
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMorphology-dependent pH-responsive release of hydrophilic payloads using biodegradable nanocarriers†
Sumit Kumar Pramanik‡
 ab,
Senne Seneca‡ab,
Martijn Petersab,
Lien D'Olieslaegerab,
Gunter Reekmansab,
Dirk Vanderzandeab,
Peter Adriaensensab and
Anitha Ethirajan*ab
ab,
Senne Seneca‡ab,
Martijn Petersab,
Lien D'Olieslaegerab,
Gunter Reekmansab,
Dirk Vanderzandeab,
Peter Adriaensensab and
Anitha Ethirajan*ab
aInstitute for Materials Research (IMO), Hasselt University, Wetenschapspark 1 and Agoralaan D, 3590 Diepenbeek, Belgium. E-mail: anitha.ethirajan@uhasselt.be
bIMEC, Associated Lab IMOMEC, Wetenschapspark 1, 3590 Diepenbeek, Belgium
First published on 31st October 2018
Abstract
The development of functional nanocarriers with stimuli-responsive properties has advanced tremendously to serve biomedical applications such as drug delivery and regenerative medicine. However, the development of biodegradable nanocarriers that can be loaded with hydrophilic compounds and ensure its controlled release in response to changes in the surrounding environment still remains very challenging. Herein, we achieved such demands via the preparation of aqueous core nanocapsules using a base-catalyzed interfacial reaction employing a diisocyanate monomer and functional monomers/polymers containing thiol and hydroxyl functionalities at the droplet interface. pH-responsive poly(thiourethane–urethane) nanocarriers with ester linkages were synthesized by incorporating polycaprolactone diol, which is susceptible to hydrolytic degradation via ester linkages, as a functional monomer in the reaction formulation. We could demonstrate that by systematically varying the number of biodegradable segments, the morphology of the nanocarriers can be tuned without imparting the efficient encapsulation of hydrophilic payload (>85% encapsulation efficiency) and its transfer from organic to aqueous phase. The developed nanocarriers allow for a fast release of hydrophilic payload that depends on pH, the number of biodegradable segments and nanocarrier morphology. Succinctly put, this study provides important information to develop pH-responsive nanocarriers with tunable morphology, using interfacial reactions in the inverse miniemulsion process, by controlling the number of degradable segments to adjust the release profile depending on the type of application envisaged.
1. Introduction
Polymeric nanocarriers, such as nanoparticles and nanocapsules, have enormous potential within various domains of nanomedicine (e.g. drug delivery, imaging, and sensing).1–3 On the one hand, they offer unlimited possibilities for the encapsulation of several useful compounds, ranging from hydrophobic and/or hydrophilic drug molecules to imaging and contrast agents.4,5 On the other hand, the functional groups on the nanocarrier surface allow for incorporating molecules to impart specific targeting functionalities and to prevent unspecific uptake by cells of the immune system (such as macrophages) by imparting the stealth effect.3,6,7 To employ nanocarriers as advanced drug delivery vehicles, the former should be biocompatible, protect its payload from the external environment (in vivo degradation), possess target-specificity, and enable controlled release of the payload.8–11 Additionally, nanocarriers that can adapt to complex microenvironments and can respond dynamically in order to release their cargo are highly desired.12 Therefore, the design of nanocarriers with tailor-made functional properties is highly envisaged for the development of advanced drug delivery systems.Stimuli-responsive nanocarriers that respond to defined triggers (such as pH, temperature, enzymes, light and redox species) have gained enormous interest because they aid in “release on demand”.1,11,13 Since variations in pH exist within different physiological environments – different organs, tissues, and cellular compartments possess mostly different pH values, as well as differences between normal and abnormal (cancer, inflammation) pathological conditions – developing nanocarriers that respond to the pH of the local environment is highly attractive.14–16 For this purpose, pH-responsive nanocarriers exploiting a release profile stimulated by the intracellular acidic environment (pH ∼ 5.5–6.0 and pH ∼ 4.5–5.0 in endosomes and lysosomes, respectively) and suppressed in the systematic circulation at physiological pH of 7.4 are of special interest.15,17 In general, polymeric nanocarriers can be made pH-responsive by incorporating linkages that are prone to hydrolytic cleavage or by introducing ionizable groups (acidic/basic) that can cause conformational changes/swelling/deswelling or solubilization of the polymer chains.
Biodegradable polymers to develop drug delivery vehicles have been extensively investigated for several decades since they can be hydrolyzed or enzymatically cleaved to form products that are metabolized or excreted under physiological conditions. To this extent, biopolymers and their derivatives (e.g. gelatin, peptides, carbohydrates such as oligo- and polysaccharides and their modified derivatives) have been used to develop biocompatible nanocarriers.3,18,19 Furthermore, the synthesis of nanocarriers from biodegradable polymers (for instance the saturated polyesters poly(lactide), poly(lactide-co-glycolide) and poly(ε-caprolactone)) have been widely investigated owing to their biocompatibility and low toxicity in vivo.20–22 Previously, it has been investigated that their degradation rates depend on several parameters such as their chemical structure, water permeability (hydrophilicity/hydrophobicity), morphology and molecular weight.20,23–25 Among the aliphatic polyesters, poly(ε-caprolactone) (PCL) – a hydrophobic and semi-crystalline polymer that has been widely used in biomedical applications – has successfully demonstrated a slow release of drugs at tumor sites.26,27 Despite the slow degradation and resorption rates of PCL,28 PCL copolymers and modified derivatives have shown improved degradation properties.29,30
Polymeric nanocarriers can be synthesized by a wide variety of methods, among which nanoprecipitation, coacervation, layer-by-layer assembly and (micro-, mini-) emulsion polymerization techniques.31–34 However, the high encapsulation efficiency of both hydrophobic and hydrophilic substances employing different polymeric matrices and its versatility (it allows for executing different polymerization reactions) renders the miniemulsion process a preferred synthesis method.34,35 Compared to hydrophobic substances, encapsulating hydrophilic substances has been shown to be very challenging to establish applications in an aqueous environment. This can be attributed mainly to the specific chemical structure of biodegradable aliphatic polyesters, which makes it challenging to encapsulate hydrophilic substances using oil-in-water emulsions.36 However, alternative approaches, such as double emulsions (w/o/w), to encapsulate hydrophilic substances have been explored in the past. The readers are referred to a review for an overview of other approaches.37 In here, the inverse miniemulsion approach offers an advantage by performing the encapsulation of hydrophilic cargo initially in an organic solvent after which the nanocarriers are transferred into an aqueous phase.35,38 Previously, successful encapsulation of hydrophilic substances has been shown by using nanoprecipitation processes or interfacial reactions such as step growth, radical or anionic polymerization in the inverse miniemulsion process.39–41
Recently, we reported on a versatile approach that allowed for the efficient encapsulation of hydrophilic payloads via an in situ inverse miniemulsion polymerization employing thiol–isocyanate reactions at the droplet interface.42 Nanocarriers with thiourethane functionality were successfully formulated at room temperature using a base catalyst and by employing different stoichiometric ratios of 1,4-butanedithiol (BDT) and toluene diisocyanate (TDI) as bifunctional monomers. Additionally, pentaerythritol tetra-3-mercaptopropionate (PETMP), a multifunctional crosslinker monomer, was used to impart potential post-surface grafting possibilities to the nanocarriers. In addition, the dispersions were easily transferable to the aqueous phase. In general, nanocapsule morphology allows for a large aqueous core to polymer ratio; hence encapsulation of larger amounts of cargo is more feasible compared to its bulk particle counterparts. Since the physicochemical property of the nanocarrier shell can be varied in order to impart interesting functionalities by the choice of precursor materials, herein we report on the synthesis of biodegradable poly(thiourethane–urethane) nanocarriers with ester linkages that can be used for controlled release of the encapsulated hydrophilic cargo in response to a pH stimulus. To achieve this objective, polycaprolactone diol (PCLD) is used as a functional starting material in the formulation along with monomer units containing thiol and isocyanate groups (Scheme 1). The incorporated PCLD segments in the shell render the polymeric nanocarrier susceptible to hydrolytic degradation via ester linkages. To our knowledge, we are the first one to document about the synthesis of functional thiourethane/urethane nanocapsules using PCLD as functional monomer. Here, it should be noted that PCL microspheres and films have shown large benefit to be used in slow degradation applications in the past.43–47 Our system is distinct as we are focusing on nanocarriers that possess a high surface to volume ratio as compared to larger (micro-)structures and possibly a different release profile. As the reaction is performed at room temperature, encapsulation of sensitive payloads, such as biomolecules and drugs, is feasible. The influence of the amount of biodegradable segments on the nanocarrier morphology as well as the release of the encapsulated materials was investigated. The time-dependent release of the payload as a function of pH was systematically investigated using the hydrophilic dye rhodamine B. As the nanocarriers are intended for drug delivery applications, the biocompatibility of the nanocarriers was also tested.
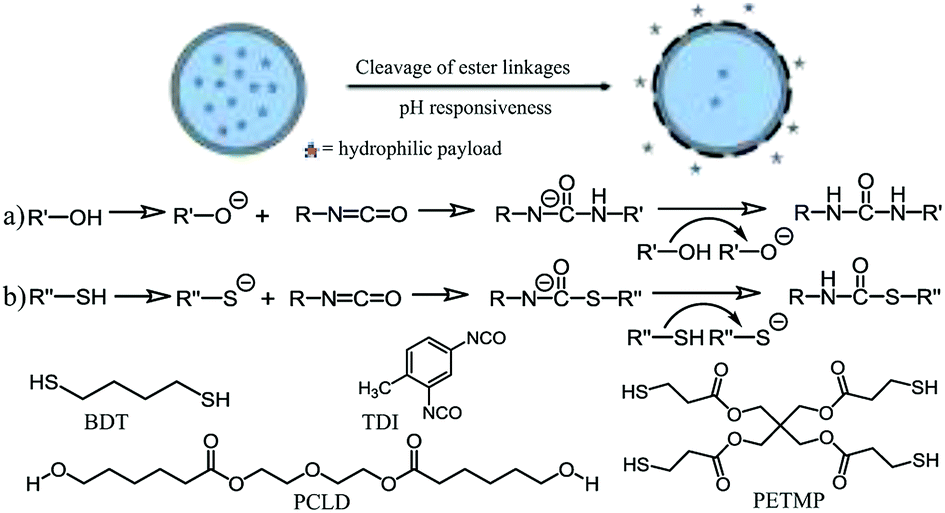 | ||
| Scheme 1 A schematic representation of the reaction mechanism between (a) hydroxyl and isocyanate groups, and (b) thiol and isocyanate groups. Both reactions are executed in the presence of a base catalyst. R′ = PCLD; R′′ = BDT or PETMP. | ||
2. Experimental
2.1 Materials
Chemicals and solvents were purchased from commercial suppliers. Cyclohexane (>99.5%), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (>99.0%), 1,4-butanedithiol (BDT) (>97%), pentaerythritol tetra-3-mercaptopropionate (PETMP), toluene diisocyanate (TDI) (95%), polycaprolactone diol (PCLD) (average molecular weight ∼ 530 g mol−1), rhodamine B (98%), triton X-100 and paraformaldehyde (PFA) were purchased from Sigma Aldrich. Potassium chloride (KCl), dimethyl sulfoxide (DMSO), penicillin/streptomycin (P/S) and sodium dodecyl sulphate (SDS) were obtained from Merck. The surfactant Hypermer B246 (a block copolymer containing polyhydroxystearic acid and poly(ethylene glycol))48,49 was kindly supplied by Croda Europe Ltd. Minimum essential medium (MEM, containing HEPES and GlutaMAX supplement) was obtained from Thermo Fisher Scientific. Iscove's Modified Dulbecco's Medium (IMDM), Dulbecco's Modified Eagle's medium (DMEM), fetal calf serum (FCS), mouse monoclonal anti-α-tubulin antibody, Alexa Fluor® 488 Donkey Anti-Mouse IgG (H + L) antibody and 4′,6-diamidino-2-phenylindole (DAPI) were bought at Life Technologies. The 1× phosphate buffered saline (PBS) was purchased at Lonza. Bovine serum albumin (BSA) was bought from US Biological and 15 μ-slide 8 wells were bought at Ibidi. The culture plates and dark plates were obtained at Greiner Bio One. HeLa cells (CCL-2) were purchased from ATCC. Water obtained from Sartorius Stedim Biotech machine was used during all experiments.2.2 Synthesis of biodegradable nanocarriers
The nanocarriers containing hydrophilic payload were prepared by an in situ reaction between different monomers at the droplet interface employing the inverse miniemulsion process. For all reactions, the continuous phase was made by heating 10 g of cyclohexane and 200 mg of Hypermer B246 at 60 °C until the surfactant was fully dissolved. An aliquot of 2.5 g was taken from the continuous phase for the additive phase. The dispersed phase contains various amounts of 1,4-butanedithiol and polycaprolactone diol; 0.35 g of 1 M KCl salt solution, 0.50 g DMSO and 2.0 mg of rhodamine B. The mixture containing the continuous phase was stirred at room temperature for 1 hour at 1000 rpm. Subsequently, it was sonicated using a Branson 450 W digital sonifier (1/4′′ tip) for 3 min (30 s pulse; 20 s pause) with an amplitude of 65%, while cooling the sample in an ice bath. An equimolar amount of TDI with respect to monomers 1,4-butanedithiol and polycaprolactone diol along with DBU (thiol/DBU molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100) was added to the additive phase. The additive phase was then dropwise introduced to the emulsion and left for stirring at room temperature for 4 hours. Finally, the emulsion was passed through a paper filter (Whatman, pore size 4–7 μm) to separate any bulk product formed from the particulate dispersion. The dispersions were then directly used for various characterizations. The redispersion of nanocapsules in the aqueous phase was performed by mixing 1.0 g of the synthesized nanocapsules/cyclohexane dispersion with 10 g of SDS water solution (0.1 wt% SDS) and stirred at 1000 rpm for 1 hour at room temperature. The resulting mixture was subjected to ultrasonication for 5 min in an ultrasonication bath at 35 kHz. The cyclohexane was evaporated afterward from an open container by stirring the mixture overnight at 1000 rpm at room temperature. Finally, the aqueous dispersion was passed through a filter paper to remove any bulk material/aggregates formed before further use in experiments. To remove excess surfactant, the redispersed samples were dialyzed multiple times using Millipore membrane tubes by centrifugation at 4 °C for 20 min at 2000 rpm. A schematic representation of the synthesis procedure has been included in the ESI (Scheme S1).†
:100) was added to the additive phase. The additive phase was then dropwise introduced to the emulsion and left for stirring at room temperature for 4 hours. Finally, the emulsion was passed through a paper filter (Whatman, pore size 4–7 μm) to separate any bulk product formed from the particulate dispersion. The dispersions were then directly used for various characterizations. The redispersion of nanocapsules in the aqueous phase was performed by mixing 1.0 g of the synthesized nanocapsules/cyclohexane dispersion with 10 g of SDS water solution (0.1 wt% SDS) and stirred at 1000 rpm for 1 hour at room temperature. The resulting mixture was subjected to ultrasonication for 5 min in an ultrasonication bath at 35 kHz. The cyclohexane was evaporated afterward from an open container by stirring the mixture overnight at 1000 rpm at room temperature. Finally, the aqueous dispersion was passed through a filter paper to remove any bulk material/aggregates formed before further use in experiments. To remove excess surfactant, the redispersed samples were dialyzed multiple times using Millipore membrane tubes by centrifugation at 4 °C for 20 min at 2000 rpm. A schematic representation of the synthesis procedure has been included in the ESI (Scheme S1).†
2.3 Determination of theoretical and experimental solid content
The experimental solid content of the dispersion was determined thermogravimetrically. To calculate the theoretical solid content, the weight of KCl solution and DMSO in the dispersed phase is taken into account and it was assumed that all monomers used in the reaction are fully converted.2.4 Dynamic light scattering (DLS)
The average size and size distribution of the nanocarriers were measured at 20 °C by DLS using a Brookhaven instruments Zetapals.2.5 Attenuated total reflectance Fourier transform infrared spectroscopy (ATR-IR)
Fourier-transform infrared (FT-IR) spectra are reported in wavenumbers (cm−1) and were recorded using a Bruker Tensor 27 Fourier transform IR spectrometer in transmission mode. The spectral region was analyzed from 4000 cm−1 to 600 cm−1.2.6 High-resolution solid-state nuclear magnetic resonance spectroscopy (NMR)
The solid-state carbon-13 CP/MAS NMR measurements were performed using methods similar to protocols described elsewhere.50,51 For the material composition analysis, solid-state carbon-13 CP/MAS NMR spectra were acquired on an Agilent VNMRS Direct Drive 400 MHz spectrometer (9.4 T wide bore magnet) equipped with a T3HX 3.2 mm probe dedicated for small sample volumes and high decoupling powers. Magic angle spinning (MAS) was performed at 12.7 kHz with ceramic rotors of 3.2 mm in diameter (34 μl rotors). Both probe (20 l min−1) and upper-barrel (30 l min−1) cooling were used to limit sample heating during the experiments. The aromatic signal of hexamethyl benzene was used to determine the Hartmann-Hahn condition (ω1H = γHB1H = γCB1C = ω1C) for cross-polarization (CP), and to calibrate the carbon chemical shift scale (132.1 ppm). Acquisition parameters used were the following: a spectral width of 50 kHz, a 90° pulse length of 2.5 μs, a spin-lock field for CP of 100 kHz, a contact time for CP of 1 ms, an acquisition time of 20 ms, a recycle delay time of 15 s and 1500–3000 accumulations. High power proton dipolar decoupling during the acquisition time was set to 100 kHz.2.7 Transmission electron microscopy (TEM)
Transmission electron microscopy (TEM) images of the nanocarrier dispersions were obtained using a TECNAI Spirit TEM from FEI, operating at an accelerating voltage of 120 kV to characterize the nanocarrier morphology. A drop of the nanocarrier aqueous dispersion (solid content of about 0.01 wt%) was drop-casted onto a carbon coated copper grid (formvar foil upon copper grids) and dried overnight under ambient conditions. No additional staining was employed.2.8 pH-dependent release kinetics
The pH-dependent release of the fluorescent dye (rhodamine B; λex = 554 nm and λem = 578 nm) from the nanocarriers as a function of time was studied using fluorescence spectroscopy. The release of dye was calculated with respect to the amount of dye encapsulated for all the samples. As the nanocarriers are insoluble, the amount of encapsulated dye of a sample was obtained by determining the unencapsulated dye amount using a previously reported procedure employing a reference sample.40 The latter was also used for determining the encapsulation efficiency of the samples. Briefly, for the reference sample, the dispersion was prepared identically using the same protocol as the rhodamine B containing respective nanocarrier dispersion except for the absence of the dye in the formulation. The obtained dispersion was redispersed in an aqueous SDS solution containing an equal amount of rhodamine B taken in the encapsulation experiment and then subjected to centrifugation. The amount of unencapsulated rhodamine B was assessed by measuring the difference in fluorescent intensities between the supernatants collected after centrifugation (6000 rpm, 30 min, 4 °C), from the reference and the respective samples. Triplicate measurements were performed, and an average was taken. The encapsulated amount is then calculated for each sample and then taken as 100% for the release studies for the respective samples.At physiological temperature (37 °C) the concentration of released rhodamine B from the capsules was determined with a fluorescence spectrometer after definite time intervals (up to 30 h) at different pH (4.0, 6.5 and 9.0). For this purpose, the redispersed samples were first adjusted to pH 6.5 and 4.0 with hydrochloric acid (0.1 M) or with sodium hydroxide (0.1 M) to pH 9.0, and then placed for incubation at a temperature of 37 °C. Here, different redispersed nanocarriers solutions were centrifuged (6000 rpm, 30 min, 4 °C), and the capsule pellet was separated from the supernatant. Then, the emission spectrum of the supernatant was measured at room temperature after dilution using deionized water (pH > 6 after dilution). The percentage of the released dye after a certain time period was calculated with respect to the respective encapsulation amount as mentioned above. Triplicate measurements were performed, and an average was taken.
2.9 Biocompatibility study using Alamar Blue assay
Alamar Blue assay measurements were performed using methods similar to a protocol described elsewhere.42,48 HeLa cells were grown in DMEM medium (supplemented with 10% FCS and 1% penicillin/streptomycin) to 80% confluency in a flat bottom 96-well plate at 37 °C and 5% CO2. After washing with 1× PBS, 100 μl of the desired concentration range (10, 50 and 100 μg mL−1) of nanocapsules in phenol red poor supplemented IMDM medium was added. Also, a positive (only supplemented IMDM medium) and negative control (100% cell death – 1:9.1 dilution in the supplemented IMDM medium of 1 g SDS in 10 mL ultrapure water solution) were included and a total of six wells was taken per condition. After 24 h incubation at 37 °C the medium was discarded and the cells were washed 3 times with 1× PBS. Next, 100 μl of a 10% Alamar Blue solution in supplemented IMDM medium was added and left to incubate for 24 h. The resulting solution was transferred to an opaque-walled dark 96-well culture plate after which the fluorescence was measured at λem = 590 nm, while being excited at λex = 570 nm, (FLUOstar OPTIMA). The experiment was executed in triplicate.
3. Results and discussion
3.1 Synthesis of nanocarriers
To establish a system for the efficient encapsulation and selective release of hydrophilic payload upon changes in the physiological environment, we designed and synthesized pH-responsive nanocapsules using PCLD as functional monomer within the formulation procedure. The well-defined aqueous core poly(thiourethane–urethane) nanocarriers with ester linkages were obtained via an interfacial polyaddition reaction of various molar ratios of BDT and PCLD to TDI using the inverse miniemulsion technique. In presence of a base catalyst (for this purpose DBU was used), strong nucleophilic thiolate and hydroxyl ions are generated that react in an equimolar ratio with the electron deficient carbonyl carbon of the isocyanate end group to form thiourethane and urethane linkages, respectively. In order to impart post-surface grafting possibilities to the nanocarriers, a multifunctional crosslinker monomer (a fixed amount of PETMP) was also used in all reactions. To ensure the stability of the obtained nanocarrier dispersion, in the dispersed phase a salt solution was used to build up an osmotic pressure of the droplets in the hydrophobic phase, as well as a fixed amount of oil-soluble block copolymer surfactant Hypermer B246 (2.0 wt% with respect to the dispersed phase). For all formulations used, the stability of the resulting dispersions was first checked optically for any visible phase separation after which the samples were directly used for further characterization. The colloidal stability, size, and polydispersity index (PDI) of the samples were studied using dynamic light scattering (DLS). The reaction efficiency was evaluated thermogravimetrically by determining the solid content of the obtained dispersion. Chemical analyses of the insoluble products were performed using attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) and high resolution solid-state 13C nuclear magnetic resonance (NMR).50,51 The characteristics of the synthesized nanocapsules, i.e. average size, size polydispersity (PDI) and solid content are summarized in Table 1. From DLS results, it was observed that most of the synthesized nanocapsules possess hydrodynamic diameters ranging between 160 and 215 nm in cyclohexane. From the values obtained, a direct relationship between the size and amount of PCLD used in the reaction can be observed up to a concentration of 1.2 mmol of PCLD; the size of the nanocapsules (sample 1–4) increases with increasing amounts of PCLD used for synthesis. Moreover, the PDI value gradually rises with increasing amount of PCLD. The larger size can possibly be attributed to the chemical structure of PCLD, which is a larger and flexible molecule compared to the other linear bifunctional monomers that were used. With two different competing reactions, unclear at this moment, one cannot exclude the role of a sterically large molecule on the interfacial polymerization reaction itself which might also affect the size and PDI.| Samplea | Dispersed phase monomer(s) | Additive phase | Size (nm)/PDI | Expt. solid content | Theo. solid content |
|---|---|---|---|---|---|
| a All the samples were prepared at room temperature and cyclohexane was used as a continuous phase. | |||||
| 1 | 4 mmol BDT, 0.2 mmol PETMP | 4.2 mmol TDI, DBU | 185/0.05 | 14.6% | 16.5% |
| 2 | 3.6 mmol BDT, 0.4 mmol PCLD, 0.2 mmol PETMP | 4.2 mmol TDI, DBU | 199/0.06 | 16.8% | 17.5% |
| 3 | 3.2 mmol BDT, 0.8 mmol PCLD, 0.2 mmol PETMP | 4.2 mmol TDI, DBU | 206/0.11 | 18.1% | 19.0% |
| 4 | 2.8 mmol BDT, 1.2 mmol PCLD, 0.2 mmol PETMP | 4.2 mmol TDI, DBU | 213/0.16 | 19.4% | 20.3% |
| 5 | 2.0 mmol BDT, 2.0 mmol PCLD, 0.2 mmol PETMP | 4.2 mmol TDI, DBU | 164/0.07 | 21.9% | 23.4% |
Surprisingly, sample 5 (which contains 1:1 BDT and a higher amount of PCLD) is relatively smaller in size. This discrepancy in the trend will be explained in the text later. Overall, the sizes observed here are comparable and similar to values previously reported for polythiourethane and polyurethane capsules.42,52 The experimental solid content, which refers to the solid content of the nanoparticle dispersion containing only particulates, excludes any large aggregates/bulk materials formed during the reaction. The theoretical solid content refers to the calculated solid content of the nanoparticle dispersion upon complete conversion of all monomers added during the reaction. As expected, the value of the solid content increases with higher amounts of PCLD used. Moreover, the measured solid contents are in agreement with previous values reported for thiourethane-based nanocapsules.42
3.2 Chemical characterization of nanocarriers
The overall success of the thiol-NCO and hydroxyl-NCO reaction is determined by the presence of the thiourethane and urethane groups in the product.53 The strong evidence provided by the ATR-IR spectra obtained from air-dried samples from the cyclohexane phase confirms the successful formation of thiourethane and urethane linkages (Fig. 1). The complete spectral range for all samples in Table 1 is presented in Fig. S1.† The characteristic peaks for isocyanate, thiol and hydroxyl groups (2280 cm−1 to 2270 cm−1, 2556 cm−1 to 2400 cm−1 and 3446 cm−1 to 3558 cm−1, respectively) have disappeared after the product formation.42 The presence of the thiourethane peak at 1675 cm−1 in Fig. 1 indicates the successful reaction between the diisocyanate and the thiol moieties,54 while the presence of N–H bending bands at 1529 cm−1 and a carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching transmittance at 1725 cm−1 for urethane indicates the successful reaction between the diisocyanate and the hydroxyl moieties of PCLD.55 The ester carbonyl group (CO) stretching transmittance of PCLD is also present at 1730 cm−1 along with the urethane carbonyl group. Moreover, it can be observed for sample 1 that the thiourethane peak becomes broadened due to the presence of urea at 1640 cm−1.55 The latter is formed as a result of the reaction between the amine (formed by isocyanate–water side reactions) and the isocyanate groups, as has been reported previously.42
O) stretching transmittance at 1725 cm−1 for urethane indicates the successful reaction between the diisocyanate and the hydroxyl moieties of PCLD.55 The ester carbonyl group (CO) stretching transmittance of PCLD is also present at 1730 cm−1 along with the urethane carbonyl group. Moreover, it can be observed for sample 1 that the thiourethane peak becomes broadened due to the presence of urea at 1640 cm−1.55 The latter is formed as a result of the reaction between the amine (formed by isocyanate–water side reactions) and the isocyanate groups, as has been reported previously.42
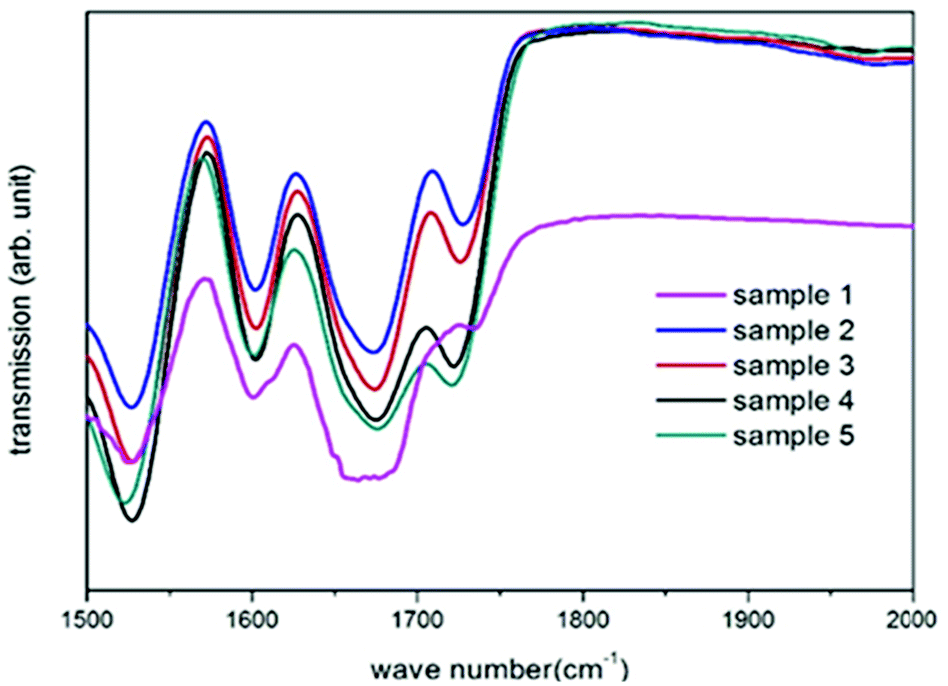 | ||
| Fig. 1 Transmission FT-IR spectra indicating the presence of thiourethane and urethane in all synthesized nanocarriers (sample 1–5). | ||
On the contrary, in samples 2–5 this characteristic peak is not observed. Interestingly, it seems like the presence of PCLD monomer inhibits the urea formation (sample 2–5) which can be attributed to one or a combination of the following reasons: (i) the presence of the hydrophobic PCLD molecules at the interface can possibly hinder the interaction between the isocyanate groups and water molecules in the dispersed phase; (ii) urea formation is only a secondary reaction where the former should compete with the primary reactions resulting in thiourethane and urethane formation.
Chemical composition analysis by solid-state 13C NMR of samples 1–5 are presented in Fig. 2. Here, the methyl group originating from the diisocyanate is assigned to the peak at 17 ppm. The peak around 62 ppm can be assigned to ether functionalities of PCLD and the peak at 70 ppm originates from the alcohol and ether functionalities of the surfactant (Hypermer B246) which is a block copolymer consisting of polyhydroxystearic acid and polyethylene glycol moieties.49 The peaks ranging from 140 ppm to 110 ppm are attributed to the carbons of the aromatic ring of TDI. The ester carbonyl carbon originating from PCLD is assigned to the peak at 173 ppm. As it can be observed, the intensity of the peak increases gradually with increasing amount of PCLD monomer. The well-defined intense peak at 166 ppm confirms the successful formation of thiourethane via the reaction between the diisocyanate and the thiol moieties.54 And the peak at 154 ppm corresponds to urethane formed by the successful reaction between the diisocyanate and the hydroxyl moieties of PCLD.56 Concomitantly, the intensity of the peak at 154 ppm gradually increases with increasing amount of PCLD monomer, which indicates an increasing amount of urethane that is formed.
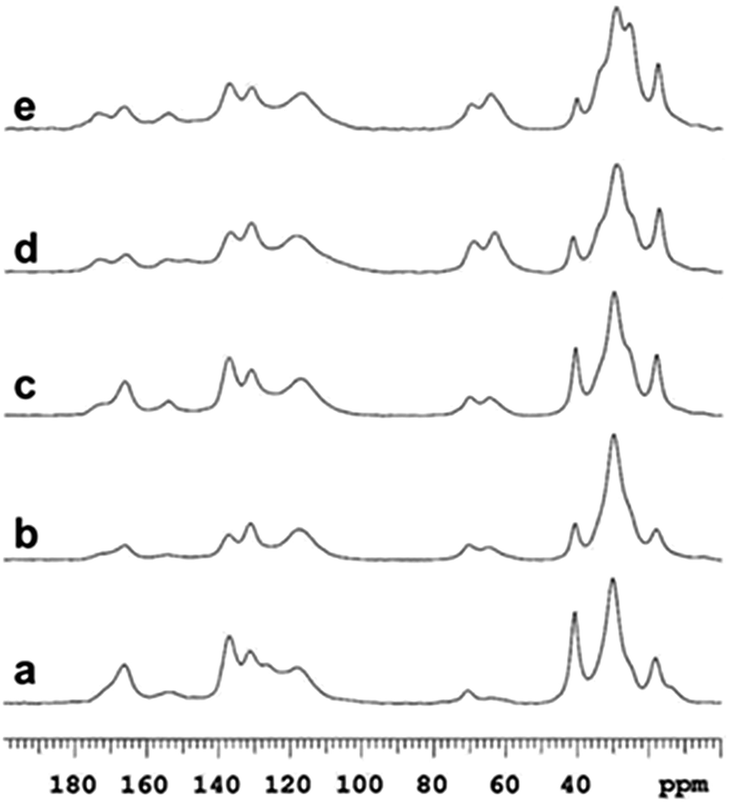 | ||
| Fig. 2 High-resolution solid-state 13C NMR data demonstrating the presence of thiourethane for sample 1 (a) and both thiourethane and urethane for samples 2 (b), 3 (c), 4 (d) and 5 (e) (see Table 1). | ||
The solid-state NMR results together with the FT-IR data thus confirm the successful incorporation of PCLD moieties in the different synthesized samples.
3.3 Morphology studies using TEM
The morphology of the nanocarriers was studied using TEM. The size distribution observed in the TEM images (Fig. 3) for all samples are comparable to the size measurements using DLS. TEM micrographs of samples 1–4 illustrate a clear core–shell nanocapsule morphology. The average shell thicknesses are 27 ± 6 nm, 38 ± 8 nm, 55 ± 12 nm and 79 ± 17 nm for samples 1, 2, 3 and 4, respectively. It was observed that with increasing amount of PCLD monomer, the thickness of the capsule shell increases (Fig. 3b–d). Since the TEM grids were prepared by air-drying small amounts of the nanoparticle dispersion (executed overnight), the size measurements via TEM are subjected to small errors because the nanocapsules are slightly deformed due to the drying process. The size distributions obtained for different samples are presented in Fig. S2† and it can be observed that the size distribution gets relatively broader, which reflects the same trend as observed in DLS measurements. However, for the highest amount of PCLD incorporated (sample 5) the nanocarriers adopt a compact solid nanoparticle morphology (Fig. 3e). As the reaction at the interface is critically dependent on the solubility of the precursor materials in the respective solvents and the interfacial tension between the different phases, the morphology and the size of the nanocarrier can be affected by the reaction conditions where the nature and the amount of used precursor materials can have a significant impact.42 This result is in agreement with DLS results in which the size measured for sample 5 is lower as compared to the other samples (1–4) that possess a nanocapsule morphology. To be useful for biomedical applications, the synthesized nanocapsules have been transferred from the cyclohexane phase to the aqueous phase. Since the other set of samples analogous to the ones listed in Table 1 will be discussed in the following sections where further studies in the aqueous phase were executed, in here only the characterization of sample 3 (from Table 1) after redispersion is presented. The redispersion process involves replacement of the organic solvent by water. The block copolymeric surfactant provided the colloidal stability of the nanocarriers in the cyclohexane phase. However, the former is not suitable for providing the colloidal stability in the aqueous phase. Therefore, in order to reduce the interfacial tension for the nanocarriers in the aqueous phase, a second surfactant (anionic surfactant, SDS) is added to the aqueous phase. The trend in the nanocarrier size and size distribution before and after redispersion in the respective phases confirms that the redispersion process was successful. The redispersed aqueous nanocapsule dispersion was colloidal stable, whereas the measured hydrodynamic diameter increased. The average size measured was 281 nm with a PDI value of 0.19, demonstrating an increase in the size of about 75 nm compared to the value measured in the organic cyclohexane phase. This size increase after redispersion is expected due to the hydration of the hydrophilic part of the block copolymeric surfactant in the aqueous phase.42 Importantly, Fig. 3f clearly illustrates that the nanocapsules (sample 3) are in fact intact after the redispersion. Overall, the results from DLS and TEM characterization clearly confirm the formation of stable aqueous dispersions and intact nanocapsules, respectively. Interestingly, our method describes an elegant interfacial reaction to synthesize pH-responsive nanocapsules and tune their morphology based on the amount of functional monomer PCLD present in the reaction formulation. | ||
| Fig. 3 TEM images of samples 1 (a), 2 (b), 3 (c), 4 (d) and 5 (e) from the organic phase. Image (f) is obtained from sample 3 after redispersion in water. Scale bar corresponds to 250 nm. | ||
3.4 pH-dependent release kinetics
Since the nanocarriers of samples 2–5 contain biodegradable segments of ester linkages that are susceptible to hydrolytic degradation, the time-dependent release of encapsulated water-soluble fluorescent dye (hydrophilic payload) as a function of pH was investigated. For these experiments, different nanocarriers (samples 1′–5′ (Table S1†)) were synthesized using rhodamine B (2 mg aqueous dye solution) as hydrophilic payload using the same protocol as employed for the samples listed in Table 1. The encapsulation efficiencies for all samples were in the range of 88–93% (Table S2† provides more information on the respective encapsulation efficiencies and loading content). The loading achieved reflects the amount of the dye included in the formulation. As the amount of dye was low with respect to the amount of polymer added for the synthesis, the loading content was also low. However, we wanted to show as a proof of concept – with a dye as hydrophilic payload – that we can achieve an efficient loading with respect to the initially incorporated amount as the theoretical and the observed values are in agreement. The encapsulation of hydrophilic compounds is of particular interest when the final purpose of the nanocarriers is envisaged for applications within a biological environment. In contrast, most of the particles based on aliphatic polyesters developed before (such as poly(lactic-co-glycolic acid), PLGA, nanoparticles) allow mostly the encapsulation of hydrophobic compounds.57–59 Moreover, as compared to nanocarriers prepared via self-assembly approaches, nanocarriers synthesized by means of the miniemulsion process have shown to allow for the highly efficient encapsulation of payload, while preventing undesirable payload release as well as the spontaneous disassembly of polymer chains.35,60In order to evaluate the optimal time period to evaluate the pH-dependent release for all samples, sample 3′ was first tested to investigate the time-dependent release of the encapsulated dye rhodamine B as a function of pH using fluorescence spectroscopy. Because sample 1′ does not contain ester linkages, it was considered as a negative control for the release studies. Aliquots of the redispersed nanocapsule samples were first adjusted to pH 6.5 and pH 4.0 with hydrochloric acid (0.1 M) and with sodium hydroxide (0.1 M) to pH 9.0, and subsequently placed in an incubator for well-defined periods at physiological temperature (37 °C). Prior to evaluation by optical studies, the size of the nanocarriers was also measured by DLS (Table S1†) in order to evaluate the effect of pH on the nanocarrier stability and size. It was found that at 37 °C and for the different pH conditions (4.0, 6.5 and 9.0) the capsule diameters and PDI increased considerably with time. The capsule diameter at pH 6.5 was increased by about 50 nm from the initial diameter (281 nm) after 8 hours and the PDI value also increased from 0.14 to 0.34. A similar trend was observed for capsules treated at pH 9.0. The capsules exposed to acidic pH (pH 4.0) demonstrated a massive change in their size (almost doubled) after 8 h, and their size distribution was also broad with PDI > 0.50. In contrast, the average size and PDI of the control capsules prepared without PCLD (sample 1′) remained almost completely unchanged. Therefore, the obtained results clearly demonstrate the disruption of the colloidal system that was synthesized with PCLD. The time-dependent release profile of rhodamine B at different pH conditions from sample 3′ are depicted in Fig. 4a. It can be observed that the capsule shells containing ester linkages (from PCLD), are labile and allow degradation of the nanocarrier shell under physiological temperature which results in a time-dependent release of the encapsulated dye with the effect being most stark at acidic conditions. The fluorescence spectroscopy measurements correlate well with the results of the DLS measurements, and confirm that the capsule shell degrades at a faster rate in acidic conditions (pH 4.0, which demonstrates the strongest release profile, and pH 6.5,61 as representative pH value for tumour cells) as compared to basic conditions (pH 9.0). For all conditions, however, a similar trend in release profile is observed where over time (in here over a time of 30 hours) more dye is being released. As next step, all samples were tested for pH-dependent release after 8 hours and the respective release profiles are summarized in Fig. 4b. A clear trend is observed for the nanocapsule samples (2′–4′), in which the samples with higher PCLD amounts demonstrate an increased dye release for all pH values. For all samples (sample 2′–5′), the release of dye is faster at acidic pH as compared to physiological and basic pH values.
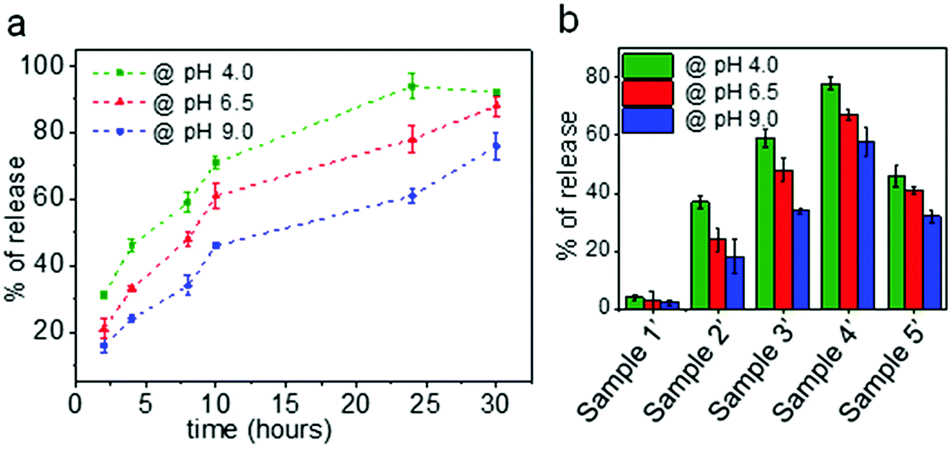 | ||
| Fig. 4 (a) Release kinetics of rhodamine B from sample 3′ at different pH values (pH 4.0, pH 6.5, and pH 9.0). (b) Percentage of rhodamine B release from different nanocarrier samples (1′–5′) at different pH conditions after 8 hours. | ||
For all samples (sample 2′–5′), the release of dye is faster at acidic pH as compared to physiological and basic pH values. However, irrespective of the amount of PCLD, the rate of release is slower from the nanoparticles (sample 5′) as compared to the nanocapsules thereby supporting the importance of nanocarrier morphology on release properties. In case of nanocapsules, the PCLD chains located in the shell are in contact with the aqueous phase as compared to the chains in the bulk of the solid particle. As the shell starts to undergo hydrolytic degradation, more chains are exposed and degradation proceeds to the core of the particle. Therefore, the release in case of the shell morphology is faster. It is also important to consider the packing and the crystallization of the PCLD chains as only less PCLD is present in the shell morphology as compared to the solid particle morphology; the packing of the chains in the latter might play a role in the slow release profile.62 All these observations clearly indicate a stimuli-responsive mechanism for the release of dye that is evidently dependent on pH, exposure time under degrading conditions and nanocarrier morphology. In contrast to previous investigations on PCL-based microspheres and films, we demonstrate here for the first time that we can design nanocarriers based on PCL with diol functionality that allow for a fast payload release of hydrophilic compounds depending on the number of degradable segments and the morphology of the nanocarriers (which is usually not the case if polycaprolactone is used as a biodegradable polymer).63,64 As compared to other commonly used biodegradable polymers such as polylactic acid, poly glycolic acid, and poly(lactic-co-glycolic acid), polycaprolactone degrades slowly due to the latter's hydrophobicity and crystallinity.65 However, based on the obtained results via our elegant interfacial formulation procedure, we have shown that the degradation profile can be tuned. Especially, based on the morphology, which we could tune by the number of degradable segments, different types of applications requiring tunable release profile can be envisaged. For instance, whereas a nanocapsule morphology offers a large aqueous core to polymer ratio encapsulating a large payload, the bulk nanoparticle can be used for applications envisaging high polymer content and slower degradation.
3.5 Biocompatibility and proof of concept for cellular uptake
To validate the potential of the nanocarriers towards biomedical applications, a biocompatibility test was performed of sample 3 using HeLa cells (as a representative cell line for cancerous cells) employing the Alamar Blue assay (Fig. S3†). No dose–response effect or significant cell toxicity (one-way ANOVA p < 0.05) was observed for the tested concentration range (100 μg mL−1) thereby demonstrating the biocompatibility of the nanocarriers. To demonstrate cellular uptake, nanocarriers encapsulating rhodamine B (sample 3′) were incubated with HeLa cells for 6 hours. The confocal microscopy image (Fig. S4†) illustrates successfully the internalization of the nanocapsules by cells, already after an exposure time of 6 hours.4. Conclusions
In summary, we have successfully developed biodegradable nanocarriers encapsulating hydrophilic payload via the inverse miniemulsion technique using a biodegradable polymer as precursor material along with monomer units containing thiol and isocyanate groups via in situ hydroxyl-NCO and thiol-NCO reactions at the droplet interface. The presence of thiourethane and urethane functionalities, as well as the incorporation of biodegradable PCLD segments, was confirmed by FT-IR spectroscopy and high-resolution 13C solid-state NMR spectroscopy. The morphology studies by TEM illustrated that the nanocarriers exhibited a core–shell as well as compact solid nanoparticle morphology depending on the amount of PCLD monomer used. The release studies of the water-soluble fluorescent dye rhodamine B, encapsulated at room temperature with high efficiency, demonstrated a time-dependent release profile for the different pH conditions evaluated. A clear trend of dye release was observed for the different nanocapsule samples in which an increased release for all pH values was evidenced by increasing PCLD content. The ester linkage of PCLD demonstrates a useful pH-sensitive degradation profile, i.e. a faster degradation rate at acidic pH and the slower rate at basic pH for all the samples. However, irrespective of the amount of PCLD, the release rate is slower for the nanoparticles compared to nanocapsules. This observation underpins the importance of nanocarrier morphology on the release profile of its cargo. Detailed cellular uptake and drug release studies are currently in progress as a follow-up of these encouraging results, thereby aiming for the development of smart advanced drug delivery vehicles. In concise, our results successfully demonstrate a stimuli-responsive mechanism for the release of dye that depends on the pH, time period for the respective degrading conditions and nanocarrier morphology. The developed system has a clear potential in opening new avenues towards the development of advanced stimuli-responsive nanocarriers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. S. is an SB PhD fellow at FWO (Research Foundation Flanders). M. P. acknowledges funding by “Agency for Innovation by Science and Technology” in Flanders. S. P. acknowledges funding by Hasselt University via “BOF incoming mobility” scheme. The authors are thankful to Prof. M. Van Bael for the access to the DLS device. HeLa cells were kindly provided by the Biophysics group of the Biomedical Research Institute (BIOMED, Hasselt University).Notes and references
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991 CrossRef CAS PubMed.
- M. A. Boerman, H. L. V. d. Laan, J. C. M. E. Bender, R. Hoogenboom, J. A. Jansen, S. C. Leeuwenburgh and J. C. M. V. Hest, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1573–1582 CrossRef CAS.
- B. Kang, T. Opatz, K. Landfester and F. R. Wurm, Chem. Soc. Rev., 2015, 44, 8301–8325 RSC.
- S. Belbekhouche, O. Mansour and B. Carbonnier, J. Colloid Interface Sci., 2018, 522, 183–190 CrossRef CAS PubMed.
- C.-K. Chen, W.-C. Law, R. Aalinkeel, Y. Yu, B. Nair, J. Wu, S. Mahajan, J. L. Reynolds, Y. Li, C. K. Lai, E. S. Tzanakakis, S. A. Schwartz, P. N. Prasad and C. Cheng, Nanoscale, 2014, 6, 1567–1572 RSC.
- H. Freichels, M. Wagner, P. Okwieka, R. G. Meyer, V. Mailander, K. Landfester and A. Musyanovych, J. Mater. Chem. B, 2013, 1, 4338–4348 RSC.
- G. Baier, D. Baumann, J. M. Siebert, A. Musyanovych, V. Mailänder and K. Landfester, Biomacromolecules, 2012, 13, 2704–2715 CrossRef CAS PubMed.
- J. K. Awino, S. Gudipati, A. K. Hartmann, J. J. Santiana, D. F. Cairns-Gibson, N. Gomez and J. L. Rouge, J. Am. Chem. Soc., 2017, 139, 6278–6281 CrossRef CAS PubMed.
- S. Kumar Pramanik, P. Losada-Pérez, G. Reekmans, R. Carleer, M. D'Olieslaeger, D. Vanderzande, P. Adriaensens and A. Ethirajan, Sci. Rep., 2017, 7, 46257 CrossRef CAS PubMed.
- H. Tian, J. Du, J. Wen, Y. Liu, S. R. Montgomery, T. P. Scott, B. Aghdasi, C. Xiong, A. Suzuki, T. Hayashi, M. Ruangchainikom, K. Phan, G. Weintraub, A. Raed, S. S. Murray, M. D. Daubs, X. Yang, X.-b. Yuan, J. C. Wang and Y. Lu, ACS Nano, 2016, 10, 7362–7369 CrossRef CAS PubMed.
- J. Song, Y. Wei, J. Hu, G. Liu, Z. Huang, S. Lin, F. Liu, Y. Mo, Y. Tu and M. Ou, Chem.–Eur. J., 2018, 24, 212–221 CrossRef CAS PubMed.
- Ö. Tezgel, A. Szarpak-Jankowska, A. Arnould, R. Auzély-Velty and I. Texier, J. Colloid Interface Sci., 2018, 510, 45–56 CrossRef PubMed.
- M. El Founi, S. M. A. Soliman, R. Vanderesse, S. Acherar, E. Guedon, I. Chevalot, J. Babin and J.-L. Six, J. Colloid Interface Sci., 2018, 514, 289–298 CrossRef CAS PubMed.
- I. Hofmeister, K. Landfester and A. Taden, Macromolecules, 2014, 47, 5768–5773 CrossRef CAS.
- I. Mellman, R. Fuchs and A. Helenius, Annu. Rev. Biochem., 1986, 55, 663–700 CrossRef CAS PubMed.
- K. Engin, D. B. Leeper, J. R. Cater, A. J. Thistlethwaite, L. Tupchong and J. D. McFarlane, Int. J. Hyperthermia, 1995, 11, 211–216 CrossRef CAS PubMed.
- A. S. E. Ojugo, P. M. J. McSheehy, D. J. O. McIntyre, C. McCoy, M. Stubbs, M. O. Leach, I. R. Judson and J. R. Griffiths, NMR Biomed., 1999, 12, 495–504 CrossRef CAS PubMed.
- A. Ethirajan, K. Schoeller, A. Musyanovych, U. Ziener and K. Landfester, Biomacromolecules, 2008, 9, 2383–2389 CrossRef CAS PubMed.
- J. Andrieu, N. Kotman, M. Maier, V. Mailänder, W. S. L. Strauss, C. K. Weiss and K. Landfester, Macromol. Rapid Commun., 2012, 33, 248–253 CrossRef CAS PubMed.
- A. Musyanovych, J. Schmitz-Wienke, V. Mailänder, P. Walther and K. Landfester, Macromol. Biosci., 2008, 8, 127–139 CrossRef CAS PubMed.
- A. Ethirajan, A. Musyanovych, A. Chuvilin and K. Landfester, Macromol. Chem. Phys., 2011, 212, 915–925 CrossRef CAS.
- R. Penjweini, S. Deville, L. D'Olieslaeger, M. Berden, M. Ameloot and A. Ethirajan, J. Controlled Release, 2015, 218, 82–93 CrossRef CAS PubMed.
- M. A. Tracy, K. L. Ward, L. Firouzabadian, Y. Wang, N. Dong, R. Qian and Y. Zhang, Biomaterials, 1999, 20, 1057–1062 CrossRef CAS PubMed.
- D. Lemoine, C. Francois, F. Kedzierewicz, V. Preat, M. Hoffman and P. Maincent, Biomaterials, 1996, 17, 2191–2197 CrossRef CAS PubMed.
- T. G. Park, W. Lu and G. Crotts, J. Controlled Release, 1995, 33, 211–222 CrossRef CAS.
- J. E. Báez, A. Martínez-Richa and A. Marcos-Fernández, Macromolecules, 2005, 38, 1599–1608 CrossRef.
- W. J. Lin, J. Biomed. Mater. Res., 1999, 47, 420–423 CrossRef CAS PubMed.
- G. Sekosan and N. Vasanthan, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 202–211 CrossRef CAS.
- L. M. Orozco-Castellanos, A. Marcos-Fernández and A. Martínez-Richa, Polym. Adv. Technol., 2011, 22, 430–436 CrossRef CAS.
- H. Kweon, M. K. Yoo, I. K. Park, T. H. Kim, H. C. Lee, H.-S. Lee, J.-S. Oh, T. Akaike and C.-S. Cho, Biomaterials, 2003, 24, 801–808 CrossRef CAS PubMed.
- P. Xue, L. Sun, Q. Li, L. Zhang, Z. Xu, C. M. Li and Y. Kang, J. Colloid Interface Sci., 2018, 509, 384–394 CrossRef CAS PubMed.
- F. Caruso, R. A. Caruso and H. Möhwald, Science, 1998, 282, 1111 CrossRef CAS PubMed.
- M. Antonietti, R. Basten and S. Lohmann, Macromol. Chem. Phys., 1995, 196, 441–466 CrossRef CAS.
- K. Landfester, A. Musyanovych and V. Mailänder, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 493–515 CrossRef CAS.
- K. Landfester, Angew. Chem., Int. Ed., 2009, 48, 4488–4507 CrossRef CAS PubMed.
- J. M. Barichello, M. Morishita, K. Takayama and T. Nagai, Drug Dev. Ind. Pharm., 1999, 25, 471–476 CrossRef CAS PubMed.
- S. Vrignaud, J.-P. Benoit and P. Saulnier, Biomaterials, 2011, 32, 8593–8604 CrossRef CAS PubMed.
- A. Ethirajan and K. Landfester, Chem.–Eur. J., 2010, 16, 9398–9412 CrossRef CAS PubMed.
- M. Antonietti and K. Landfester, Prog. Polym. Sci., 2002, 27, 689–757 CrossRef CAS.
- J. M. Siebert, G. Baier, A. Musyanovych and K. Landfester, Chem. Commun., 2012, 48, 5470–5472 RSC.
- E.-M. Rosenbauer, M. Wagner, A. Musyanovych and K. Landfester, Macromolecules, 2010, 43, 5083–5093 CrossRef CAS.
- S. Kuypers, S. K. Pramanik, L. D'Olieslaeger, G. Reekmans, M. Peters, J. D'Haen, D. Vanderzande, T. Junkers, P. Adriaensens and A. Ethirajan, Chem. Commun., 2015, 51, 15858–15861 RSC.
- T. K. Dash and V. B. Konkimalla, Mol. Pharm., 2012, 9, 2365–2379 CrossRef CAS PubMed.
- X. L. Lu, Z. J. Sun, W. Cai and Z. Y. Gao, J. Mater. Sci.: Mater. Med., 2008, 19, 395–399 CrossRef CAS PubMed.
- M.-H. Huang, A.-H. Chou, S.-P. Lien, H.-W. Chen, C.-Y. Huang, W.-W. Chen, P. Chong, S.-J. Liu and C.-H. Leng, J. Biomed. Mater. Res., Part B, 2009, 90, 832–841 CrossRef PubMed.
- Z. Li and B. H. Tan, Mater. Sci. Eng., C, 2014, 45, 620–634 CrossRef CAS PubMed.
- N. Nasongkla, X. Shuai, H. Ai, B. D. Weinberg, J. Pink, D. A. Boothman and J. Gao, Angew. Chem., Int. Ed., 2004, 43, 6323–6327 CrossRef CAS PubMed.
- S. K. Pramanik, S. Sreedharan, H. Singh, N. H. Green, C. Smythe, J. A. Thomas and A. Das, Chem. Commun., 2017, 53, 12672–12675 RSC.
- N. Yasarawan and J. S. van Duijneveldt, Soft Matter, 2010, 6, 353–362 RSC.
- P. Adriaensens, L. Storme, R. Carleer, J. Gelan and F. E. Du Prez, Macromolecules, 2002, 35, 3965–3970 CrossRef CAS.
- R. Mens, P. Adriaensens, L. Lutsen, A. Swinnen, S. Bertho, B. Ruttens, J. D'Haen, J. Manca, T. Cleij, D. Vanderzande and J. Gelan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 138–145 CrossRef CAS.
- N. Jagielski, S. Sharma, V. Hombach, V. Mailänder, V. Rasche and K. Landfester, Macromol. Chem. Phys., 2007, 208, 2229–2241 CrossRef CAS.
- M. Rogulska, A. Kultys and E. Olszewska, J. Therm. Anal. Calorim., 2013, 114, 903–916 CrossRef CAS.
- J. D. Flores, J. Shin, C. E. Hoyle and C. L. McCormick, Polym. Chem., 2010, 1, 213–220 RSC.
- F. Tiarks, K. Landfester and M. Antonietti, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2520–2524 CrossRef CAS.
- M. Sumi, Y. Chokki, Y. Nakai, M. Nakabayashi and T. Kanzawa, Die Makromolekulare Chemie, 1964, 78, 146–156 CrossRef CAS.
- B. K. Lee, Y. Yun and K. Park, Adv. Drug Delivery Rev., 2016, 107, 176–191 CrossRef CAS PubMed.
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3, 1377–1397 CrossRef CAS PubMed.
- J. Zhao, H. Yang, J. Li, Y. Wang and X. Wang, Sci. Rep., 2017, 7, 18014 CrossRef PubMed.
- Y. Zhao, L.-P. Lv, S. Jiang, K. Landfester and D. Crespy, Polym. Chem., 2015, 6, 4197–4205 RSC.
- A. Som, S. Bloch, J. E. Ippolito and S. Achilefu, Sci. Rep., 2016, 6, 27803 CrossRef CAS PubMed.
- Y. Yao, D. Xu, C. Liu, Y. Guan, J. Zhang, Y. Su, L. Zhao, F. Meng and J. Luo, RSC Adv., 2016, 6, 97684–97693 RSC.
- C.-W. Ou, C.-H. Su, U. S. Jeng and S.-h. Hsu, ACS Appl. Mater. Interfaces, 2014, 6, 5685–5694 CrossRef CAS PubMed.
- W. Badri, K. Miladi, S. Robin, C. Viennet, Q. A. Nazari, G. Agusti, H. Fessi and A. Elaissari, Pharm. Res., 2017, 34, 1773–1783 CrossRef CAS PubMed.
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602–2663 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Characteristics of rhodamine B loaded nanocarriers, FT-IR analysis, size distribution, Alamar Blue assay and cellular uptake of nanocarriers. See DOI: 10.1039/c8ra07066k |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |