
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of α,β-unsaturated esters of perfluoropolyalkylethers (PFPAEs) based on hexafluoropropylene oxide units for photopolymerization†
Céline
Bonneaud
 b,
Mélanie
Decostanzi
b,
Julia
Burgess
a,
Giuseppe
Trusiano
c,
Trevor
Burgess
a,
Roberta
Bongiovanni
c,
Christine
Joly-Duhamel
b and
Chadron M.
Friesen
*a
b,
Mélanie
Decostanzi
b,
Julia
Burgess
a,
Giuseppe
Trusiano
c,
Trevor
Burgess
a,
Roberta
Bongiovanni
c,
Christine
Joly-Duhamel
b and
Chadron M.
Friesen
*a
aTrinity Western University, Department of Chemistry, 7600 Glover Road, Langley, British Columbia V2Y 1Y1, Canada. E-mail: chad.friesen@twu.ca
bIngénierie et Architectures Macromoléculaires, Institut Charles Gerhardt, Ecole Nationale Supérieure de Chimie de Montpellier (UMR5253-CNRS), 240 Avenue Prof Emile Jeanbrau, 34296 Montpellier Cedex 5, France
cDepartment of Applied Science and Technology, Politecnico di Torino, 10128 Torino, Italy
First published on 21st September 2018
Abstract
α,β-unsaturated esters are usually synthesized for polymer applications. However, the addition of maleate (cis-configuration) to a fluorinated moiety is challenging due to its potential isomerization during esterification. Various synthetic routes were attempted and led to very low conversion or side-products. The immiscibility of both reagents combined with an easy isomerization or attack on the double bond were potential explanations. In this paper, the synthesis of maleates oligo(hexafluoropropylene oxide) is reported by Steglich esterification and the reaction conditions are discussed depending on the molecular weight of the fluorinated moieties. After UV-curing, hydrophobic polymers were obtained by copolymerization with vinyl ethers by electron acceptor–donor systems.
Introduction
α,β-unsaturated esters are very useful in polymer applications.1–3 Two configurations of α,β-unsaturated esters are conceivable: the cis-configuration (maleate) and the trans-configuration (fumarate) (Fig. 1). In this paper, we focused on the mechanism and synthesis of maleates containing fluorinated moieties. Indeed, the synthesis of the corresponding fumarates can then easily be transposed but the maleates are inclined towards isomerization and consequently their synthesis is more challenging. In the past, maleate monomers have been successfully copolymerized with a variety of monomers such as vinyl acetate,1,4,5 vinyl ether,2 epoxy3 or even hydrophobic types.6 They have been seen in applications such as latex films,1,5 polyelectrolytes6 or cooperative complexation of cyclodextrins.7 It is also well known that photopolymerization is a fast and eco-friendly process; using UV-light at room temperature with maleates contributes to this environmentally friendly chemical process. The copolymerization of vinyl ethers with maleates was accomplished under UV-light by electron acceptor–donor (AD) systems. These systems proved to form an alternated copolymer by utilizing [AD] properties and had the appropriate stoichiometric ratios.8,9 Under UV-curing, maleates usually showed a faster reactivity than the more thermodynamically stable fumarates.2 Nonetheless, the opposite effect was observed10 and another study will be dedicated to the comparison of their kinetics. In comparison to the common (meth)acrylic systems, their polymerization is less sensitive to oxygen.11,12 The use of α,β-unsaturated esters with fluorinated chains is almost absent from the literature.13–15 However, novel outstanding properties such as high hydrophobicity and oleophobicity can be obtained thanks to the fluorine atoms. The aim of our work was to bond monoalkyl maleates to long fluorinated moieties. Perfluoropolyalkylethers (PFPAEs) are non-toxic fluorinated long chains16 which demonstrate interesting properties in many high technology areas such as aerospace, aeronautic (seals, gaskets) or automotive industries.17 | ||
| Fig. 1 Cis- and trans- configurations of α,β-unsaturated esters. | ||
These PFPAEs based on structural units such as –(CF2O)–, –(CF2CF2O)–, –(CF2CF2CF2O)– and –(CF(CF3)CF2O)– showed high chemical and thermal inertness, low surface energy and flammability, and excellent ageing and weather resistances.18 Oligo(hexafluoropropylene) (oligo(HFPO)) products obtained by the anionic ring-opening reaction of hexafluoropropylene oxide with cesium fluoride were used in this work. Two different molecular weights of oligo(HFPO) were studied (Mw ∼ 1250 g mol−1 and Mw ∼ 2000 g mol−1). Due to the high withdrawing effect of the fluorine atoms on its long chain, the nucleophilicity of these oligomers is particularly low. They also inherently have the characteristic of being insoluble in many organic solvents and are only soluble in specific fluorinated solvents. Conversely, the highly polar maleates are soluble in many organic solvent types but are insoluble in fluorinated solvents. After the synthesis of the monoalkyl maleates with different substituents was achieved, different synthetic methods were conceivable and are discussed for the esterification of the fluorinated alcohols to the remaining carboxylic acid group of the maleate. After their synthesis, the photopolymerization was carried out to create highly hydrophobic polymers.
Experimental
Materials
Maleic anhydride, methanol, ethanol, propan-2-ol, pentanol, benzyl alcohol, phenol, tert-butyl alcohol, tert-amyl alcohol, thionyl chloride, triethylamine, carbonyldiimidazole, Amberlyst-15 hydrogen form (strongly acidic, cation exchanger, dry), dicyclohexylcarbodiimide, 4-dimethylamino pyridine, 2-hydroxy-2-methylpropiophenone and dichloromethane were purchased from Sigma Aldrich. Triethylamine and thionyl chloride were distilled before use. Triethylamine and trifluorotoluene were kept under activated molecular sieves (3A). The different alcohols were previously dried by using MgSO4. 1,1,1,3,3-pentafluorobutane was purchased from Alfa Aesar. The 1250 g mol−1 oligo(HFPO) methylene alcohol was prepared from Krytox® acyl fluoride. The 1250 Krytox® acyl fluoride and the 2000 g mol−1 Krytox® methylene alcohol were kindly provided by E. I. du Pont de Nemours and Company.Analysis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The sample was previously diluted in methoxyperfluorobutane (3M’s Novec™ HFE-7100) in a GC vial.
:50 1% LiCl in MeOH and 2% perfluorocinnamic acid dissolved in 50:50 MeOH:methoxynonafluorobutane (3 M HFE-7100). A 1 μL solution was then pipetted on to a ground steel plate, dried, and irradiated for a minimum of 5000 shots.
:1. The sample was previously diluted in methoxyperfluorobutane (3M’s Novec™ HFE-7100) in a GC vial.
:50 1% LiCl in MeOH and 2% perfluorocinnamic acid dissolved in 50:50 MeOH:methoxynonafluorobutane (3 M HFE-7100). A 1 μL solution was then pipetted on to a ground steel plate, dried, and irradiated for a minimum of 5000 shots.
Synthesis of the monoalkyl maleates
In a general procedure, 10 mmol of maleic anhydride (981 mg) were dissolved in 20 mmol of the corresponding alcohol (2 eq.). The mixture was stirred at room temperature or heated between 45 °C and 55 °C. The different reaction times and temperatures are reported for each compound. The alcohol was then removed under high vacuum or the product was purified by flash chromatography if specified.Monomethyl maleate 1a (R.T., 5 h, yield = 96%): 1H NMR (400 MHz, CDCl3, 25 °C, δ): 3.90 (s, CH3OCO, 3H), 6.43 (dd, –CH = CHCOOH–, 2H, 3JH–H = 12.7 Hz and 35.2 Hz), 13C NMR (100 MHz, CDCl3, 25 °C, δ): 53.8 (CH3OCO–), 129.0 (–CH = CHCOOH–), 137.0 (–CHCOOH), 164.3 (CH3CH2OCO–), 168.4 (–COOH), GC-MS, 70 eV, m/z: 41.1 (17), 43.1 (34), 45 (34), 54 (24), 55 (10), 59 (11), 72 (17), 99 (100), 100 (16), FT-IR (ATR) νmax (cm−1): 819.6, 856.2, 1165.6, 1222.5, 1439.6, 1633.1, 1727.90.
Synthesis of maleate oligo(HFPO) Mw ∼ 2000 g mol−1
To a solution of oligo(HFPO) alcohol (Mw = 2000 g mol−1, 2 g, 1 mmol), monoalkyl maleate (2 eq.) and DMAP (0.1 eq.) in 1,1,1,3,3-pentafluorobutane (15 mL), 2.2 eq. of DCC in DCM (10 mL) were added dropwise at 0 °C during 30 min. After 5 min of reaction, the ice bath was removed. The conversion of the reaction was followed by 19F NMR. After 20 min, the reaction was stopped. The reaction mixture was filtrated and concentrated under vacuum. A flash column chromatography by solid deposit was performed. The solvents were removed under vacuum to afford oil products. The products were analyzed by NMR, GC-MS, MALDI-TOF and IR.Methyl maleate oligo(HFPO) 2a (purified by flash chromatography 10:90 EtOAc:pentane, yield = 55%): 1H NMR (400 MHz, C6D6, 25 °C, δ): 3.63 (s, –COOCH3, 3H), 4.64–4.78 (m, HFPO–CH2O–, 2H), 6.20 (dd, –CH = CH-cis, 2H, 3JH–H = 11.9 Hz and 30.7 Hz), 13C NMR (100 MHz, C6D6, 25 °C, δ): 50.7 (CH3OCO–), 59.8 (–COOCH2Rf), 127.5 (–CH = CHCOOCH2–), 131.2 (–CHCOOCH2Rf), 163.1 (–COOCH3), 164.6 (RfCH2COO–), 19F NMR (376 MHz, C6D6, 25 °C, δ): −135.2 (dR-Sq, –CF(CF3)CH2Rh), GC-MS, 70 eV, m/z: 68.9 (55), 84.9 (14), 99.9 (16), 113 (100), 118.9 (14), 146.9 (17), 149.9 (26), 168.9 (73), MALDI-TOF, [M + Li]+: 1765.5, 1931.8, 2098.0, 2263.2, 2429.5, FT-IR (ATR) νmax (cm−1): 982.4, 1126.4, 1230.2, 1646.0, 1743.7.
Synthesis of maleate oligo(HFPO) Mw ∼ 1250g mol−1
To a solution of oligo(HFPO) alcohol (Mw = 1250 g mol−1, 938 mg, 0.75 mmol), monoalkyl maleate (2 eq.) and DMAP (0.1 eq.) in 1,1,1,3,3-pentafluorobutane (10 mL) and DCM (30 mL), 2.2 eq. of DCC in DCM (10 mL) were added dropwise at 0 °C during 20 min. After 5 min of reaction, the ice bath was removed. The conversion of the reaction was followed by 19F NMR. After 15 min, the reaction was stopped. The reaction mixture was filtrated and concentrated under vacuum. After filtration onto silica and then onto Celite®, the solvents were removed under vacuum to afford clear colorless oil products.Methyl maleate oligo(HFPO) 3a (yield = 72%): 1H NMR (400 MHz, C6D6, 25 °C, δ): 3.77 (s, –OCH3, 3H), 4.76–4.89 (m, HFPO–CH2O–, 2H), 6.32 (dd, –CH = CH-cis, 2H, 3JH–H = 11.9 Hz and 34.3 Hz), 6.87 (dd, –CH = CH-trans, 2H, 3JH–H = 15.9 Hz and 28.7 Hz, 4%), 13C NMR (100 MHz, C6D6, 25 °C, δ): 51.0 (CH3OCO–), 60.0 (–COOCH2Rf), 127.5 (–CH = CHCOOCH2–), 131.5 (–CHCOOCH2Rf), 163.3 (–COOCH3), 165.0 (RfCH2COO–), 19F NMR (376 MHz, C6D6, δ): −135.3 (dR-Sq, –CF(CF3)CH2Rh), GC-MS, 70 eV, m/z: 69.1 (32), 85.1 (13), 99 (12), 100.1 (12), 113.1 (100), 150.1 (11), 169 (54), FT-IR (ATR) νmax (cm−1): 979.6, 1117.8, 1226.6, 1645.8, 1742.3.
Synthesis of vinyl ether oligo(HFPO) Mw ∼ 1250g mol−1
To a solution of Krytox Acid Fluoride (2 g) in 5 mL of previously dried trifluorotoluene with 363 μL of dry triethylamine (1.5 eq.), 155 μL of ethylene glycol vinyl ether (1 eq.) in 5 mL of dry trifluorotoluene was added dropwise at 0 °C. After 5 min, the ice bath was removed and the reaction mixture was left to stir at room temperature during 14 h. The solvent and volatiles were removed under vacuum. The crude was then washed with water (5×) and acetone (3×). The solvent traces were then removed under vacuum.4 (yield = 38%) 1H NMR (400 MHz, C6D6, 25 °C, δ): 3.86 (br, –CH2CH2OCH = CHaHb, 2H), 3.94 (dd, –CH2CH2OCH = CHaHb, 1H, 3JH–H = 6.7 and 2.1 Hz), 4.09 (d, –CH2CH2OCH = CHaHb, 1H, 3JH–H = 14.5 Hz), 4.52 (br, –CH2CH2OCH = CHaHb, 2H), 6.35 (dd, –CH2CH2OCH = CHaHb, 1H, 3JH–H = 6.7 and 14.4 Hz), 13C NMR (100 MHz, C6D6, 25 °C, δ): 64.0 (–CH2CH2OCH = CH2), − 65.9 (–CH2CH2OCH = CH2), 85.8 (–CH2CH2OCH = CH2), 150.7 (–CH2CH2OCH = CH2), 158.6 (–COOCH2CH2OCH = CH2), 19F NMR (376 MHz, C6D6, 25 °C, δ): −133.1 (dR-Sq, –CF(CF3)CH2Rh), FT-IR (ATR) νmax (cm−1): 979.1, 118.8, 1227.1, 1622.0, 1788.4.
Results and discussion
Synthesis of the starting monoalkyl maleates
The different monoalkyl maleates were synthesized by the ring-opening reaction of maleic anhydride with the corresponding alcohol. Poor-nucleophilic alcohols such as phenol, tert-butanol, tert-amyl alcohol did not permit the ring-opening reaction under mild conditions as expected. Indeed, the withdrawing mesomeric effect of the phenol or the high steric hindrance of the tert-butyl and tert-amyl explained their low nucleophilicity. Thus, benzyl alcohol was used as an alternative to create a hindered bulk on one side of the maleate. All the products were synthesized with a good yield (Scheme 1).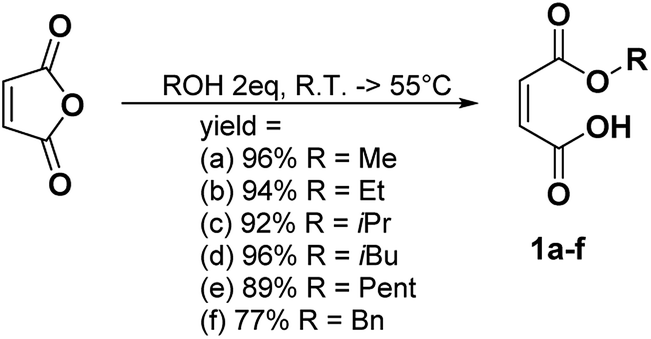 | ||
| Scheme 1 Synthesis scheme of the monoalkyl maleates. | ||
Synthesis of the disubstituted maleates
Four different synthetic strategies were attempted: by direct esterification, by using thionyl chloride, by using carbonyldiimidazole (CDI) and by Steglich esterification. No conversion or very low conversion was observed by direct esterification even in the presence of catalysts. Isomerization was highlighted by using thionyl chloride. In addition, the bonding of the in situ-formed imidazole into the double bond of the maleate by using CDI prohibited the use of any of these synthetic routes (See ESI† for further information).By Steglich esterification
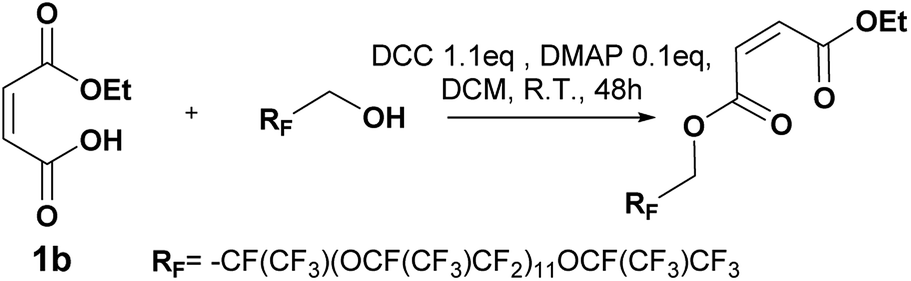
Carbonyldiimidazole is a “greener” method for esterification of the fluorinated alcohols to the maleates, but due to the nucleophilicity of the in situ-formed imidazole, a non-nucleophilic base, dicyclohexylcarbodiimide (DCC), was used with 4-dimethylaminopyridine (DMAP) as catalyst to carry out the Steglich esterification instead (Scheme 2).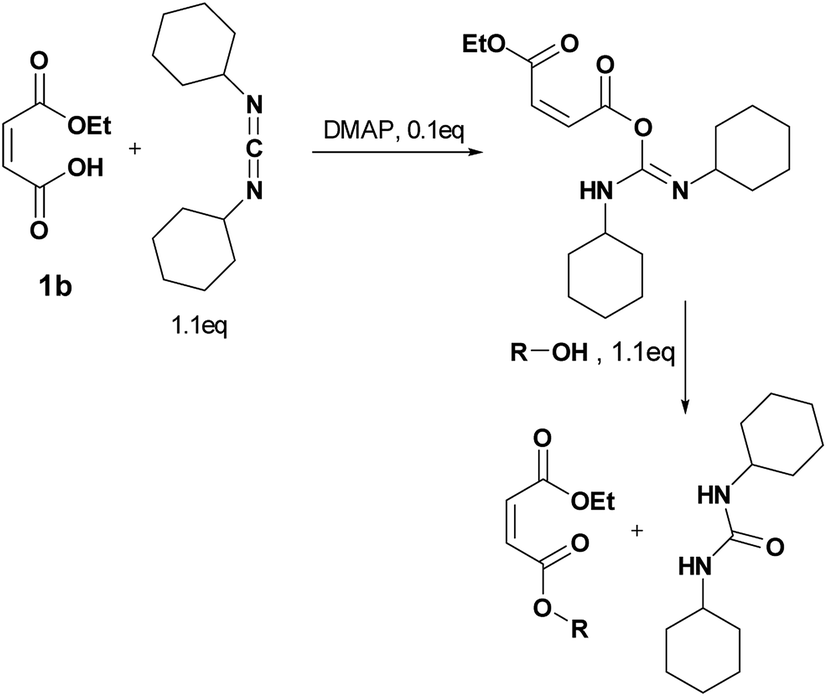 | ||
| Scheme 2 Scheme synthesis of the esterification of maleate by Steglich esterification. | ||
In a common procedure, 2–4 equivalents of alcohol were used to achieve good yields.19 More recent procedures used only a slight excess of the carboxylic acid or the alcohol with a high efficiency of reactivity.20–22 Before working on the long fluorinated oligo(HFPO) methylene alcohol, experiments were carried out on hydrogenated or partially fluorinated alcohols to determine if the desired product could be obtained. Different alcohols were used as model molecules: methanol, 1-pentanol, 1-octanol and 2,2,3,3,4,4,5,5-octafluoro-1-pentanol. In any case, a complete conversion of the starting maleate was observed. However, side-products were also present (Table 1). The presence of the trans-product was reported in small quantity. Indeed, in the literature, it was reported for this maleate product a chemical shift for the two protons of the double bond of 6.24 ppm23,24 whereas the chemical shift for the fumarate product was 6.85 ppm.24,25 As the cis and trans-products did not show the same reactivity in copolymerization,2,10 a control of the reaction time is essential.
| Product | Structure | Methanol (%mol) | 1-pentanol (%mol) | 1-octanol (%mol) | 2,2,3,3,4,4,5,5-octafluoropentanol (%mol) |
|---|---|---|---|---|---|
| a 3 h of reaction (calculated using benzophenone as internal reference). | |||||
| Cis-product |
|
65 | 70 | 71 | 70 |
| Trans-product |

|
8 | 4 | 6 | 4 |
| 1,3,5-Trisubstituted hydantoin |

|
18 | 15 | 10 | 22 |
| Addition on α-ester |

|
9 | 11 | 13 | 4 |
| Conversion of the starting maleate 1b |

|
100 | 100 | 100 | 100 |
A product from the addition of the alcohol onto the double bond was also observed. It was clearly identified thanks to the presence of three doublets of doublets at 2.43, 3.51 and 4.32 ppm. The three signals were correlated in 1H–1H COSY (as observed for the carbonyldiimidazole reaction). Nonetheless, the lower nucleophilicity of 2,2,3,3,4,4,5,5-octafluoro-1-pentanol underwent a low percentage of this side-product (Fig. 2 & Table 1). Three doublets of doublets were also observed at 2.85, 3.0 and 4.04 ppm and were correlated in 1H–1H COSY. However, these signals did not correspond to the addition onto the α-position of the acid as assumed at the beginning but to the formation of another side product: 1,3,5-trisubstituted hydantoin.26,27 This product was prepared by an intramolecular reaction from the in situ activated carboxylic acid (I) due to the electrophilic centers of the double bond. Followed by a rearrangement called O → N acyl migration on (II), the corresponding 1,3,5-trisubstituted hydantoin (III) was formed (Scheme 3). It has to be noticed that the intramolecular reaction is always in competition with the nucleophilic substitution between the nucleophile (ROH, RNH2, RCOOH, etc.) and the in situ activated carboxylic acid with DCC. By using α,β-unsaturated carboxylic acids, the team of Volonterio et al. showed the synthesis of 1,3,5-trisubstituted hydantoins (III) by a one-pot domino condensation/aza-Michael addition/O → N acyl migration of symmetric and asymmetric carbodiimides. Then a part of the alcohol did not react and remained in the final mixture. The hydantoin products were confirmed thanks to the three doublets of doublets as well as the triplets of triplets from the hydrogens of the cyclohexyl in α-position of the amines (Fig. 2). As the nucleophilicty of 2,3,3,4,4,5,5-octafluoro-1-pentanol is lower, more hydantoin product was formed. Concerning the octanol, the higher nucleophilicity allowed more formation of the desired product instead of hydantoin without a large amount of addition product to the α-ester.
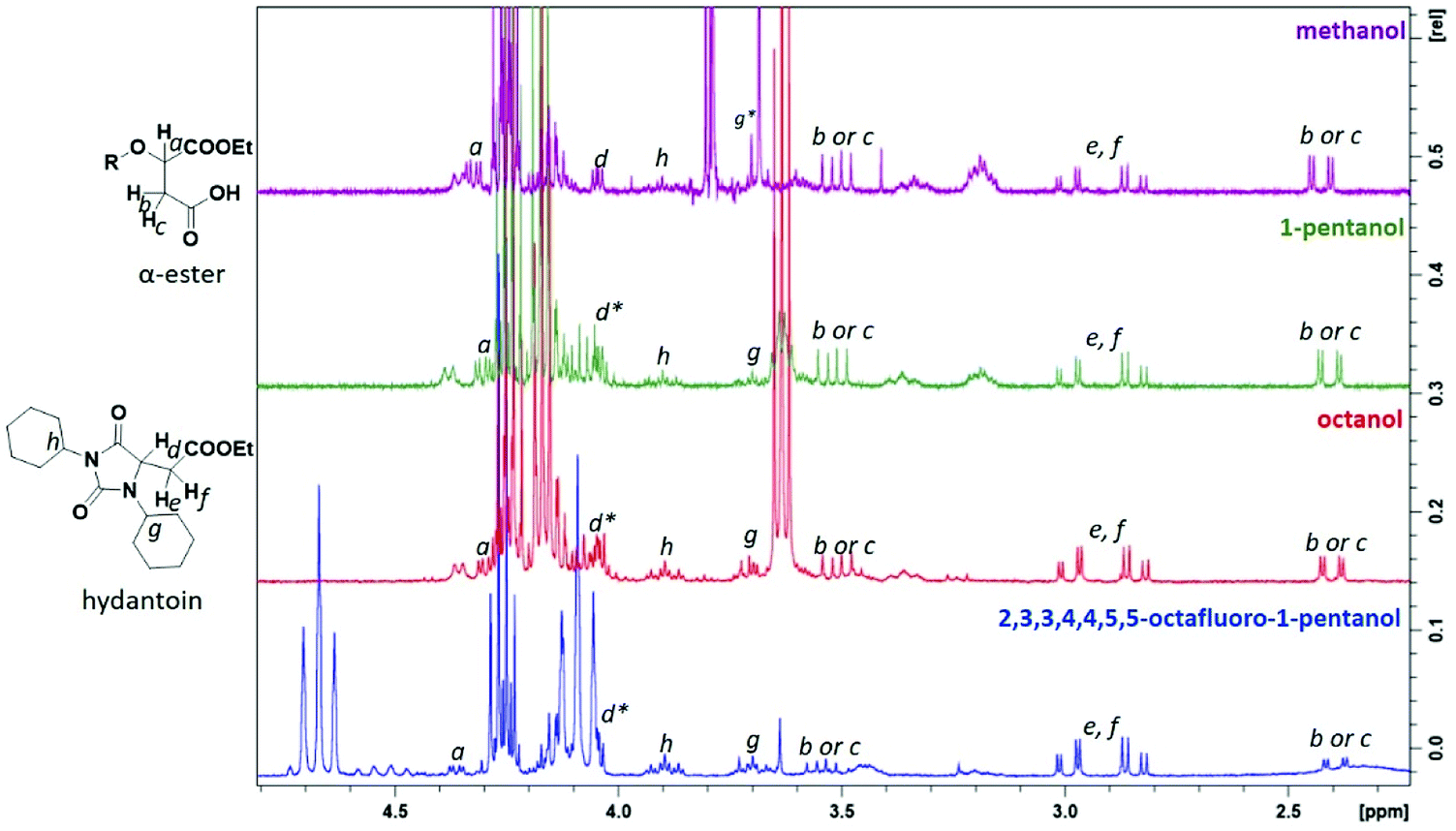 | ||
| Fig. 2 1H NMR spectra of the side-products from Steglich esterification: (1) the addition onto the double bond of the maleate, (2) the formation of hydantoin (* signal overlapped). | ||
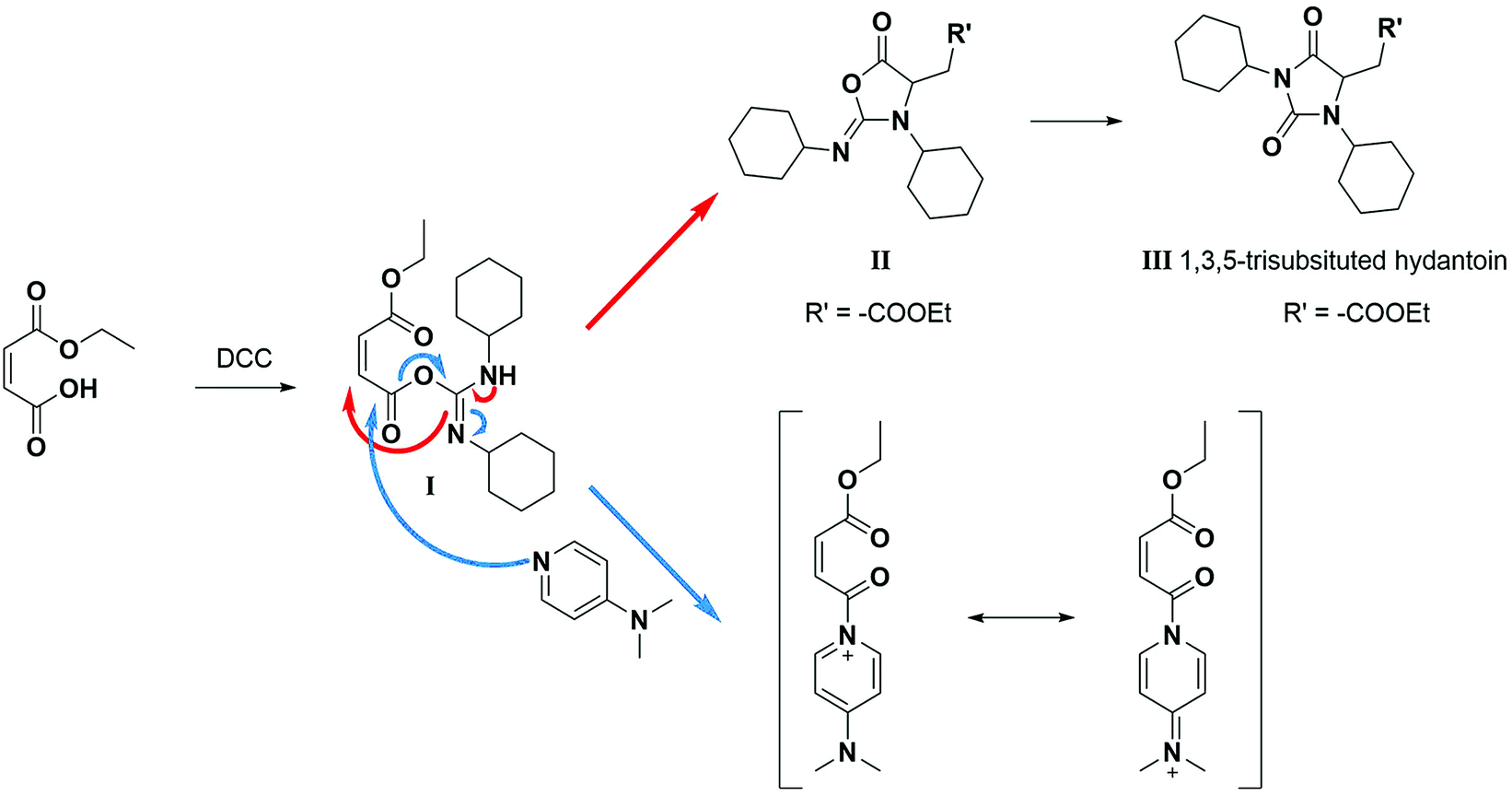 | ||
| Scheme 3 Reaction scheme of the formation of hydantoin using DCC in absence of any nucleophilic reagent.26 | ||
When the similar reaction was carried out in absence of any alcohol (Scheme 3), the 1,3,5-trisubstituted hydantoin was confirmed. Another product was found and the chemical shifts were typical of the diester maleate: one triplet from the CH3–, one quadruplet from the –CH2– and one singlet from the hydrogen of the double bonds (See ESI†). The assumed product was most likely the compound I, which was stable enough to be recovered. DMAP could also have attacked the carbonyl by following the Steglich mechanism (Scheme 3) and that would explain the low quantity of dicyclohexyl urea found. Besides, in presence of 1 equivalent of DMAP instead of using it as catalyst (0.1 eq.), no hydantoin was formed after 24 h and dicyclohexyl urea was formed in larger quantities. Moreover, for the previous reactions, even if a slight excess (1.1 eq.) of the starting alcohol was used, a significant peak of this reagent was present at the end of the reaction in comparison to the complete conversion of the maleate. Furthermore, even by taking into account the percentage of non-used alcohol due to the hydantoin reaction, the percentage of alcohol was still higher than expected. By using 2 equivalents of maleate and DCC, the starting alcohol was fully consumed as well as the maleate. The use of 2 equivalents of maleate increased the conversion of the alcohol to the desired product. The conversion of the alcohol was also dependent on its nucleophilicity undergoing either the formation of hydantoin in presence of a poor nucleophilic alcohol or the formation of the α-ester for a good nucleophile. In our case, oligo(HFPO) alcohol was not assumed to attack onto double bond due to the very low nucleophilicity of the long fluorinated chain. However, the 1,3,5-trisubstituted hydantoin is highly expected to be formed if our alcohol does not show fast enough reaction kinetics.
Synthesis of maleate PFPAEs by steglich esterification
 | ||
| Scheme 4 Synthesis scheme of maleate oligo(HFPO) using DCC and DMAP. | ||
The first synthesis was carried out on the highest molecular weight (Mw ∼ 2000 g mol−1). This oligomer showed the lowest solubility in the organic solvent and was expected to show the lowest nucleophilicity. The procedure followed the Steglich esterification in Scheme 5 to synthesize six different maleates by using a mixture 60:40 1,1,1,3,3-pentafluorobutane:DCM. The disappearance of the starting alcohol was followed by 19F NMR and confirmed in less than 20 min. These oligo(HFPO) products have been purified by column chromatography for the first time. The purification mainly permitted the removal of any hydrogenated organic molecules such as the excess of DCC, starting monoalkyl maleate, DMAP, 1,3,5-trisubstituted hydantoin and the in situ formed dicyclohexyl urea. Thus, the products could be analyzed by 1H, 13C, 19F NMR, GC-MS, FTIR and MALDI-TOF to confirm their structure. The doublet of doublet around 6.1–6.25 ppm with a 3JH–H lower than 12 Hz confirmed the cis product. In addition, the maleate end group was supported by GC/MS with the correct fragments, by IR with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C band at 1645 cm−1 as well as by MALDI-TOF thanks to the desired molecular weights [M + Li]+. However, as they are not soluble in pentane and ethyl acetate except in high dilution it explains the low yields. Besides, a higher percentage of isomerization was detected for longer reaction times as well as a percentage of the starting alcohol. The reaction was then stopped as soon as the product was confirmed by 19F NMR.
C band at 1645 cm−1 as well as by MALDI-TOF thanks to the desired molecular weights [M + Li]+. However, as they are not soluble in pentane and ethyl acetate except in high dilution it explains the low yields. Besides, a higher percentage of isomerization was detected for longer reaction times as well as a percentage of the starting alcohol. The reaction was then stopped as soon as the product was confirmed by 19F NMR.
 | ||
| Scheme 5 Reaction scheme of Steglich esterification with oligo(HFPO) methylene alcohol Mw ∼ 2000 g mol−1. | ||
On contrary to the longest chain, the oligo(HFPO) (Mw ∼ 1250 g mol−1) showed a highest percentage of the starting alcohol after purification. It was noticed that after 15 min of addition, no starting alcohol was observed. Overtime, a side-reaction caused a reversal of the reaction, leaving the starting alcohol. By using a higher dilution ([CMw∼1250 g mol−1] = 0.02 g mL−1 instead of [CMw∼2000 g mol−1] = 0.08 g mL−1) and a mixture 20:80 1,1,1,3,3-pentafluorobutane:DCM, no starting alcohol was observed after 48 h (Scheme 6). However, for the same reaction stopped after 15 min (after confirmation by 19F NMR of absence of starting alcohol) and after column chromatography, 20% of the starting alcohol was observed. We believe the product reverted back to the starting alcohol due to the pH and large excess of the silica. Besides, the longer fluorinated chained alcohol created higher steric hindrance, protecting itself from removal.
 | ||
| Scheme 6 Reaction scheme of Steglich esterification on oligo(HFPO) alcohol Mw ∼ 1250 g mol−1. | ||
 | ||
| Fig. 3 Structure of the vinyl ether oligo(HFPO) 4 and the photoinitiator PI. | ||
For a stoichiometric amount of maleate/vinyl ether with 4% w/w of PI, a complete conversion of the vinyl ether was obtained in less than 40 s. (Fig. 4) No homopolymerization of 4 was observed under UV-light after 500 s and in presence of PI (4% w/w). The IR band (CC) of the vinyl ether did not overlap with other structures, therefore, kinetics was done only on the vinyl ether band at 1622 cm−1. Surprisingly, in comparison with literature, a partial homopolymerization of 2a was observed in presence of PI but proved to be much slower than its copolymerization (See ESI†). Besides, the vinyl ether 4 showed quantitative conversion and since a stoichiometric ratio was used, no homopolymerization of the maleate could have happened or only in small quantities within 40 s. No clear effect was observed due to the chain length (2a and 3a) for the polymerization kinetics. In absence of photoinitiator, no conversion of the monomers was observed. In addition, the presence of air was not a key factor in polymerization as the final conversion was reached in the same amount of time (See ESI†). The degradation temperature T5% was mainly dependent on the chain length (i.e. 1250 g mol−1 and 2000 g mol−1). Concerning oligo(HFPO) with Mw ∼ 1250 g mol−1, the addition of a substituent (maleates or vinyl ether) increased the degradation temperature in comparison to the starting alcohol. However, for the higher molecular weight, no changes were observed except for the benzyl maleate 2f due to the more stable phenyl group. The copolymers showed a similar T5% but demonstrated slower degradation kinetics than the monomers: 174 °C and 200 °C for 3a:4 and 2a:4 respectively. The glass transition temperatures were also linked with the length of the fluorinated chain: around −75 °C and −68 °C, respectively for 1250 g mol−1 and 2000 g mol−1. Compound 2f showed a slightly higher Tg due to the reduced chain flexibility. Regarding the copolymers, only the glass transition temperature of the fluorinated phase was detected (−69 °C and −68 °C for 3a:4 and 2a:4 respectively). Highly hydrophobic polymers (i.e. 109° and 111° for 3a:4 and 2a:4 respectively) were obtained, as reported for acrylic photopolymers containing PFPAEs' (see ESI†).28,29
 | ||
| Fig. 4 Photopolymerization profiles of 3a:4 and 2a:4 in presence of photoinitiator (4% w/w). | ||
Conclusions
The synthesis of fluorinated maleates was successfully carried out by Steglich esterification. Indeed, the high reactivity of the α,β-unsaturated esters indicated that other side products are possible. However, other routes to esterification were not conceivable. It is noted that long fluorinated moieties decreased the nucleophilicity of the hydroxyl group but reduces the formation of other side-products. Further considerations would be required to fully understand the reactivity of the α,β-unsaturated esters in presence of DCC, DMAP and a nucleophile. In addition, thanks to the fluorinated groups, highly hydrophobic polymers were made by UV-curing. An electron acceptor/donor system (maleate/vinyl ether) showed a complete conversion in less than 40 s for both tested maleate monomers. Further studies will be carried out for the optimization of the photopolymerization kinetics. The choice of photoinitiator, its concentration as well as the influence of the substituent will be investigated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement No. 690917 – PhotoFluo; Natural Sciences and Engineering Research Council of Canada (NSERC), Discovery Grants Program RGPIN-2015-05513 and Ministère de l’Education supérieure et de la recherche.Notes and references
- J. P. S. Farinha, J. Wu, M. A. Winnik, R. Farwaha and J. Rademacher, Macromolecules, 2005, 38, 4393–4402 CrossRef CAS.
- J. V. Crivello and T. M. McGrath, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4726–4736 CrossRef CAS.
- F. R. Mustata and I. G. Bicu, Ind. Eng. Chem. Res., 2008, 47, 1355–1364 CrossRef CAS.
- H. Lee, J. W. Pack, W. Wang, K. J. Thurecht and S. M. Howdle, Macromolecules, 2010, 43, 2276–2282 CrossRef CAS.
- J. Wu, J. K. Oh, J. Yang, M. A. Winnik, R. Farwaha and J. Rademacher, Macromolecules, 2003, 36, 8139–8147 CrossRef CAS.
- E. Di Cola, N. Plucktaveesak, T. A. Waigh, R. H. Colby, J. S. Tan, W. Pyckhout-Hintzen and R. K. Heenan, Macromolecules, 2004, 37, 8457–8465 CrossRef CAS.
- D. Taura, A. Hashidzume, Y. Okumura and A. Harada, Macromolecules, 2008, 41, 3640–3645 CrossRef CAS.
- C. Decker, C. Bianchi, D. Decker and F. Morel, Prog. Org. Coat., 2001, 42, 253–256 CrossRef CAS.
- R. Auvergne, R. Saint-Loup, C. Joly-Duhamel, J. J. Robin and B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1324–1335 CrossRef CAS.
- L. Pichavant, C. L. Guillermain and X. Coqueret, Biomacromolecules, 2010, 11, 2415–2421 CrossRef CAS PubMed.
- C. Decker and D. Decker, Polymer, 1997, 38, 2229–2237 CrossRef CAS.
- T. Haraldsson, M. Johansson and A. Hult, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2810–2816 CrossRef CAS.
- C. Y. Huang and A. G. Doyle, J. Am. Chem. Soc., 2015, 137, 5638–5641 CrossRef CAS PubMed.
- N. Kaplaneris, A. Bisticha, G. N. Papadopoulos, D. Limnios and C. G. Kokotos, Green Chem., 2017, 19, 4451–4456 RSC.
- N. Yoshino, M. Morita, A. Ito and M. Abe, J. Fluorine Chem., 1995, 70, 187–191 CrossRef CAS.
- G. Malinverno, G. Pantini and J. Bootman, Food Chem. Toxicol., 1996, 34, 639–650 CrossRef CAS PubMed.
- C. M. Friesen and B. Améduri, Prog. Polym. Sci., 2018, 81, 238–280 CrossRef CAS.
- D. W. Smith, S. T. Iacono and S. S. Iyer, Handbook of Fluoropolymer Science and Technology, Wiley, 2014 Search PubMed.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed. Engl., 1978, 17, 522–524 CrossRef.
- A. Sethi, A. Bhatia, D. Shukla, A. Kumar, R. Sonker, R. Prakash and G. Bhatia, J. Mol. Struct., 2012, 1028, 88–96 CrossRef CAS.
- X. WAng, Q. Ye, J. Song, C. M. Cho, C. He and J. Xu, RSC Adv., 2015, 5, 4547–4553 RSC.
- E. Baysak, H. Durmaz, U. Tunca and G. Hizal, Macromol. Chem. Phys., 2017, 218, 1600572 CrossRef.
- U. Gross, P. Koos, M. O'Brien, A. Polyzos and S. V. Ley, Eur. J. Org. Chem., 2014, 2014(29), 6418–6430 CrossRef CAS.
- P. Nolis, A. Roglans and T. Parella, J. Magn. Reson., 2005, 173, 305–309 CrossRef CAS PubMed.
- T. L. Troyer, H. Muchalski, K. B. Hong and J. N. Johnston, Org. Lett., 2011, 13, 1790–1792 CrossRef CAS PubMed.
- A. Volonterio, C. R. De Arellano and M. Zanda, J. Org. Chem., 2005, 70, 2161–2170 CrossRef CAS PubMed.
- T. Marcelli, F. Olimpieri and A. Volonterio, Org. Biomol. Chem., 2011, 9, 5156–5161 RSC.
- A. Vitale, R. Bongiovanni and B. Ameduri, Chem. Rev., 2015, 115, 8836–8866 CrossRef PubMed.
- A. Vitale, M. Quaglio, S. Marasso, A. Chiodoni, M. Cocuzza and R. Bongiovanni, Langmuir, 2013, 29, 15711–15718 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H, 13C, IR, GC-MS spectra for compounds 1a–f, 2a–f, 3a–b, 4a, 19F, TGA, DSC spectra for 2a–f, 3a–b, 4a and MALDI-TOF for 2a–f. IR spectra for the kinetics of photopolymerization. See DOI: 10.1039/c8ra06354k |
| This journal is © The Royal Society of Chemistry 2018 |