
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe sequential structure of tripyridiniumylporphyrin pendants in water-soluble copolymers and their association behaviour with tetrasulfonatophenylporphyrin guests: UV-vis absorption and fluorescence emission spectra study
Ke-Wei Ding *ab,
Tao-Qi Lib,
Zhong-Xue Geab,
Jian-Hua Bub and
Ying Liub
*ab,
Tao-Qi Lib,
Zhong-Xue Geab,
Jian-Hua Bub and
Ying Liub
aState Key Laboratory of Fluorine & Nitrogen Chemicals, Xi'an 710065, China. E-mail: dkw204@163.com
bXi'an Modern Chemistry Research Institute, Xi'an 710065, China
First published on 19th October 2018
Abstract
A novel cationic tripyridiniumylporphyrin monomer, 5-[4-[2-(acryloyloxy)ethoxy]phenyl]-l0,l5,20-tris(N-methyl-4-pyridiniumyl)porphyrinate zinc(II) (ZnTrMPyP), was synthesized, and its self-aggregation in water was studied by UV-vis absorption. The monomer was copolymerized with acrylamide in water and DMSO, respectively, to prepare the water-soluble polymers P-W and P-D. The aggregation behaviour of the copolymers in aqueous solution was investigated by UV-vis absorption and fluorescence emission spectra. The polymer P-D displayed very similar absorption and emission spectra to those of ZnTrMPyP in water, indicating that the polymer chains in P-D have no significant effect on the aggregate structure of ZnTrMPyP in aqueous media. In comparison, two new absorption bands appeared in the Q band range of polymer P-W and its fluorescence spectra red shifted and the fluorescence quantum yield decreased obviously. These characteristics remained unchanged even in a good solvent for the monomer, suggesting that a new aggregation structure for the porphyrin pendants fixed by the covalent bond was formed. According to the different dispersed states of the porphyrin monomer in water and DMSO, the porphyrin pendants should distribute randomly in the P-D polymer chains while having micro-blocky sequences in polymer P-W. The association behaviour between the copolymers and tetra(p-sulfonatophenyl)porphyrin, TSPP, bearing opposite charged substituents were studied by absorption and emission Spectra and further analyzed by the Benesi–Hildebrand and the Stern–Volmer methods. The results showed that relatively discrete porphyrin pendants in P-D formed a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric complex with TSPP and both static and dynamic mechanisms were active in this quenching process, while the tightly associated porphyrin pendants in P-W interacted with TSPP as an entirety and static quenching was dominant in this process. This observation was in accordance with their sequential structure. The polymer P-W has a wider absorption range and higher absorption intensity in the long wavelength region than the porphyrin monomer, which can more efficiently absorb light to accomplish light harvesting in water.
:1 stoichiometric complex with TSPP and both static and dynamic mechanisms were active in this quenching process, while the tightly associated porphyrin pendants in P-W interacted with TSPP as an entirety and static quenching was dominant in this process. This observation was in accordance with their sequential structure. The polymer P-W has a wider absorption range and higher absorption intensity in the long wavelength region than the porphyrin monomer, which can more efficiently absorb light to accomplish light harvesting in water.
1 Introduction
Porphyrin architectures can be used to construct functional molecular devices and mimic natural systems, particularly light harvesting antenna.1,2 In living systems, water is not only a universal solvent, but it also acts as a surrounding medium for biological macromolecules.3 For future applications, water should be considered a harmless solvent to conduct other photoredox reactions driven by visible light. Therefore, investigating porphyrin assemblies and their photophysical properties in aquatic environments is of fundamental importance.Recently, numerous porphyrin aggregates induced by association in heterogeneous systems, such as dendrimers,4 LB films,5 micelles6–10 and so on11–13 have attracted much attention. Hydrophobically associated water-soluble polymers could provide a heterogeneous model to investigate the aggregation behaviour of porphyrin units in an aquatic environment.14–20 The most common approach to prepare water-soluble copolymers with porphyrin pendants is to copolymerize the porphyrin monomer with water-soluble monomers. Linking an active polymerizable acrylate group to a porphyrin molecule bearing both hydrophobic and hydrophilic moieties could prepare an amphiphilic porphyrin monomer. This type of porphyrin monomer has the capacity to self-organize in aqueous solutions depending on the balance between electrostatic repulsion interactions and hydrophobic associative interactions. Thus, there are two important architectural parameters for amphiphilic porphyrin pendants in water-soluble copolymers: the self-association of the amphiphilic porphyrin monomer and the relative position between the porphyrin monomer and the water-soluble monomer. The combined action of multiple factors in such a copolymer would help create novel porphyrin architectures with special properties. The porphyrin pendants could also serve as optical probes due to their sensitive spectroscopic ability to reveal the aggregation structure of this complex system.
Heteroaggregates constructed by different porphyrins or porphyrin/phthalocyanine species via coordination bond,21–23 H bond,24,25 electrostatic interaction26,27 and so on have been widely studied in order to mimic some photoelectric functions in vivo. In view of the advantages of amphiphilic water-soluble copolymers in preparing porphyrin aggregates in an environment more closely to the natural system, investigation of their association behaviour with other porphyrin guests and construction of noncovalent heteroaggregates with a novel structure is a meaningful and challenging target. In this work, a novel cationic porphyrin monomer, ZnTrMPyP (Scheme 1), containing a pyridine quaternary ammonium, was synthesized, and its association behaviour in water was studied by UV-vis absorption. Based on this association behaviour, amphiphilic water-soluble copolymers with different sequential structures were prepared and their association behaviour with TSPP was also investigated by absorption spectra.
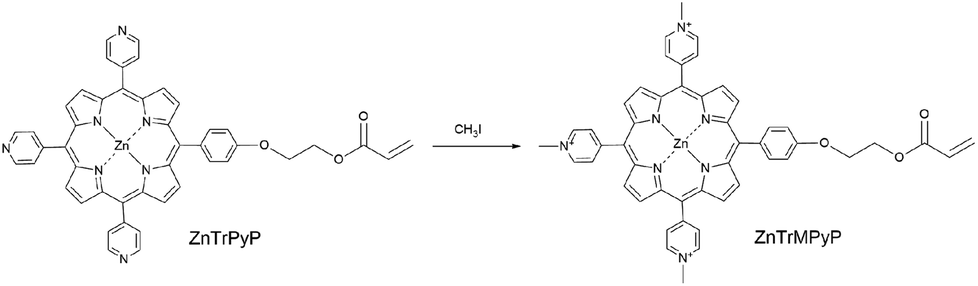 | ||
| Scheme 1 Preparation of ZnTrMPyP. | ||
2 Experiment
2.1 Materials
5-[4-[2-(Acryloyloxy)ethoxy]phenyl]-l0,l5,20-tris(4-pyridyl)porphyrinate zinc(II) (ZnTrPyP) was synthesized according to the ref. 18. All reagents not explicitly referenced were obtained from commercial sources and used as received. Iodomethane and tetra(p-sulphonic phenyl)porphyrin (TSPP) were purchased from Alfa Aesar company. Acrylamide (AM) was purchased from Jiangxi Changjiu Biochemical Engineering Corporation. 2,2′-Azobis (isobutyronitrile) (AIBN), 2,2′-azobis(2-methylpropionamidine)dihydrochloride (AAPH), N,N-dimethyl formamide (DMF), dimethyl sulfoxide (DMSO) and other A.R. grade reagents were obtained from Beijing Chemical Works. The aqueous solution was prepared by deionized water.2.2 Synthesis of porphyrin monomer 5-[4-[2-(acryloyloxy)ethoxy]phenyl]-l0,l5,20-tris(N-methyl-4-pyridiniumyl) porphyrinate zinc(II) (ZnTrMPyP)
ZnTrPyP (0.025 g, 0.03 mmol) was dissolved in a solution of DMF (35 mL). Iodomethane (0.4 mL, 6.4 mmol) was added to the solution and the reaction mixture was stirred at room temperature for 4 h. After removal of excess iodomethane and DMF under reduced pressure, the residue was purified by recrystallization (methanol/ether) to give ZnTrMPyP as a purple solid (0.026 g, 99%). 1H NMR (400 MHz, DMSO) δ 9.59 (d, J = 6.6 Hz, 6H, 2,6-pyridinium), 9.26 (s, 4H, β-pyrrole), 9.17 (8, 4H, β-pyrrole), 9.12 (d, J = 6.5 Hz, 6H, 3,5-pyridinium), 8.25 (d, J = 8.5 Hz, 2H, 2,6-phenyl), 7.55 (d, J = 8.6 Hz, 2H, 3,5-phenyl), 6.47 (dd, J = 17.3, 1.4 Hz, 1H, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), 6.34 (dd, J = 17.3, 10.2 Hz, 1H, CH–), 6.06 (dd, J = 10.2, 1.4 Hz, 1H, CH2), 4.83 (s, 9H, N+–Me), 4.64 (t, J = 4.8 Hz, 2H, CH2), 4.56 (t, J = 4.8 Hz, 2H, CH2). MS (ESI) calcd for M+ 838.2468, found 838.2582.
), 6.34 (dd, J = 17.3, 10.2 Hz, 1H, CH–), 6.06 (dd, J = 10.2, 1.4 Hz, 1H, CH2), 4.83 (s, 9H, N+–Me), 4.64 (t, J = 4.8 Hz, 2H, CH2), 4.56 (t, J = 4.8 Hz, 2H, CH2). MS (ESI) calcd for M+ 838.2468, found 838.2582.
2.3 Preparation of copolymers
The copolymerization of ZnTrMPyP with acrylamide (AM) was carried out in dimethyl sulfoxide (DMSO) and water, and the resultant copolymers were denoted as P-D and P-W, respectively (Table 1, Fig. 1). ZnTrMPyP (0.017 g, 0.02 mmol) and AM (0.284 g, 4.00 mmol) was dissolved in dimethyl sulfoxide (8 mL) and water (8 mL) at room temperature. AIBN (0.0065 g, 0.04 mmol) was added to the solution of dimethyl sulfoxide, whereas AAPH (0.0108 g, 0.04 mmol) was added to the solution of water as an initiator. The polymerization was performed in sealed tubes at 60 °C (AIBN) and 55 °C (AAPH) for 24 h after purging with nitrogen for 20 min.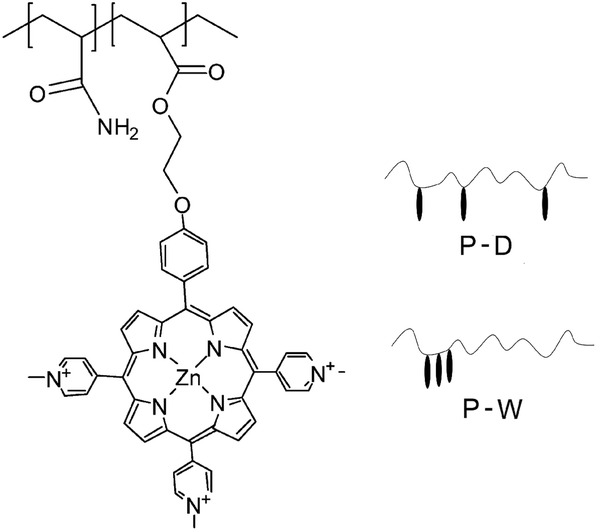 | ||
| Fig. 1 Chemical structures of polymer P(ZnTrMPyP-AM). | ||
The copolymer P-D displayed very similar UV-vis absorption spectra to the corresponding porphyrin monomer in an H2O–DMSO (1/9 v/v) mixed solvent. The content of the porphyrin units, in the copolymer P-D, can be estimated from the quantitative absorption spectra in the H2O–DMSO (1/9 v/v) mixed solvent, setting the molar extinction coefficient of the Soret band for ZnTrMPyP as a standard. The intensity of the Soret band in the copolymer P-W decreased greater than that of the monomer ZnTrMPyP, and the Q1 band in both copolymer P-W and monomer ZnTrMPyP remain nearly unchanged in the H2O–DMSO (1/9 v/v) mixed solvent. Therefore, the content of porphyrin units in the copolymer P-W was estimated from the quantitative absorption spectra in the H2O–DMSO (1/9 v/v) mixed solvent setting the εM of the Q1 band for ZnTrMPyP as a standard. The molecular weight of the polymer was investigated by Static Light Scattering (SLS) in water. The results are shown in Table 1. Compared to that in feed, the proportion of porphyrins in polymer was considerably small, which shows that the polymerization rate of the porphyrin monomer is much lower than that of AM. The lower polymerization rate may be ascribed to the steric repulsion of the porphyrin rings.
2.4 Explanation of data analysis
:1 stoichiometric complex, the Benesi–Hildebrand equation can be defined as follows:| 1/(A − A0) = 1/[(Ac − A0)KaC] + 1/(Ac − A0) | (1) |
| F0/F = 1 + Ksv [Q] | (2) |
A static quenching mechanism due to the formation of non-fluorescent complex abides by the Stern–Volmer equation:
| F0/F = 1 + K [Q] | (3) |
The equation is similar with that in dynamic quenching mechanism, but the K, the slope of this typically linear plot, is defined as the formation constant of the complex between the fluorophore and the quencher.
The plot of F0/F versus [Q] is linear if either quenching mechanism is dominant. However, deviations from linearity can occur as a result of both static and dynamic mechanisms being active in the quenching process, resulting in upward curvature of the Stern–Volmer plot.
2.5 Measurement
1H NMR spectrum was obtained on a Bruker DPX400 spectrometer. Mass spectrum was carried out on Buker APEX-IV. Static light scattering (SLS) measurement of copolymers was performed by the DAWN EOS (Wyatt). FT-IR spectra were obtained from Excalibur 3100 (Varian, USA) infrared spectrometer in KBr disks. UV-vis absorption spectrum was recorded with a Hitachi UV-3900 UV-vis spectrophotometer. Fluorescence emission spectrum was performed on a Hitachi F-4500 fluorescence spectrophotometer. 1 mm colorimetric ware was applied in the test of spectroscopic properties of sample with high concentration.3 Results and discussion
3.1 Association behaviour of the porphyrin monomer ZnTrMPyP
Table 2 lists the steady-state absorption spectral data for the monomer ZnTrMPyP in various solvents. The UV-vis spectrum of ZnTrMPyP in DMSO showed an intense Soret band, with an absorption maximum at 439.5 nm and relatively weak Q-bands at approximately 565 and 610 nm. However, when ZnTrMPyP was dissolved in water, DMF or methanol, the Soret band became broad and the molar extinction coefficient diminished drastically. According to exciton theory, broadening of the Soret band indicates weak electronic interactions exist between ZnTrMPyP in these solvents. This is because the porphyrin monomer ZnTrMPyP is comprised of both a hydrophobic tetrapyrrole macrocycle and hydrophilic pyridine quaternary ammonium cations in the molecular architecture. This imparts a high capacity for self-organization depending on the balance between repulsive and associative interactions. Consequently, ZnTrMPyP self-aggregates in water, DMF and methanol. Nevertheless, the spectroscopic properties of ZnTrMPyP exhibit no notable changes or mutations in water, even if the concentration is at saturation (approximately 1.2 × 10−2 mol L−1). This indicates that the association of the porphyrin monomer, ZnTrMPyP, in water is weak and does not change abruptly in the range of dissolubility.| Soret band | Q band | εQ1/εSoret | |||
|---|---|---|---|---|---|
| λ (nm) | Half-bandwidth (nm) | ε × 10−5 (mol−1 L cm−1) | λ (nm) | ||
| DMSO | 439.5 | 23 | 2.14 | 565, 610 | 0.090 |
| H2O | 435.5 | 31 | 1.41 | 564, 611 | 0.091 |
| DMF | 441.5 | 34 | 1.42 | 568, 619 | 0.088 |
| Methanol | 440.0 | 35 | 1.48 | 565, 613 | 0.092 |
For further insight into the solvent-induced self-association of ZnTrMPyP, Soret absorption bands were monitored at different water/DMSO ratios. As shown in Fig. 2, when the content of water increased from 0% to 100%, the maximum absorption wavelength of the Soret band shifted from 439.5 nm to 435.5 nm (inset (A) in Fig. 2), and the half-bandwidth increased from 23 nm to 31 nm at the same time (inset (B) in Fig. 2). Furthermore, this change resulted in isosbestic points in the UV-vis titration spectra, shown as arrows in Fig. 2, which suggests that there is equilibrium between the aggregated species and the monomeric species in the water–DMSO mixture. The rising baseline indicates that the system became heterogeneous to some extent due to the formation of the aggregated species of ZnTrMPyP. Therefore, DMSO could be considered a good solvent for ZnTrMPyP, while water is a poor solvent for this system.
 | ||
| Fig. 2 UV-vis spectra of ZnTrMPyP in a mixture of water/DMSO. Inset (A) and (B) show changes in the Soret absorption peaks and half-bandwidth, respectively, upon the content of water in the solvent mixtures. | ||
Inspired by the increased baseline in Fig. 2, dynamic light scattering (DLS) was used to characterize the formation of the ZnTrMPyP aggregates. For the solution of ZnTrMPyP in DMSO, DLS signals were not observed. In dilute solutions of ZnTrPyP in water, DLS signals were hardly observed as well. Fig. 3 shows the hydrodynamic radius (RH) of the ZnTrPyP aggregates measured by DLS in water when the concentration was above 5.0 × 10−4 mol L−1. As shown in Fig. 3, the average RH value shifts to a larger value at increased concentrations. This observation demonstrates that the aggregates of ZnTrMPyP were formed and that the aggregates extend to a larger scale as the concentration increases.
 | ||
| Fig. 3 DLS size distribution functions for aggregates of ZnTrMPyP in water at various concentrations. | ||
3.2 Absorption spectra of the copolymers P(ZnTrMPyP-AM)
Fig. 4(A) illustrates the steady-state absorption spectra of the copolymer P-D in an aqueous solution at a variety of concentrations. Some absorption spectral data are listed in Table 3. The intense Soret band is at 439.5 nm, and relatively weak Q-bands are approximately 566 and 613 nm. Except for minor redshifting, the polymer P-D displays a very similar UV-vis absorption spectrum to that of ZnTrMPyP in water, and the spectra remains the same over a wide concentration range. This indicated that the polymer chains in P-D have no significant effect on the aggregate structure of ZnTrMPyP in aqueous media. Methylation of the pyridine moieties reduces the hydrophobicity of porphyrin and gives rise to electrostatic repulsion interactions between the porphyrin pendants. It also inhibits the formation of N–H⋯N(py) intermolecular hydrogen bands. Therefore, neither H-aggregates nor slipped-cofacial dimers, as reported in our earlier work, were formed in the aqueous solution of polymer P-D. | ||
| Fig. 4 UV-vis absorption spectra of the copolymers (A) P-D and (B) P-W in aqueous solution at various concentrations of 1 × 10−4, 2 × 10−4, 3 × 10−4 and 4 × 10−4 mol L−1. | ||
| Polymer | Absorption band (nm) | Half-bandwidth of Soret band | εQmax/εSoret | ||||
|---|---|---|---|---|---|---|---|
| Soret | Q1 | Q2 | Q3 | Q4 | |||
| P-D | 439.5 | 566.0 | 613.5 | 33.5 | 0.09 | ||
| P-W | 438.0 | 569 | 616 | 636 | 778 | 54.0 | 0.18 |
However, the absorption spectra of the polymer P-W in aqueous solution changed notably, as shown in Fig. 4(B). The Soret band was no longer symmetric, and its half-bandwidth increased to 54 nm and the maximum absorption wavelength shifted to 438.0 nm. Two new bands, labeled Q3 and Q4, appeared in the Q band range, among which Q3 was dominant. Moreover, the relative intensity of the Q band over the Soret band increased greatly, which can be seen from that the value of εQ3/εSoret is 0.18 in P-W, while the value of εQ1/εSoret is 0.09 in P-D. The dramatic distinction in absorption spectra of P-W and P-D suggests that the aggregation state of porphyrin pendants in them is different.
Some studies20,31,32 have reported the appearance of new Q bands in porphyrin aggregates. However, the origin of these new bands has not been ascertained in detail. It is said that the distinctive enhancement of Q bands could be attributed to an intensity transfer from a strong B band to the Q band region,33 which is mediated by an excitonic coupling between B and Q transition dipoles. The appearance of new Q bands and the intensity transfer from the B band to Q bands of porphyrin pendants in copolymer P-W is in accord with the above situations. It is obvious that the porphyrin pendants in copolymer P-W form aggregates with a certain structure by virtue of the polymer chains.
It is well known that sunlight has high intensity in the range of 400–900 nm. The polymer P-W expands the absorption limitation of the porphyrin from 650 nm to 850 nm and enhances the absorption coefficient in the long wavelength region. The absorption property of polymer P-W is optimized to the wavelength of the solar spectrum; therefore, the polymer P-W can more efficiently absorb light to accomplish light harvesting in water.
3.3 Emission and excitation spectra of the copolymers P(ZnTrPyP-AM)
Emission spectra of the polymers P-D and P-W in water were broad peaks, and the peak shape was not affected by the excitation wavelength and the concentration of polymer. Fig. 5 illustrates the normalized fluorescence spectra of ZnTrMPyP, P-D and P-W in water. It can be seen that fluorescence spectra of ZnTrMPyP and P-D were very similar, confirming further the random polymer chain has little influence on the aggregation state of porphyrin pendants in P-D. However, the fluorescence spectrum of P-W red shift and the fluorescence quantum yield decreased obviously, indicating that further aggregation occured. This further aggregation was not caused by hydrophobic association, but by sequence distribution on polymer chain. | ||
| Fig. 5 Normalized fluorescence emission spectra of monomer ZnTrMPyP, polymer P-D and P-W in water. | ||
Fluorescence excitation spectra of the polymers P-D and P-W in aqueous solution were also established and further compared with their absorption spectra. As shown in Fig. 6, the excitation spectrum of P-D was similar with its absorption spectrum shown in Fig. 4(A), while that of P-W was significantly different from its absorption spectrum. There was only one intensive excitation peak around 638 nm in Q bands region of P-W, which corresponding to the newly appeared peak Q3 in absorption spectrum. The difference of fluorescence excitation spectra further demonstrated that porphyrin aggregates in the polymer P-W have different energy level structures with those in P-D.
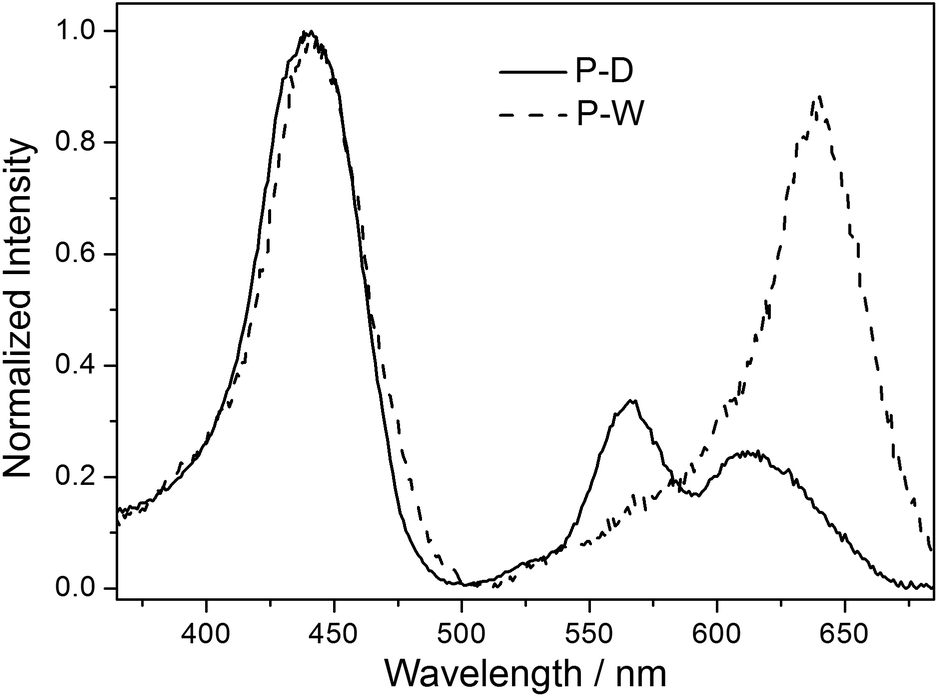 | ||
| Fig. 6 Normalized fluorescence excitation spectra of P-D and P-W in water (emission wavelength = 700 nm). | ||
3.4 Microstructure of the copolymers P(ZnTrMPyP-AM)
The differences in absorption, emission and excitation spectra between P-D and P-W were directly related to the polymer microstructure (Fig. 1). It is well known that the initial monomer disperse states affected the distribution of the monomer in the copolymer. In the P-D polymerization system, the monomers AM and ZnTrMPyP distributed uniformly in DMSO. During the polymerization, the initiator AIBN initiated any kind of monomers encountered in the solution randomly, i.e., either AM or ZnTrMPyP, to form propagating radicals of AM and porphyrin molecules until they were terminated. Under these polymerization conditions, the porphyrin pendants were distributed randomly in the P-D polymer chains.According to the results of 3.1, the ZnTrMPyP aggregates formed by self-association prevailed in water. In each of the aggregates, the vinyl groups were localized in a finite volume, and if one encountered the growing macroradical headgroup, chain propagation may occur by a “zip” mechanism. A whole sequence of the monomer molecules within an aggregate would then be expected to build into the copolymer, leading to micro-blocks.34–36 The propagating radicals polymerized with the AM monomer after departing from the aggregates of ZnTrMPyP. These steps were repeated until the propagating radicals were terminated. Therefore, P-W had micro-blocky sequences of cationic porphyrin units in the copolymer chains. Although the exact sequence distribution was unable to be detected by NMR or IR, due to the number of hydrophobic monomers being too small, this sequence structure made the porphyrin pendants form compact aggregates that were revealed by the change observed in the absorption spectra, as shown in Fig. 4(B).
To further examine the previous conclusion, the absorption spectra of P-W in water and 1H2O:9DMSO mixture (a good solvent for ZnTrMPyP), were compared in Fig. 7. If the changes in absorption spectra of P-W were originated from the porphyrin aggregates formed via some intermolecular interactions, the addition of 1H2O:9DMSO mixture could destroy these interactions, and the spectra would become similar to that of the monomer. In contrast, as Fig. 7 shows, subtle changes in spectra suggested that the porphyrin aggregates in polymer P-W were fixed by a covalent bond. This was consistent with the micro-blocky sequences of the cationic porphyrin units in the polymer chains.
 | ||
| Fig. 7 UV-vis absorption spectra of P-W in water and H2O–DMSO (1/9 v/v) mixture. | ||
3.5 Interaction of the copolymers P(ZnTrMPyP-AM) with TSPP
The different sequential structure of the porphyrin pendants in the polymer could influence the association behaviour between the porphyrin pendants and other guest materials. To further confirm the sequential structure of porphyrin pendants in the polymer, we chose tetra(p-sulfonato phenyl)porphyrin, TSPP, bearing oppositely charged substituents as the guest material and studied the association behaviour by the absorption and emission spectra.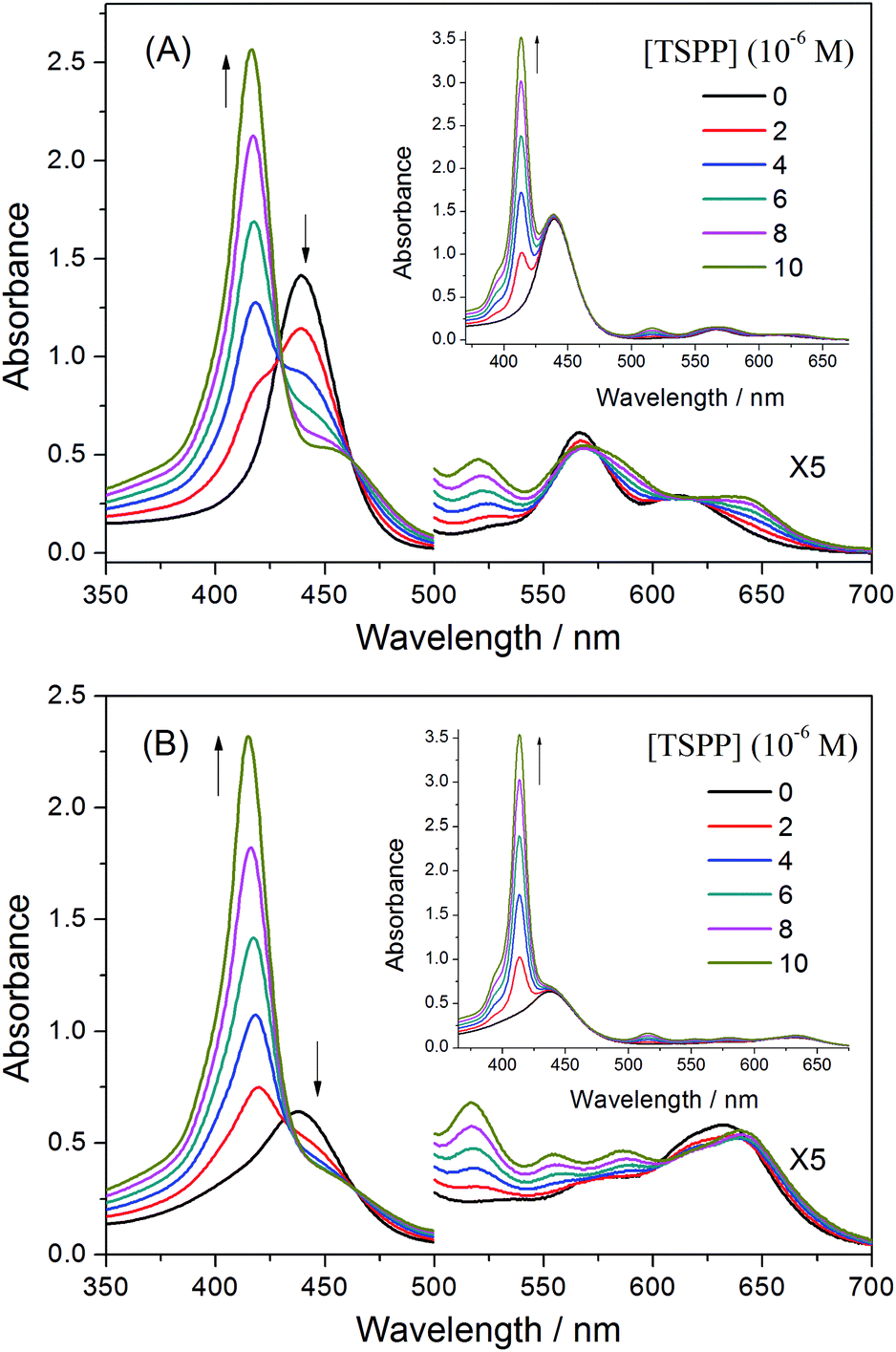 | ||
| Fig. 8 UV-vis spectra of (A) P-D and (B) P-W at 2 × 10−4 g mL−1 with added TSPP concentrations of 0, 2 × 10−6, 4 × 10−6, 6 × 10−6, 8 × 10−6, 1 × 10−5 mol L−1 in water. Insets (A) and (B) show the sum of absorption spectra of (A) P-D and (B) P-W and TSPP. | ||
Fig. 8(B) shows the spectral changes for an aqueous solution of P-W upon titration with a solution of TSPP. Upon the addition of TSPP, the Soret band of P-W at 435.5 nm decreases, while the absorption at 417, 517.5 and 555 nm appear with isosbestic points at 433, 464, 604 and 640 nm, respectively. The resulting spectra are also different from the sum of the absorption spectra for P-W and TSPP, in which no isosbestic points appeared and the increasing peak at 413.5 nm is the Soret band of TSPP, as shown in the insert picture of Fig. 8(B). This is similar with that of P-D and TSPP. However, the decrease of the Soret band of P-W is obviously weak, which may originate from the different sequential structure of the porphyrin pendants on the polymer chains.
We applied the Benesi–Hildebrand method for the determination of the association constant between the ZnTrMPyP pendants in the copolymers and TSPP. When an aqueous solution of the copolymers P-D and P-W were titrated by TSPP, the increasing absorption peak originated from both the formed complex and the dissociative TSPP, while the decrease of absorption at 440 nm was entirely caused by the formation of the ZnTrMPyP–TSPP complex. Therefore, changes in absorbance (ΔA = A0 − A) for the Benesi–Hildebrand equation were collected at 440 nm. Fig. 9 shows the plot of 1/(A0 − A) versus 1/[TSPP] in the titration experiment. Linear fits were obtained for the P-D and TSPP systems. This suggests that the ZnTrMPyP pendants in polymer P-D form a 1:1 stoichiometric complex with TSPP. From the linear fits, the calculated value of the association constant is 1.25 × 104 M−1. However, for P-W and TSPP systems, the Benesi–Hildebrand plots deviate from a straight line. This indicates that ZnTrMPyP pendants in polymer P-W do not associate with TSPP in a 1:1 stoichiometry.
 | ||
| Fig. 9 Benesi–Hildebrand plot from the absorption spectra of polymers P(ZnTrPyP-AM) in water with gradual addition of TSPP. | ||
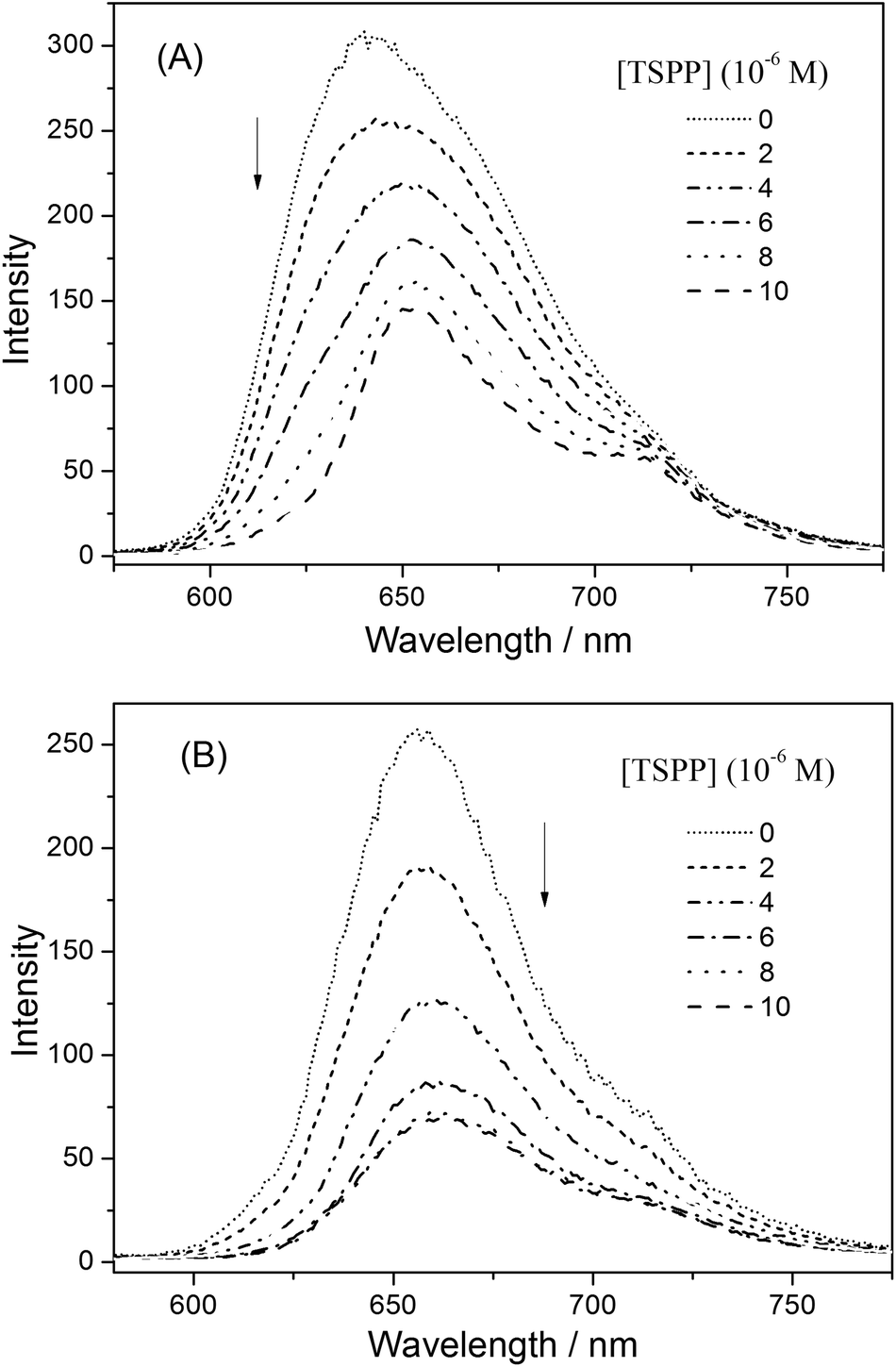 | ||
| Fig. 10 The fluorescence emission spectra of (A) P-D and (B) P-W at 2 × 10−4 g mL−1 with added TSPP concentrations of 0, 2 × 10−6, 4 × 10−6, 6 × 10−6, 8 × 10−6, 1 × 10−5 mol L−1 in water. | ||
The fluorescence quenching kinetics of the polymers P(ZnTrPyP-AM) with TSPP were further investigated by the Stern–Volmer equation and the results were given in Fig. 11. For the polymer P-W, the Stern–Volmer plot had a linear relationship when the TSPP was in the lower concentration range and the slope was about 3.1 × 105 M−1. If this process was treated as dynamic quenching, the quenching constant kq could be calculated to be 2.4 × 1014 M−1 s−1 according to Ksv = kqτ0 where τ0 = 1.3 ns. But the magnitude order of this quenching constant was much higher than that of the diffusion-controlled process, so the quenching process of the polymer P-W by TSPP could be considered as static quenching and the slope (3.1 × 105) represents the association constant between ZnTrMPyP aggregates and TSPP. When the concentration of TSPP continued to increase, the plot of F0/F versus [TSPP] showed a downward curvature, which may be caused by the emission of TSPP induced by energy transfer from ZnTrMPyP. For the polymer P-D, the Stern–Volmer plot of F0/F versus [TSPP] showed an upward curvature over the whole concentration range of TSPP, indicating both static and dynamic mechanisms were active in this quenching process.
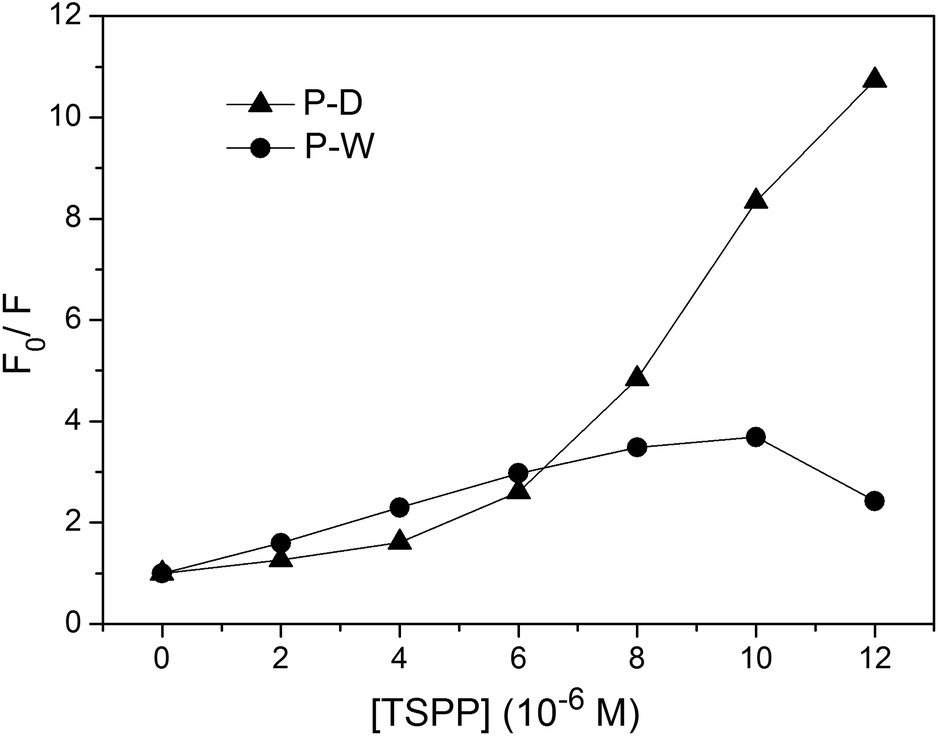 | ||
| Fig. 11 The Stern–Volmer plot for quenching process of the polymers P(ZnTrPyP-AM) by TSPP. The fluorescence wavelength is 615 nm for P-D and 660 nm for P-W. | ||
:1 stoichiometry complex with increasing the concentration of TSPP, as shown in the Scheme 2. So in the quenching process of P-D by TSPP, the dynamic mechanism is primary at low concentration of TSPP while static mechanism is primary as the concentration of TSPP increases. On the other hand, ZnTrMPyP pendants in P-W associate with each other tightly, and they may only further interact with TSPP as an entirety. So the addition of TSPP caused a little change in absorption spectra of P-W in water. The associate constant between ZnTrMPyP entirety, which with more significant electro positivity, and TSPP is large enough and static quenching is dominant in this quenching process. This was in accordance with that the polymer P-D has randomly distributed ZnTrMPyP pendants while the polymer P-W has micro-blocky ZnTrMPyP pendants. The investigation on the association behaviour between cationic porphyrin pendants with different sequential structures in water-soluble copolymers and anionic porphyrin guests in this work is helpful to construct hetero porphyrin aggregates with certain structure.
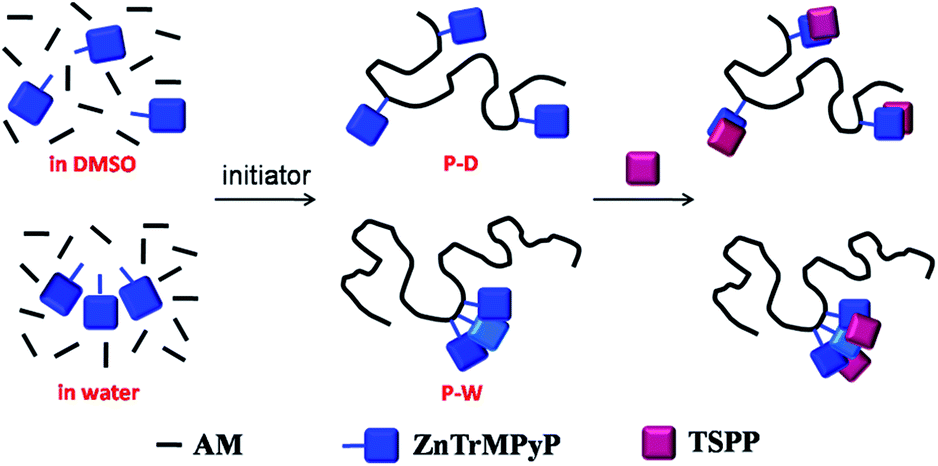 | ||
| Scheme 2 Association behaviour between porphyrin pendants on the co-polymers and TSPP. | ||
4 Conclusions
The novel cationic tripyridiniumylporphyrin monomer ZnTrMPyP and its corresponding copolymers with AM, named P-W and P-D, respectively, were prepared. Electronic interactions between ZnTrMPyP molecules in water were observed. The polymer P-D displayed very similar absorption and emission spectra to those of ZnTrMPyP in water, whereas in the polymer P-W, two new absorption bands appeared in the Q band range and its fluorescence spectra red shift and the fluorescence quantum yield decreased obviously. The drastic differences in absorption and emission spectra were because that the porphyrin pendants distribute randomly in P-D while having micro-blocky structures in P-W. Further Benesi–Hildebrand and Stern–Volmer investigations showed that relatively discrete porphyrin pendants in P-D could form a 1:1 stoichiometric complex with TSPP and both static and dynamic quenching were active in their association process, while the tightly associated porphyrin pendants in P-W interacted with TSPP as an entirety and static quenching was dominant. The polymer P-W has a wider absorption range and higher absorption intensity in the long wavelength region than its porphyrin monomer, which can more efficiently absorb light in order to accomplish light harvesting in water.
Conflicts of interest
There are no conflicts to declare.Notes and references
- I. Beletskaya, V. S. Tyurin, A. Y. Tsivadze, R. Guilard and C. Stern, Chem. Rev., 2009, 109, 1659–1713 CrossRef CAS PubMed.
- A. Satake and Y. Kobuke, Tetrahedron, 2005, 61, 13–41 CrossRef CAS.
- M. J. Tait and F. Franks, Nature, 1971, 230, 91–94 CrossRef CAS PubMed.
- P. M. R. Paulo and S. M. B. Costa, Photochem. Photobiol. Sci., 2003, 2, 597–604 RSC.
- I. Prieto, J. M. Pedrosa, M. T. Martin-Romero, D. Mobius and L. Camacho, J. Phys. Chem. B, 2000, 104, 9966–9972 CrossRef CAS.
- P. J. Gonçalves, L. P. F. Aggarwal, C. A. Marquezin, A. S. Ito, L. De Bonib, N. M. Barbosa Neto, J. J. Rodrigues Jr, S. C. Zílio and I. E. Borissevitch, J. Photochem. Photobiol., A, 2006, 181, 378–384 CrossRef.
- S. C. M. Gandini, V. E. Yushmanov, I. E. Borissevitch and M. Tabak, Langmuir, 1999, 15, 6233–6243 CrossRef CAS.
- L. MonsúScolaro, C. Donato, M. Castriciano, A. Romeo and R. Romeo, Inorg. Chim. Acta, 2000, 300–302, 978–986 CrossRef.
- D. M. Togashi, S. M. B. Costa, A. J. F. N. Sobral and A. M. A. R. Gonsalves, J. Phys. Chem. B, 2004, 108, 11344–11356 CrossRef CAS.
- S. M. Andrade, C. Teixeira, D. M. Togashi, S. M. B. Costa and A. J. F. N. Sobral, J. Photochem. Photobiol., A, 2006, 178, 225–235 CrossRef CAS.
- M. Ali and S. Pandey, J. Photochem. Photobiol., A, 2009, 207, 28–31 CrossRef.
- J.-J. Wu, H.-L. Ma, H.-S. Mao, Y. Wang and W.-J. Jin, J. Photochem. Photobiol., A, 2005, 173, 296–300 CrossRef CAS.
- D. Kuciauskas, J. Kiskis, G. A. Caputo and V. Gulbinas, J. Phys. Chem. B, 2010, 114, 16029–16035 CrossRef CAS PubMed.
- Y. Morishima, K. Saegusa and M. Kamachi, Macromolecules, 1995, 28, 1203–1207 CrossRef CAS.
- Y. Morishima, H. Aota, K. Saegusa and M. Kamachi, Macromolecules, 1996, 29, 6505–6509 CrossRef CAS.
- Y. Morishima, K. Saegusa and M. Kamachi, J. Phys. Chem., 1995, 99, 4512–4517 CrossRef CAS.
- M. Nowakowska, A. Karewicz, M. Klos and S. Zapotocny, Macromolecules, 2003, 36, 4134–4139 CrossRef CAS.
- K. W. Ding, F. Wang and F. P. Wu, J. Photochem. Photobiol., A, 2011, 220, 64–69 CrossRef CAS.
- K. W. Ding, F. Wang and F. P. Wu, Chin. J. Polym. Sci., 2012, 30, 63–71 CrossRef CAS.
- F. Wang, K. W. Ding and F. P. Wu, Dyes Pigm., 2011, 91, 199–207 CrossRef CAS.
- R. K. Kumar and I. Goldberg, Angew. Chem., Int. Ed., 1998, 37, 3027–3030 CrossRef CAS PubMed.
- R. T. Stibrany, J. Vasudevan, S. Knapp, J. A. Potenza, T. Emge and H. J. Schugar, J. Am. Chem. Soc., 1996, 118, 3980–3981 CrossRef CAS.
- R. Takahashi and Y. Kobuke, J. Am. Chem. Soc., 2003, 125, 2372–2373 CrossRef CAS PubMed.
- J. L. Sessler, B. Wang and A. Harriman, J. Am. Chem. Soc., 1995, 117, 704–714 CrossRef CAS.
- H. Ohkawa, A. Takayama, S. Nakajima and H. Nishide, Org. Lett., 2006, 8, 2225–2228 CrossRef CAS PubMed.
- A. Miura, Y. Shibata, H. Chosrowjan, N. Mataga and N. Tamai, J. Photochem. Photobiol., A, 2006, 178, 192–200 CrossRef CAS.
- M. D. Napoli, S. Nardis, R. Paolesse, M. Graca, H. Vicente, R. Lauceri and R. Purrello, J. Am. Chem. Soc., 2004, 126, 5934–5935 CrossRef PubMed.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- W. B. Person, J. Am. Chem. Soc., 1965, 87, 167–170 CrossRef CAS.
- B. Roy, M. Ghosh, P. Mukherjee, S. Chowdhury, B. C. Gupta, K. Majhi and S. Sinha, Spectrochim. Acta, Part A, 2018, 188, 311–317 CrossRef CAS PubMed.
- A. Nakano, T. Yamazaki, Y. Nishimura, I. Yamazaki and A. Osuka, Chem.–Eur. J., 2000, 6, 3254–3271 CrossRef CAS.
- T. S. Balaban, M. Linke-Schaetzel, A. D. Bhise, N. Vanthuyne and C. Roussel, Eur. J. Org. Chem., 2004, 2004, 3919–3930 CrossRef.
- J. Zimmermann, U. Siggel, J.-H. Fuhrhop and B. Roder, J. Phys. Chem. B, 2003, 107, 6019–6021 CrossRef CAS.
- D. G. Peiffer, Polymer, 1990, 31, 2353–2360 CrossRef CAS.
- D. N. Schulz, J. J. Kaladas, J. J. Maurer, J. Bock, S. J. Pace and W. W. Schulz, Polymer, 1987, 28, 2110–2115 CrossRef CAS.
- A. Hill, F. Candau and J. Selb, Macromolecules, 1993, 26, 4521–4532 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |