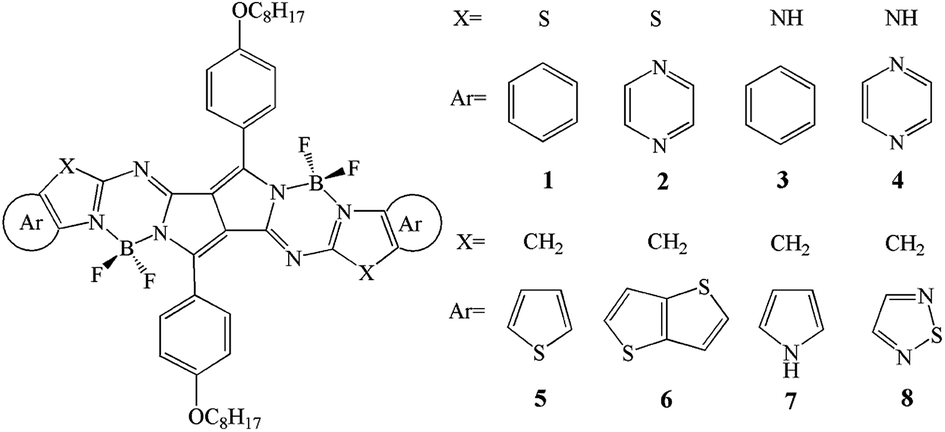
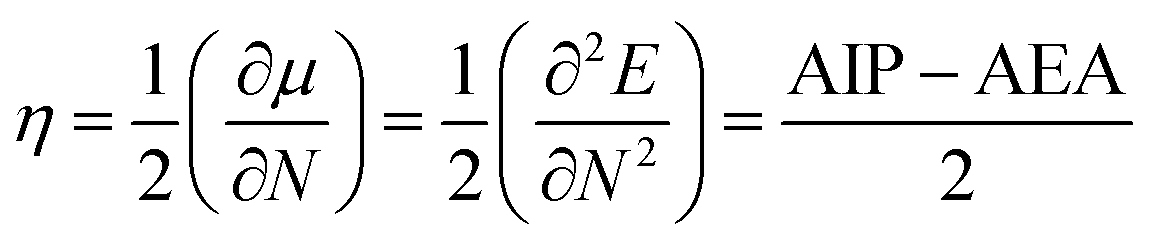DOI:
10.1039/C8RA06940A
(Paper)
RSC Adv., 2018,
8, 33659-33665
Rational design of near-infrared dyes based on boron dipyrromethene derivatives for application in organic solar cells†
Received
20th August 2018
, Accepted 20th September 2018
First published on 2nd October 2018
Abstract
With the aim to further improve the light-absorption efficiency of organic solar cells (OSCs), we have designed a series of novel pyrrolopyrrole boron dipyrromethene (BODIPY) derivatives by replacing the sulfur atom and introducing different fused aromatic heterocycle end-caps. The optical, electronic, and charge transporting properties of the designed molecules have been systematically investigated by applying density functional theory (DFT) and time-dependent DFT (TD-DFT) methodologies. The calculated the frontier molecular orbital (FMO) energies and spectral properties showed that the designed molecules exhibit narrower band gaps and strong absorption in the red/near-infrared (NIR) region, which led to the higher light-absorbing efficiency. Furthermore, the calculated reorganization energies show that the designed molecules are expected to be promising candidates for hole and/or electron transport materials. The results reveal that the designed molecules can serve as high-efficiency red/NIR-active donor materials as well as hole and/or electron transport materials in OSC applications.
Introduction
In recent years, near-infrared (NIR) dyes (with a wavelength about 700 nm) have received considerable attention owing to their wide range of applications, such as organic solar cells (OSCs), photodynamic therapy, two-photon absorption, and nonlinear optics.1 Accordingly, various NIR dyes have been reported.2 Among them, boron dipyrromethene (BODIPY) derivatives are very promising NIR dyes because of their excellent properties such as high molar extinction coefficient, good photo and chemical stability, and excellent light-harvesting ability.3 Furthermore, a donor–acceptor conjugation structure (D–A) is a general way to develop novel OSC materials with a low band gap because this construct enables the energy and electrons to transfer after excitation with light.4 It has been reported that D–A construct NIR dyes of BODIPY exhibit high charge separation and recombination efficiencies.5 In addition, BODIPY derivatives possess low-lying frontier molecular orbitals (FMO) energies, including the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO).6 The deep HOMO of donor materials assures a relatively high open circuit potential (Voc). With the aim to harvest more sunlight, the energy gaps of the designed donor materials should been decreased, which results in an increase in the short circuit current density (Jsc).7 Therefore, these outstanding optoelectronic properties of BODIPY derivatives demonstrate that the materials containing BODIPY units are expected to be promising candidates for donor materials with narrow band gap and excellent light-harvesting ability for OSCs. Nowadays, to improve the NIR-active BODIPY dyes, many strategies have been used to red-shift the low-energy absorption band to capture NIR solar photons through a variety of chemical modifications, such as extending the π-system, ring fusion/annelation, introduction of substituents, replacement of the fluorine atoms by other groups or the meso carbon by nitrogen (cf. aza-BODIPY).8,9 It is noticeable that the solar energy is concentrated mostly in the NIR region. Therefore, we will focus on the light-trapping structures of BODIPY derivatives to get the best absorption of solar energy.
With the aim to explore a new class of NIR-active dyes for improving the efficiency of OSCs, a series of novel NIR-active dyes based pyrrolopyrrole BODIPY have been designed by replacing sulfur atom and introduction of different fused aromatic heterocycles end-cappers (Scheme 1). Furthermore, in order to get a further insight on the relationship between topologic structure and optical as well as charge transporting properties, we investigated the optical, electronic, and charge transporting properties of designed molecules. This may provides a demonstration for the rational design of promising candidate for high efficiency OSCs. The FMOs (EHOMO and ELUMO) energies, the HOMO–LUMO gaps (Eg), the absorption and fluorescence spectra as well as the reorganization energy were calculated by applying density functional theory (DFT) and time-dependent DFT (TD-DFT) methodology.
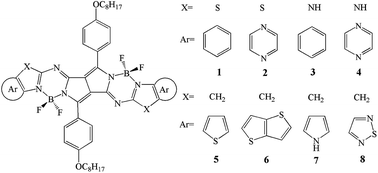 |
| | Scheme 1 Molecular structures of the investigated molecules. | |
Computational methods
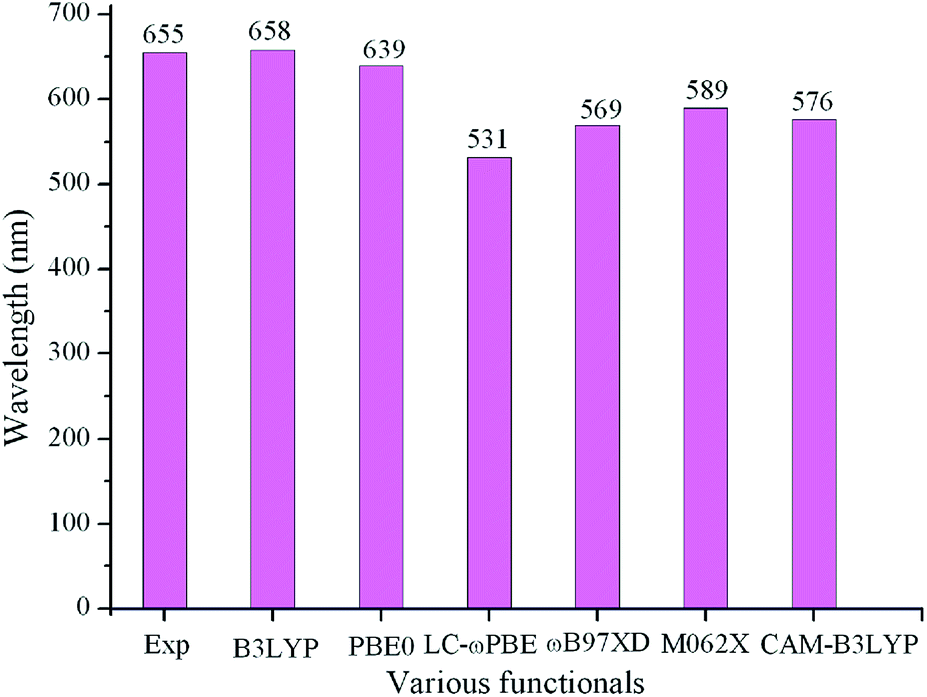
Geometry optimization for the ground state (S0) and the lowest singlet excited state (S1) were performed with the DFT and TD-DFT method using Gaussian 09 code,10 respectively. The harmonic vibrational frequency calculations obtained at the same theoretical level were used to confirm that the optimized molecules are stable structures. Based on the optimized geometries in the S0 and S1, the absorption and fluorescent spectra of the compounds under investigation were predicted using the TD-DFT method, respectively. In order to evaluate the reliability of the selected approach, various functionals including B3LYP,11 PBE0,12 LC-ωPBE,13 ωB97XD,14 M062X,15 and CAM-B3LYP16 were employed to optimize the geometries of the parent molecule 1 in S0 and S1 states. On the basis of the optimized geometry for S0 and S1 states, the absorption and fluorescence were predicted using the polarized continuum model (PCM)17 in the chloroform solvent. The longest wavelengths of absorption and fluorescence (λabs and λfl) in chloroform as well as the experimental data are given in Table S1 in the ESI.† The λabs values for various functionals are plotted in Fig. 1. As showing Fig. 2 and Table S1,† among the various levels of theory tested, the calculated λabs and λfl at B3LYP/6-31G(d,p) (658 and 697 nm) level provided a better agreement with the experimental values (655 and 672 nm)9 than those obtained with other levels of theory, with the deviation of 3 and 25 nm, respectively. With the aim to testify a further the validity of the selected approach, we take a pyrrolopyrrole aza-BODIPY analogue (PPB) as an example because it possesses a similar structure to our designed molecules. The sketch map of the structure of PPB is shown in Fig. S2 in the ESI.† The predicted longest λabs and λfl in chloroform at different theory levels as well as the experimental data18 are given in Table S2 in ESI.† It also suggested that the B3LYP functional is well suitable to our designed molecules. Therefore, B3LYP functional is reasonable to investigate the current system. All calculations in this paper were performed using the 6-31G(d,p) basis set. 0–0 transition energies ΔE0–0 were also obtained for all species using the B3LYP functional. The 0–0 transition energy is the energy difference between the optimized S1 and S0 states, including the corresponding zero point energy difference of those two states.
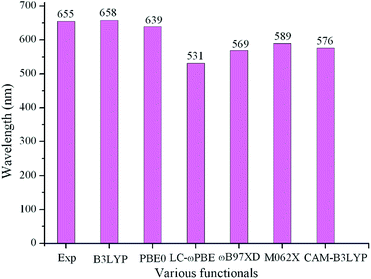 |
| | Fig. 1 Calculated absorption wavelengths (λabs) of 1 in chloroform using various functionals, together with the experimental results. | |
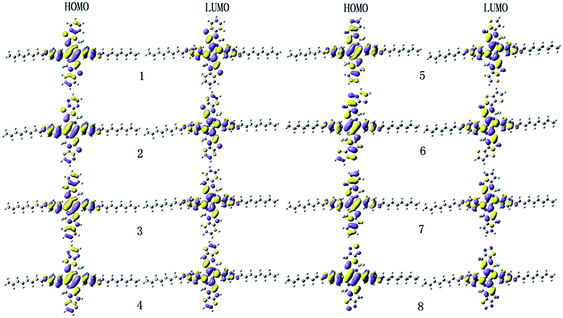 |
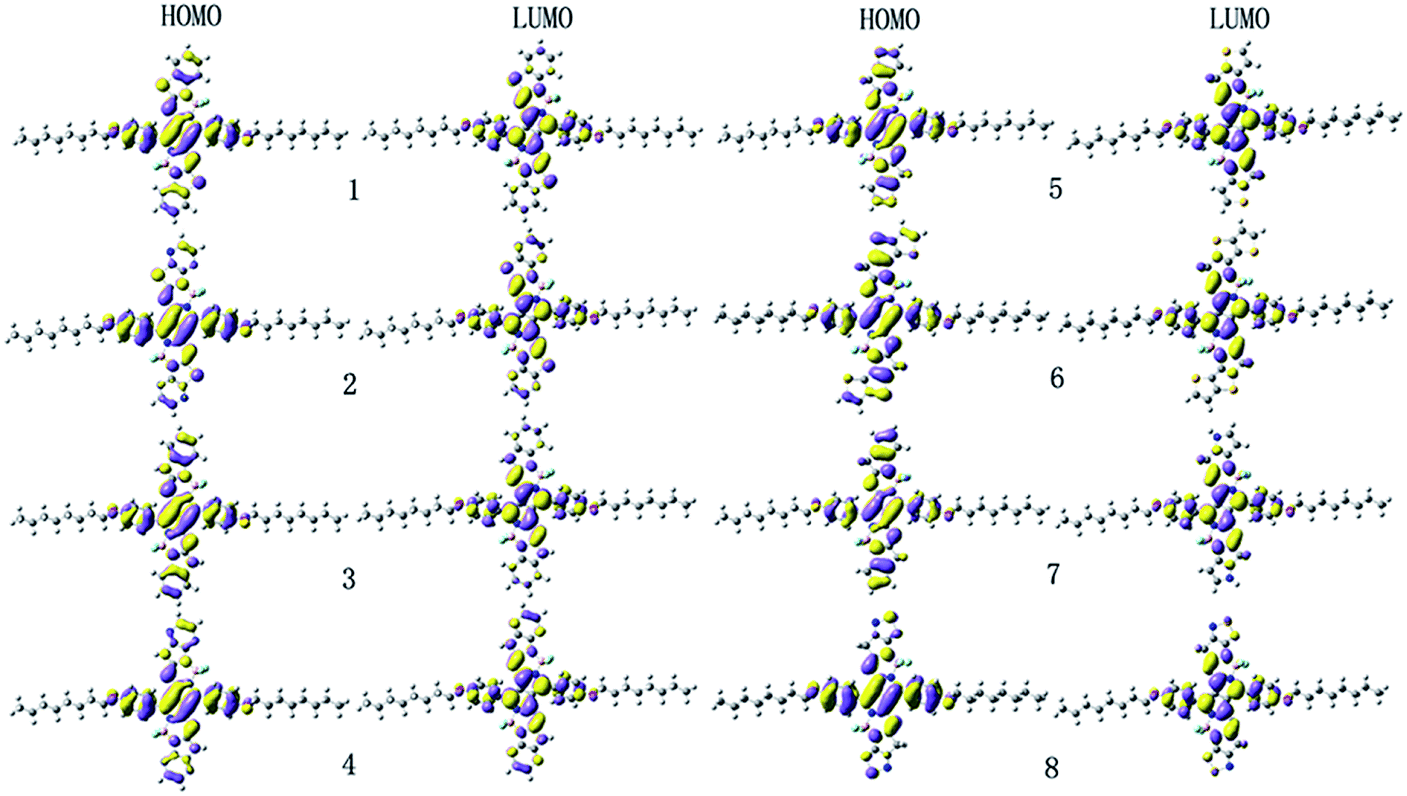
| | Fig. 2 The electronic density contours of the frontier orbital for the studied compounds at the B3LYP/6-31G(d,p) level. | |
According to the Marcus theory,19 the reorganization energy (λ) is the most important parameter for effective transport. The lower the reorganization energy values, the higher the charge transfer rate. In this work, we only pay attention to the internal reorganization energy because the external reorganization energy in organic crystals are much smaller than their counterparts and can be neglected.20 The internal reorganization energy can be expressed as:
| | |
λ = λ1 + λ2 = (E±0 − E±±) + (E0± − E00)
| (1) |
Here,
E±0 and
E±± are the energies of the charged states with the optimized neutral and charged geometry, respectively.
E0± and
E00 represent the energy of the neutral species with the optimized geometry of the charged and neutral species, respectively. The reorganization energy for electron (
λe) and hole (
λh) were calculated directly from the adiabatic potential energy surfaces at the B3LYP/6-31G(d,p) level.
21 This method can evaluate the internal reorganization energy accurately.
22
The stability is the most important criteria to determine the nature of devices for photoelectric materials. On the basis of the molecular orbital theory, the absolute hardness (η) was applied to explore the stability of the designed molecules. The η values of the designed molecules were calculated using equation:23
| |
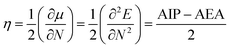 | (2) |
Here,
μ and
N represent the chemical potential and total electron number, respectively. AIP and AEA are the adiabatic ionization potential and electron affinity, which were determined by AIP =
Ecr −
Ep and AEA =
Ep −
Ear, respectively, here,
Ep,
Ecr and
Ear correspond to the energies of the neutral molecule and its cation and anion radical generated after electron transfer, respectively.
Results and discussion
Frontier molecular orbitals level and band gaps
It is noteworthy that the FMOs energies (EHOMO and ELUMO) and band gaps (Eg) are closely related to optical, electronic, and carrier transport properties. The Cartesian coordinates of the designed molecules for the S0 and S1 states are listed in Tables S2 and S3 in the ESI,† respectively. The calculated values of EHOMO, ELUMO, and Eg are given in Table 1. In order to gain insight into the optical, electronic, and carrier transport properties, the distribution patterns of the HOMOs and LUMOs in S0 for 1–8 are plotted in Fig. 2. As visualized in Fig. 2, both the HOMOs and LUMOs of 1–8 exhibited π orbital features. Additionally, since the planar and rigid conjugated molecular geometry of the designed compounds, their HOMOs and LUMOs spread over the whole molecule. As shown in Table 1, the EHOMO values of 1–8 decreases in the order of 7 > 3 > 6 > 5 > 1 > 4 > 2 > 8, while the corresponding values of ELUMO are in the sequence of 3 > 7 > 1 > 5 > 4 > 6 > 2 > 8. Therefore, the Eg values are in the order of 3 > 4 > 1 > 5 > 2 ≈ 8 > 7 > 6. One can find that both the EHOMO and ELUMO values of 3 and 7 increase, while the corresponding values of 2, 4 and 8 decrease compared with those of 1, respectively. However, for 5 and 6, the EHOMO values increase, whereas the corresponding values of ELUMO decrease compared with those of 1, respectively. This shows that the molecules with replacing of nitrogen (3) and with replacing of carbon and pyrrole aromatic end-capper (7) can increase the EHOMO and ELUMO values compared with those of 1, respectively. Molecules with pyrazine end-capper (2) and with replacing of nitrogen and pyrazine end-capper (4) and with replacing of carbon atom and with1,2,5-thiadiazole end-capper (8) have lower the EHOMO and ELUMO values than those of 1, respectively. For molecules with replacing of carbon atom and with thiophene (5) and thieno[3,2-b]thiophene (6) end-cappers, they exhibit higher EHOMO and lower ELUMO values than those of 1, respectively. It implies that the replacing of nitrogen (3 and 4) can increase Eg values slightly, whereas molecule with pyrazine end-capper (2) and molecules with replacing of carbon and thiophene (5), thieno[3,2-b]thiophene (6), pyrrole (7), and 1,2,5-thiadiazole (8) end-capper possesses narrow Eg. The above results indicates that the replacing sulfur atom and introduction of aromatic heterocyclic groups as end-capper can broaden absorption spectrum and improve absorptions in the NIR region.
Table 1 The calculated FMOs energies EHOMO and ELUMO, and HOMO–LUMO gaps Eg (eV) of 1–8 at the B3LYP/6-31G(d,p) level
| Species |
EHOMO |
ELUMO |
Eg |
| 1 |
−5.09 |
−3.00 |
2.09 |
| 2 |
−5.34 |
−3.30 |
2.04 |
| 3 |
−4.87 |
−2.65 |
2.22 |
| 4 |
−5.18 |
−3.04 |
2.14 |
| 5 |
−5.06 |
−3.02 |
2.04 |
| 6 |
−5.04 |
−3.07 |
1.97 |
| 7 |
−4.71 |
−2.72 |
1.99 |
| 8 |
−5.54 |
−3.50 |
2.04 |
Absorption and fluorescence spectra
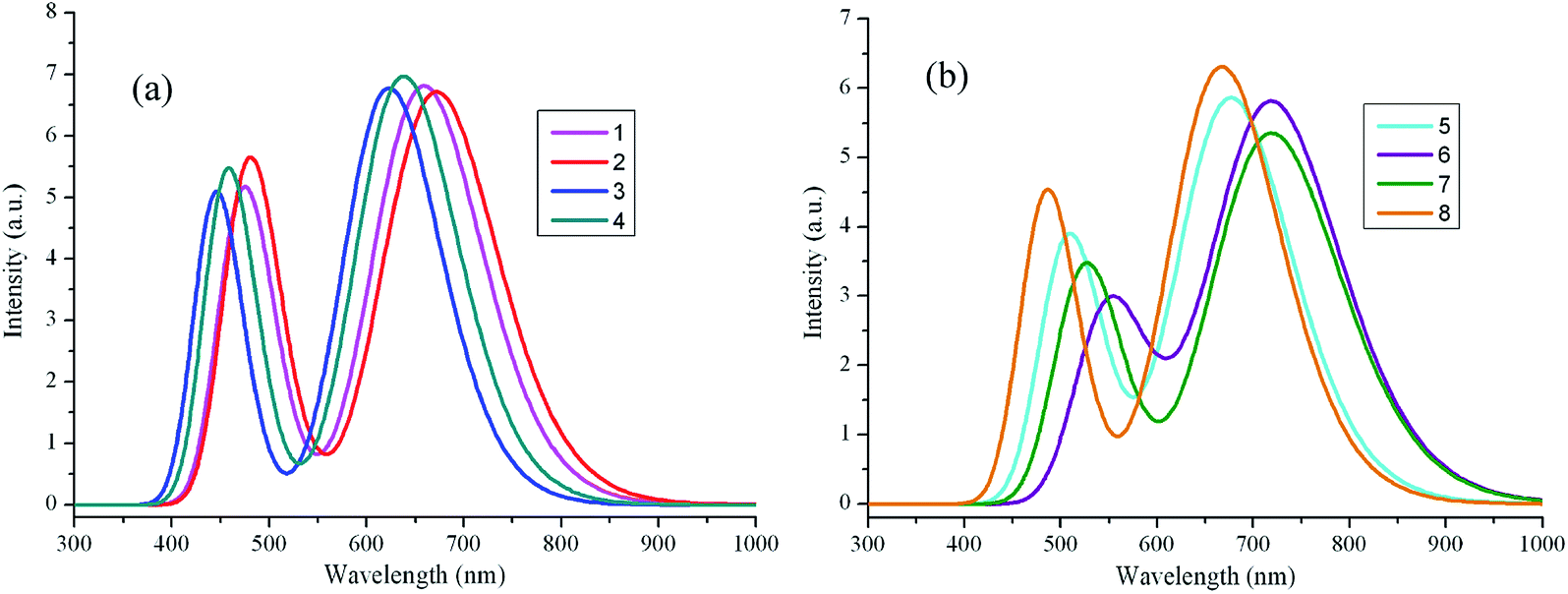
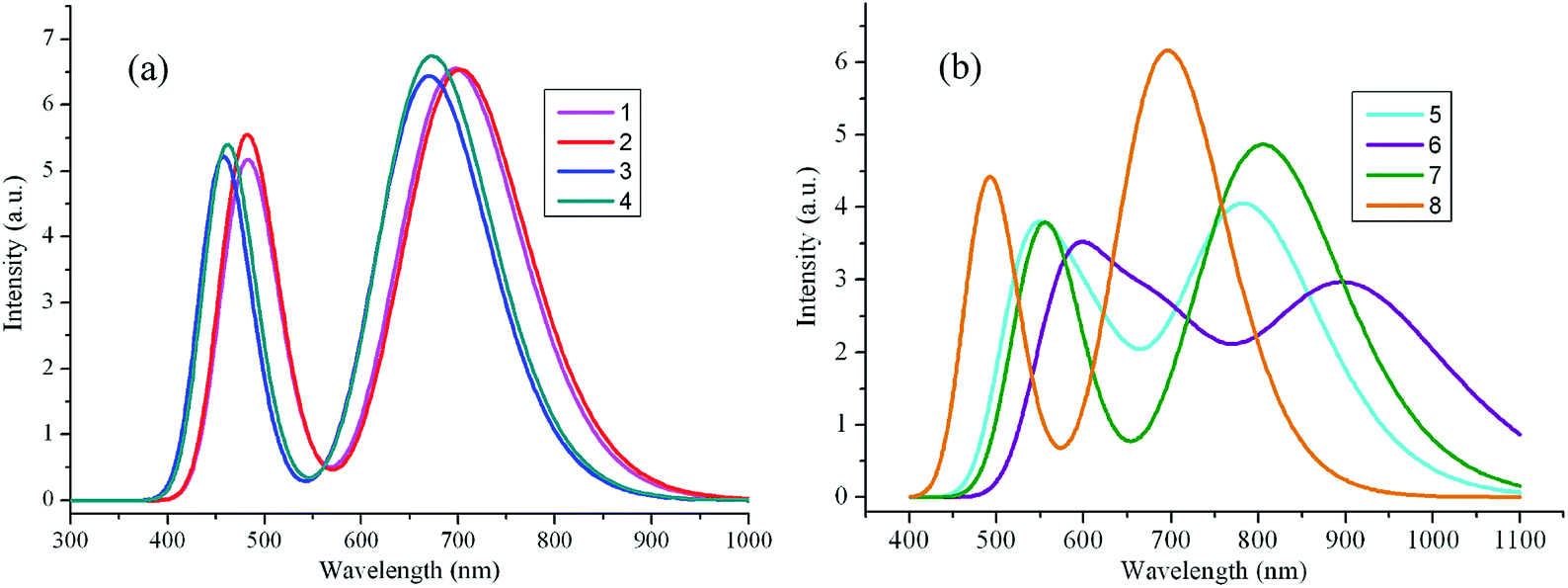
The maximum absorption wavelengths λabs (nm), vertical excitation energies E (eV), 0–0 transitions ΔE0–0 (eV), oscillator strength f, the main assignments, and the absorption region R for 1–8 in chloroform are listed in Table 2. R corresponds to the difference of the longest and shortest wavelength values with oscillator strength larger than 0.01 considering the first fifteen excited states. The corresponding λabs and f of the first fifteen excited states for the designed compounds are listed in Table S3 in ESI.† The simulated absorption and fluorescence spectra of 1–8 in chloroform are shown in Fig. 3 and 4 respectively, which were plotted by using the GaussSum 1.0 program.24 As can be seen in Table 2, the absorption maximum of designed molecules are assigned to the S0 → S1 electronic transitions, which mainly originates from HOMOs → LUMOs excitations. The λabs of 2 and 5–8 exhibit bathochromic shifts 14, 19, 61, 60, and 9 nm (317, 427, 1290, 1270, and 205 cm−1) compared with that of the parent compound 1, respectively. However, the λabs of 3 and 4 show hypsochromic shifts 35 and 20 nm (853 and 476 cm−1) compared with that of the parent compound 1, respectively. This can be explained by the fact that the molecular structures and nature of the substituent groups. The designed compounds possess planar and rigid conjugated molecular geometry. The HOMOs → LUMOs excitations results in transfer of electron density from the two aromatic end-capper fragments to dimeric aza-BODIPY centrals for the designed π-conjugated ladder molecules (see Fig. 2). It is worth noting that the photophysical properties of intramolecular charge transfer are highly dependent on the electron donor/acceptor strength.25 Therefore, replacing of nitrogen (3 and 4) and carbon (5–8) in five-membered heterocyclic ring affects the intramolecular charge transfer. The electron-donating ability of nitrogen is lower, while the corresponding ability of carbon is higher than that of sulfur, respectively. Accordingly, the electron can move more fluently from the electron donor to the electron acceptor. It indicates that the molecular π-conjugation in 5–8 becomes higher than that of 1, which led to the bathochromic shift compared with the parent compound 1 in the absorption spectra for 5–8. On the contrary, the lower electron-donating ability of nitrogen prevents the electron movement from the electron donors to the electron acceptors for 3 and 4. As a consequence, the λabs of 3 and 4 exhibit hypsochromic shifts compared with that of 1. The different molecular structure between 1 and 2 is only the end-capper groups (see Scheme 1). So, the λabs of 2 has slight bathochromic shift compared with 1. Furthermore, the introduction of different aromatic end-capper groups also affects the absorption spectra. For the molecules with replacing of carbon (5–8), the order of electron-donating ability of end-capper groups is thieno[3,2-b]thiophene (6) > 1H-pyrrole (7) > thiophene (5) > 1,2,5-thiadiazole (8), which is in agreement with their λabs values. For 1 and 2 as well as 3 and 4, the λabs of 2 and 4 are larger than those of 1 and 3 respectively, which may be attributed to the electron donor nature for the lone pair electrons of two nitrogen's in pyrazine rings for 2 and 4. Therefore, the λabs values of 1–8 are in the sequence of 6 > 7 > 5 > 2 > 8 > 1 > 4 > 3, which is almost in agreement with the corresponding reverse order of their Eg values. Furthermore, we find in Table 2 that the designed molecules possess large oscillator strengths. The oscillator strengths values of 2–8 are almost equal to that of the parent compound 1. The large oscillator strengths can enhance the light-absorption efficiency,26 which led to improvement of short-circuit current JSC for OSCs. At the same time, the designed molecules have large absorption region R (330–399 nm). The R values of 2–4 are slightly smaller, whereas the corresponding values of 5–8 are larger than that of parent compound 1. It suggests that the replacing of nitrogen (3 and 4) in five-membered heterocyclic ring results in slight smaller R values, while the replacing of carbon (5–8) in five-membered heterocyclic ring lead to the increase of R values compared with parent molecule 1. Moreover, in order to maximize the power conversion efficiencies of OSCs, it is necessary that a good overlap between the absorption spectrum of the designed molecules and the solar emission spectrum, particularly in NIR region. As mention above, the absorption bands of the designed molecules were red shifted, which is beneficial for the higher light-absorbing efficiency. These results imply that the designed compounds have strong absorption in NIR region and are expected to be the promising candidates for donor materials in OSCs applications.
Table 2 Predicted λabs (in nm), E (in eV), ΔE0–0 (in eV), f, R (in nm), and main assignments for 1–8 in chloroform at the TD-B3LYP/6-31G(d,p) level, along with available experimental data
| Species |
λabs |
E |
ΔE0–0 |
f |
Assignment |
R |
| Experimental data in chloroform were taken from ref. 9. |
| 1 |
658 |
1.883 |
1.836 |
0.94 |
H → L (0.71) |
362 |
| 2 |
672 |
1.846 |
1.794 |
0.93 |
H → L (0.71) |
359 |
| 3 |
623 |
1.990 |
1.919 |
0.93 |
H → L (0.71) |
340 |
| 4 |
638 |
1.944 |
1.881 |
0.96 |
H → L (0.71) |
330 |
| 5 |
677 |
1.832 |
1.794 |
0.81 |
H → L (0.71) |
364 |
| 6 |
719 |
1.725 |
1.622 |
0.80 |
H → L (0.70) |
395 |
| 7 |
718 |
1.725 |
1.627 |
0.74 |
H → L (0.71) |
399 |
| 8 |
667 |
1.859 |
1.805 |
0.87 |
H → L (0.71) |
364 |
| Expa |
655 |
1.893 |
1.680 |
|
|
|
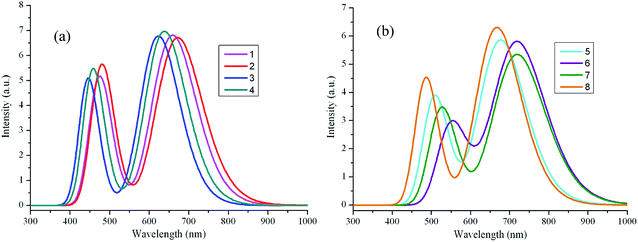 |
| | Fig. 3 The calculated absorption spectra of the investigated molecules (value of full width at half maximum is 3000 cm−1). (a) Molecules 1–4; (b) molecules 5–8. | |
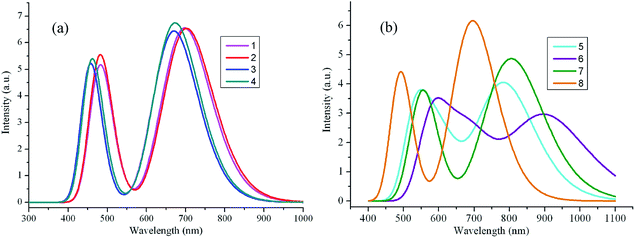 |
| | Fig. 4 The calculated fluorescence spectra of the investigated molecules (value of full width at half maximum is 3000 cm−1). (a) Molecules 1–4; (b) molecules 5–8. | |
To assess the impact of the vertical approximation on the theoretically determined absorption maxima, ΔE0–0 energies were obtained using the B3LYP method. The 0–0 transition ΔE0–0 of the parent molecule 1 provided a better agreement with the experimental value9 (1.680 eV) than that obtained with vertical excitation energy E; the deviations are 0.156 and 0.203 eV, respectively. It is well-known that the 0–0 transition energies are more reliable and accurate than that raw vertical transition energy compared with the experimental data.27 It has been shown that the vertical excitation energies E are slightly larger than those of ΔE0–0 values, the deviations are 0.038–0.103 eV. In most cases, charge transfer is reflected in underestimations of the TD-DFT excitation energies.28 However, the studied species do not exhibit significant charge transfer (see Fig. 2). In the present work the use of B3LYP functional leads to a slightly overestimation of the vertical excitation energies.
Table 3 collected the longest fluorescence wavelengths λfl (in nm) main assignments, and the oscillator strength f for 1–8 in chloroform. From Table 3, one can find that the fluorescence of the designed molecules are assigned to the S1 → S0 electronic transitions and LUMOs → HOMOs transitions play a dominant role. As their absorption, the λfl of 2 and 5–8 show bathochromic shifts, while the corresponding values of 3 and 4 exhibit hypsochromic shifts compared with that of the parent compound 1. Furthermore, The Stokes shifts of 1–8 are 39, 30, 47, 35, 107, 184, 87, and 29 nm (851, 636, 1126, 815, 2016, 2834, 1506, and 645 cm−1), respectively. The oscillator strengths f values of 2–4, 7, and 8 are almost equal to that of 1, while the corresponding value of 5 and 6 are slightly smaller than that of 1, respectively.
Table 3 Predicted λfl (in nm), f, and main assignments of 1–8 in chloroform at the TD-B3LYP/6-31G(d,p) level, along with available experimental data
| Species |
λfl |
f |
Assignment |
| Experimental data in chloroform were taken from ref. 9. |
| 1 |
697 |
0.90 |
L → H (0.71) |
| 2 |
702 |
0.90 |
L → H (0.71) |
| 3 |
670 |
0.89 |
L → H (0.71) |
| 4 |
673 |
0.93 |
L → H (0.71) |
| 5 |
784 |
0.55 |
L → H (0.70) |
| 6 |
903 |
0.40 |
L → H (0.70) |
| 7 |
805 |
0.67 |
H → L (0.71) |
| 8 |
696 |
0.85 |
H → L (0.71) |
| Expa |
672 |
|
|
The results displayed in Tables 2 and 3 reveal that the introduction of different aromatic heterocyclic end-capper and/or replacing sulfur atom can tune absorption and fluorescence spectra of the designed molecules. It indicates that the designed molecules have smaller band gaps with extended absorption and fluorescence spectra skewed toward NIR region, which is beneficial improving the efficiency of OSCs.
Reorganization energy
Table 4 collected the calculated reorganization energies for hole (λh) and electron (λe) of 1–8. The computed λe values of the designed molecules are smaller than that of the typical electron transport material tris(8-hydroxyquinolinato)aluminum(III) (Alq3, λe = 0.276 eV).29 It displays that the electron transfer rates of the designed molecules might be higher than that of Alq3. For the computed λh, the values of 5–7 are smaller than that of the typical hole transport material N,N′-diphenyl-N,N′-bis(3-methylphenyl)-(1,1′-biphenyl)-4,4′-diamine (TPD, λh = 0.290 eV),30 whereas the corresponding values of 1–4 and 8 are larger than that of TPD. It indicates that the hole transfer rates of 5–7 might be higher, while the corresponding hole transfer rates 1–4 and 8 might be lower than that of TPD. Reorganization energies are estimated according to the eqn (1). The lower the energy values of cation/anion species, the lower the λh/λe values. For the designed compounds possess planar and rigid conjugated molecular geometry, the electron spread over the whole molecule for anion species, which led to the lower energies value of anion species. Therefore, the designed compounds have lower λe values and are expected to be the promising candidates for electron transport materials. The cation species are generated after electron transfer. As mentioned above, the molecular π-conjugation in 5–7 exhibit higher than that of other molecules, which result in the easier for charge transfer. On the other hand, for 1–4 and 8, the lower molecular π-conjugations prevent the charge transfer, which result in the higher energies for cation species. Accordingly, 1–4 and 8 have large λh values than those of 5–7 and TPD. The calculated λh and λe values are in the order of 4 > 2 > 8 > 1 > 3 > 5 > 7 > 6 and 3 > 1 > 4 ≈ 6 > 5 > 8 > 7 > 2, respectively. Above results indicates that molecules with pyrazine end-capper (2) and with replacing of nitrogen and pyrazine end-capper (4) and with replacing of carbon atom and 1,2,5-thiadiazole end-capper (8) can increase the λh values, while molecules with replacing of nitrogen (3) and with replacing of carbon and thiophene (5), thieno[3,2-b]thiophene (6), pyrrole (7) end-capper decrease the λh values compared with that of 1, respectively. For the computed λe, the designed molecules can decrease the λe values except 3 can increases the λe value compared with that of 1, respectively. It suggests that 5–7 are expected to be the promising candidates for hole as well as electron transport materials, whereas 1–4 and 8 can serve as electron transport materials only.
Table 4 Calculated molecular λe and λh (both in eV) of 1–8 at the B3LYP/6-31G(d,p) level
| Species |
λh |
λe |
| 1 |
0.300 |
0.239 |
| 2 |
0.332 |
0.219 |
| 3 |
0.298 |
0.259 |
| 4 |
0.336 |
0.235 |
| 5 |
0.259 |
0.233 |
| 6 |
0.200 |
0.235 |
| 7 |
0.231 |
0.227 |
| 8 |
0.331 |
0.229 |
Ionization potential, electron affinity and stability properties
As we know, the AIP and AEA are the very important properties to shed light on the charge transfer behavior for organic materials. The lower AIP and higher AEA are the fundamental parameters for effective electron and hole transport, respectively.26 The calculated AIP, AEA, and η of all systems are collected in Table 5. It is clear from Table 5 that the AIP values show the decreasing sequence of 8 > 2 > 4 > 1 > 5 > 6 > 3 > 7. On the other hand, the AEA values are predicted in decreasing order of 8 > 2 > 6 > 4 > 1 > 5 > 7 > 3. Molecules 1, 4 and 8 can increase both the AIP and AEA values, while molecules 3, 5 and 7 decrease both the AIP and AEA values compared with those of 1, respectively. For molecules 6, its AIP and AEA values are smaller and larger than those of 1, respectively. It is noticeable that the introduction of different aromatic heterocyclic end-capper and/or replacing sulfur atom can affect the AIP and AEA of the designed molecules.
Table 5 Calculated molecular AIP, AEA, and η (all in eV) of 1–8 at the B3LYP/6-31G(d,p) level
| Species |
AIP |
AEA |
η |
| 1 |
5.951 |
2.119 |
1.916 |
| 2 |
6.193 |
2.413 |
1.890 |
| 3 |
5.745 |
1.750 |
1.996 |
| 4 |
6.047 |
2.134 |
1.956 |
| 5 |
5.946 |
2.088 |
1.929 |
| 6 |
5.884 |
2.177 |
1.853 |
| 7 |
5.616 |
1.784 |
1.916 |
| 8 |
6.418 |
2.580 |
1.919 |
Generally, the absolute hardness η can be used to evaluate the stability of materials. The results displayed in Table 5 show that the η values of 2–5 are larger than the value of 1, and the corresponding value of 7 is equal to that of 1, respectively. The η value of 6 is smaller slightly than the value of 1. It suggests that the introduction of different aromatic heterocyclic end-capper and/or replacing sulfur atom do not significantly affect the stability of the designed molecules.
Conclusions
In this article, we have designed a series of novel pyrrolopyrrole BODIPY molecules by replacing sulfur atom and introduction of different fused aromatic heterocycles end-cappers. The optical, electronic, and charge transporting properties of designed molecules have been systematically investigated by applying DFT and TD-DFT methodology. The calculated the FMOs energies and absorption spectra properties turned out that the λabs of 2 and 5–8 exhibit bathochromic shifts whereas 3 and 4 show hypsochromic shifts compared with original molecule 1. The designed compounds exhibit narrower band gaps and strong absorption in red/NIR region, which led to the higher light-absorbing efficiency. Furthermore, the calculated reorganization energies show that 5–7 are expected to be the promising candidates for hole as well as electron transport materials, whereas 1–4 and 8 can serve as electron transport materials only. Finally, the stability of the designed molecules does not significantly affected by the introduction of different end-capper or replacing sulfur atom. Hence, the designed molecules can serve as high-efficiency red/NIR-active donor materials as well as hole and/or electron transport materials in OSCs applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial supports from the NSFC (No. 21563002), the Natural Science Foundation of Inner Mongolia Autonomous Region (No. 2015MS0201), and the Research Program of Sciences at Universities of Inner Mongolia Autonomous Region (No. NJZZ235) are gratefully acknowledged.
Notes and references
-
(a) J. Roncali, Acc. Chem. Res., 2009, 42, 1719–1730 CrossRef CAS PubMed;
(b) A. Poirel, A. de Nicola and R. Ziessel, Org. Lett., 2012, 14, 5696–5699 CrossRef CAS PubMed;
(c) L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., Int. Ed., 2014, 53, 2119–2123 CrossRef CAS PubMed;
(d) J. Fabian, H. Nakazumi and M. Matsuoka, Chem. Rev., 1992, 92, 1197–1226 CrossRef CAS;
(e) A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC;
(f) T. Li, J. Wang, H. Chen, P. Cheng, S. Huang, Y. Lin, H. Yu and X. Zhan, Dyes Pigm., 2017, 137, 553–559 CrossRef CAS.
-
(a) H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128–6185 RSC;
(b) G. Qian and Z. Y. Wang, Chem.–Asian J., 2010, 5, 1006–1029 CrossRef CAS PubMed.
-
(a) A. Sutter, P. Retailleau, W. C. Huang, H. W. Lin and R. Ziessel, New J. Chem., 2014, 38, 1701–1710 RSC;
(b) H. Zhao, B. Wang, J. Liao, H. Wang and G. Tan, Tetrahedron Lett., 2013, 54, 6019–6022 CrossRef CAS.
-
(a) C. Dou, X. Long, Z. Ding, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 1436–1440 CrossRef CAS PubMed;
(b) S. Li, W. Liu, M. Shi, J. Mai, T. K. Lau, J. Wan, X. Lu, C. Z. Li and H. Chen, Energy Environ. Sci., 2016, 9, 604–610 RSC;
(c) C. Zhan, X. Zhang and J. Yao, RSC Adv., 2015, 5, 93002–93026 RSC;
(d) Y. Lin, J. Wang, Z. G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- M. E. El-Khouly, S. Fukuzumi and F. D'Souza, ChemPhysChem, 2014, 15, 30–47 CrossRef CAS PubMed.
-
(a) H. Lu, J. Mack, Y. Yang and Z. Shen, Chem. Soc. Rev., 2014, 43, 4778–4823 RSC;
(b) W. Liu, J. Yao and C. Zhan, RSC Adv., 2015, 5, 74238–74241 RSC.
-
(a) Z. Zhang, Z. Zhou, Q. Hu, F. Liu, T. P. Russell and X. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 6213–6219 CrossRef CAS PubMed;
(b) S. Loser, S. J. Lou, B. M. Savoie, C. J. Bruns, A. Timalsina, M. J. Leonardi, J. Smith, T. Harschneck, R. Turrisi, N. Zhou, C. L. Stern, A. A. Sarjeant, A. Facchetti, R. P. H. Chang, S. I. Stupp, M. A. Ratner, L. X. Chen and T. J. Marks, J. Mater. Chem. A, 2017, 5, 9217–9232 RSC.
-
(a) C. Zhang, J. Zhao, S. Wu, Z. Wang, W. Wu, J. Ma, S. Guo and L. Huang, J. Am. Chem. Soc., 2013, 135, 10566–10578 CrossRef CAS PubMed;
(b) N. F. Montcada, B. Pelado, A. Viterisi, J. Albero, J. Coro, P. D. L. Cruz, F. Langa and E. Palomares, Org. Electron., 2013, 14, 2826–2832 CrossRef CAS;
(c) J. You, L. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C. C. Chen, J. Gao, G. Li and Y. Yang, Nat. Commun., 2013, 4, 1446 CrossRef PubMed;
(d) S. Brück, C. Krause, R. Turrisi, L. Beverina, S. Wilken, W. Saak, A. Lutzen, H. Borchert, M. Schiek and J. Parisi, Phys. Chem. Chem. Phys., 2014, 16, 1067–1077 RSC.
- S. Shimizu, T. Iino, A. Saeki, S. Seki and N. Kobayashi, Chem.–Eur. J., 2015, 21, 2893–2904 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery Jr, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 09, Gaussian, Inc, Wallingford, CT, 2009 Search PubMed.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- Y. Tawada, T. Tsuneda, S. Yanagisawa, T. Yanai and K. Hirao, J. Chem. Phys., 2004, 120, 8425–8433 CrossRef CAS PubMed.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
-
(a) M. Cossi, V. Barone, R. Cammi and J. Tomasi, J. Phys. Chem. Lett., 1996, 255, 327–335 CrossRef CAS;
(b) D. Fan, Y. Yi, Z. Li, W. Liu, Q. Peng and Z. Shuai, J. Phys. Chem. A, 2015, 119, 5233–5240 CrossRef CAS PubMed.
- S. Shimizu, T. Iino, Y. Araki and N. Kobayashi, Chem. Commun., 2013, 49, 1621–1623 RSC.
-
(a) R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS;
(b) R. A. Marcus, Annu. Rev. Phys. Chem., 1964, 15, 155–196 CrossRef CAS.
-
(a) D. L. Cheung and A. Troisi, J. Phys. Chem. C, 2010, 114, 20479–20488 CrossRef CAS;
(b) S. Di Motta, E. Di Donato, F. Negri, G. Orlandi, D. Fazzi and C. Castiglioni, J. Am. Chem. Soc., 2009, 131, 6591–6598 CrossRef CAS PubMed.
- M. E. Köse, W. J. Mitchell, N. Kopidakis, C. H. Chang, S. E. Shaheen, K. Kim and G. Rumbles, J. Am. Chem. Soc., 2007, 129, 14257–14270 CrossRef PubMed.
- J. Sancho-García, Chem. Phys., 2007, 331, 321–331 CrossRef.
-
(a) R. G. Pearson, J. Am. Chem. Soc., 1985, 107, 6801–6806 CrossRef CAS;
(b) M. S. Stark, J. Phys. Chem. A, 1997, 101, 8296–8301 CrossRef CAS.
- N. M. O'Boyle and J. G. Vos, GaussSum 1.0, Dublin City University, Dublin, 2003 Search PubMed.
- W. Huang, X. Zhang, L. Ma, C. Wang and Y. Jiang, Chem. Phys. Lett., 2002, 352, 401–407 CrossRef CAS.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- D. Jacquemin, A. Planchat, C. Adamo and B. J. Mennucci, J. Chem. Theory Comput., 2012, 8, 2359–2372 CrossRef CAS PubMed.
- A. Dreuw and M. Head-Gordon, J. Am. Chem. Soc., 2004, 126, 4007–4016 CrossRef CAS PubMed.
- B. C. Lin, C. P. Cheng, Z. Q. You and C. P. Hsu, J. Am. Chem. Soc., 2005, 127, 66–67 CrossRef CAS PubMed.
- N. E. Gruhn, D. A. da Silva Filho, T. G. Bill, M. Malagoli, V. Coropceanu, A. Kahn and J. L. Bredas, J. Am. Chem. Soc., 2002, 124, 7918–7919 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1: Calculated the λabs and λfl of parent molecule 1 by various methods with 6-31G(d,p) basis set, along with available experimental data, Table S2: Calculated the λabs and λfl of PPB by various methods with 6-31G(d,p) basis set, along with available experimental data, Table S3: The absorption wavelengths λabs and f of the first fifteen excited states for the designed compounds, Table S4: The Cartesian coordinates of 1–8 in S0 at the B3LYP/6-31G(d,p) level, Table S5: The Cartesian coordinates of 1–8 in S1 at the TD-B3LYP/6-31G(d,p) level, Fig. S1: Geometries of PPB. See DOI: 10.1039/c8ra06940a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*