
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClear distinction between CAC and CMC revealed by high-resolution NMR diffusometry for a series of bis-imidazolium gemini surfactants in aqueous solutions†
Kosma Szutkowski *a,
Żaneta Kołodziejskab,
Zuzanna Pietralikb,
Igor Zhukovc,
Andrzej Skrzypczakd,
Katarzyna Maternad and
Maciej Kozakb
*a,
Żaneta Kołodziejskab,
Zuzanna Pietralikb,
Igor Zhukovc,
Andrzej Skrzypczakd,
Katarzyna Maternad and
Maciej Kozakb
aNanoBioMedical Centre, Adam Mickiewicz University in Poznań, Umultowska 85, PL61614 Poznań, Poland. E-mail: kosma_sz@amu.edu.pl; Tel: +48 618295230
bDepartment of Macromolecular Physics, Faculty of Physics, Adam Mickiewicz University in Poznań, Umultowska 85, PL61614 Poznań, Poland
cInstitute of Biochemistry and Biophysics, Polish Academy of Sciences, Pawińskiego 5a, PL02106 Warsaw, Poland
dInstitute of Chemical Technology and Engineering, Faculty of Chemical Technology, Poznań University of Technology, Berdychowo 4, PL60965 Poznań, Poland
First published on 22nd November 2018
Abstract
The aggregation behavior in the transition region was studied for a series of dicationic surfactants 3,3′-[α,ω-(dioxaalkane)]bis(1-dodecylimidazolium)dichlorides with varied spacer length from two to twelve carbon atoms. We employed Nuclear Magnetic Resonance diffusometry and Bayesian DOSY analysis to obtain the aggregate size distribution in the transition region. The critical concentrations CC were independently obtained from surface tension, electric conductivity, UV-Vis and NMR methods. The micelle aggregation numbers were estimated from the self-diffusion coefficients and were independently confirmed using steady-state fluorescence quenching. The morphology of the aggregates was characterized by small-angle scattering of synchrotron radiation and molecular dynamics simulations. The obtained CC values are identified as critical aggregation concentrations CAC. A broad transition region was observed, and stable micelles were obtained at much higher concentrations than CAC. The accurate CMC values could not be identified for the systems in the study. We indicated that the distribution of aggregate size becomes small and the system becomes homogeneous at much larger concentrations than CAC (typically 15–20 mM). The existence of a slow exchange between two environments, an aggregate and aqueous environment, was confirmed by 1H NMR and 2D HSQC NMR spectroscopy.
1 Introduction
Gemini surfactants at the molecular level consist of a dimer of two monomeric surfactants. The gemini molecule contains two aliphatic chains, two hydrophilic heads, and a spacer group.1 Two hydrophobic tails can be either symmetric (m–s–m) or asymmetric (n–s–m), where m, n and s denotes two different chains and the spacer accordingly.The properties of gemini surfactants, such as the critical micelle concentration CMC, wetting, foaming, and solubility are significantly better than those of the monomeric counterparts.2,3 Accordingly, because considerably lower concentrations can be applied, gemini surfactants are proposed to be better for technological and biological purposes such as cosmetics formulations, oil recovery, detergency, nanotechnology, catalysis as well as drug delivery.4–15 The last one, namely, the drug delivery is of special concern. It involves the application of the micellar solutions, vesicles or lyotropic liquid crystals solutions as colloidal drug carrier systems. For those purposes the stability of the system is of utmost importance to provide stable and repeatable adsorption, carrying and release of a drug.16
Gemini surfactants are extremely surface active at the CMC (two-fold decrease of the surface tension of water). However, the lower values of CMC do not usually say much about the probable morphology of the aggregates except the fact that the broad transition region from monomers to stable aggregates is observed in the case of small micelles.17 It seems that they do not follow simple geometric packing behavior concerning the size and generally the spherical shape of the micelles.1,17 Instead, it is expected they would behave much like natural surfactants, e.g., phospholipids, and will tend to create bilayers.18 Moreover, recent coarse-grained simulation investigations confirmed the complexity of morphologies of gemini surfactants.19
The critical micelle concentration (CMC) defines the point at which amphiphilic molecules assemble into larger spherical aggregates, whereas critical aggregation concentration (CAC) determines the concentration at which premicellar aggregates are formed. The CMC value can be learned from the slope of the surface tension isotherm. The smaller the value of CMC the better solubilization properties of hydrophobic species in aqueous solutions. While the solubilization capacity of aliphatic chains or hydrophobic proteins is proportional to the surfactant's chain length, the values of CMC of gemini surfactants do not always follow this dependency.20,21 Such a response means that some physical properties of gemini surfactants with varying tail/spacer lengths are not necessarily linear. The reason for that is the molecular rearrangements of monomer molecules and preaggregation phenomena such as the existence of the sub-micellar aggregates in the solution.19 In general, the surfactant molecules may exist in the so-called premicellar state as dimers, tetramers and larger aggregates.1,22 Molecules of gemini surfactants may rearrange, bend and induce self-coiled structures given that the spacer is flexible enough. The molecular rearrangement is a three-stage process, and preassembly process precedes the sub-micellar state.1 The phenomenon of pre-micelle formation for normal and gemini surfactants below CMC concentration was studied recently by NMR diffusometry and relaxometry.23,24
The aggregate morphologies of gemini surfactants are more diverse than those observed for their monomeric counterparts. The determination of the morphology at the transition region (above CMC) can be useful for specific applications where narrow size distribution of micelles is desired such as the transmembrane peptide structure elucidation.25
We would like to expand the analysis of pre-micellization phenomenon for a whole family of bis-imidazolium cationic gemini surfactants with a varied spacer length. We have derived the critical concentrations values without naming them directly as either CAC or CMC. The main experimental technique was diffusion NMR spectroscopy.26–28 Furthermore we have applied complementary techniques such as surface tension isotherms, electric conductivity, UV-Vis absorption, steady-state fluorescence quenching (SSFQ), 2D nuclear Overhauser spectroscopy (NOESY), 2D heteronuclear single-quantum correlation NMR (HSQC) as well as small-angle scattering of synchrotron radiation (SR-SAXS). Our primary objective was the quantitative analysis of aggregate size distribution from the distribution of diffusion coefficients employing Bayesian Diffusion Ordered SpectroscopY Transformation (BDT NMR) in the concentration dependent transition region where surfactant molecules assemble from monomers into larger aggregates.
2 Experimental
2.1 Synthesis and sample preparation
A series of dicationic surfactants 3,3′-[α,ω-(dioxaalkane)]bis(1-dodecylimidazolium)dichlorides denoted as C12JCR, where R is the number of methylene groups in the hydrophobic part of the spacer (Table 1). The chemical structure of synthesized compounds is presented in Fig. 1. The surfactants were synthesized according to previously reported procedure.29 The purity and the structure of surfactants were validated using 1H and 13C NMR.| Gemini surfactant name | Short name | Spacer name |
|---|---|---|
| 3,3′-[α,ω-(dioxaethane)]bis(1-dodecylimidazolium)dichloride | C12JC2 | C2 |
| 3,3′-[α,ω-(dioxabutane)]bis(1-dodecylimidazolium)dichloride | C12JC4 | C4 |
| 3,3′-[α,ω-(dioxahexane)]bis(1-dodecylimidazolium)dichloride | C12JC6 | C6 |
| 3,3′-[α,ω-(dioxaoctane)]bis(1-dodecylimidazolium)dichloride | C12JC8 | C8 |
| 3,3′-[α,ω-(dioxadecane)]bis(1-dodecylimidazolium)dichloride | C12JC10 | C10 |
| 3,3′-[α,ω-(dioxadodecane)]bis(1-dodecylimidazolium)dichloride | C12JC12 | C12 |
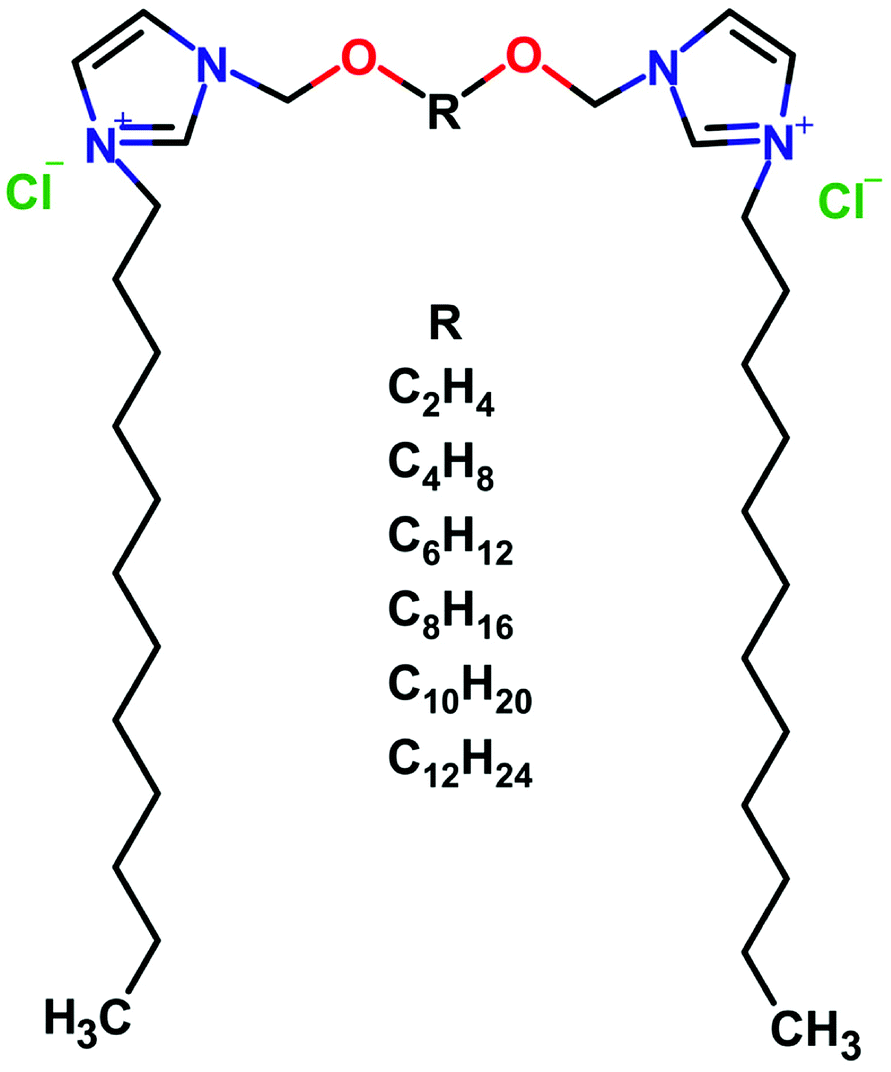 | ||
| Fig. 1 The schematic representation of dicationic gemini surfactants 3,3′-[α,ω-(dioxaalkane)]bis(1-dodecylimidazolium)dichlorides with varied length of the spacer R. | ||
2.2 Surface tension isotherms
The surface tension was determined using the pendant drop method. The measurements were carried out by the use of a Drop Shape Analysis System DSA100 (Krüss GmbH, Germany, accuracy ±0.01 mN m−1), at 25 °C. The temperature was controlled using a Fisherbrand FBH604 thermostatic bath (Fisher, Germany, accuracy ±0.1 °C). The pendant drop method is a widely used technique to measure the surface tension between gas–liquid and liquid–liquid interfaces. We have obtained the values of the surface tension by fitting the Young–Laplace equation to a shape of the drop captured by a digital camera suspended at the end of a capillary tube. The detection of a drop edge from the digital image yielded in a set of geometrical points describing the shape. The Young–Laplace equation for an axisymmetric interface was solved for given initial parameters; then the best fit was obtained by minimizing a summation of the squared distances between the experimental points and the theoretical drop profile.302.3 Electric conductivity
Electric conductivity measurements were carried out using pHenomenal CO 3100L conductometer (VWR, Pennsylvania) equipped with a 2-pole graphite probe and the available conductivity range from 0.001 μS cm−1 to 1000 mS cm−1. We have obtained the conductivity values by the titration of surfactant in the concentration range between 0 μ and 5 mM. We have determined the critical concentrations CC as the point of the inflection.2.4 UV-Vis absorption and steady-state fluorescence quenching
The spectrophotometric method for the determination of the critical concentrations of gemini surfactants is based on the tautomerization reaction of benzoylacetone (1-phenyl-1,3-butanedione, BZA, Sigma-Aldrich Poland). BZA was dissolved in dioxane (5 mg ml−1) and then diluted in water to 140 mM concentration. The BZA solution and the surfactant were mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The final concentration of the BZA sample was around 70 mM, and dioxane was 1% wt. The UV-Vis spectra were recorded in the 220–360 nm range using Jasco V-650 spectrophotometer. The resolution was 1 nm and scanning speed 100 nm min−1. A quartz 10 mm long cuvette was used. All measurements were carried out at a temperature of 25 ± 1 °C.
:1 ratio. The final concentration of the BZA sample was around 70 mM, and dioxane was 1% wt. The UV-Vis spectra were recorded in the 220–360 nm range using Jasco V-650 spectrophotometer. The resolution was 1 nm and scanning speed 100 nm min−1. A quartz 10 mm long cuvette was used. All measurements were carried out at a temperature of 25 ± 1 °C.
The SSFQ experiments were performed using Jasco FP 6300 for a 10 μM solution of pyrene with rising concentrations of a quencher Coumarin 153 (Sigma-Aldrich Poland) dissolved in ethanol and diluted with water to the final ethanol concentration of 2% wt. according to the procedure given by Turro and Yekta.31 The excitation band was λ = 320 nm. We have analyzed the intensity of fluorescence at λ = 383 nm. The aggregation numbers Nagg were obtained from the slope of the ln(I0/IC153) vs. the coumarin concentration (μM) using the following equation: Nagg = (Csurf − CMC) × 1000 × Cmic, where Cmic = 1/slope.
2.5 Bayesian DOSY NMR
The diffusion coefficients of aqueous surfactant solutions in D2O were measured by using Agilent DD2 600 MHz NMR spectrometer at 21 °C. Two probes were used: OneNMR liquid probe 1H-X (z-gradient 60 Gs cm−1) and DOTY DSI-1374 (modified to generate z-gradient up to 2800 Gs cm−1). The convection compensated DPFGDSTE (Pulse Field Gradient Double Stimulated Echo) pulse sequence was applied.32 The diffusion time Δ was long enough to obtain a 90% of the signal attenuation, for OneNMR probe the Δ time was typically varied between 50 and 500 ms. The magnetic field gradient duration δ was set to 3 ms. Initially, the values of average diffusion coefficients of surfactants were obtained by VNMRJ 4.2 software using the dependence of integral amplitudes over 0–4 ppm signal vs. the gradient amplitude and fitting the Stejskal–Tanner equation. Apart from normal analysis, we have utilized a semi 2D Diffusion Ordered SpectroscopY displays (DOSY). The distributions of the diffusion coefficients were obtained using MestReNova v12.02 (Mestrelab Research S.L.) and Bayesian DOSY transformation (BDT). Similarly to other authors, we did not perform an automated BDT transform.33 Initially, we have applied the exponential apodization with 10 Hz resolution to decrease the signal-to-noise of PGSE data. Next, auto phase and auto baseline corrections (Whittaker Smoother method) were involved. The baseline in the first spectrum in PGSE experiments is usually slightly distorted due to small spin echo shifts which generally leads to an artificial increase in the value of the diffusion coefficient. Finally, BDOSY displays were obtained using the BDT method with the resolution factor 1. The number of points in the diffusion dimension was 256 to smoother the diffusion profiles. We have investigated the possible dependence of the distribution of diffusion coefficients on the diffusion times using DOTY DSI-1374 2800 Gs cm−1 diffusion probe. For C12JC2 and C12JC4 samples, we have carried out experiments for diffusion times between 20 and 500 ms.We have estimated the aggregation numbers Na from the volume ratios Vh/Vmon, where Vh is the hydrodynamic volume of an aggregate and Vmon is the estimated hydrodynamic volume of single surfactant molecule. The apparent hydrodynamic volume is 4/3πRh3, where Rh is an effective hydrodynamic radius. The hydrodynamic radius can be obtained using well known Einstein–Stokes–Sutherland–Smoluchowski (also known as Einstein–Smoluchowski) formula Rh = kBT/6πηD, which relates the self-diffusion coefficient D with medium viscosity η, an absolute temperature T and Boltzmann constant kB. The viscosity of D2O at 294 K and experimentally obtained diffusion coefficients were used for further calculations. The hydrodynamic volumes of monomers Vmon were obtained from the self-diffusion coefficients measured at the lowest concentrations.
2.6 1H–13C HSQC NMR
The two-dimensional 1H–13C HSQC (Heteronuclear Single Quantum Correlation spectroscopy) experiments were conducted on Agilent DD2 600 NMR spectrometer equipped with three channels and 1H13C/15N/31P Penta probe head with an inverse detection of 1H. The experimental datasets were collected as 2048 × 32 complex points (acquisition times 85 ms and 8 ms) in 1H and 13C dimensions respectively, using States-TPPI quadrature detection.34 Each trace was averaged over 32 scans. All spectra were referenced to external sodium 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) and processed with NMRPipe software.352.7 Small-angle X-ray scattering of synchrotron radiation (SR-SAXS)
SAXS scattering experiments were performed using the synchrotron radiation (λ = 0.09919 nm) on the BM29 SAXS Beamline36 at ESRF (Grenoble, France). We have investigated the SAXS curves for two surfactant concentrations (15.2 and 42 mM) using Pilatus3 S 1M detector (active area: 981 × 1043 pixels) (Dectris, Switzerland). The typical sample volume was 30 μl. Samples were injected into the capillary flow cell at 15 °C, using a robotic auto-sampling system. The sample-to-detector distance was fixed at 2.867 m. The momentum transfer axis (s-range) was calibrated using silver behenate (where s = 4πsinθ/λ).37 The SR-SAXS data were recorded within the s-axis range from 0.06 to 4.2 nm−1. For each sample, 10 successive frames were accumulated (exposition time 10 s per frame). The recorded SAXS data were analyzed and processed using the program PRIMUS from the ATSAS package.38
3 Results and discussion
3.1 Surface tension isotherms
In this part of the work, we have decided to use a notion of the critical concentration (CC) instead of naming the obtained concentrations as CAC or CMC directly. The surface tension isotherms are shown in Fig. 2. The CC values decrease with an increasing spacer length, and the highest value of CC was obtained for the surfactant with the shortest spacer (C2). Accordingly, the CC values decrease with an increasing spacer length from 0.71 mM for C12JC2 down to 0.26 mM for C12JC8 respectively. Further increase of the number of methylene groups does not promote substantial decrease of the CC values. Although it can be noticed that C12JC12 surfactant seems to decrease surface tension more effectively. With this in mind, the surface tension observed at the lowest concentration for C8, and C10 spacers are around 65 mN m−1 while the corresponding value of the surface tension for the C12 spacer drops down to 55 mN m−1. The values of the surface tension at CC, γCMC, increase with the spacer length from 37 mN m−1 for C2 spacer up to 42.5 mN m−1 for the C12 spacer.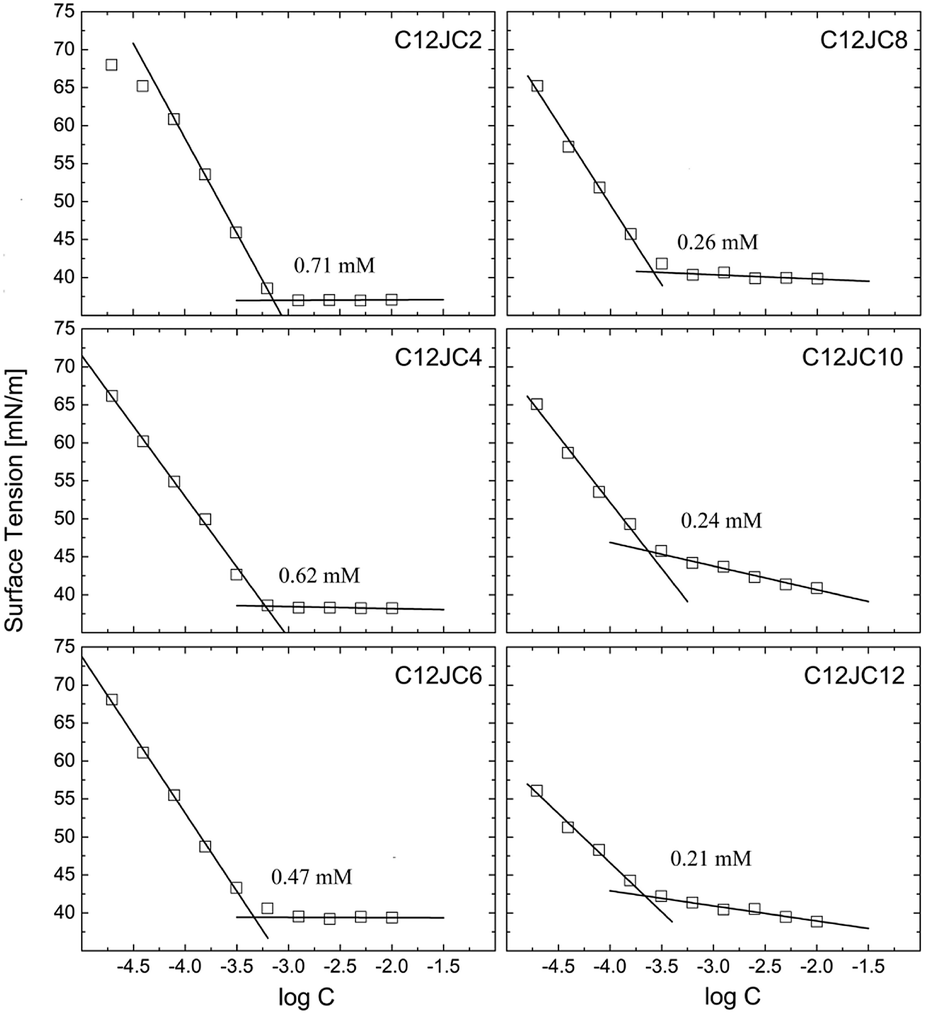 | ||
| Fig. 2 The surface tension isotherms for C12JC2–C12JC12 gemini surfactants. The solid lines represent linear fits. | ||
Furthermore, the values of the surface area of a single molecule also increase with an increasing spacer length from 3 × 10−19 m2 for C2 up to 8 × 10−19 m2 for C12. It must be noted, that the values of the molecular area obtained from the Gibbs isotherm, usually do not agree with the values obtained from the neutron reflectivity studies.39 We expect that differences between CC values obtained for C8, C10, and C12 spacers will be higher. In contrast to monomeric surfactants, long-tailed gemini surfactants may have problems with immediate aligning at the interface. The lower surface activity than expected for gemini surfactants was observed for longer chains.1 Under those circumstances, the resulting CMC values derived from the surface tension might be underestimated. For example, for xylylene diphosphate and stilbene spacer gemini surfactants for which surface tension experiments may give either underestimated or overestimated CMC values.1 An alternative explanation may be provided assuming that the Gibbs adsorption equation, which describes the dependence of the surface tension vs. surfactant concentration is dependent on the ionic state of the surfactant, described as the n parameter. Commonly accepted values of n for non-ionic single chain surfactant are 1 and 2 for cationic and anionic surfactants respectively. Regardless, it is still not obvious which value should be applied for gemini surfactants, either 2 or 3.40 It is even more complicated since neutron reflectometry showed that n is not constant and can vary between 2 and 3 with respect not only to the spacer length but also to the concentration, assuming the same tail lengths.39 We provide the summary of the obtained values of the CC, surface tension at CC (γCC), the surface excess Γ, molar area A, and free energy of adsorption of a single molecule ΔGads in Table S1.†
3.2 Electric conductivity
The dependence of the specific conductivity σ on the concentration for C12JC2–C12JC12 surfactants is shown in Fig. 3. The transition of the conductivity at the CC is not very sharp although the fitting procedure gave relatively good agreement of the CC with the results of the surface tension isotherms. The CC values decreased from 0.7 mM for C2 spacer down to 0.18 mM for C12. Similarly to the surface tension results, the differences between the values of the critical concentrations derived for C12JC8, C12JC10, and C12JC12 surfactants were small.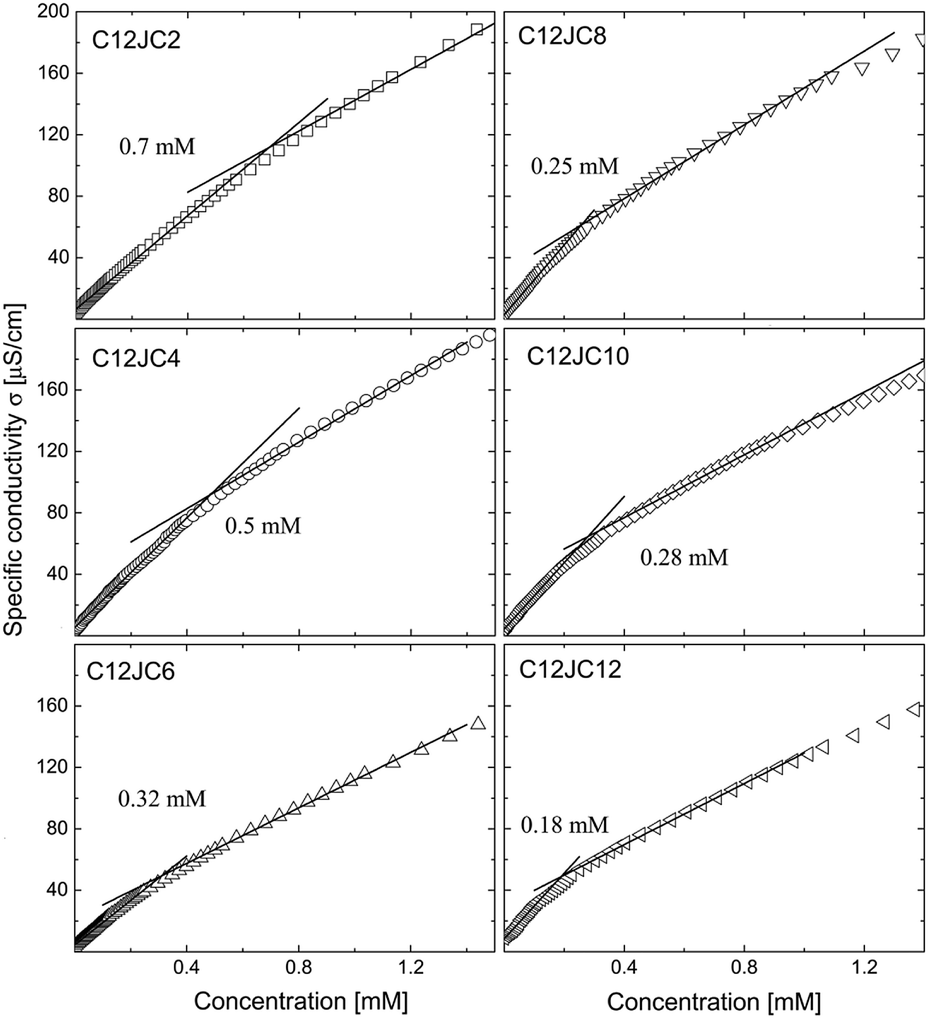 | ||
| Fig. 3 The dependence of specific conductivity σ obtained for C12JC2–C12JC12 gemini surfactants. The solid lines represent linear fits. | ||
3.3 Absorbance of BZA studied by UV-Vis
The dependence of the A310/A250 absorbance obtained for the C12JC2–C12JC12 gemini surfactants with BZA in the UV-Vis range is shown in Fig. 4. Additionally, we provide the exemplary UV-Vis absorption spectra of the bare C12JC2 and C12JC12 surfactants in Fig. S1.† The inset figures in Fig. 4 represent the dependence of the UV-Vis spectra in the 240–360 nm range obtained for each specific concentration. To determine the critical concentration CC (CAC or CMC) we utilize the fact that BZA can exist in ketonic and enolic tautomeric forms.41 While in the pure aqueous solutions BZA mainly exists in the ketonic form, the non-polar environment such as the hydrophobic interior of a micelle transforms BZA into the enolic form. Accordingly, the method is susceptible to the polarity of the solvent.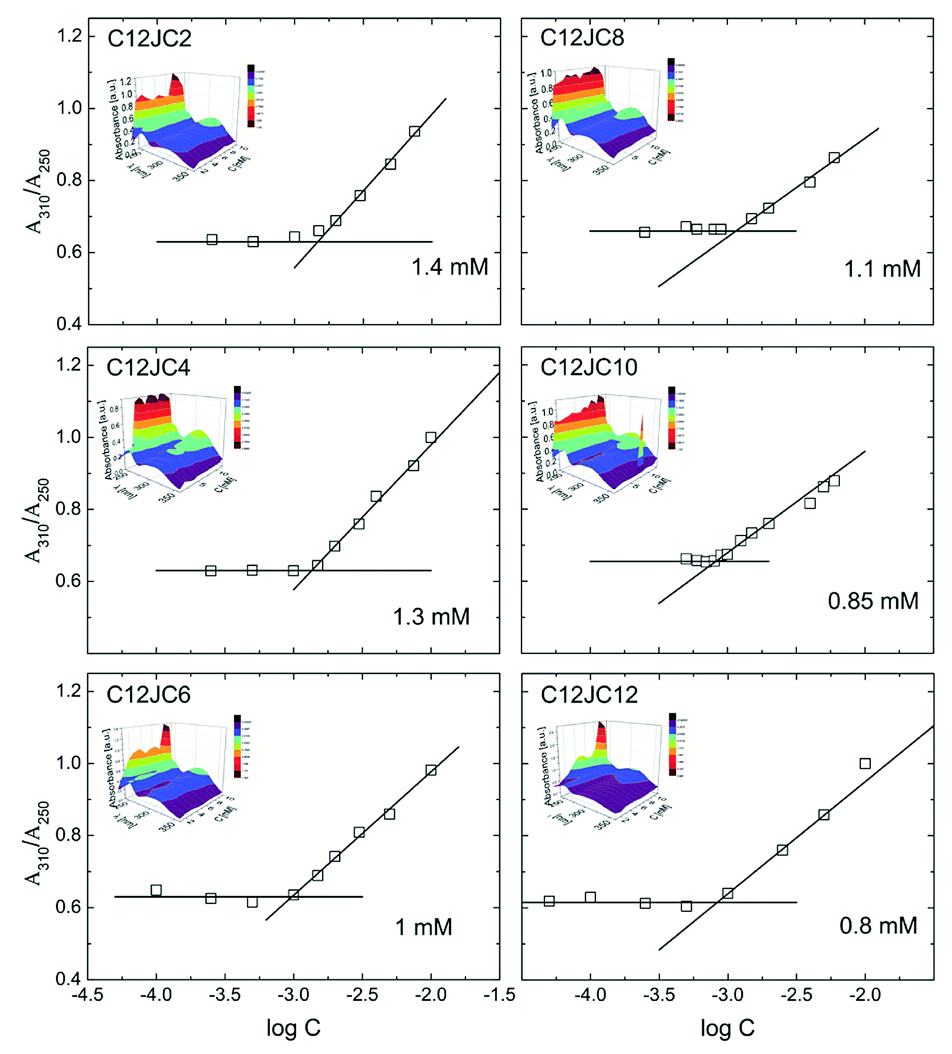 | ||
| Fig. 4 Absorbance of BZA tautomeric forms as a function of concentration and spacer length. | ||
The maximum absorption of the enolic form of BZA is observable at λ = 310 nm while the ketonic form is observable at λ = 250 nm. In our case, at high concentrations (above 10 mM) the absorption peak from the surfactant imidazole group is overlapped with the absorption band of the ketonic form (Fig. S1†). Therefore it was not possible to measure this ratio for a broader concentration range. The position of the imidazole peak is placed at around 213 nm, and it slightly changes with the spacer length albeit the maximum shift was around 1 nm between spacer length 2 and 12. Accordingly, we have assumed that this minor change is not affecting the A310/A250 ratio, at least at lowest concentrations.
The primary conclusion from the results of the BZA tautomerization reaction is that the obtained CC values are larger than those obtained from surface tension isotherms as well as electric conductivity. Subsequently, for C2 spacer the value of the CC is twice as large (1.4 mM) as one obtained from surface tension isotherm and electric conductivity (0.7 mM). This difference is even more significant for longer spacers C8–C12 where the values of CC are four times higher (ca. 0.8 mM instead of 0.2 mM). This difference may arise from the fact that the method involving BZA is not sensitive to small aggregates since the aggregates just above CC are still too small to mimic the non-polar environment and capture BZA molecules for a sufficiently long time to observe a difference. Once sufficiently large aggregates are developed, their interior efficiently mimic the non-polar solvent. Accordingly, the ratio between enolic and ketonic form will be varied what will be immediately detectable using the UV-Vis absorption spectra.
In the next part we provide diffusion NMR results in order to estimate the size of the aggregates.
3.4 Self-diffusion studied by PGSE NMR
We show the dependence of self-diffusion coefficients for a set of C12JC2–C12JC8 surfactants in the concentration range 0.05–20 mM in Fig. 5. The values of self-diffusion coefficients D were derived from the BDOSY displays in such a way that a single diffusion coefficient is resolved when the distribution of D is narrow. Accordingly, two limiting values of the diffusion coefficients are plotted at each concentration point to pronounce the distribution width. To clarify this approach, we show exemplary BDOSY spectra for C12JC6 sample in Fig. 6 in which the distribution of D is shown on the vertical axis. The selected 2D DOSY displays for three selected concentrations of all six surfactants are provided in the ESI Fig. S2.† Those three concentrations represent diffusion results below CC, just above CC and for the highest concentrations to show the distinctive differences between D distributions.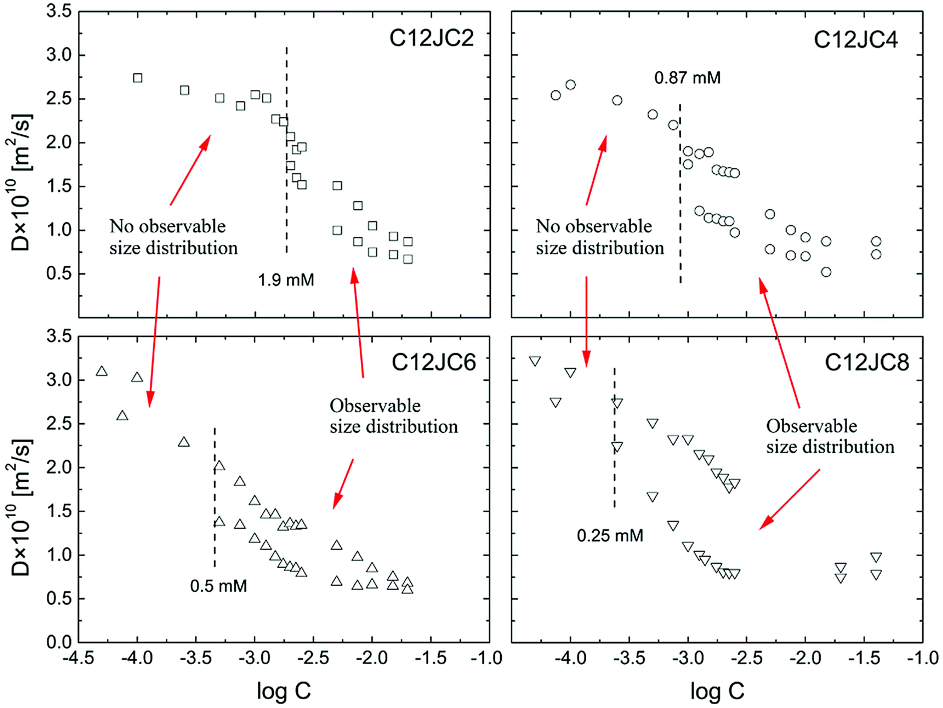 | ||
| Fig. 5 The concentration dependence of the self-diffusion coefficients D obtained from BDOSY 2D displays and distribution of diffusion coefficients for C12JC2–C12JC8 gemini surfactants in D2O. | ||
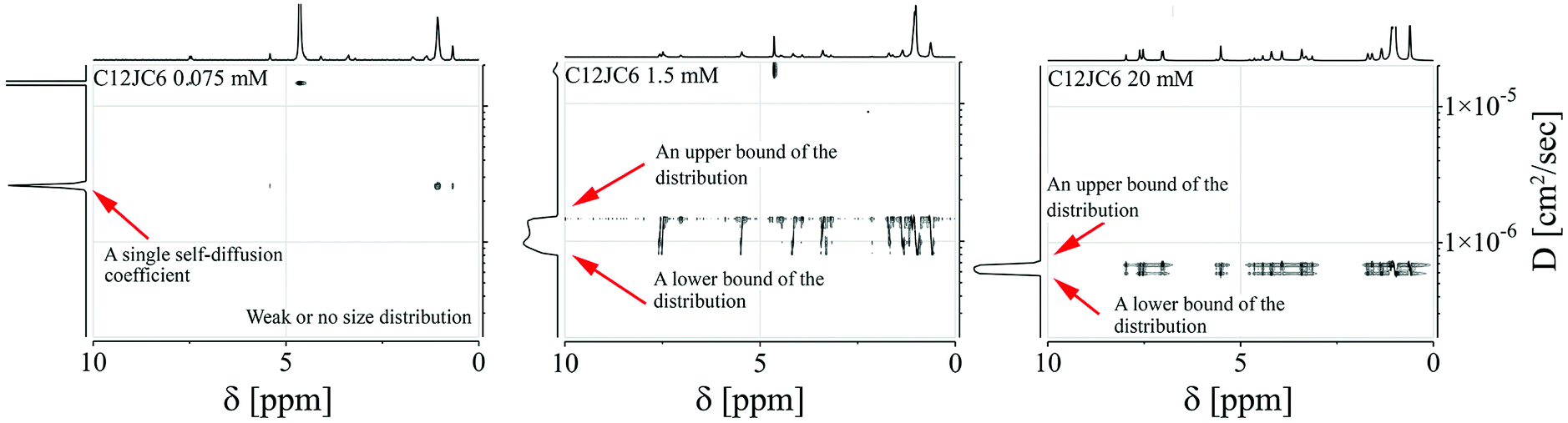 | ||
| Fig. 6 The selected BDOSY spectra for C12JC6. | ||
We found it difficult to measure the self-diffusion coefficients for C12JC10 and C12JC12 at the lowest concentrations due to low signal-to-noise ratio. Accordingly, we did not capture the transition region for C12JC10 and C12JC12 surfactants by diffusion NMR, therefore, we show only BDOSY plots for several concentrations in Fig. S2.† The critical concentrations were indicated in Fig. 5 as dashed lines where each line indicate the critical concentration. The CC value divides the dependence of the self-diffusion coefficient vs. the concentration into two distinctive areas. Simply speaking, the first region (below CC) is characterized by a single diffusion coefficient while the other one (above CC) demonstrates the distribution of diffusion coefficients. Those will be further presented and discussed below.
Nonetheless, we should expect that some mixed composition of surfactant molecules exists in the concentration region below CC.42 By mixed compositions we mean, monomers, dimers, trimers which undergo fast exchange between selected forms. This fast exchange effectively averages out the diffusion process in the NMR time-scale (from milliseconds to seconds). The existence of such mixed forms such as dimers, trimers and larger aggregates of gemini surfactants has been predicted theoretically by evaluation of Potential Mean Force (PMF) between two gemini molecules.42 The interaction between two monomers is both, energy (ΔU) and entropy driven (−TΔS) although it cannot be determined unambiguously which of the contributions prevails since it is highly dependent on the chemical composition.42 What is interesting the existence of dimeric forms was predicted even for monomeric surfactants.43
Still, in this regime, a single-exponential decay is observed in the FT-PGSE data what is shown in Fig. S3.† Additionally, we show in Fig. S4† a linear dependence of the normalized integral amplitude of CH3, lnA vs. g2 to show that indeed a single diffusion coefficient is resolved. Also Bayesian DOSY (BDOSY) analysis did not resolve any distribution of self-diffusion coefficients below CC.
What is interesting, the molecules with longer spacers seem to diffuse faster at lowest concentrations. The values of the self-diffusion coefficients for C6 and C8 spacers were around 3 × 10−10 m2 s−1. This finding, e.g., why surfactant molecules with longer spacers may diffuse faster, can be explained by employing Molecular Dynamics simulations. In the Fig. S5 (ESI)† we provide the rough analysis of the MD trajectory obtained for all the surfactants during 22 ns of MD simulation. At the beginning of the simulation (0–4 ns), the surface area per molecule is highest for the surfactants with longer spacers, but then after molecules find its minimum conformational energy, the longer spacer can enhance the flexibility and self-coiling, thus achieving smaller solvent-accessible areas than those with the shorter spacers. As a result, the Stokes drag force is weaker and results in the faster self-diffusion process.
A vs. g2, where A is an integral of the selected spectral line of the NMR spectrum and g in an amplitude of the gradient pulse in the FT-PGSE NMR experiment, becomes non-linear and multi-component. This is a typical behavior observed in the polydisperse systems.44,45 An example of the non-linear dependence for 2 mM C12JC2 sample is shown in Fig. S6 and 7.† Correspondingly, BDOSY analysis resolves multicomponent diffusion which herein we simply describe as the distribution of diffusion coefficients D. This observation either implies that the timescale is too short or we indeed observe a distribution of aggregates of various sizes. From the perspective of the inter-particle mean collision time, PGSE NMR experiment probes the process of self-diffusion in a vast time scale. The slow-exchange will be discussed later.Another interesting behavior upon an increase of the surfactant concentration is that the distribution width of the diffusion coefficients is changing. Namely, the difference between an upper and a lower bound of the diffusion coefficient is decreasing (Fig. 6). To emphasize this finding and to clarify this conclusion we provide an exemplary distribution of D for C12JC2 5 mM solution in Fig. S7† where Ddist is well resolved. Conversely, at higher concentrations the distribution is smaller since the system is more uniform and homogeneous. We provide additional data for the aforementioned finding in Fig. S8† (40 mM solution of C12JC4). All in all, the distribution of diffusion coefficients is much smaller at higher concentrations for all surfactants in the study which means that stable micelles are obtained at relatively high concentrations concerning initially determined CCs. This is an important finding which implications will be discussed in the further part.
It is also important to realize that at higher concentrations (around 40 mM and more), the self-diffusion coefficient might be affected by the non-spherical shape of a micelle. This will be addressed in the further part of the paper (see SR-SAXS data).
3.5 The diffusion transition curve. An impact of the obstruction factor and aggregation on the self-diffusion coefficients
At this stage we should discuss an impact of the obstruction factor on the self-diffusion coefficients.46 A simple collision-like interaction between micelles can develop the dependence of the diffusion coefficient vs. concentration. The model was initially developed for colloidal suspension of spherical PMMA latex particles. The change of the obstructing volume from 0% to 50% can decrease the diffusion coefficient by 30%, while the obstructing volume equal to 5% is practically negligible. In turn, we observe a three-fold decrease of self-diffusion coefficients which cannot be explained by the obstruction volume effect. A raw estimation of the volume fraction is following. The solvent accessible volume of a C12JC2 single molecule derived from Yasara software is around 1884 Å3 (1.88 × 10−27 m3). For 20 mM solution the total volume of the surfactant in 1 l of the solution is 2.27 × 10−5 m3. The volume fraction ϕ for 20 mM solution is around 2.3%. We estimate that at this volume fraction the self-diffusion coefficient might be decreased by around 1% which is less than the experimental error. Therefore the decrease of the diffusion coefficients, in this case, cannot be explained only by the excluded volume effect. We can also estimate an impact of the aggregation on the collision rate using the Smoluchowski equation kT: ki = ρ(2RikT/3ηR0), where the rate of collisions ki is the function of the interaction length Ri, hydrodynamic radius R0 of a micelle, viscosity η and thermal energy.47 The characteristic time between collisions τi is 1/ki. If we assume that the surfactant molecules undergo a simple ballistic type of motion, then the probability of collision is proportional to the concentration of molecules and the diffusion coefficient. Without aggregation, an increasing surfactant concentration will decrease the interaction length Ri and increase the collision rate ki but if we assume that at constant concentration, aggregates appear such as dimers, trimers, this in fact will decrease the collision rate as well as increase the hydrodynamic radius R0 and decrease the self-diffusion coefficient D. Accordingly, any aggregation will decrease the time between the collisions τi and an impact of the inter-aggregate interaction will be smaller. In other words, the premicellar aggregation may compensate an effect of an increased concentration in a wide concentration range so that the obstruction effect is compensated. This reasoning in one dimension is following. A simple dimerization will increase the interaction length by factor of 2. At the same time the hydrodynamic radius is increased from 1 (monomer) to 1.26 × R0 (dimer). Assuming that the hydrodynamic radius is much smaller than interaction length R0 ≪ Ri, Ri(n) = Rin and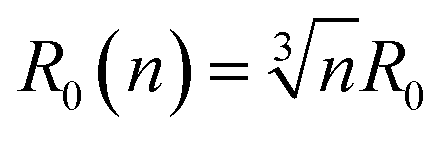 we can derive a simple correction factor for the collision time τi in one dimension vs. the number of molecules in the spherical aggregate n:
we can derive a simple correction factor for the collision time τi in one dimension vs. the number of molecules in the spherical aggregate n:
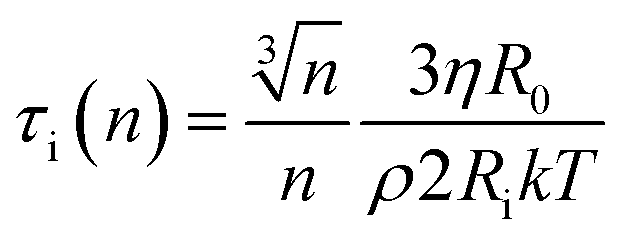 | (1) |
From this equation it is clear that aggregation decreases the collision rate in non-linear manner while the impact of the concentration 1/Ri is linear.
The decrease of the self-diffusion coefficient with an increasing concentration may arise due to interactions between micelles in such a way that mutual (Dynamic Light Scattering DLS) and self-diffusion coefficients are impaired. As a result, the hydrodynamic radii are under or overestimated due to intermicellar interactions. For example, Kato and Seimiya studied the C12E6 non-ionic surfactant between 25–170 mM solutions.47 A similar result was obtained a long time ago by Nilsson and Lindman, where in turn, the self-diffusion of water was affected by the excluded volume effects and obstruction factors.48 We did not observe any change in the self-diffusion coefficient of water. In our case, the concentrations at which we observe the decrease of the self-diffusion coefficients vs. concentration are much lower than those for monomeric surfactants and excluded volume effects play the minor role.
Samples C12JC2 and C12JC6 are characterized by the broad transition region which indicates that the initial size of the aggregates is rather small.17 At higher concentrations, around 20 mM, stable micelles are formed, with narrow size distribution. The transition region spans over 15 mM of the concentration range. The origin of the micellization transition curve and the existence of the distribution of the diffusion coefficients can be explained by the monomer-micelle equilibrium and the law of mass action.43 The following relation describes the equilibrium between cationic surfactants and micelles:
| nL+ + mX− ⇌ M(n − m)+, | (2) |
lnCMC), which describes the change of the Gibbs free energy change per monomer can be correctly used for such polydisperse systems.
Just to show the different behavior of traditional surfactants, we provide the results obtained for cetrimonium bromide (CTAB) and its analog gemini surfactant: 1,4-bis(N,N-dimethyl-N-tetradecylammonium)butane dibromide (Fig. S9 and S10,† to be published). For these types of surfactants, we did not observe the distribution of diffusion coefficients due to fast exchange between monomers and aggregates. The exemplary BDOSY spectra for 10 mM aqueous solution of 1,4-bis(N,N-dimethyl-N-tetradecylammonium)butane dibromide is shown in the ESI (Fig. S11, to be published).† While ammonium based surfactants show no signs of size distribution, the imidazole-based surfactants the exchange between monomers and aggregates is somewhat slow in the NMR timescale.
3.6 The slow-exchange and non-averaged dipole–dipole interactions above the critical concentrations
It is a well established fact that surfactant molecules undergo fast exchange between an aggregate and aqueous environment. This process should average out direct dipolar couplings between nuclear spins on the NMR spectra as well as self-diffusion coefficients in the NMR time-scale.28,46Whereas it is generally accepted that the fast exchange is typically observed in the surfactant systems, the slow exchange has not been reported yet. The first indication of a slow exchange is the multi-component self-diffusion process and most likely the size distribution of aggregates. The second indication of the slow-exchange comes from an analysis of the NMR spectra. In fact, we also observe the effects of non-averaged direct dipolar couplings between chains, linkers and imidazole groups. We believe that this deserves a separate discussion.
In the ESI (Fig. S12†) we show the dependence of the 1H NMR spectra vs. C12JC2 surfactant concentration where slow-exchange between an aggregate and aqueous environment results in an appearance of a new resonance lines visible at around 7.6–7.7 ppm (imidazole protons). Further investigation of the interactions between imidazole groups within the aggregates was studied using heteronuclear single quantum coherence spectroscopy (HSQC). The 1H–13C HSQC NMR spectra are shown in Fig. 7. When two aromatic rings are in the close vicinity, some resonance structures e.g., electron densities, are more favorable than others due to Π–Π stacking interactions. The overall charge of the imidazole ring is 1, but broken charge symmetry leads to alteration of 13C chemical shifts which can be observed for samples above CC, 20 mM C12JC2 (Fig. 7b), 2 mM C12JC10 (Fig. 7d) and 2 mM C12JC12 (Fig. 7e). The 13C chemical shift difference for C12JC2 was around 1.5 ppm, while for C12JC10 and C12JC12 surfactants this difference was more substantial (around 3 ppm) due to stronger attractive Π–Π interactions. The chemical shift difference means that internal packing and relative orientation of imidazole groups in aggregates are not the same for all lengths of the spacers and they are affected by charge density of the imidazole ring.
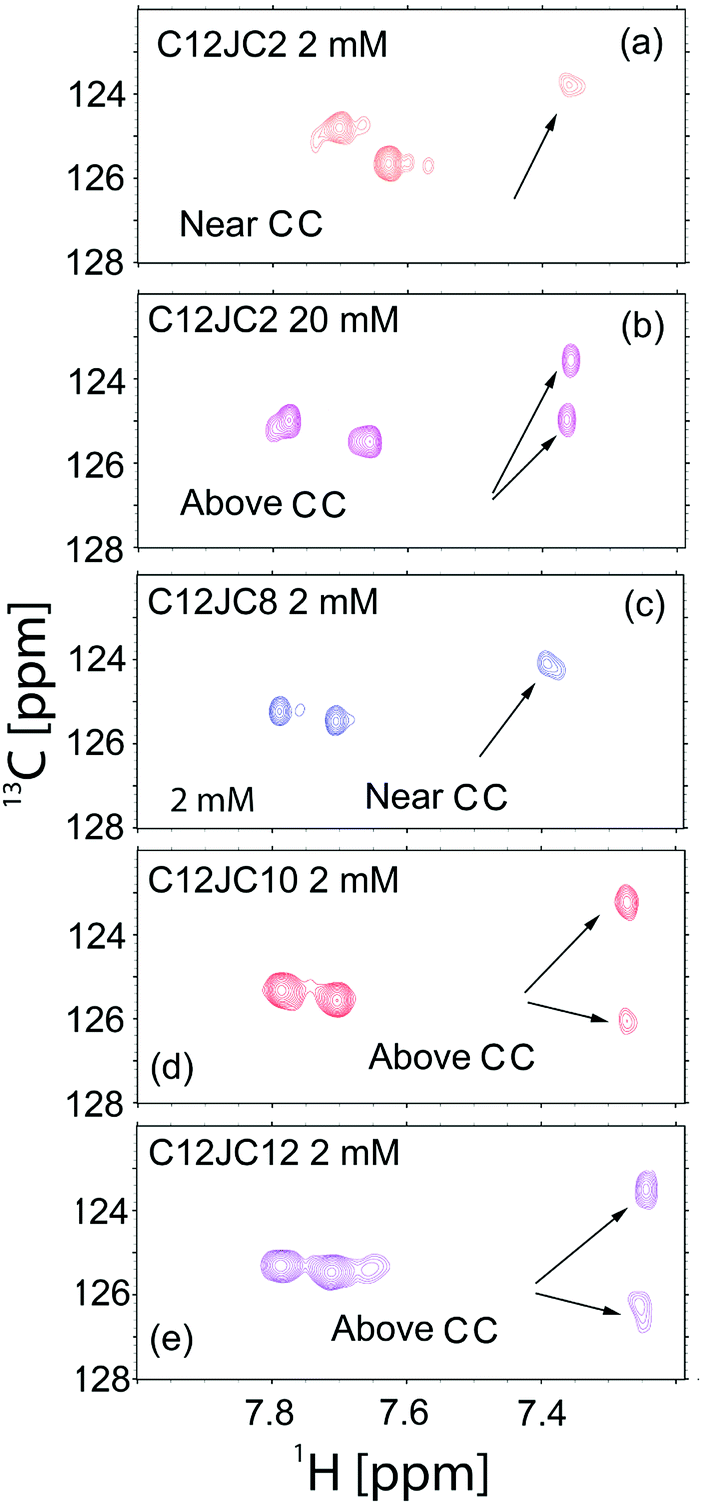 | ||
| Fig. 7 HSQC NMR spectra obtained for selected samples. (a) C12JC2 near CMC concentration (2 mM), (b) C12JC2 above CMC (20 mM), (c) C12JC8 above CMC (2 mM), (d) C12JC10 above CMC (2 mM), (e) C12JC12 above CMC (2 mM). | ||
Also 2D NOESY NMR spectra confirmed that NOE (Nuclear Overhauser Effect) cross-peaks appear at concentrations higher than CC (Fig. S13†). As expected NOE peaks did not develop at low concentrations around 1 mM. Instead, they develop at concentrations 2 mM and higher which confirms previous findings about the aggregation, especially those from BZA absorption studied by UV-Vis. Similar findings are obtained by means of the FTIR spectroscopy. The results for all surfactants in the study for 20 mM solutions are provided in the ESI (Fig. S14).† The frequency shift (Fig. S14a–c†) vs. the spacer length is observed which indicates a higher level of the order in the structures formed by surfactants, especially by those with the longer spacers. Other authors have shown previously that spacers longer than 12 methylene groups incorporate into the hydrophobic micellar core formed by side chains.49 Therefore dependence obtained for asymmetric stretching vibrations (Fig. S14c†) may indicate that linker is in the proximity of alkyl chains. As a result, the CH2 groups from the spacer are closer to each other. Such situation would allow minimizing the hydrophobic surface of the aggregates and a higher level of the organization due to the more contracted linker as self-assembly is driven by hydrophobic, electrostatic and van der Waals interactions.
3.7 The estimation of aggregation numbers Na
Once we obtain the values of self-diffusion coefficients it is interesting to estimate the size of the aggregates. In this case we can use a well known Einstein–Stokes–Sutherland–Smoluchowski relation (see experimental section). The results of the estimated aggregation numbers are shown in Fig. 8.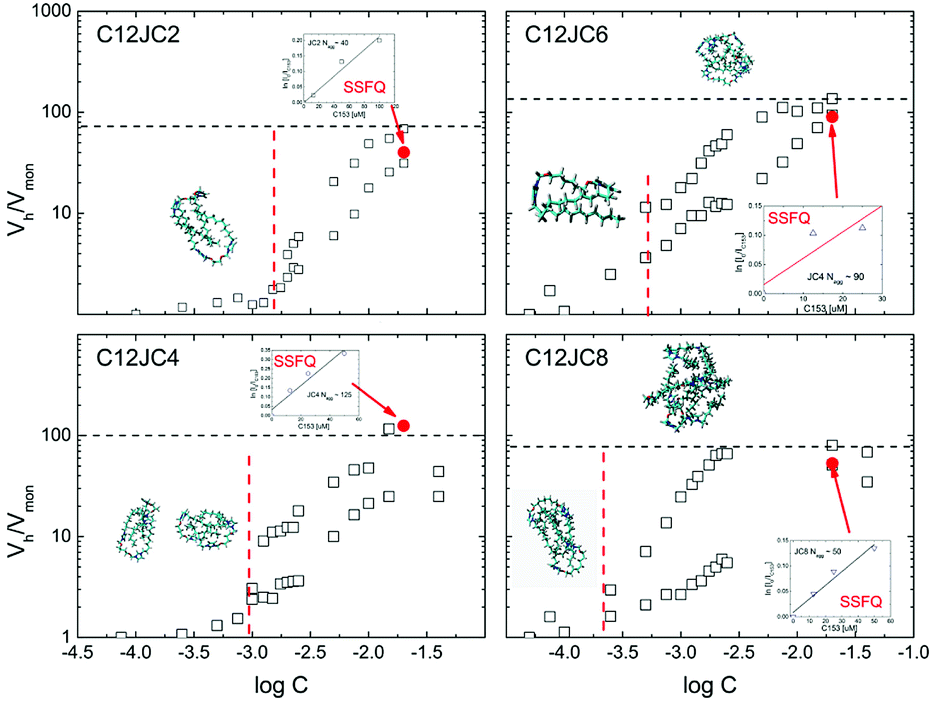 | ||
| Fig. 8 The estimation of aggregation numbers Nagg based on the ratio of hydrodynamic volumes Vh/Vmon for C12JC2–C12JC8 surfactants. The points indicated by arrows represent the aggregation numbers Nagg obtained independently from the Steady State Fluorescence Quenching (SSFQ) (inset figures). | ||
As it has been shown in the experimental section, the ratio of hydrodynamic volumes Vh/Vmon is only a rough estimation of the aggregation number. Despite the obvious simplification such as a spherical shape of the aggregate, it is proven that this approach may provide qualitative information about the molecular size.50
The points indicated by the arrows denote the aggregation numbers obtained from the conventional pyrene quenching SSFQ experiment. Raw SSFQ data are presented in the ESI Fig. S15.† The aggregation numbers predicted from NMR are, surprisingly, in a good agreement with SSFQ at least at given concentration regime (Table S2†).
The information about the shape of aggregates can be determined from the SR-SAXS. The results can give us a substantial and independent confirmation of the shape of the aggregates. For this purpose we have analyzed samples with two concentrations 15.2 and 42 mM. The scattering curves are shown in Fig. 9a and b. A shift towards lower s values is a result of increasing aggregation numbers for surfactants with longer spacer lengths. For these curves, we observe a single, symmetric and broad (within s-range from 0.5 to 3 nm−1) diffraction peak of the maximum in s = 1.61 nm−1 (for C12JC2). However, with the increasing length of the spacer, an additional narrower peak at s = 0.7 nm−1 is appearing in the scattering curve. This peak is shifted with an increasing length of the spacer group towards lower s-values (from s = 0.73 nm−1 for C12JC6 to s = 0.62 nm−1 for C12JC12). The SAXS data confirm that spherical micelles are developed at 15 mM concentration albeit with some size distribution for surfactants with short spacers. We attempted to fit the theoretical micelle model using SASfit package. The only model that correctly fit the scattering curves was double layer (vesicle) mode with size distribution (data not shown). Even though this result seems to confirm the spherical shape of the aggregates, this part of our work needs further investigation. For very high concentrations of ca. 42 mM, the deviations from the spherical shape appear even for spacer lengths above C4. These deviations of the symmetry of the scattering peak indicates the formation of the micelles of elongated shape and also formation of larger aggregates coexisting with micelles.
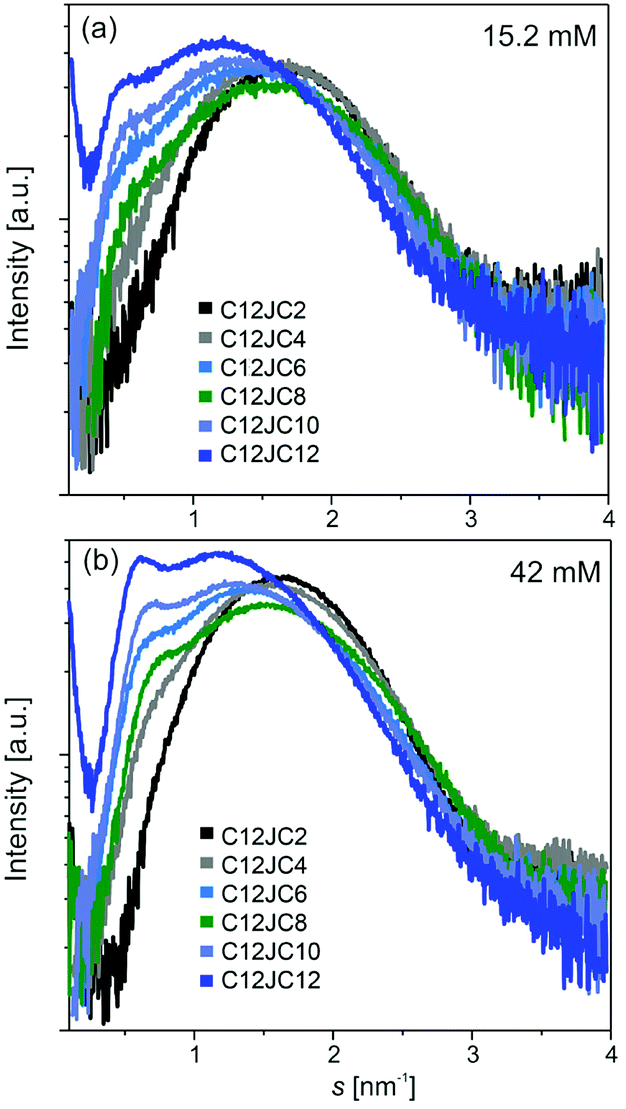 | ||
| Fig. 9 Small-angle scattering of synchrotron radiation (SR-SAXS) curves obtained for two concentrations of C12JC2–C12JC12 gemini surfactants. | ||
In order to extend our analysis, the size and shape of small aggregates was predicted using Molecular Dynamics simulations. From the 40 ns long MD simulation we have chosen exemplary aggregate configurations. These are shown in Fig. 10. The most stable configuration is the tail-to-tail orientation of two surfactant molecules. Higher order aggregates are less stable albeit clearly observable. Similar result was recently obtained by coarse-grained simulations where small gemini aggregates appear in the very short time regime.19 On the course of the simulation all types of aggregates appear such as monomers (1×), dimers (2×) as well as higher order aggregates. We were unable to obtain micelles using traditional MD simulations. Those would require coarse-grained approach and simulation times reaching 1 μs such as one showed by Want et al.19
 | ||
| Fig. 10 The selected aggregates captured from the MD simulations. The number of molecules in the aggregates are depicted as 1× for monomers, 2× for dimers, 3× for trimers, 4× for tetramers and 5× for pentamers. | ||
3.8 The comparison of critical concentrations obtained by various methods. The implications of confusing CMC with CAC
The obtained CC values are summarized in the Table 2. The CC values are comparable in overall, albeit, the differences between various techniques are noticeable. It seems that most accurate techniques are based on the Surface Tension (ST) and the electric conductivity. While these two methods are the industry standards and the most widely spread techniques, they are not sensitive to the aggregation in such a way that no distinction between CMC and CAC can be provided. The observed discrepancies indicate that the CC values obtained from surface tension and electric conductivity, are in fact the critical aggregation concentrations CACs. As concerning the spectrophotometric UV-Vis method, we measure the critical concentration at the point at which aggregates are large enough to trap the BZA molecules effectively. In other words. In the case of the studied surfactants, we cannot deliberately say that either CMC or CAC is measured using UV-Vis. It is neither CAC, nor CMC concentration.| CC [mM] | C12JC2 | C12JC4 | C12JC6 | C12JC8 | C12JC10 | C12JC12 |
|---|---|---|---|---|---|---|
| Surface tension | 0.71 | 0.62 | 0.47 | 0.26 | 0.24 | 0.21 |
| Electric conductivity | 0.7 | 0.5 | 0.32 | 0.25 | 0.28 | 0.18 |
| Self-diffusion (NMR) | 1.2–1.9 | 0.87 | 0.5 | 0.25 | — | — |
| Spectrophotometry | 1.4 | 1.3 | 1 | 1.1 | 0.85 | 0.8 |
There are several major implications from this type of confusion. The most obvious one is related to the thermodynamic parameters directly calculated from the CMC such as Gibbs free energy change per monomer ΔG. If CMC is confused with CAC the obtained ΔG values are most likely meaningless. This is not the first indication that ST data may provide some discrepancies, same holds for surface excess values which have to be correlated with other experimental methods.51
The second implication is related to the applications of gemini surfactants where the researcher assumes that traditional micelles exist. Most likely they don't exist near claimed CMC value derived upon a single technique (STI or conductivity). This is extremely important in many fields, such as new detergents, cosmetics and pharmaceutical product development. For these applications, often mixed systems of polymers and surfactants are used, and the CAC value determines the point at which stable blends are formed.52,53 Therefore, establishing new methods to reliably evaluate the CAC and CMC values is necessary for synthesizing novel series of improved amphiphiles and their characterization.52,54,55
Moreover, studding the aggregation process is essential not only in case of surfactants; but also polymers,56 proteins and peptides.57 CAC values are also meaningful in some specific applications like in case of amphiphilic molecules for membrane protein stabilization at high dilutions.58 Also, it was proven that above CAC the viscosity of surfactant solution decreases significantly,53 which could probably inhibit the unfavourable process of protein aggregation called amyloidogenesis, which leads to neurodegenerative disorders.59
Additionally, determination of CAC and CMC values for gemini surfactants is essential for optimization of their usage as delivery systems for nucleic acid in gene therapy.60,61 They are extremely efficient at condensing DNA and RNA molecules and therefore can be used at low concentrations, what reduces the adverse cytotoxicity effects. Those properties are attributed to low CMC of gemini surfactants. However, some reports suggest that surfactants can interact and condensed DNA at concentration much lower than CMC values, but higher than CAC.62 Above the CAC, polar heads of surfactant molecules associate with the phosphate groups of DNA,62,63 whereas above CMC entire micelles are interacting with DNA forming larger complexes.63 Consequently, precise determination of both CAC and CMC values is crucial for application of surfactants as vehicles in gene therapy, as it allows to maximize their efficiency while maintaining the cytotoxicity at lowest level possible.
We have also studied some standard systems such as well known CTAB surfactant. In that case NMR diffusion can clearly resolve CMC at around 1 mM. Why this is not CAC in this case? The concentration dependence of NMR spectra does reveal a characteristics of a fast exchange process (data not shown). Interestingly NMR diffusion data indicate that system consist of a stable micelles at around 3 mM (Fig. S9†). At the same time ST method is not sensitive due to fully occupied water–air interface.51
4 Conclusions
The obtained CC values correspond to the critical aggregation concentrations CACs. The CAC values obtained from the surface tension isotherms follow the linear decrease up to the C8 spacer. It seems that further increase of the spacer length is not followed by a decrease of the CAC values. Interestingly the surface activity at the lowest concentrations is decreased for C12JC12 surfactant in comparison to the surfactants with shorter spacers.The UV-Vis results indicate that aggregates above CAC do not capture BZA molecules effectively, since they are too small to act as a hydrophobic environment. Above CMC the distribution of diffusion coefficients is considerably broader. The upper limit is related to unimer molecules, while the lower limit characterizes the size of the largest aggregates in the solution. The estimated aggregation numbers, obtained from the diffusion NMR, suggest that surface tension and electric conductivity methods are somewhat sensitive to the pre-micellization process where small aggregates appear. The dependence of the aggregation upon increased surfactant concentration seems to be semi-continuous above CAC, and no geometrical criteria are met to create a spherical micelle. The size of the aggregates appears to be controlled by the collision probability which is proportional to the concentration and diffusivity. The later one is inversely proportional to the size of the aggregates and the lower diffusivity have to be compensated by higher concentrations.
We draw several findings from the NMR diffusion results. The values of CAC are decreasing from around 2 mM for C12JC2 down to 0.25 mM for C12JC8. This general tendency is in agreement with the surface tension and electric conductivity results. More interesting is the comparison with UV-Vis result, for which CAC values are much higher than those obtained from the surface tension and electric conductivity. Most likely, just above the CAC concentration, the micelles are too small to accommodate the BZA molecules. The aggregation numbers obtained from the diffusion NMR seem to support the elevated CC values obtained from UV-Vis. Namely, the aggregates at concentrations slightly larger than CAC are still small. The general impact of the spacer length on the CAC concentrations is in agreement with surface tension isotherms and electric conductivity results where the CAC decreases with the spacer length. Most likely the criterion for the appearance of the distribution of diffusion coefficients is tangible but not sufficient. The more precise determination of CAC from diffusion NMR would require titration experiments at the concentrations near values of CAC derived from BDOSY experiments but below the concentration at which a distinctive distribution of diffusion coefficient appears.
So far we have studied only one family of gemini surfactants and for sure, the ΔG obtained using ST method are not accurate. It is even more complicated since no CMC value can be clearly determined due to continuous-like aggregation. It is important to realize that 1H spectra should be recorded too in order to estimate whether the exchange rate of the surfactant molecules between two environments: (1) an aggregate and (2) aqueous environment is slow in the NMR time-scale. The slow exchange will likely generate new peaks in the NMR spectrum. The existence of the slow exchange seem to be a necessary condition to determine whether diffusion NMR provide us with CAC or CMC value. In the case of the fast-exchange, one should apply a standard model for an averaged self-diffusion coefficient.
The accurate CMC values cannot be identified for the systems in the study. However we can indicate that the distribution of aggregate size becomes small and the system becomes homogeneous at concentration larger than 15–20 mM. Accordingly, if one plans this type of imidazole-based gemini surfactants for biological applications, great care must be taken concerning the aggregate size distribution.
Authors contributions
K. S. designed the concept of the work, wrote the manuscript, prepared the figures, analyzed all the data including all aspects of the NMR diffusion experiments as well as performed and analyzed MD simulations. Z. P. was responsible for sample preparation as well as FTIR, UV-Vis and electric conductivity experiments and critical revision of the manuscript. Z. K. performed UV-Vis and SSFQ experiments. I. Z. performed HSQC NMR experiments. AS synthesized gemini surfactants. K. M. performed surface tension experiments. M. K. performed and analysed SR-SAXS experiments and contributed to the critical revision of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research project has been financed by the funds from the National Science Centre (Poland) granted on the basis of decision no. DEC-2011/01/B/ST5/00846. K. S. thanks to the grant H2020-INFRAIA-2016–2017 under research grant EUSMI – European infrastructure for spectroscopy, scattering and imaging of soft matter, contract number GA731019, funded under H2020-EU.1.4.1.2.RIA. Z. K. acknowledges Grant Preludium – 2014/15/N/ST5/02003. K. S. also thanks to Prof. Peter Stilbs from the Royal Institute of Technology (KTH) in Stockholm for valuable discussion concerning the BDOSY NMR analysis and slow exchange phenomenon. We acknowledge the European Synchrotron Radiation Facility for provision of synchrotron radiation facilities and we would like to thank beamline staff for assistance in using beamline BM29.References
- F. Menger and C. Littau, J. Am. Chem. Soc., 1993, 115, 10083–10090 CrossRef CAS.
- G. Van Biesen and C. S. Bottaro, J. Chromatogr. A, 2007, 1157, 437–445 CrossRef CAS PubMed.
- G. Van Biesen and C. S. Bottaro, J. Chromatogr. A, 2008, 1180, 171–178 CrossRef CAS PubMed.
- M. Grzegorzek and K. Majewska-Nowak, Sep. Purif. Technol., 2018, 195, 1–11 CrossRef CAS.
- M. Moradi, Y. Yamini, J. Kakehmam, A. Esrafili and M. Ghambarian, J. Chromatogr. A, 2011, 1218, 3945–3951 CrossRef CAS PubMed.
- R. Xu, Z. Zhang, L. Wang, N. Yin and X. Zhan, Environ. Sci. Pollut. Res., 2018, 1–10 Search PubMed.
- K. Urum, T. Pekdemir and M. Çopur, J. Colloid Interface Sci., 2004, 276, 456–464 CrossRef CAS PubMed.
- M. Zou, J. Dong, G. Yang and X. Li, Phys. Chem. Chem. Phys., 2015, 17, 10265–10273 RSC.
- M. Murawska, M. Wiatr, P. Nowakowski, K. Szutkowski, A. Skrzypczak and M. Kozak, Radiat. Phys. Chem., 2013, 93, 160–167 CrossRef CAS.
- E. Amirthalingam, M. Rodrigues, L. Casal-Dujat, A. C. Calpena, D. B. Amabilino, D. Ramos-López and L. Pérez-García, J. Colloid Interface Sci., 2015, 437, 132–139 CrossRef CAS PubMed.
- R. Zana, Adv. Colloid Interface Sci., 2002, 97, 205–253 CrossRef CAS PubMed.
- R. Zana, J. Colloid Interface Sci., 2002, 248, 203–220 CrossRef CAS PubMed.
- E. Sikorska, D. Wyrzykowski, K. Szutkowski, K. Greber, E. A. Lubecka and I. Zhukov, J. Therm. Anal. Calorim., 2016, 123, 511–523 CrossRef CAS.
- M. Dobies, J. Izykowska, M. Wilkowska, A. Wozniak-Braszak, K. Szutkowski, A. Skrzypczak, S. Jurga and M. Kozak, J. Phys. Chem. C, 2017, 121, 11839–11850 CrossRef CAS.
- Z. Pietralik, J. R. Kumita, C. M. Dobson and M. Kozak, Colloids Surf., B, 2015, 131, 83–92 CrossRef CAS PubMed.
- S. Javadian and J. Kakemam, J. Mol. Liq., 2017, 242, 115–128 CrossRef CAS.
- F. M. Menger and J. S. Keiper, Angew. Chem., Int. Ed., 2000, 39, 1906–1920 CrossRef PubMed.
- F. Reiss-Husson and V. Luzzati, J. Phys. Chem., 1964, 68, 3504–3511 CrossRef CAS.
- P. Wang, S. Pei, M. Wang, Y. Yan, X. Sun and J. Zhang, Phys. Chem. Chem. Phys., 2017, 19, 4462–4468 RSC.
- X. K. Jiang, W. Q. Fan and Y. Z. Hui, J. Am. Chem. Soc., 1984, 106, 7202–7205 CrossRef CAS.
- M. Sikirić, I. Šmit, L. Tušek-Božić, V. Tomašić, I. Pucić, I. Primožic and N. Filipović-Vinceković, Langmuir, 2003, 19, 10044–10053 CrossRef.
- M. J. Vold, Langmuir, 1992, 8, 1082–1085 CrossRef CAS.
- K. Maiti, P. K. Sen and B. Pal, J. Mol. Liq., 2018, 251, 238–248 CrossRef CAS.
- M. Popova and D. Raev, Appl. Magn. Reson., 2018, 49, 619–630 CrossRef CAS.
- A. R. Choudhury, E. Sikorska, J. van den Boom, P. Bayer, Ł. Popenda, K. Szutkowski, S. Jurga, M. Bonomi, A. Sali and I. Zhukov, et al., PLoS One, 2015, 10, e0135455 CrossRef PubMed.
- O. Söderman and P. Stilbs, Prog. Nucl. Magn. Reson. Spectrosc., 1994, 26, 445–482 CrossRef.
- O. Söderman, P. Stilbs and W. S. Price, Concepts Magn. Reson., Part A, 2004, 23, 121–135 CrossRef.
- P. Stilbs, J. Colloid Interface Sci., 1982, 87, 385–394 CrossRef CAS.
- W. Andrzejewska, Z. Pietralik, M. Taube, A. Skrzypczak and M. Kozak, Polimery, 2014, 59, 569–574 CrossRef CAS.
- D. B. Thiessen, D. J. Chione, C. B. McCreary and W. B. Krantz, J. Colloid Interface Sci., 1996, 177, 658–665 CrossRef CAS.
- N. J. Turro and A. Yekta, J. Am. Chem. Soc., 1978, 100, 5951–5952 CrossRef CAS.
- M. Nilsson, A. M. Gil, I. Delgadillo and G. A. Morris, Anal. Chem., 2004, 76, 5418–5422 CrossRef CAS PubMed.
- D. Šaman and E. Kolehmainen, Magn. Reson. Chem., 2015, 53, 256–260 CrossRef PubMed.
- D. Marion, M. Ikura, R. Tschudin and A. Bax, J. Magn. Reson., 1989, 85, 393–399 CAS.
- F. Delaglio, S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer and A. Bax, J. Biomol. NMR, 1995, 6, 277–293 CrossRef CAS PubMed.
- P. Pernot, A. Round, R. Barrett, A. De Maria Antolinos, A. Gobbo, E. Gordon, J. Huet, J. Kieffer, M. Lentini, M. Mattenet, C. Morawe, C. Mueller-Dieckmann, S. Ohlsson, W. Schmid, J. Surr, P. Theveneau, L. Zerrad and S. McSweeney, J. Synchrotron Radiat., 2013, 20, 660–664 CrossRef CAS PubMed.
- T. Huang, H. Toraya, T. Blanton and Y. Wu, J. Appl. Crystallogr., 1993, 26, 180–184 CrossRef CAS.
- M. V. Petoukhov, D. Franke, A. V. Shkumatov, G. Tria, A. G. Kikhney, M. Gajda, C. Gorba, H. D. Mertens, P. V. Konarev and D. I. Svergun, J. Appl. Crystallogr., 2012, 45, 342–350 CrossRef CAS PubMed.
- Z. Li, C. Dong and R. Thomas, Langmuir, 1999, 15, 4392–4396 CrossRef CAS.
- C. V. Nguyen, T. V. Nguyen and C. M. Phan, Colloids Surf., A, 2015, 482, 365–370 CrossRef CAS.
- A. Domínguez, A. Fernández, N. González, E. Iglesias and L. Montenegro, J. Chem. Educ., 1997, 74, 1227 CrossRef.
- K.-J. Jeong and A. Yethiraj, J. Phys. Chem. B, 2018, 122(13), 3259–3265 CrossRef CAS PubMed.
- P. Mukerjee, Adv. Colloid Interface Sci., 1967, 1, 242–275 CrossRef.
- M. Röding, S. J. Bradley, N. H. Williamson, M. R. Dewi, T. Nann and M. Nydén, PLoS One, 2016, 11, e0155718 CrossRef PubMed.
- N. H. Williamson, M. Nydén and M. Röding, J. Magn. Reson., 2016, 267, 54–62 CrossRef CAS PubMed.
- B. Jönsson, H. Wennerström, P. Nilsson and P. Linse, Colloid Polym. Sci., 1986, 264, 77–88 CrossRef.
- T. Kato and T. Seimiya, J. Phys. Chem., 1986, 90, 3159–3167 CrossRef CAS.
- P. G. Nilsson and B. Lindman, J. Phys. Chem., 1984, 88, 4764–4769 CrossRef CAS.
- I. Badea, R. Verrall, M. Baca-Estrada, S. Tikoo, A. Rosenberg, P. Kumar and M. Foldvari, J. Gene Med., 2005, 7, 1200–1214 CrossRef CAS PubMed.
- N. Kuzmina, S. Moiseev, V. Krylov, V. Yashkir and V. Merkulov, J. Anal. Chem., 2014, 69, 953–959 CrossRef CAS.
- P. X. Li, R. K. Thomas and J. Penfold, Langmuir, 2014, 30, 6739–6747 CrossRef CAS PubMed.
- K. C. Tam and E. Wyn-Jones, Chem. Soc. Rev., 2006, 35, 693–709 RSC.
- S. Guillot, M. Delsanti, S. Désert and D. Langevin, Langmuir, 2003, 19, 230–237 CrossRef CAS.
- V. A. Burilov, G. A. Fatikhova, M. N. Dokuchaeva, R. I. Nugmanov, D. A. Mironova, P. V. Dorovatovskii, V. N. Khrustalev, S. E. Solovieva and I. S. Antipin, Beilstein J. Org. Chem., 2018, 14, 1980 CrossRef CAS PubMed.
- W. Loh, C. Brinatti and K. C. Tam, Biochim. Biophys. Acta, Gen. Subj., 2016, 1860, 999–1016 CrossRef CAS PubMed.
- F. Aydin, X. Chu, G. Uppaladadium, D. Devore, R. Goyal, N. S. Murthy, Z. Zhang, J. Kohn and M. Dutt, J. Phys. Chem. B, 2016, 120, 3666–3676 CrossRef CAS PubMed.
- M. Novo, S. Freire and W. Al-Soufi, Sci. Rep., 2018, 8, 1783 CrossRef PubMed.
- M. Serra-Batiste, J. Tolchard, F. Giusti, M. Zoonens and N. Carulla, Front. Mol. Biosci., 2018, 5, 38 CrossRef PubMed.
- W. Gospodarczyk and M. Kozak, RSC Adv., 2017, 7, 10973–10984 RSC.
- T. Ahmed, A. O. Kamel and S. D. Wettig, Nanomedicine, 2016, 11, 289–306 CrossRef CAS PubMed.
- Z. Pietralik, A. Skrzypczak and M. Kozak, ChemPhysChem, 2016, 17, 2424–2433 CrossRef CAS PubMed.
- S. Husale, W. Grange, M. Karle, S. Bürgi and M. Hegner, Nucleic Acids Res., 2008, 36, 1443–1449 CrossRef CAS PubMed.
- P. Yadava, D. Buethe and J. A. Hughes, ACS Symp. Ser., 2006, 198–216 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07081d |
| This journal is © The Royal Society of Chemistry 2018 |