DOI:
10.1039/C8RA07352J
(Paper)
RSC Adv., 2018,
8, 35753-35758
Fast and safe synthesis of micron germanium in an ammonia atmosphere using Mo2N as catalyst
Received
3rd September 2018
, Accepted 7th October 2018
First published on 19th October 2018
Abstract
Here, we reported a new method for fast and safe synthesis of a micron germanium (Ge) semiconductor. The Ge was successfully prepared from mixed GeO2 with a low amount of MoO3 by the NH3 reduction method at 800 °C for an ultra-short time of 10 min. XRD patterns show that the Ge has a tetragonal structure. SEM images show that the size of the Ge particles is from 5 μm to 10 μm, and so it is on the micron scale. UV-visible diffuse reflectance spectroscopy shows that the Ge has good light absorption both in the ultraviolet and visible regions. The formation of Ge mainly goes through a two-step conversion in the NH3 flow. Firstly, GeO2 is converted to Ge3N4, and then Ge3N4 is decomposed to generate Ge. The comparison experiments of MoO3 and Mo2N demonstrate that Mo2N is the catalyst for the Ge synthesis which improves the Ge3N4 decomposition. The presented fast and safe synthesis method of Ge has great potential for industrialization and the proposed Mo2N boosting the Ge3N4 decomposition has provided significant guidance for other nitride decomposition systems.
1 Introduction
With the development of advanced electronic information industries, semiconductor materials have attracted widespread attention due to their unique optical and electrical properties.1–6 Ge as an important semiconductor material with stable chemical properties and obvious non-metallic properties has many advantages, such as non-toxicity, biocompatibility, electrochemical stability, current microelectronics compatibility, etc.7–13 At present, Ge has extensive and important applications in fields such as semiconductors, aerospace measurement and control, nuclear physics detection, optical fiber communication, infrared optics, solar cells, chemical catalysts, and biomedicine.14–20 Ge is one of the most dispersed elements and there is almost no concentrated Ge deposit in the Earth's crust. Thus, it is particularly important to find suitable methods to enrich, prepare and purify Ge.
The synthetic method of Ge can be divided into physical and chemical methods. The physical methods mainly include chemical vapor deposition,21 gas phase pyrolysis,22 plasma technology,23–25 sputtering,26,27 etching,28,29 laser ablation30 and so on. However, these technological processes need extreme temperatures and pressures and are expensive. Chemical methods mainly include room temperature reduction of a GeCl4/Br4 precursor,31,32 high temperature reduction of a GeI2/I4 precursor,33,34 a one-step synthesis method,35,36 an electrodeposition method37–41 and so on. In the industry, rough Ge is commonly prepared by the reduction of GeO2 in hydrogen flow at 650–680 °C, and then Ge with high purity is obtained by chemical gasification and decomposition of rough Ge. However, in the process of preparing rough Ge, the produced hydrogen is a dangerous gas due to its large range of the explosion limit. In addition, a leak of hydrogen is not easy to discover due to it being colorless and tasteless. So, it is necessary to develop and improve a safe and low-cost method to produce rough Ge.
Ammonia is an important chemical raw material, which has important applications in industrial and agricultural production. At the same time, ammonia is also a reducing gas capable of reducing various metal oxides.42–45 Here, we successfully prepared micron Ge material in ammonia atmosphere using GeO2 as raw material with some MoO3 in 10 min. The transformation process from GeO2 to Ge and the molybdenum based catalyst were determined. The synthesis method of Ge has the advantages of safe, fast and low cost, which is beneficial to industrial production.
2 Experimental
2.1 Synthesis of Ge
All the chemical agents are from China National Pharmaceutical Group Corporation. GeO2 as raw material and a small amount of MoO3 are mixed and milled in a mortar for 30 minutes. The mixed samples are the calcined in an ammonia flow of 100 mL min−1 at different temperature and for different time. The synthesis of Ge in H2 flow is same as that in NH3 flow except H2 instead of NH3.
Also, in this paper A/B means A mixed with B. For example, Mo2N/GeO2 means Mo2N mixed with GeO2.
2.2 Characterization
The structure of the as-prepared Ge was determined by X-ray diffraction (D/MAX2500, Rigaku, Japan) with a Cu-Kα radiation at a voltage of 4 kV at room temperature. The ultraviolet-visible diffuse reflectance spectra (UV-vis DRS) were obtained on a spectrometer (U-4100), and BaSO4 was used for the corrected base line. The size and morphology were examined by scanning electron microscopy (SEM, JSM7500F) with the accelerating voltage of 0.5–30 kV and transmission electron microscopy (TEM, F20) with the accelerating voltage of 120 kV.
3 Results and discussion
3.1 Synthesis of Ge in NH3 flow using Mo based catalyst
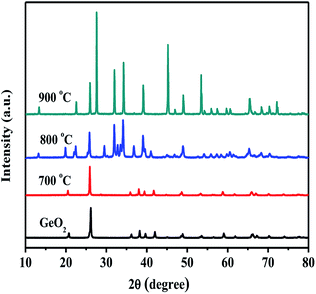
Fig. 1 shows the XRD patterns of GeO2 calcined at different temperatures in an ammonia atmosphere. The XRD of the GeO2 is also present for comparison. When the reaction temperature is lower than 800 °C, GeO2 (PDF # 36-1463) still remain unchanged. At 800 °C, all GeO2 changed into Ge3N4 (PDF # 38-1374). At 900 °C, a part of Ge (PDF # 04-0545, 2θ = 27.5°) generates which indicates a small part of Ge3N4 decomposes at overhigh temperature.
 |
| | Fig. 1 XRD patterns of commercial GeO2 and GeO2 calcined in NH3 flow at different temperatures for 2 h. | |
Fig. 2 shows the XRD patterns of 5.0 wt% MoO3/GeO2 calcined in NH3 flow at different temperatures for 2 h. It can be seen that when the reaction temperature is at or above 800 °C, almost all the GeO2 are changed into Ge (PDF # 04-0545). Comparing with Fig. 1, the introduction of the Mo element decreases the reaction temperature of Ge generation over 100 °C.
 |
| | Fig. 2 XRD patterns of commercial GeO2 and samples of 5.0 wt% MoO3/GeO2 calcined in NH3 flow at different temperatures for 2 h. | |
Fig. 3 shows XRD patterns of 5.0 wt% MoO3/GeO2 calcined in NH3 flow at 800 °C for different time. The process of Ge generation is ultra fast and almost all the GeO2 can be reduced to Ge in 10 min. Fig. 4 shows the XRD pattern of MoO3/GeO2 with different mass ratio calcined in ammonia flow. When the amount of MoO3 is less than 5.0%, the reaction product is Ge and Ge3N4 (e.g. the sample of 2.0 wt% MoO3/GeO2). When the amount of MoO3 is 5.0%, almost all the GeO2 is converted to Ge. In view of the fact of that GeO2 changed into Ge3N4 (Fig. 1, 800 °C) without any catalyst, we inferred the process Ge generation is that first the GeO2 transfers into Ge3N4, and then the Ge3N4 decomposes into Ge with the Mo base catalyst. This ultra fast method of Ge generation using Mo based catalyst has major advantages of low cost and high efficiency.
 |
| | Fig. 3 XRD patterns of commercial GeO2, 5.0 wt% MoO3/GeO2 and 5.0 wt% MoO3/GeO2 calcined in NH3 flow at 800 °C for different time. | |
 |
| | Fig. 4 XRD patterns of GeO2 and MoO3/GeO2 with different MoO3 loading mass ratio calcined in NH3 flow at 800 °C for 0.5 h. | |
3.2 Determination of the catalyst of Ge generation
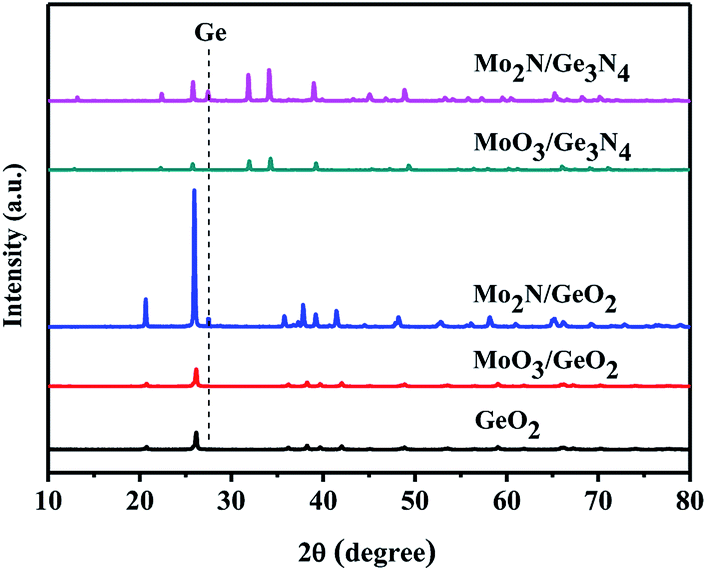
It is well known the molybdenum oxide will change into molybdenum nitride in NH3 atmosphere at high temperature. So, it is necessary to judge which Mo species play the catalytic role in the Ge generation. Fig. 5 shows XRD patterns of MoO3 calcined in the NH3 flow at different temperatures. At 600 °C, some MoO3 (PDF # 35-0609) is reduced into MoO2 (PDF # 32-0671). When the temperatures reach at and above 700 °C, all MoO3 is converted to Mo2N (PDF # 25-1366). In Fig. 2, Ge is generated at 800 °C from MoO3/GeO2. At this moment (800 °C), the MoO3 had been reduced into Mo2N (Fig. 5, 800 °C), and the GeO2 had been reduced into Ge3N4 (Fig. 1, 800 °C). These results indicate that the Mo2N generated by MoO3 acts as a catalyst and improves the decomposition of the Ge3N4 into Ge.
 |
| | Fig. 5 XRD patterns of MoO3 and MoO3 calcined in NH3 flow at different temperatures for 2 h. | |
In order further determine the origination (Mo2N or MoO3) of the catalysis role in Ge generation, the calcination experiments in nitrogen flow using GeO2 and Ge3N4 as raw materials have been designed. Fig. 6 shows the XRD patterns of Mo2N/GeO2, MoO3/GeO2, Mo2N/Ge3N4 and MoO3/Ge3N4 calcined in N2 atmosphere at 800 °C for 2 h. It can be seen that there are all a small amount of Ge generated by the addition of Mo2N whether the raw material is GeO2 or Ge3N4. However, by the addition of MoO3, there is no Ge generation for both MoO3/GeO2 and MoO3/Ge3N4. These again demonstrate that Mo2N plays the catalysis role in Ge generation, whereas MoO3 is not the catalyst.
 |
| | Fig. 6 XRD patterns of GeO2, 5.0% Mo2N/GeO2, 5.0% MoO3/GeO2, 5.0% Mo2N/Ge3N4 and 5.0% MoO3/Ge3N4 calcined in N2 atmosphere at 800 °C for 2 h. | |
3.3 Comparison with the industrial preparation method of Ge in H2 flow
In order to compare our preparation method with industrial method, GeO2 is reduced into Ge in hydrogen atmosphere has been carried out. Fig. 7 shows the XRD patterns of GeO2 calcined in hydrogen flow at different temperatures for 4 hours. At or over 500 °C, Ge is generated in H2 flow, and 650 °C is chosen generally in the industry. Fig. 8 shows the XRD pattern of GeO2 calcined in hydrogen flow at 500 °C for different time. Ge can be produced in the H2 flow in 2 hours. So, compared with the industrial method in the H2 flow, our preparation method in the NH3 flow with Mo2N catalyst has the advantages of ultra short reaction time (10 min vs. 2 h) and more safety (NH3 vs. H2), though the reaction temperature is higher (800 °C vs. 650 °C). We also performed the experiment which the GeO2 was calcined in H2 flow at 800 °C for the further contrast. The Fig. 9 shows the XRD pattern of GeO2 calcined in hydrogen flow at 800 °C for different time. The GeO2 was converted into Ge in 10 min under the hydrogen atmosphere at 800 °C. It is similar to that of in the NH3 flow reduction. However, calcining GeO2 in an ammonia atmosphere means more secure.
 |
| | Fig. 7 XRD patterns of GeO2 calcined in hydrogen atmosphere at different temperatures for 4 h. | |
 |
| | Fig. 8 XRD patterns of GeO2 calcined in H2 flow at 500 °C for different time. | |
 |
| | Fig. 9 XRD patterns of GeO2 calcined in H2 flow at 800 °C for different time. | |
3.4 The physical properties of prepared Ge
Fig. 10 shows the morphologies of the raw material GeO2 and the prepared Ge in NH3 flow with different magnifications. The GeO2 particles (Fig. 10a–c) are uniform and show sharp angular shapes. The prepared Ge in NH3 flow (Fig. 10d–f) shows a smooth surface and some agglomeration, indicating the structure changes from GeO2 to Ge. The size of Ge is between 5 μm and 10 μm. Fig. 11 shows the HRTEM of prepared Ge in NH3 flow, the lattice fringes of Ge with interplanar distances of 0.237 nm is indexed to the (111) planes of Ge.
 |
| | Fig. 10 SEM images of the raw material of GeO2 (a–c) and the Ge (d–f) prepared from 5% MoO3/GeO2 in NH3 flow at 800 °C for 2 h. | |
 |
| | Fig. 11 HRTEM of Ge prepared in the NH3 flow from GeO2 mixed with 5.0% MoO3 at 800 °C for 2 h. | |
In order to further explore the morphology changes in the formation process of Ge, SEM as shown in Fig. 12 has been carried out for the samples prepared from MoO3/GeO2 for different times and with different MoO3 mass ratios. Compared the Fig. 12b with Fig. 12a, it is found the rod-shaped material (Fig. 12a) decreases and the large smooth-surfaced Ge (Fig. 12b) increases with the reaction time prolonging. Associate with XRD patterns in Fig. 4, the rod-shaped material can be inferred as Ge3N4. Compared the Fig. 12c with Fig. 12b, it can be seen that there is hardly any Ge3N4 particles in the Fig. 12c, indicating that almost all Ge3N4 transforms into Ge with the increase of MoO3. Here, the MoO3 had been converted into Mo2N due to in the NH3 flow at 800 °C (Fig. 5, 800 °C). Fig. 12 again demonstrates the process of Ge generation goes through two stages. Firstly, GeO2 is reduced to Ge3N4, and then Ge3N4 decomposed into Ge by the catalysis of Mo2N.
 |
| | Fig. 12 SEM images of samples prepared in the NH3 flow from 2.0% MoO3/GeO2 at 800 °C for 0.5 h (a), 2.0% MoO3/GeO2 at 800 °C for 2 h (b) 5% MoO3/GeO2 at 800 °C for 2 h (c). | |
Fig. 13 shows the UV-vis diffuse reflectance spectra of MoO3/GeO2 calcined in the NH3 flow with different mass ratios of MoO3. The raw material of GeO2 show a strong light absorption at ultraviolet region with the absorption edge of 218 nm, which means it is a large gap semiconductor. With the amount of MoO3 increasing from 0 to 5.0%, the light absorption region gradually extends from ultraviolet region to visible region and the absorption strength gradually increases. The Ge prepared from 5.0% MoO3/GeO2 shows the most strongest light absorption in the range from 200 nm to 800 nm. The light absorption curve of the Ge prepared in NH3 flow is same as that prepared in H2 flow except the absorption strength. The difference of absorption strength can be attributed to the small amount of Mo2N catalyst.
 |
| | Fig. 13 The UV-visible diffuse reflectance spectra of MoO3/GeO2 with different mass ratios of MoO3 calcined in NH3 flow at 800 °C for 2 h. The spectra of GeO2 and Ge prepared in the H2 flow at 650 °C for 4 h are presented for comparison. | |
4 Conclusions
A fast, low cost and safe method for the Ge synthesis has been reported by the NH3 reduction of GeO2 using a Mo2N catalyst. When GeO2 and 5.0% MoO3 is mixed and nitrided in NH3 flow at 800 °C, the Ge is formed in 10 min. The prepared Ge shows micron scale particles size and good light absorption both in the ultraviolet and visible region. The reaction mechanism of Ge generation carries out a two-stage process. GeO2 is converted into Ge3N4 firstly, and then Ge3N4 is decomposed into Ge with the Mo2N catalyst. Mo2N instead of MoO3 is demonstrated the catalyst for the Ge generation by two proofs. One is that MoO3 is converted into Mo2N at 800 °C in the NH3 flow. Another is that a small part of Ge is produced from both the mixed Mo2N/GeO2 and the Mo2N/Ge3N4 calcined in N2 flow, whereas there is no Ge generated from both MoO3/GeO2 and the MoO3/Ge3N4. Finally, the method for Ge preparation in NH3 flow has been compared with the industrial method in H2 flow. Our presented method has the advantages of fast and safety and has great potential for industrialization. The proposed Mo2N boosting the Ge3N4 decomposition has guiding significance to other nitride decomposition system.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is funded by the Natural Science Foundation of Ningxia Province of China (NZ17038), National First-rate Discipline Construction Project of Ningxia (Chemical Engineering and Technology NXYLXK2017A04), Major Innovation Projects for Building First-class Universities in China's Western Region (ZKZD2017003) and the National Natural Science Foundation of China (NSFC, 21263018).
Notes and references
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- Y. Wang and N. Herron, J. Phys. Chem., 1991, 95, 525–532 CrossRef CAS.
- L. Zhang and M. Jaroniec, Appl. Surf. Sci., 2017, 430, 2–17 CrossRef.
- I. D. Samuel and G. A. Turnbull, Chem. Rev., 2007, 107, 1272–1295 CrossRef CAS PubMed.
- H. A. Qayyum, M. F. Al-Kuhaili, S. M. A. Durrani, T. Hussain and S. H. A. Ahmad, J. Alloys Compd., 2018, 747, 374–384 CrossRef CAS.
- Q. Yin and C. L. Hill, Joule, 2017, 1, 645–646 CrossRef.
- P. Reiss, M. Carrière, C. Lincheneau, L. Vaure and S. Tamang, Chem. Rev., 2016, 116, 10731–10819 CrossRef CAS PubMed.
- A. M. Derfus, W. Chan and S. N. Bhatia, Nano Lett., 2004, 4, 11–18 CrossRef CAS PubMed.
- L. Ye, K. T. Yong, L. Liu, I. Roy, R. Hu, J. Zhu, H. Cai, W. C. Law, J. Liu, K. Wang, J. Liu, Y. Liu, Y. Hu, X. Zhang, M. T. Swihart and P. N. Prasad, Nat. Nanotechnol., 2012, 7, 453–458 CrossRef CAS PubMed.
- S. Hayashi, M. Ito and H. Kanamori, Solid State Commun., 1982, 44, 75–79 CrossRef CAS.
- J. Tauc, R. Grigorovici and A. Vancu, Phys. Status Solidi, 1966, 15, 627–637 CrossRef CAS.
- D. Carolan, Prog. Mater. Sci., 2017, 90, 128–158 CrossRef CAS.
- Z. L. Hu, S. Zhang, C. J. Zhang and G. L. Cui, Coord. Chem. Rev., 2016, 326, 34–85 CrossRef CAS.
- L. Zhang, B. G. Zhang, B. C. Pan and C. W. Wang, Appl. Surf. Sci., 2017, 422, 247–256 CrossRef CAS.
- S. V. Grayli, A. Ferrone, L. Maiolo, A. De Iacovo, A. Pecora, L. Colace, G. W. Leach and B. Bahreyni, Sens. Actuators, A, 2017, 263, 341–348 CrossRef CAS.
- W. S. Chen, B. C. Chang and K. L. Chiu, J. Environ. Chem. Eng., 2017, 5, 5215–5221 CrossRef CAS.
- J. P. Wright, L. J. Harkness-Brennan, A. J. Boston, D. S. Judson, M. Labiche, P. J. Nolan, R. D. Page, F. Pearce, D. C. Radford, J. Simpson and C. Unsworth, Nucl. Instrum. Methods Phys. Res., Sect. A, 2018, 892, 84–92 CrossRef CAS.
- Y. Matsuura, Curr. Appl. Phy., 2017, 17, 1465–1468 CrossRef.
- J. Biedrzycki, K. Tarnowski and W. Urbańczyk, Opto-Electron. Rev., 2018, 26, 57–62 CrossRef.
- D. D. Liu, H. S. Liu, C. H. Jiang, J. Leng, Y. M. Zhang, Z. H. Zhao, K. W. Zhuang, Y. G. Jiang and Y. Q. Ji, Thin Solid Films, 2015, 592, 292–295 CrossRef CAS.
- Ö. M. Dag, A. Kuperman and G. A. Ozin, Adv. Mater., 1994, 6, 147–150 CrossRef.
- C. R. Stoldt, M. A. Haag and B. A. Larsen, Appl. Phys. Lett., 2008, 93, 43125 CrossRef.
- R. Gresback, Z. Holman and U. Kortshagen, Appl. Phys. Lett., 2007, 91, 335 CrossRef.
- Z. C. Holman and U. R. Kortshagen, Langmuir, 2009, 25, 11883–11889 CrossRef CAS PubMed.
- Z. C. Holman, C. Y. Liu and U. R. Kortshagen, Nano Lett., 2010, 10, 2661–2666 CrossRef CAS PubMed.
- S. Hayashi, M. Fujii and K. Yamamoto, Jpn. J. Appl. Phys., 1989, 28, L1464–L1466 CrossRef CAS.
- Y. Maeda, N. Tsukamoto, Y. Yazawa, Y. Kanemitsu and Y. Masumoto, Appl. Phys. Lett., 1991, 59, 3168–3170 CrossRef CAS.
- T. I. Kamins, D. A. A. Ohlberg, R. S. Williams, W. Zhang and S. Y. Chou, Appl. Phys. Lett., 1999, 74, 1773–1775 CrossRef CAS.
- G. Kartopu, A. V. Sapelkin, V. A. Karavanskii and R. Turan, J. Appl. Phys., 2008, 103, 113518 CrossRef.
- S. Ngiam, K. F. Jensen and K. D. Kolenbrander, J. Appl. Phys., 1994, 76, 8201–8203 CrossRef CAS.
- D. Carolan and H. Doyle, J. Mater. Chem. C, 2014, 2, 3562–3568 RSC.
- D. Carolan and H. Doyle, J. Nanomater., 2015, 16, 1–9 Search PubMed.
- N. Shirahata, J. Solid State Chem., 2014, 214, 74–78 CrossRef CAS.
- E. Muthuswamy, A. S. Iskandar, M. M. Amador and S. M. Kauzlarich, Chem. Mater., 2013, 25, 1416–1422 CrossRef CAS.
- I. I. Dimitri, J. F. Bondi and R. E. Schaak, Chem. Mater., 2010, 22, 6103–6108 CrossRef.
- D. J. Xue, J. J. Wang, Y. Q. Wang, X. Sen, Y. G. Guo and L. J. Wan, Adv. Mater., 2011, 23, 3704–3707 CrossRef CAS PubMed.
- N. K. Mahenderkar, Y. C. Liu, J. A. Koza and J. A. Switzer, ACS Nano, 2014, 8, 9524–9530 CrossRef CAS PubMed.
- A. Lahiri, S. Z. E. Abedin and F. Endres, J. Phys. Chem. C, 2012, 116, 17739–17745 CrossRef CAS.
- C. Y. Cummings, P. N. Bartlett, D. Pugh, G. Reid, W. Levason, M. M. Hasan, A. L. Hector, J. Spencer, D. C. Snith and S. Marks, ChemElectroChem, 2016, 3, 726–733 CrossRef CAS.
- A. Lahiri, A. Willert, S. Z. E. Abedin and F. Endres, Electrochim. Acta, 2014, 121, 154–158 CrossRef CAS.
- A. Lahiri and F. Endres, J. Electrochem. Soc., 2017, 164, D597–D612 CrossRef CAS.
- Q. Feng, W. Zhao and S. Wen, J. Alloys Compd., 2018, 744, 301–309 CrossRef CAS.
- A. Sánchez and M. Martín, J. Cleaner Prod., 2018, 178, 325–342 CrossRef.
- M. F. Ezzat and I. Dincer, Appl. Energy, 2018, 219, 226–239 CrossRef CAS.
- K. Sakuragi, K. Igarashi and M. Samejima, Polym. Degrad. Stab., 2018, 148, 19–25 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Dekang Li,
Xiaoyan Wang and
Keying Lin
*,
Dekang Li,
Xiaoyan Wang and
Keying Lin