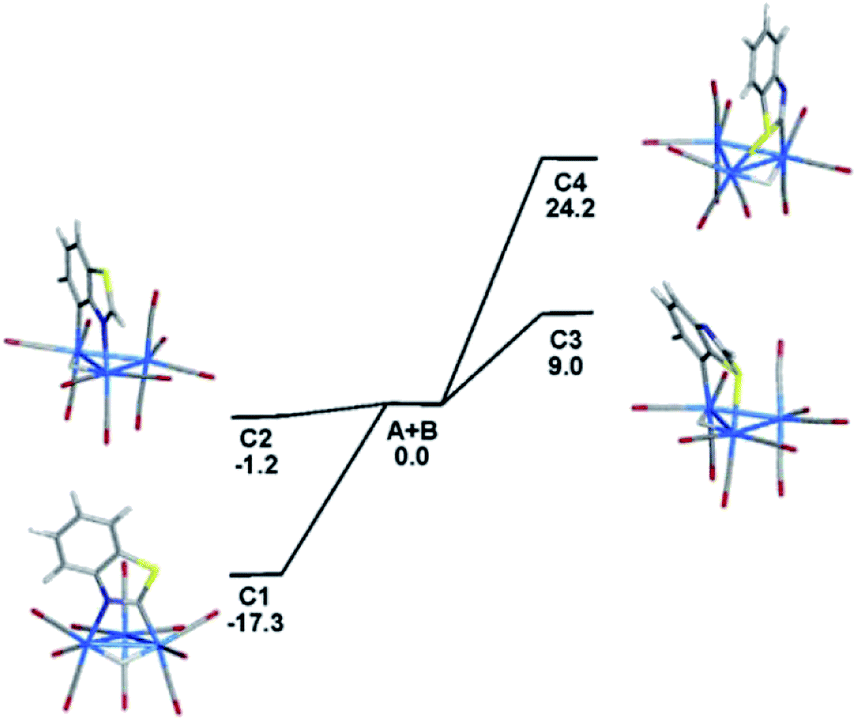 | ||
| Fig. 1 Optimized metalation products C1–C4 from the reaction of 1,2-Os3(CO)10(MeCN)2 (A) with benzothiazole (B). Energy values (ΔG) are in kcal mol−1 relative to A and B. The optimized structures for A, B, and free MeCN are not shown. | ||
Regioselective PPh3 addition and DFT evaluation of the different isomers of 2
Treatment of HOs3(CO)10(μ-1,2-N,C-η1,κ1-C7H4NS) (1) with Me3NO (one equivalent) at room temperature in the presence of PPh3 affords the monosubstituted cluster HOs3(CO)9(PPh3)(μ-1,2-N,C-η1,κ1-C7H4NS) (2) in crude yields on the order of 80%. Accompanying this reaction is the disubstituted cluster HOs3(CO)8(PPh3)2(μ-1,2-N,C-η1,κ1-C7H4NS) (3) in 5–10% yield. Control experiments using pure 2 confirm the stepwise nature of the substitution with 3 produced in high yield (>85% crude) when equimolar amounts of 2 and PPh3 are treated with Me3NO (one equivalent). Scheme 4 outlines the course of these reactions where the depicted products of 2 and 3 are based on the crystallographically determined structures (vide infra). Both new clusters were isolated by chromatography over silica gel and characterized in solution by IR and NMR spectroscopy, and the solid-state structure of each product was established by single-crystal X-ray diffraction analysis.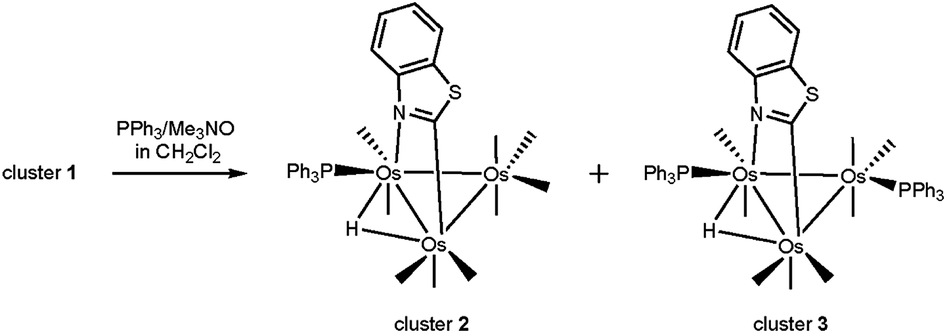 | ||
| Scheme 4 Reaction of 1 with PPh3 to give 2 and 3. Only the major product in each substitution step is shown. | ||
The asymmetric unit of 2 contains two independent Os3 clusters along with a molecule of CH2Cl2 and water. Since the two structures do not show any significant structural differences, we will discuss only one of these crystallographically independent molecules whose structure is depicted in Fig. 2. Selected bond distances and angles are listed in the figure caption. The molecular core consists of an approximate isosceles triangle of osmium atoms [Os(1)–Os(2) 2.9843(2), Os(2)–Os(3) 2.8795(3) and Os(1)–Os(3) 2.8533(3) Å] ligated by nine carbonyls and a PPh3 ligand, along with an edge-bridging hydride that shares the same Os(1)–Os(2) vector as the metalated benzothiazolate ligand. The edge-bridged Os–Os bond is the longest of the three metal–metal bonds. The phosphine ligand occupies an equatorial coordination site at the nitrogen-bound osmium and lies cis to the bridging hydride. The Os(1)–P(1) [2.3888(11) Å], Os(1)–N(1) [2.138(4) Å], and Os(2)–C(10) [2.086(5) Å] bond distances involving the ancillary ligands are similar to those reported by us in related trisosmium clusters.14,18,20 However, the locus of PPh3 substitution in 2 is quite different from that displayed in the 4-methylquinolinate analogues HOs3(CO)9(PPh3)[μ-1,2-η1,κ1-C9H5(CH3)N] and HOs3(CO)9[P(OMe)3][μ-1,2-η1,κ1-C9H5(CH3)N].14 The pnictogen ligand in these latter derivatives occupies an equatorial site at the non-metalated osmium atom. The different regiochemistry for phosphine substitution in the current cluster that contains a π-excessive heterocycle versus our earlier report on the Os3 cluster with a π-deficient heterocycle is striking and underscores the importance of the metalated platform in controlling the outcome of the reaction. We examined the bonding in 2 by DFT, and the optimized structure (D1) is shown alongside the experimentally determined structure in Fig. 2. The computed structure of D1 shows good agreement with the solid-state structure.
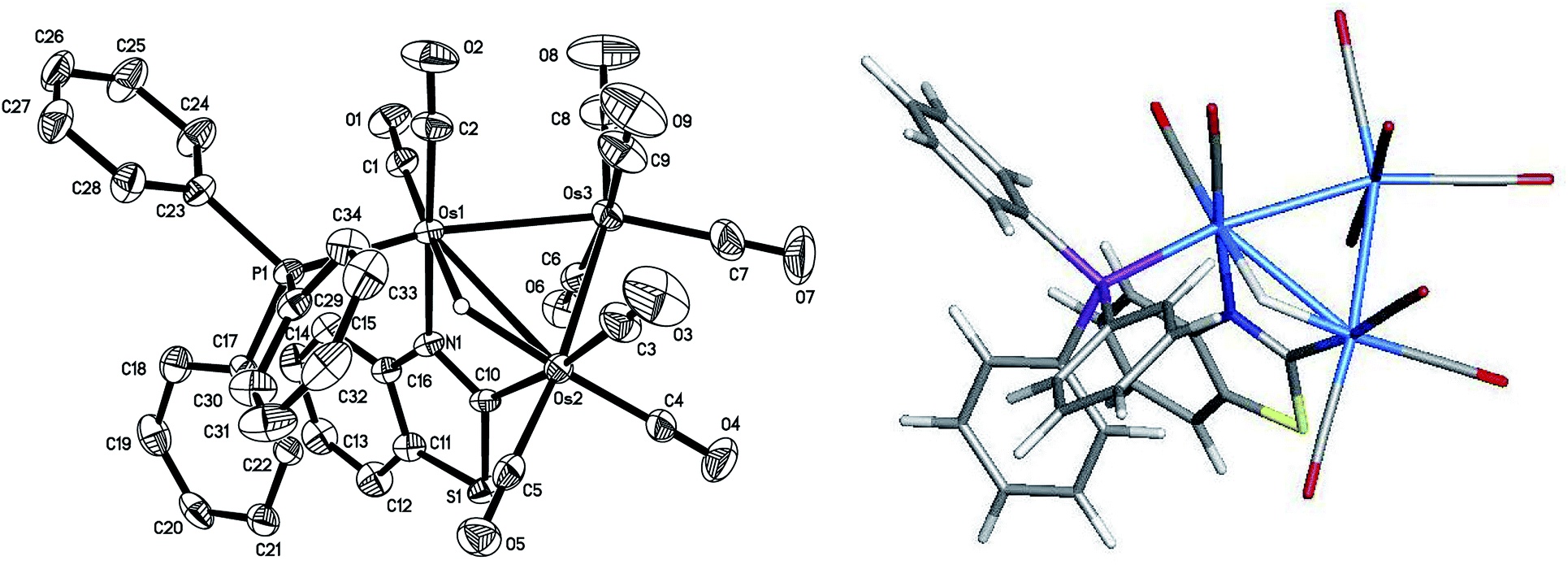 | ||
| Fig. 2 ORTEP drawing of the molecular structure of 2 (left) showing 50% probability thermal ellipsoids and the DFT-optimized structure of D1 (right). Selected interatomic distances (Å) and angles (°) for 2: Os(1)–Os(2) 2.9843(3), Os(2)–Os(3) 2.8795(3), Os(1)–Os(3) 2.8533(3), Os(1)–N(1) 2.138(4), Os(2)–C(10) 2.086(5), Os(1)–P(1) 2.3888(12), N(1)–C(10) 1.331(6), P(1)–Os(1)–Os(2) 105.08(3), P(1)–Os(1)–Os(3) 163.06(3), P(1)–Os(1)–N(1) 90.49(10), Os(1)–N(1)–C(10) 109.6(3), C(16)–N(1)–C(10) 113.7(4), N(1)–C(10)–Os(2) 116.6(3), C(5)–Os(2)–Os(1) 114.99(13). | ||
The 1H and 31P NMR spectra recorded for 2 indicate the presence of two clusters with the dominant species present in >95% yield; the amount of the minor species varies but never exceeds 5%. The major product shows a high-field hydride at δ −14.21 (J 11.6) and a 31P singlet δ 11.8 at room temperature, while the minor species reveals a hydride doublet and 31P singlet at δ −15.80 (J 12.0) and δ 1.98, respectively. VT NMR measurements over the temperature range 298–223 K showed no change in the relative intensities of these species, allowing us to rule out a dynamic exchange of the hydride and PPh3 ligand between the two species on the NMR time scale. This latter premise was further corroborated by 1H and 31P EXSY NMR experiments conducted at 253 K.
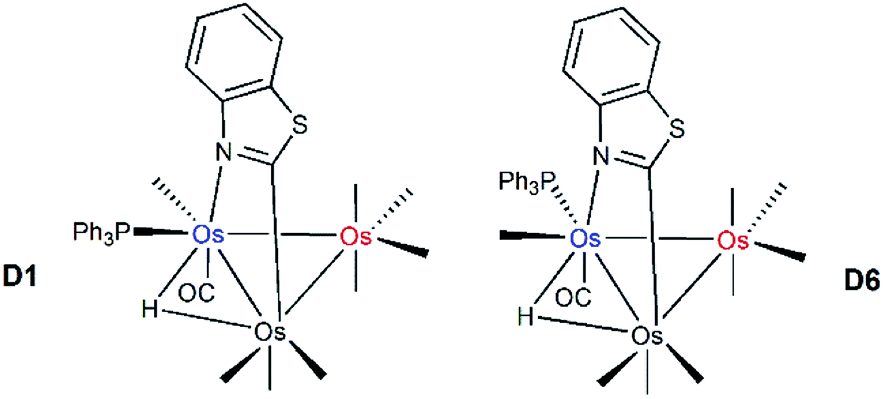
The identity of the two species in the NMR spectra of 2 and the relative stability of the two products were addressed through electronic structure calculations on the likely monophosphine derivatives of 2. We restricted our calculations to products where the two edge-bridging ligands share a common Os–Os vector due to their enhanced stability compared to isomers that contain two Os–Os bonds with a single edge-bridging ligand.5e,21 And concerning the disposition of the PPh3 group in HOs3(CO)9(PPh3)(μ-1,2-N,C-η1,κ1-C7H4NS), we have examined only those isomers that possess an equatorial phosphine since axially disposed phosphine ligands experience an energetic penalty of 10–15 kcal mol−1 due to unfavorable van der Waals interactions between the phosphine and the pair of axial ligands on the adjacent osmium centers.22 Not counting species D1, five additional structures exist with an equatorial PPh3 ligand, and these are depicted in Fig. 3. The structure for D2 is 2.5 kcal mol−1 less stable than D1 and the PPh3 ligand is situated cis to the hydride at the C-metalated osmium atom. Species D3 and D4 represent a pair of stereoisomers where the PPh3 occupies one of the two equatorial sites at the Os(CO)3P moiety. The energy difference between D3 and D4 is negligible, and both species lie at least 3.5 kcal mol−1 above the thermodynamically favored species D1. Phosphine substitution at the alternative equatorial sites associated with the hydride-bridged Os–Os bond furnishes species D5 and D6. The ancillary PPh3 ligand in these two species adopts a distal orientation with respect to the edge-bridging hydride and each species exhibits a nearly linear P–Os–H arrangement of atoms based on the computed bond angles of 167° (D5) and 175° (D6).
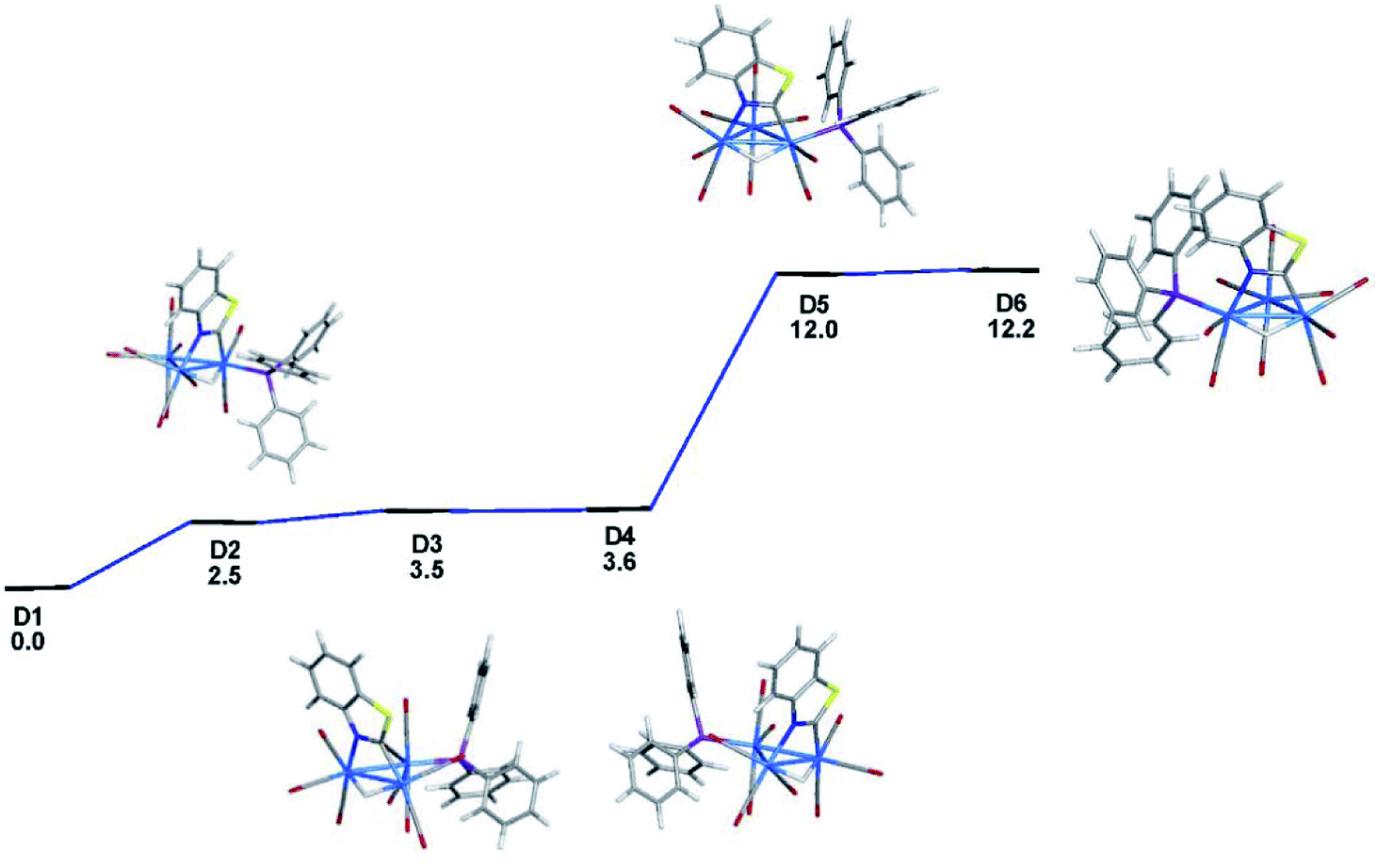 | ||
| Fig. 3 DFT-optimized structures and energy ordering for the different equatorial-substituted PPh3 isomers of HOs3(CO)9(PPh3)(μ-1,2-N,C-η1,κ1-C7H4NS) (D1–D6). Energy values are ΔG in kcal mol−1 relative to D1. Fig. S2† shows the ChemDraw pictures of species D1–D6. | ||
The origin for the computed energy difference in the D1 and D6 stereoisomers undoubtedly derives from a combination of steric and electronic effects at each Os3 product. The observed substitution pattern adopted by the ancillary benzothiazolate, phosphine, and hydride groups largely governs the relative stability of the different isomers D1–D6. Adverse interactions emanating from the phosphine and the heterocyclic auxiliary in D6 vis-à-vis D1 shape the forces that energetically differentiate this pair of stereoisomers. The site of PPh3 substitution in D6 places the pnictogen ligand near the benzoannulated portion of the heterocyclic moiety, which promotes a 0.043 Å elongation of the  –N bond in D6; coupled with this perturbation, is a canting of the CO, the one trans to the heterocyclic nitrogen, away from the PPh3 ligand. The van der Waals interactions serve to reduce the OC–
–N bond in D6; coupled with this perturbation, is a canting of the CO, the one trans to the heterocyclic nitrogen, away from the PPh3 ligand. The van der Waals interactions serve to reduce the OC– –N bond angle from 176° in D1 to 166° in D6. Accompanying this perturbation is an increase in the
–N bond angle from 176° in D1 to 166° in D6. Accompanying this perturbation is an increase in the  –
– –CO bond angle from 89° (D1) to 93° (D6) and a 0.1 Å increase in the
–CO bond angle from 89° (D1) to 93° (D6) and a 0.1 Å increase in the  –
– bond distance in D6. The defining color labels associated with these structural changes are illustrated in Scheme 5. Analysis of the D2 and D5 stereoisomers reveals similar trends at the C-metalated osmium atom.
bond distance in D6. The defining color labels associated with these structural changes are illustrated in Scheme 5. Analysis of the D2 and D5 stereoisomers reveals similar trends at the C-metalated osmium atom.
 | ||
| Scheme 5 Color code used in the discussion of the steric perturbation of the PPh3 group on the cluster core in D1 and D6. | ||
In terms of an operational electronic effect in D1 and D6, the geometrical arrangement of the participant P, Os, and H atoms will influence the net stability of the orbitals involved in the four-centered, four-electron (4c,4e) interaction here. Within this delocalized bonding model, the hydride may be treated as proton that remains associated with the electron-rich Os–Os bond. The “proton” will experience enhanced stabilization if it can interact with additional electron density in an orbital of suitable energy and proper symmetry. The orbital interactions between a hydride(s) and a metal–metal bond in polynuclear systems have been successfully analyzed within a 3c,2e bonding model.23 And we build on this concept by confirming the thermodynamic preference for products that have the phosphine positioned syn to the bridging hydrogen. Here electron density contributed by the P–Os bond helps stabilize the bridging hydrogen when the phosphine adopts a site syn to the hydride-bridged Os–Os vector. In this scenario, the P– –Os atoms display an obtuse angle, a feature underscored by numerous structural studies involving related trimetallic clusters where the P–
–Os atoms display an obtuse angle, a feature underscored by numerous structural studies involving related trimetallic clusters where the P– –Os bond angle ranges from 105–117°.14,16–18,24 The computed P–
–Os bond angle ranges from 105–117°.14,16–18,24 The computed P– –Os bond angle in D1 is 109°. Assuming a fixed cluster polyhedron, expansion of the P–
–Os bond angle in D1 is 109°. Assuming a fixed cluster polyhedron, expansion of the P– –Os bond angle in D1 effectively permutes the PPh3 ligand to the other equatorial site at the nitrogen-substituted osmium center. This progression yields D6 and is accompanied by a 100° increase in the P–
–Os bond angle in D1 effectively permutes the PPh3 ligand to the other equatorial site at the nitrogen-substituted osmium center. This progression yields D6 and is accompanied by a 100° increase in the P– –Os bond angle. The nearly linear arrangement of the P–
–Os bond angle. The nearly linear arrangement of the P– –Os atoms in D6 effectively eliminates any through-bond P–
–Os atoms in D6 effectively eliminates any through-bond P– interaction with the bridging hydrogen that remains tethered to the original
interaction with the bridging hydrogen that remains tethered to the original  –Os vector. The stabilizing orbital effect promulgated by the ancillary phosphine when syn disposed to the bridging hydride in D1 is not unlike many organic reactions where secondary orbital interactions serve to govern the reaction outcome.25 Syn positioning of the phosphine to the hydride-bridged metal–metal bond in other clusters does not always represent the thermodynamically favored regiochemistry for this pair of ligands.14 We suspect that in such cases any beneficial orbital effect involving the hydrogen and the electrons in the P–Os bond is counterbalanced by extant steric perturbations within the molecule. The hydride regiochemistry displayed in this genre of metal clusters cannot be generalized with certainty at this time and must be evaluated on a case by case basis.
–Os vector. The stabilizing orbital effect promulgated by the ancillary phosphine when syn disposed to the bridging hydride in D1 is not unlike many organic reactions where secondary orbital interactions serve to govern the reaction outcome.25 Syn positioning of the phosphine to the hydride-bridged metal–metal bond in other clusters does not always represent the thermodynamically favored regiochemistry for this pair of ligands.14 We suspect that in such cases any beneficial orbital effect involving the hydrogen and the electrons in the P–Os bond is counterbalanced by extant steric perturbations within the molecule. The hydride regiochemistry displayed in this genre of metal clusters cannot be generalized with certainty at this time and must be evaluated on a case by case basis.
We have successfully analyzed the course of hydride addition to the metalated heterocycle in a series of heterocyclic-bridged Os3 clusters earlier by taking into account the charge distribution in the starting material.7b,11a–c Accordingly, we have examined the charge distribution in C1 to help shed light on the isomeric products formed in the reaction that gives cluster 2. Scheme 6 shows the NPA atomic charges for the osmium atoms in C1. The three osmium atoms carry a net negative charge and the nitrogen-substituted osmium is the least electron rich of the three osmium atoms (Q = −1.02), followed by the C-metalated osmium center whose Q was computed as −1.21. The metal center in the Os(CO)4 moiety exhibits the greatest negative charge (−1.38). The data from the NPA calculations support, insomuch as charge control is responsible, the formation of D1 as the major monophosphine product. The presence of D2 as the other computed component in the reaction mixture is in keeping with PPh3 association with the second most electropositive osmium center in C1. Finally, the NPA charge for the edge-bridging hydride was positive with a Q of 0.096.26
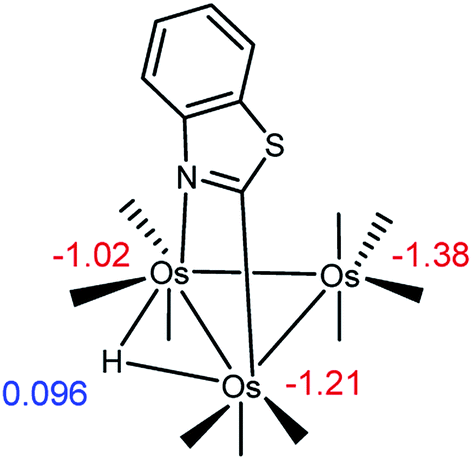 | ||
| Scheme 6 Natural population analysis (NPA) charges for the osmiums and bridging hydride in C1. | ||
Formation of HOs3(CO)8(PPh3)2(μ-1,2-N,C-η1,κ1-C7H4NS) (3) and stereoselective tripodal rotation at the Os(CO)3P moiety
The reaction of PPh3 with 2 in the presence of Me3NO affords 3, which exists as a pair of discernible isomers in solution (298 K) by NMR spectroscopy. The structure of the major isomer was established by X-ray diffraction analysis. Fig. 4 displays the ORTEP diagram of the molecular structure of 3 with selected bond distances and angles listed in the figure caption. 3 consists of a triangular array of osmium atoms [Os(1)–Os(2) 2.9638(2), Os(2)–Os(3) 2.9066(2), and Os(1)–Os(3) 2.8651(2) Å] where the mean Os–Os bond distance is 2.9118 Å. The Os(1)–Os(2) vector is the longest edge and it is bridged by the hydride and benzothiazolate ligands. Eight carbonyls and two PPh3 ligands complete the coordination sphere of this cluster. As in 2, one of the two PPh3 ligands is bound to the nitrogen-bound osmium [Os(1)] while the second phosphine [P(2)] is coordinated to the Os(3) atom at an equatorial site syn to the Os(2) center. The mean Os–P bond distance in 3 is 2.3564 Å and not unlike that Os–P distance found in 2 [2.3888(11) Å]. The Os–N [2.143(4) Å] and Os–C(9) [2.076(5) Å] bond distances are unremarkable compared with those bond lengths reported in 1 and related clusters.4a,14,17,18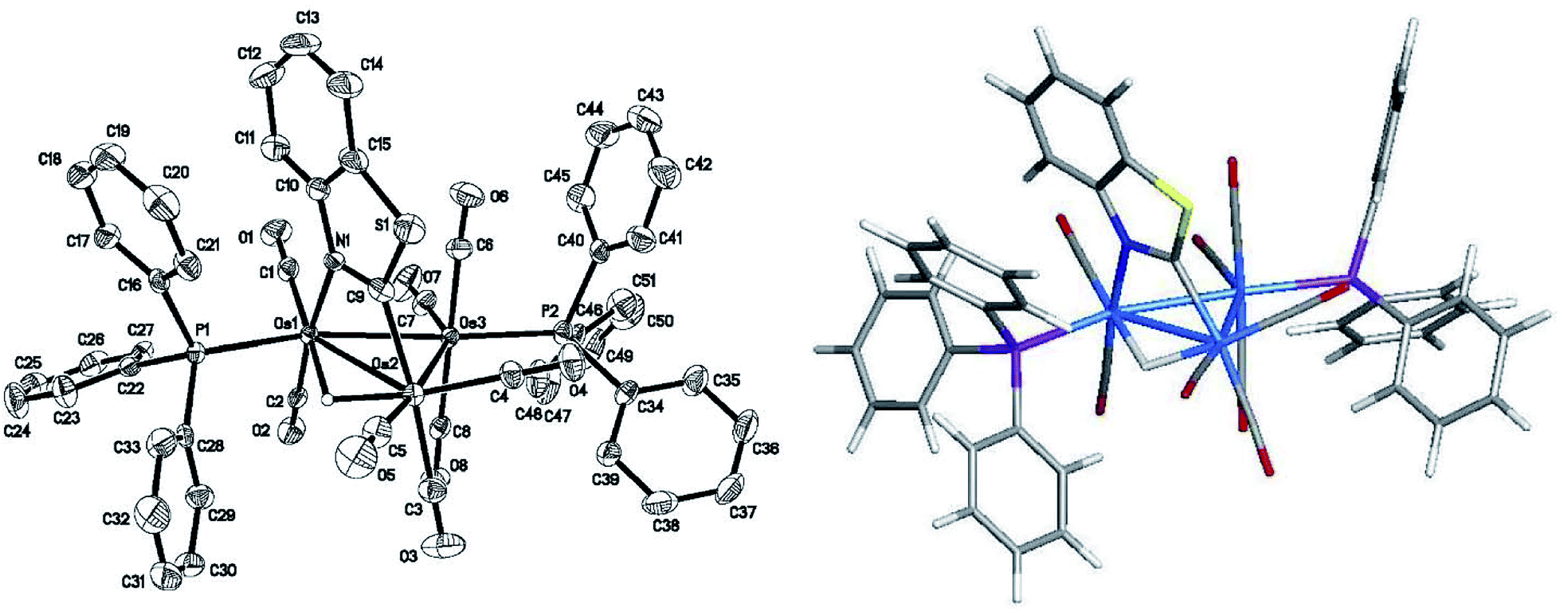 | ||
| Fig. 4 ORTEP drawing of the molecular structure of 3 (left) showing 50% probability thermal ellipsoids and the DFT-optimized structure of E1 (right). Selected interatomic distances (Å) and angles (°) for 3: Os(1)–Os(2) 2.9638(2), Os(2)–Os(3) 2.9066(2), Os(1)–Os(3) 2.8651(2), Os(1)–N(1) 2.143(4), Os(2)–C(9) 2.076(5), Os(3)–P(2) 2.3550(11), Os(1)–P(1) 2.3578(11), N(1)–C(10) 1.408(6), N(1)–C(9) 1.310(6), C(9)–S(1) 1.739(5), Os(2)–Os(1)–Os(3) 59.793(6), Os(1)–Os(2)–Os(3) 58.414(6), Os(1)–Os(3)–Os(2) 61.791(6), P(1)–Os(1)–Os(3) 166.31(3), P(1)–Os(1)–Os(2) 107.16(3), P(1)–Os(1)–N(1) 91.29(10), P(2)–Os(3)–Os(1) 175.57(3), P(2)–Os(3)–Os(2) 116.74(3), N(1)–C(9)–Os(2) 117.3(3), C(9)–Os(2)–Os(1) 66.21(13), C(9)–Os(2)–Os(3) 86.24(12), Os(1)–N(1)–C(9) 108.9(3). | ||
The 1H and 31P NMR spectra of the major species in solution are consistent with the solid-state structure. Here a diagnostic hydride doublet at δ −13.74 (J 11.6) confirms the coupling of the edge-bridging hydride to one of the two phosphine groups in the major isomer of 3. The magnitude of the 2JPH is similar to that recorded in 2, allowing us to confidently assign the coupling to the geminal phosphine defined by the P(1) atom in Fig. 4. The 31P{1H} NMR spectrum exhibits two broad singlets at δ 8.9 and 3.9 for the PPh3 ligands. At room temperature, the minor species (<3%) shows a weak hydride at δ −15.38 (J 12.5) while the 31P resonances were not visible. In order to probe the relationship between the two isomers, we examined 3 by VT NMR spectroscopy over the temperature range 323–223 K. Lowering the temperature to 223 K reveals the presence of four hydride doublets centered at δ −13.73, −13.80, −13.90, and −15.45 in the 1H NMR spectrum (Fig. S3†). Consistent with this change in the 1H NMR spectrum is the observation of eight broad singlets at δ 2.6, 3.5, 4.2, 4.5, 6.3, 6.8, 8.8, and 10.2 in the 31P NMR spectrum (Fig. S3 and S4†). Collectively, these data confirm the existence of four isomeric products for 3 whose relationship was established by EXSY measurements at 223 K. The EXSY data confirm the dynamical exchange between the two hydrides representing the minor species (δ −13.73 with δ −15.45); hydride interconversion between the two species (δ −13.80 with δ −13.90) associated with the major isomer was also verified. No hydride exchange between the major and minor hydride species was observed. Similar exchange behavior for the pair of 31P resonances at δ 10.2 and 4.2 with those at δ 6.8 and 3.5 for the major species was found. The pairwise exchange of the 31P resonances at δ 8.8 and 4.5 with those at δ 6.6 and 2.6 define the exchanging pairs of resonances in the minor isomeric species.
The identity of the isomers present in the NMR spectra of 3 is best addressed by electronic structure calculations, and here we employed the solid-state structure of 3 as the starting point for our studies. The geometry-optimized structure of species E1, which is depicted alongside the X-ray diffraction structure in Fig. 4, exhibits good agreement with the experimental structure. Fig. 5 shows the ground-state energy ordering for the different species based on 3 that possess equatorial phosphines and where one of the PPh3 ligands is positioned cis to the edge-bridging hydride. The latter structural feature is required in order to reproduce the 2JPH coupling of ∼12 Hz in the 1H NMR spectra displayed in Fig. S3.† As with cluster 2, the optimized structure based on the X-ray diffraction structure of 3 (species E1) represents the thermodynamically favored isomer. E2 is a stereoisomer of E1, and it lies 0.9 kcal mol−1 above the latter species. The principal difference between E1 and E2 concerns the disposition of the newly added phosphine relative to the two available equatorial sites at the Os(CO)3P moiety. In the case of E1, the two phosphine groups display a synclinal orientation about the Os–Os bond common to both ligands, while they exhibit an anticlinal arrangement in E2.27 Species E3 and E4 are stereoisomers that originate from D2 and exhibit a substitution pattern similar to that found in species E1 and E2, with synclinal E3 1.6 kcal mol−1 more stable than anticlinal E4. Species E5 and E6 are isomers of E1–E4, and they may be viewed as the products of hydride migration from its common locus in E1 and E2 to the adjacent Os–Os bond in E5 and E6, respectively. The hydride migration that gives E5 and E6 is accompanied by a 7.4 and 9.1 kcal mol−1 loss in energy, respectively.28
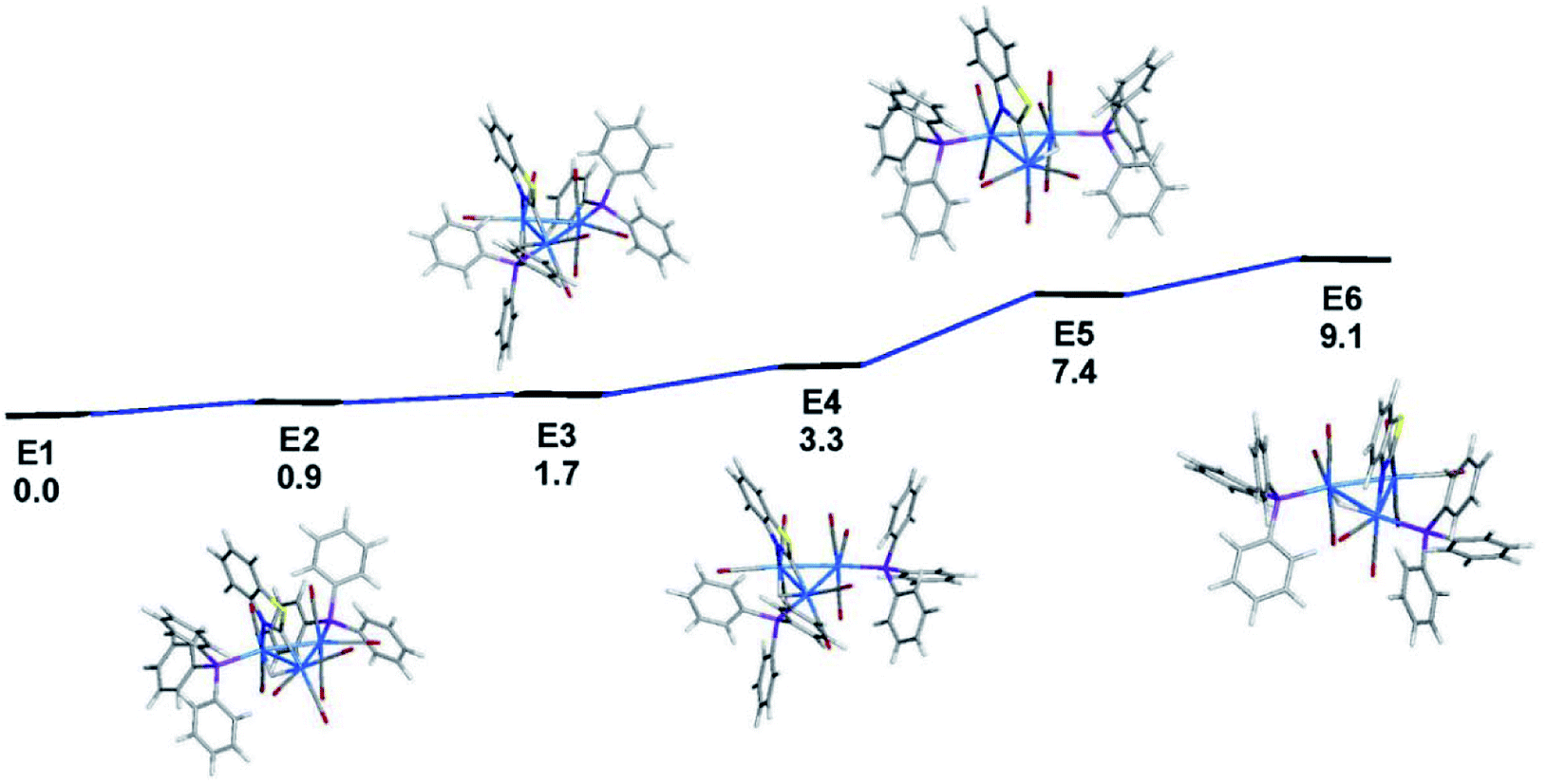 | ||
| Fig. 5 DFT-optimized structures and energy ordering for selected isomers based on cluster 3. Energy values are ΔG in kcal mol−1 relative to E1. Fig. S5† shows the ChemDraw pictures of E1–E6. | ||
The dynamic NMR behavior exhibited by the major component of 3 may be rationalized by an intramolecular exchange between E1 ⇌ E2. The equilibration of these stereoisomers is rapid at room temperature and proceeds via a tripodal permutation involving the PPh3 ligand and two CO groups at the Os(CO)3P center. Scheme 7 shows the rotational direction available to the two fragment rotors capable of promoting isomerization. Two of the three rotor substituents in both fragments include the equatorial CO and PPh3 groups. When the axial CO trans to the benzothiazolate ligand is used as the third rotor group (endo route), a stereoselective clockwise rotation of the fragment rotor was computationally confirmed. In this process the  syn the heterocycle remains unchanged. The participation of the
syn the heterocycle remains unchanged. The participation of the  in the tripodal rotation proceeds via a counterclockwise manner (exo route) and this leads to unfavourable van der Waals interactions between the heterocycle and the two migrating carbonyls (
in the tripodal rotation proceeds via a counterclockwise manner (exo route) and this leads to unfavourable van der Waals interactions between the heterocycle and the two migrating carbonyls ( and
and  ). In both tripodal rotations, the PPh3 group avoids the bridging benzothiazolate ligand by adopting an orientation coincident to the plane defined by the three metals. No evidence for a turnstile rotation involving the Os(CO)3P moiety was found, and these data are consistent with our earlier NMR studies that have demonstrated facile tripodal rotation at the Os(CO)3L vertices in a variety of heterocyclic-bridged clusters.8d,29
). In both tripodal rotations, the PPh3 group avoids the bridging benzothiazolate ligand by adopting an orientation coincident to the plane defined by the three metals. No evidence for a turnstile rotation involving the Os(CO)3P moiety was found, and these data are consistent with our earlier NMR studies that have demonstrated facile tripodal rotation at the Os(CO)3L vertices in a variety of heterocyclic-bridged clusters.8d,29
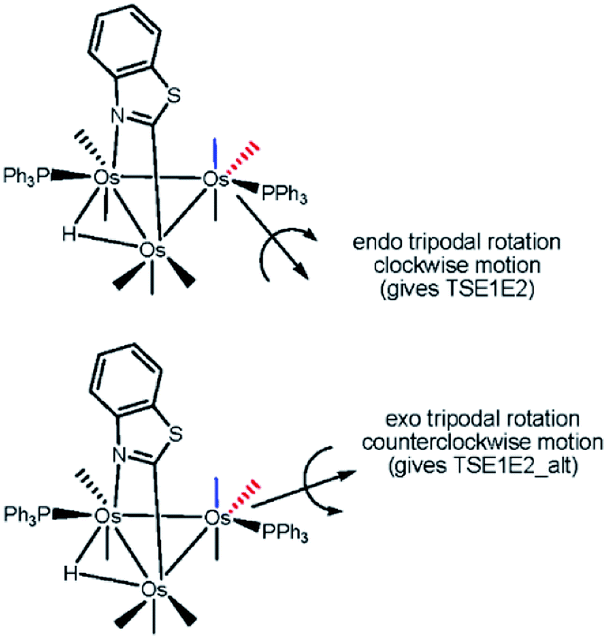 | ||
| Scheme 7 Endo and exo tripodal rotation pathways in E1. | ||
The optimized transition structures for TSE1E2 and TSE1E2_alt and the computed barriers for the tripodal rotation that promote the E1 ⇌ E2 exchange are displayed in Fig. 6. Both activation barriers are low and in concert with the VT NMR behavior recorded for 3. The endo tripodal rotation is favored by 1.2 kcal mol−1 over the exo rotation process. Fig. S6† provides a detailed view of the permutation process as a function of the ancillary ligands at the Os(CO)3P moiety.
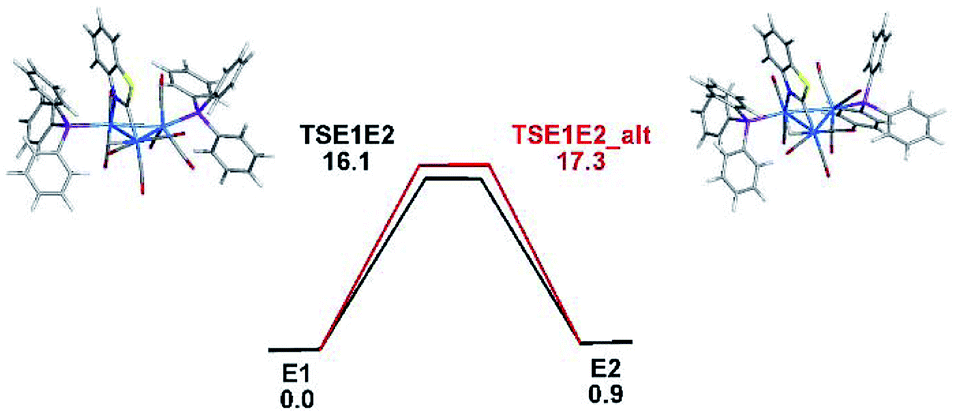 | ||
| Fig. 6 Free-energy profile for the E1 ⇌ E2 isomerization through endo (TSE1E2) and exo (TSE1E2_alt) tripodal rotations. Energy values are in kcal mol−1 relative to E1. | ||
The dynamical exchange between the minor stereoisomers E3 ⇌ E4 follows a similar sequence with the endo tripodal rotation again favored over the exo rotation (Fig. S7†). The computed ΔΔG‡ for the TSE3E4 and TSE3E4_alt is 2.0 kcal mol−1 (Fig. S8†). Here the endo rotation proceeds from E3 by a counterclockwise rotation of groups associated with the fragment rotor. The change in the rotation direction in E3 ⇌ E4 from that found in E1 ⇌ E2 is dictated by the synclinal disposition of the phosphines in E3.
Computational analysis of hydride migration in the species E1 ⇌ E5 and E2 ⇌ E6
Hydride mobility in metal clusters is a well-established phenomenon, and the dynamical exchange of a hydride(s) between different metal–metal bonds can occur rapidly on the NMR time scale.29b,30 Since hydride migration in E1 to the adjacent Os–Os bond in E5 could account for the NMR behavior displayed by 3, we investigated this scenario by DFT. Indeed, a suitable transition structure (TSE1E5) was found for the concerted migration of hydride through the interior of the cluster core by way of an interstitial-like hydride.31,32 The optimized structure for TSE1E5 is depicted in Fig. 7. The barrier computed for this process is 19.4 kcal mol−1, confirming that this hydride migration is not competitive to the tripodal rotation associated with TSE1E2 that lies 3.3 kcal mol−1 lower in energy.29a The migration of the hydride in E2 to give E6 was also examined through a series of step-scan calculations. Multiple attempts were made to find a suitable transition structure, but we were unable to successfully model this transformation with a species possessing an acceptable imaginary frequency and reasonable energy.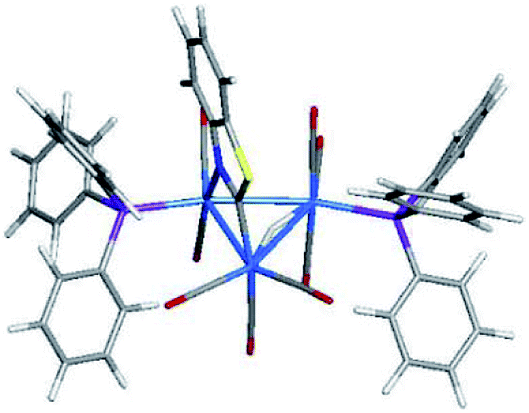 | ||
| Fig. 7 DFT-optimized structure for TSE1E5. | ||
Conclusions
DFT calculations on the possible metalated heterocycles from the reaction of benzothiazole and 1,2-Os3(CO)10(MeCN)2 have confirmed the greater stability of the 1,2- and 1,7-N,C coordination isomers over the corresponding S,C-functionalized species. The reactivity of the 1,2-N,C isomer, HOs3(CO)10(μ-1,2-N,C-η1,κ1-C7H4NS) (1), in PPh3 substitution has been examined through a combined experimental/computational approach. The high regioselectivity experimentally found in each substitution step site has been successfully analyzed by electronic structure calculations. DFT calculations on the fluxional NMR behavior exhibited by 3 reveal a stereoselective tripodal rotation at the Os(CO)3(PPh3) moiety where rotation of the fragment rotor proceeds distal to the metalated heterocycle. The relative stabilities of the different isomers of 2 and 3 have been determined by DFT, and the computed species reconciled with those products observed in solution by NMR spectroscopy. The ability to functionalize the benzothiazolate ligand in 1 with soft organic nucleophiles and hydride ligand are planned. These studies will allow us to chart the directing effect that the 1,2-N,C coordination mode of the benzothiazolate group in 1 has, if any, versus our earlier studies based on the 1,7-N,C isomer.Experimental
General and instrumentation
All the reactions were carried out under a nitrogen atmosphere using standard Schlenk techniques unless otherwise stated. Reagent grade solvents were dried by the standard methods and freshly distilled prior to use. The benzothiazole, PPh3, and Me3NO·2H2O used in these studies were purchased from Aldrich Chemical Co.; the former chemicals were used as received. Me3NO·2H2O was dried by azeotropic distillation using benzene and a Dean–Stark distillation unit. HOs3(CO)10(μ-1,2-N,C-η1,κ1-C7H4NS) (1) was prepared from 1,2-Os3(CO)10(MeCN)2 and benzothiazole according to literature procedures.6c,15 The separation of clusters 2 and 3 were performed by TLC in the air on 0.5 mm silica gel 60 Å F254 glass plates. Infrared spectra were recorded on a Shimadzu IR Prestige-21 spectrophotometer, and the VT 1H and 31P NMR spectra were recorded on a Bruker DPX 400 spectrometer. All NMR chemical shift and coupling constant data are reported in δ units and Hz, respectively. Elemental analyses were performed by the Microanalytical Laboratory of Wazed Miah Research Centre at Jahangirnagar University.Reaction of HOs3(CO)10(μ-1,2-N,C-η1,κ1-C7H4NS) (1) with PPh3
To a CH2Cl2 solution (20 mL) of HOs3(CO)10(μ-1,2-N,C-η1,κ1-C7H4NS) (1) (100 mg, 0.101 mmol) and PPh3 (27 mg, 0.101 mmol) was added 7 mL of CH2Cl2 containing Me3NO (8 mg, 0.106 mmol) dropwise over a period of 30 min. The resultant solution was stirred for another 30 min, during which time the solution color changed from yellow to deep orange. The reaction mixture was then filtered through a short silica column (4 cm) to remove any excess Me3NO, and the solvent was removed under reduced pressure. The residue was purified by chromatography over silica gel using a mixture of cyclohexane/CH2Cl2 (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, v/v) as the eluent. Five bands were isolated, the first of which corresponded to unreacted 1 (12 mg). The second and third bands afforded HOs3(CO)9(PPh3)(μ-1,2-N,C-η1,κ1-C7H4NS) (2) (55 mg, 41%) and HOs3(CO)8(PPh3)2(μ-1,2-N,C-η1,κ1-C7H4NS) (3) (15 mg, 10%) as orange crystals after recrystallization from n-hexane/CH2Cl2 at −4 °C. The contents of the fourth and fifth bands were too small for complete characterization. Data for 2: anal. calcd for C34H20NO9Os3PS: C, 33.47; H, 1.65; N, 1.15. Found: C, 33.21; H, 1.79; N, 1.13%. IR (ν(CO), CH2Cl2): 2090s, 2051s, 201s, 1998s, 1979sh, 1967m, 1940w cm−1. 1H NMR (CDCl3, 25 °C): δ 7.49 (d, J 6.8, 1H), 7.23 (m, 15H), 6.94 (m, 2H), 6.80 (d, J 7.6, 1H), −14.21 (d, J 11.6, 1H). 31P{1H} NMR (CDCl3, 25 °C): δ 11.8 (s). Data for 3: anal. calcd for C51H35NO8Os3P2S·CH2Cl2: C, 40.57; H, 2.42; N, 0.91. Found: C, 41.16; H, 2.48; N, 1.03. IR (ν(CO), CH2Cl2): 2065m, 2060m, 2023s, 1993s, 1967m, 1952s cm−1. 1H NMR (CDCl3, 25 °C): δ 7.50 (m, 8H), 7.37 (m, 11H), 7.25 (m, 12H), 6.92 (m, 3H), −13.74 (d, J 11.6, 1H). 31P{1H} NMR (CDCl3, 25 °C): δ 8.9 (s), 3.9 (s).
:3, v/v) as the eluent. Five bands were isolated, the first of which corresponded to unreacted 1 (12 mg). The second and third bands afforded HOs3(CO)9(PPh3)(μ-1,2-N,C-η1,κ1-C7H4NS) (2) (55 mg, 41%) and HOs3(CO)8(PPh3)2(μ-1,2-N,C-η1,κ1-C7H4NS) (3) (15 mg, 10%) as orange crystals after recrystallization from n-hexane/CH2Cl2 at −4 °C. The contents of the fourth and fifth bands were too small for complete characterization. Data for 2: anal. calcd for C34H20NO9Os3PS: C, 33.47; H, 1.65; N, 1.15. Found: C, 33.21; H, 1.79; N, 1.13%. IR (ν(CO), CH2Cl2): 2090s, 2051s, 201s, 1998s, 1979sh, 1967m, 1940w cm−1. 1H NMR (CDCl3, 25 °C): δ 7.49 (d, J 6.8, 1H), 7.23 (m, 15H), 6.94 (m, 2H), 6.80 (d, J 7.6, 1H), −14.21 (d, J 11.6, 1H). 31P{1H} NMR (CDCl3, 25 °C): δ 11.8 (s). Data for 3: anal. calcd for C51H35NO8Os3P2S·CH2Cl2: C, 40.57; H, 2.42; N, 0.91. Found: C, 41.16; H, 2.48; N, 1.03. IR (ν(CO), CH2Cl2): 2065m, 2060m, 2023s, 1993s, 1967m, 1952s cm−1. 1H NMR (CDCl3, 25 °C): δ 7.50 (m, 8H), 7.37 (m, 11H), 7.25 (m, 12H), 6.92 (m, 3H), −13.74 (d, J 11.6, 1H). 31P{1H} NMR (CDCl3, 25 °C): δ 8.9 (s), 3.9 (s).
Conversion of 2 to 3
To a Schlenk tube containing 2 (30 mg, 0.024 mmol) and PPh3 (7 mg, 0.027 mmol) in 20 mL of CH2Cl2 was added a CH2Cl2 solution (5 mL) of Me3NO (2 mg, 0.027 mmol) over a period of 15 min with the aid of a dropping funnel. The reaction mixture was then stirred at room temperature for 2 h, after which time TLC analysis confirmed the consumption of the starting cluster and the presence of desired product 3. Chromatographic workup, followed by recrystallization, afforded 3 (26 mg, 75%).X-ray crystallography
Single crystals of 2 and 3 suitable for single crystal X-ray diffraction analysis were grown by slow diffusion of hexane into a CH2Cl2 solution containing each product. A suitable single crystal of each cluster was mounted on an Agilent Super Nova dual diffractometer (Agilent Technologies Inc., Santa Clara, CA) using a Nylon Loop and the diffraction data were collected at 150(2) K using Mo-Kα radiation (λ = 0.71073). Unit cell determination, data reduction, and absorption corrections were carried out using CrysAlisPro.33 The structures were solved with the ShelXS34 structure solution program using Direct Method and refined with the ShelXL34 refinement package using Least Squares minimization within the Olex2 (ref. 35) graphical user interface. Non-hydrogen atoms were refined anisotropically, and hydrogen atoms (except those directly bonded to metals) were included using a riding model. The bridging hydride ligand in 2 and 3 was found in the difference map and freely refined. Additional details of data collection and structure refinement are given in Table 1.| 2 | 3 | |
|---|---|---|
| a R1 = Σ||Fo| − |Fc||/Σ|Fo|.b wR2 = {Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]}1/2. | ||
| CCDC entry no | 1833188 | 1833187 |
| Cryst system | Triclinic | Monolinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| a, Å | 11.0561(2) | 18.8799(6) |
| b, Å | 16.0438(3) | 15.0410(3) |
| c, Å | 21.7248(5) | 18.7314(5) |
| α, deg | 100.9456(19) | 90 |
| β, deg | 92.7106(19) | 117.376(4) |
| γ, deg | 90.9327(17) | 90 |
| V, Å3 | 3777.99(15) | 4723.5(3) |
| Mol formula | C69H42Cl2N2O19Os6P2S2 | C51H35NO8Os3P2S |
| Fw | 2541.20 | 1454.40 |
| Formula units per cell (Z) | 2 | 4 |
| Dcalcd (mg m−3) | 2.234 | 2.045 |
| λ (Mo Kα), Å | 0.71073 | 0.71073 |
| μ (mm−1) | 10.287 | 8.218 |
| F (000) | 2348 | 2744 |
| Total reflections | 61519 |
74577 |
| Independent reflections | 14819 |
9274 |
| Data/res/paras | 14819/0/936 |
9274/0/599 |
| GOF on F2 | 1.076 | 1.071 |
| R1a [I ≥ 2σ(I)] | 0.0234 | 0.0242 |
| wR2b (all data) | 0.0569 | 0.0592 |
| Δρ(max), Δρ(min) (e Å−3) | 1.79, −1.52 | 2.60, −1.89 |
Computational methodology and modeling details
All DFT calculations were performed with the Gaussian 09 package of programs36 using the B3LYP hybrid functional. This functional is comprised of Becke's three-parameter hybrid exchange functional (B3)37 and the correlation functional of Lee, Yang, and Parr (LYP).38 Each osmium atom was described with the Stuttgart–Dresden effective core potential and SDD basis set,39 and the 6-31G(d’) basis set40 was employed for all remaining atoms.All reported geometries were fully optimized, and analytical Hessian was evaluated at each stationary point to determine whether the geometry was an energy minimum (no negative eigenvalues) or a transition structure (one negative eigenvalue). Unscaled vibrational frequencies were used to make zero-point and thermal corrections to the electronic energies, and the resulting free energies are reported in kcal mol−1 relative to the specified standard. Intrinsic reaction coordinate (IRC) calculations were performed on all transition-state structures in order to establish the reactant and product species associated with each transition-state structure. The geometry-optimized structures have been drawn with the JIMP2 molecular visualization and manipulation program.41
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SEK thanks the Ministry of Education, the Government of the Peoples' Republic of Bangladesh, and the University Grants Commission of Bangladesh for sponsorship and financial support of this work. MGR acknowledges The Robert A. Welch Foundation through Grant B-1093 for generous research support. The DFT calculations were performed at UNT through CASCaM, which is an NSF-supported facility (CHE-1531468).Notes and references
- (a) A. Eisenstadt, C. M. Giandomenico, M. F. Frederick and R. M. Laine, Organometallics, 1985, 4, 2033 CrossRef; (b) R. H. Fish, T.-J. Kim, J. H. Bushweller, R. K. Rosen and J. W. Dupon, Organometallics, 1986, 5, 2193 CrossRef.
- E. Rosenberg and R. Kumar, Dalton Trans., 2012, 41, 714 RSC.
- S. E. Kabir, D. S. Kolwaite, E. Rosenberg, L. G. Scott, T. McPhillips, R. Duque, M. Day and K. I. Hardcastle, Organometallics, 1996, 15, 1979 CrossRef.
- (a) E. Arcia, D. S. Kolwaite, E. Rosenberg, K. Hardcastle, J. Ciurash, R. Duque, R. Gobetto, L. Milone, D. Osella, M. Botta, W. Dastrú, A. Viale and I. Fiedler, Organometallics, 1998, 17, 415 CrossRef; (b) R. Smith, E. Rosenberg, K. I. Hardcastle, V. Vazquez and J. Roh, Organometallics, 1999, 18, 3519 CrossRef.
- (a) A. K. Raha, S. Ghosh, M. M. Karim, D. A. Tocher, N. Begum, A. Sharmin, E. Rosenberg and S. E. Kabir, J. Organomet. Chem., 2008, 693, 3613 CrossRef; (b) S. Ghosh, M. N. Uddin, N. Begum, G. M. G. Hossain, K. A. Azam and S. E. Kabir, J. Chem. Crystallogr., 2010, 40, 572 CrossRef; (c) M. I. Hossain, S. Ghosh, G. Hogarth, G. M. G. Hossain and S. E. Kabir, J. Organomet. Chem., 2011, 696, 3036 CrossRef; (d) S. Ghosh, M. R. Al-Mamun, G. M. G. Hossain and S. E. Kabir, Inorg. Chim. Acta, 2011, 378, 307 CrossRef; (e) S. A. Begum, M. A. H. Chowdhury, S. Ghosh, D. A. Tocher, M. G. Richmond, E. Rosenberg and S. E. Kabir, J. Organomet. Chem., 2017, 849–850, 337 CrossRef.
- (a) S. E. Kabir, D. S. Kolwaite, E. Rosenberg, K. Hardcastle, W. Cresswell and J. Grindstaff, Organometallics, 1995, 14, 3611 CrossRef; (b) B. Bergman, R. Holmquist, R. Smith, E. Rosenberg, J. Ciurash, K. Hardcastle and M. Visi, J. Am. Chem. Soc., 1998, 120, 12818 CrossRef; (c) M. J. Abedin, B. Bergman, R. Holmquist, R. Smith, E. Rosenberg, J. Ciurash, K. Hardcastle, J. Roe, V. Vazquez, C. Roe, S. Kabir, B. Roy, S. Alam and K. A. Azam, Coord. Chem. Rev., 1999, 190–192, 975 CrossRef.
- (a) E. Rosenberg, S. E. Kabir, M. J. Abedin and K. I. Hardcastle, Organometallics, 2004, 23, 3982 CrossRef; (b) D. G. Musaev, T. Nowroozi-Isfahani, K. Morokuma, J. Abedin, E. Rosenberg and K. I. Hardcastle, Organometallics, 2006, 25, 203 CrossRef; (c) E. Rosenberg, J. Abedin and K. I. Hardcastle, J. Cluster Sci., 2012, 23, 901 CrossRef.
- (a) A. B. Din, B. Bergman, E. Rosenberg, R. Smith, W. Dastru, R. Gobetto, L. Milone and A. Viale, Polyhedron, 1998, 17, 2975 CrossRef; (b) E. Rosenberg, M. J. Abedin, D. Rokhsana, D. Osella, L. Milone, C. Nervi and J. Fiedler, Inorg. Chim. Acta, 2000, 300–302, 769 CrossRef; (c) S. E. Kabir, K. M. A. Malik, H. S. Mandal, M. A. Mottalib, M. J. Abedin and E. Rosenberg, Organometallics, 2002, 21, 2593 CrossRef; (d) E. Rosenberg, M. J. Abedin, D. Rokhsana, A. Viale, W. Dastru, R. Gobetto, L. Milone and K. Hardcastle, Inorg. Chim. Acta, 2002, 334, 343 CrossRef; (e) S. T. Beatty, B. Bergman, E. Rosenberg, W. Dastru, R. Gobetto, L. Milone and A. Viale, J. Organomet. Chem., 2000, 593–594, 226 CrossRef.
- (a) E. Rosenberg, F. Spada, K. Sugden, B. Martin, L. Milone, R. Gobetto, A. Viale and J. Fiedler, J. Organomet. Chem., 2003, 668, 51 CrossRef; (b) D. Colangelo, A. L. Ghiglia, A. R. Ghezzi, M. Ravera, E. Rosenberg, F. Spada and D. Osella, J. Inorg. Biochem., 2005, 99, 505 CrossRef PubMed.
- (a) C. Nervi, R. Gobetto, L. Milone, A. Viale, E. Rosenberg, D. Rokhsana and J. Fiedler, Chem.–Eur. J., 2003, 9, 5749 CrossRef PubMed; (b) E. Rosenberg, D. Rokhsana, C. Nervi, R. Gobetto, L. Milone, A. Viale, J. Fiedler and M. A. Botavina, Organometallics, 2004, 23, 215 CrossRef; (c) E. Rosenberg, F. Spada, K. Sugden, B. Martin, R. Gobetto, L. Milone and A. Viale, J. Organomet. Chem., 2004, 689, 4729 CrossRef.
- (a) M. A. Mottalib, N. Begum, S. M. T. Abedin, T. Akter, S. E. Kabir, M. A. Miah, D. Rokhsana, E. Rosenberg, G. M. G. Hossain and K. I. Hardcastle, Organometallics, 2005, 24, 4747 CrossRef; (b) D. G. Musaev, T. Nowroozi-Isfahani, K. Morokuma and E. Rosenberg, Organometallics, 2005, 24, 5973 CrossRef; (c) T. Nowroozi-Isfahani, D. G. Musaev, K. Morokuma and E. Rosenberg, Inorg. Chem., 2006, 45, 4963 CrossRef PubMed.
- (a) B. C. Gates, Catalytic Chemistry, J. Wiley and Sons, New York, 1992, p. 409 Search PubMed; (b) See symposium: Mechanism of HDS/HDN Reactions, Prepr. ACS Div. of Pet. Chem. 1988, vol. 33, p. 638 Search PubMed; (c) R. H. Fish, in Aspects of Homogeneous Catalysts, ed. R. Ugo, Kluwer Academics, Dordrecht, The Netherlands, 1990, pp. 65–83 Search PubMed.
- (a) S. D. Gray, D. P. Smith, M. S. Bruck and D. E. Wigley, J. Am. Chem. Soc., 1992, 114, 5462 CrossRef; (b) S. D. Gray, P. A. Fox, M. S. Kingsborough, M. S. Bruci and D. E. Wigley, Prepr.–Am. Chem. Soc., Div. Pet. Chem., 1993, 38, 706 Search PubMed; (c) I. Horvath and R. D. Adams, Prog. Inorg. Chem., 1985, 33, 127 Search PubMed; (d) A. Eisenstadt, C. M. Giandomenico, M. F. Frederick and R. M. Laine, Organometallics, 1985, 4, 2033 CrossRef; (e) R. M. Laine, New J. Chem., 1987, 11, 543 Search PubMed; (f) R. D. Adams, J. E. Babin, J. T. Tanner and J. A. Wolfe, J. Am. Chem. Soc., 1990, 112, 3426 CrossRef; (g) S. E. Kabir, M. Day, M. Irving, T. McPhillips, H. Minassian, E. Rosenberg and K. I. Hardcastle, Organometallics, 1991, 10, 3997 CrossRef.
- M. B. Hursthouse, S. E. Kabir, K. M. A. Malik, M. Tesmer and H. Vahrenkamp, J. Organomet. Chem., 1998, 568, 133 CrossRef.
- N. Begum, M. K. Islam, S. E. Kabir and M. M. Hassan, J. Bangladesh Acad. Sci., 2005, 29, 145 Search PubMed.
- K. M. Uddin, S. Ghosh, A. K. Raha, G. Hogarth, E. Rosenberg, A. Sharmin, K. I. Hardcastle and S. E. Kabir, J. Organomet. Chem., 2010, 695, 1435 CrossRef.
- N. Begum, A. J. Deeming, M. K. Islam, S. E. Kabir, D. Rokhsana and E. Rosenberg, J. Organomet. Chem., 2004, 689, 2633 CrossRef.
- K. A. Azam, R. Dilshad, S. E. Kabir, K. Khatoon, L. Nessa, M. M. Rahman, E. Rosenberg, M. B. Hursthouse, K. M. A. Malik and A. J. Deeming, Dalton Trans., 1996, 1731 RSC.
- J. A. Joule and K. Mills, Heterocyclic Chemistry, Wiley, Hoboken, NJ, 2010 Search PubMed.
- K. A. Azam, M. B. Hursthouse, S. A. Hussain, S. E. Kabir, K. M. A. Malik, M. M. Rahman and E. Rosenberg, J. Organomet. Chem., 1998, 559, 81 CrossRef.
- M. A. H. Chowdhury, S. Rajbangshi, A. Rahaman, L. Yang, V. N. Nesterov, M. G. Richmond, S. M. Mobin and S. E. Kabir, J. Organomet. Chem., 2015, 779, 21 CrossRef.
- (a) B. F. G. Johnson, J. Lewis, E. Nordlander and P. R. Raithby, Dalton Trans., 1996, 755 RSC; (b) X. Zhang, S. Kandala, L. Yang, W. H. Watson, X. Wang, D. A. Hrovat, W. T. Borden and M. G. Richmond, Organometallics, 2011, 30, 1253 CrossRef; (c) M. K. Hossain, S. Rajbangshi, A. Rahaman, M. A. H. Chowdhury, T. A. Siddiquee, S. Ghosh, M. G. Richmond, E. Nordlander, G. Hogarth and S. E. Kabir, J. Organomet. Chem., 2014, 760, 231 CrossRef.
- (a) R. Bau, R. G. Teller, S. W. Kirtley and T. F. Koetzle, Acc. Chem. Res., 1979, 12, 176 CrossRef; (b) R. Hoffmann, B. E. R. Schilling, R. Bau, H. D. Kaesz and D. M. P. Mingoss, J. Am. Chem. Soc., 1978, 100, 6088 CrossRef; (c) S. K. Brayshaw, J. C. Green, N. Hazari and A. S. Weller, Dalton Trans., 2007, 1781 RSC.
- (a) R. D. Adams, N. M. Golembeski and J. P. Selegue, Inorg. Chem., 1981, 20, 1242 CrossRef; (b) R. D. Adams, Z. Dawoodi, D. F. Foust and B. E. Segmuller, Organometallics, 1983, 2, 315 CrossRef; (c) A. J. Deeming, Y. Fuchita, K. Hardcastle, K. Henrick and M. McPartlin, Dalton Trans., 1986, 2259 RSC; (d) K. A. Azam, M. B. Hursthouse, S. A. Hussain, S. E. Kabir, K. M. A. Malik, M. Rahman and E. Rosenberg, J. Organomet. Chem., 1998, 559, 81 CrossRef; (e) M. Day, D. Espitia, K. I. Hardcastle, S. E. Kabir, E. Rosenberg, R. Gobetto, L. Milone and D. Osella, Organometallics, 1991, 10, 3550 CrossRef; (f) S. Monira, S. Afrin, K. A. Azam, M. K. Hossain, D. A. Tocher, S. Ghosh, S. Rajbangshi, S. E. Kabir and G. Hogarth, J. Organomet. Chem., 2015, 799–800, 281 CrossRef.
- (a) T. A. Albright, J. K. Burdett and M. H. Whangbo, Orbital Interactions in Chemistry, Wiley-Interscience, New York, 1985 Search PubMed; (b) I. Fleming, Molecular Orbitals and Organic Chemical Reactions, Hoboken, New Jersey, 2010 CrossRef.
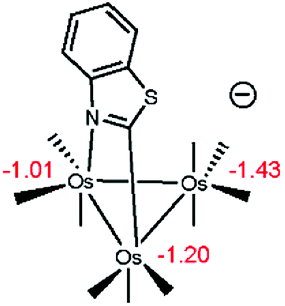
- Since metal hydrides may be viewed within an amphoteric construct, we have performed NPA calculations on the anionic cluster of C1 that is formed on deprotonation of the hydride. Here we employed the 6-31+G(d’) basis set for the main group atoms in our calculations which were first performed on C1 as a benchmark. The charges computed for the three metals and the hydride ligand in C1 using the added diffuse function mimic those Q values quoted in Scheme 6. The charges computed for the anionic cluster show little change, as illustrated in the below structure, and reinforce the more important influence the heterocycle plays in directing the ligand substitution chemistry.
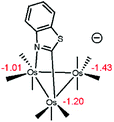 .
. - (a) L. C. Cross and W. Klyne, Appendix, Section E., Pure Appl. Chem., 1976, 45, 11 Search PubMed; (b) E. L. Eliel, S. H. Wilen and L. N. Mander, Stereochemistry of Organic Compounds, Wiley, New York, 1994 Search PubMed.
- The structural isomer containing adjacent equatorial phosphines at the edge-bridged Os–Os bond and cis to the hydride (E7, not shown) was also examined computationally and found to be 9.3 kcal mol−1 less stable than E1. The orientation of the two PPh3 groups in E7 should furnish a hydride triplet in the 1H NMR spectrum, allowing us to exclude this species as a participant isomer..
- (a) M. Day, D. Espitia, K. I. Hardcastle, S. E. Kabir, E. Rosenberg, R. Gobetto, L. Milone and D. Osella, Organometallics, 1991, 10, 3550 CrossRef; (b) E. Rosenberg and R. Kumar, J. Cluster Sci., 2014, 25, 239 CrossRef.
- (a) E. Band and E. L. Muetteries, Chem. Rev., 1978, 78, 639 CrossRef; (b) B. F. G. Johnson, Transition Metal Clusters, Wiley, New York, 1980 Search PubMed; (c) E. Rosenberg, Polyhedron, 1989, 8, 383 CrossRef.
- For reports on the hydride fluxionality involving edge-bridge ⇌ interstitial migration, see: (a) T. Eguchi, B. T. Heaton, R. Harding, K. Miyagi, G. Longoni, J. Nahring, N. Nakamura, H. Nakayama, T. A. Pakkanen, J. Pursianen and A. K. Smith, Dalton Trans., 1996, 625 RSC; (b) T. Eguchi and B. T. Heaton, Dalton Trans., 1996, 3523 Search PubMed.
- For DFT studies where interstitial hydrides have been computed as reactive intermediates (ground-state species) or transition structures in hydride isomerizations, see: (a) M. J. Hossain, S. Rajbangshi, M. M. M. Khan, S. Ghosh, G. Hogarth, E. Rosenberg, K. I. Hardcastle, M. G. Richmond and S. E. Kabir, J. Organomet. Chem., 2014, 767, 185 CrossRef; (b) M. A. H. Chowdhury, M. R. Haque, S. Ghosh, S. M. Mobin, D. A. Tocher, G. Hogarth, M. G. Richmond, S. E. Kabir and H. W. Roesky, J. Organomet. Chem., 2017, 836–837, 68 CrossRef; (c) M. M. M. Khan, M. M. Alam, S. Ghosh, A. Rahaman, D. A. Tocher, M. G. Richmond, S. E. Kabir and H. W. Roesky, J. Organomet. Chem., 2017, 843, 75 CrossRef; (d) M. A. Alvarez, M. E. García, D. García-Vivó, M. A. Ruiz and A. Toyos, Dalton Trans., 2017, 46, 15317 RSC.
- CrysAlisPro; Oxford Diffraction, Yarnton, England, 2015 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef.
- (a) M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866 CrossRef; (b) S. P. Watch and C. W. Bauschlicher, J. Chem. Phys., 1983, 78, 4597 CrossRef.
- (a) G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193 CrossRef; (b) G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081 CrossRef.
- (a) JIMP2, version 0.091, a free program for the visualization and manipulation of molecules, M. B. Hall and R. F. Fenske, Inorg. Chem., 1972, 11, 768 CrossRef; (b) J. Manson, C. E. Webster and M. B. Hall, Texas A & M University, College Station, TX, 2006: http://www.chem.tamu.edu/jimp2/index.html.
Footnote |
| † Electronic supplementary information (ESI) available: Atomic coordinates of all DFT-optimized stationary points, ChemDraw pictures, and spectroscopic data. CCDC 1833188 and 1833187 contain the supplementary crystallographic data for compounds 2 and 3, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra07400c |
| This journal is © The Royal Society of Chemistry 2018 |