
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceColor tuning and white light emission by codoping in isostructural homochiral lanthanide metal–organic frameworks†
Wenbo Wanga,
Ruiying Wangb,
Yafang Gea and
Benlai Wu *a
*a
aCollege of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou 450001, P. R. China. E-mail: wbl@zzu.edu.cn; Tel: +86 0371 67783126
bSchool of Chemical Engineering, Henan Vocational College of Applied Technology, Zhengzhou 450042, P. R. China
First published on 18th December 2018
Abstract
Four lanthanide-based homochiral metal–organic frameworks (Ln-HMOFs), {[Ln2(HL)2(H2O)4]·2Cl·5H2O}n [Ln = Gd (1), Eu (2), Tb (3) and Dy (4)], have been synthesized through solvothermal reactions of chiral ligand (S)-5-(((1-carboxyethyl)amino)methyl)isophthalic acid (H3L) with corresponding LnCl3·6H2O. They are binodal (3,6)-connected frameworks with kgd nets based on binuclear cluster units and zwitterionic (HL)2− linkers. Considering the isostructuralism of these Ln-HMOFs as well as the blue emission of compound 1 and the strong typical Eu3+ and Tb3+ emissions of compounds 2 and 3, single-phase mixed-lanthanide HMOFs have been prepared by doping of Ln3+ into the Ln-HMOFs to modulate light-emitting color. Interestingly, the bimetallic doped Eu/Tb-HMOFs [(EuxTb1−x)2(HL)2(H2O)4]·2Cl·5H2O display a fluent change of light-emitting color among green, yellow, orange, orange-red, and red by adjusting the doping concentration of Eu3+ ions into the Tb-HMOF. Very importantly, the trimetallic doped Eu/Gd/Tb-HMOF [(Eu0.1388Gd0.6108Tb0.2504)2(HL)2(H2O)4]·2Cl·5H2O emits white light upon excitation at 355 nm, whose emission can also be switched between different colors when excited with different ultraviolet light. Furthermore, the fluorescence response of Tb-HMOF to various usual metal ions, and especially fluorescent sensing behaviours to Fe3+, Cr3+ and Al3+ have been preliminarily investigated.
Introduction
Further motivated by a longing for greener, safer and energy efficient light-emitting materials, the exploration of solid-state luminescent materials through various approaches including metal-doped or hybrid inorganic materials,1–3 organic molecules,4 polymers,5 nanomaterials,6 and metal complexes1 has evolved rapidly in recent years. Lanthanide metal–organic frameworks (Ln-MOFs) owing to their tunable structure diversities,7,8 attractive photophysical properties, and potential applications in biomedical imaging,9 lighting,10 and display devices7 make them promising candidates for the development of high-efficiency light-emitting materials. As is well known, lanthanide ions feature excellent luminescence properties such as long lifetime, large Stokes shift and narrow-band but the weak light absorption caused by Laporte forbidden f–f transitions makes the direct lanthanide excitation inefficient.11 However, Ln-MOFs incorporating Ln3+ with judiciously selected organic linkers which can participate in efficient light absorption and ligand-to-metal energy-transfer processes through the so-called “antenna effect” can possibly overcome this paucity.12–14 Additionally, the self-assemblies of varied Ln3+ ions with the same organic bridges under the same reaction conditions always result in isostructural Ln-MOFs due to very similar coordination environments of Ln3+ ions, which provides a facile platform for color tuning and white light emission through the doping of different Ln3+ ions into isostructural MOFs without disruption of the original structure.15–19 Currently, aromatic polycarboxylate ligands have been well explored in the formation of Ln-MOFs luminescence materials, not only because their carboxyl moieties can adopt multiple coordination modes to give high-dimensional unique structures with good thermal stability, but also because they can increase light absorption, provide blue color sources, and act as excellent sensitizers to activate Ln3+ ions.20–24Homochiral metal–organic frameworks (HMOFs) have attracted special attention owing to their fascinating structures and intriguing potential applications in asymmetric catalysis and enantioselective separation.25–33 In contrast, studies on lanthanide-based homochiral metal–organic frameworks (Ln-HMOFs) as multicolor luminescence materials are still quite limited at present, although they have unique optical properties, such as nonlinear optics, circular dichroism and chiral polarized photoemission,16,34–37 which could further expand its application in luminescent materials. Herein, (S)-5-(((1-carboxyethyl)amino)methyl)isophthalic acid (H3L, Scheme 1), a typical aromatic polycarboxylate linker being synthesized and used in our recent fabrication of novel transition metal HMOFs where H3L ligand displays various existing forms and versatile coordination models and also transmits its chirality into the whole frameworks,38 was selected as a chiral bridge and sensitizer to construct Ln-HMOFs. We report the preparation of isostructural Ln-HMOFs, {[Ln2(HL)2(H2O)4]·2Cl·5H2O}n [Ln = Gd (1), Eu (2), four Tb (3) and Dy (4)], and a series of single-phase mixed-lanthanide HMOFs analogues. More importantly, color tuning and white light-emission can be easily attained by carefully adjusting the relative concentration of lanthanum ions in the resulting 2D Ln-HMOFs. Furthermore, crystal structure, circular dichroism, luminescent mechanism, color tuning, and cation sensing have been investigated in detail.
 | ||
| Scheme 1 Schematic representation of chiral ligand H3L and the coordination modes of its zwitterionic (HL)2− in 1–4. | ||
Experiment
Materials and general procedures
All the reagents were of analytical grade and obtained from commercial sources without further purification. Enantiopure ligand (S)-5-(((1-carboxyethyl)amino)methyl)isophthalic acid (H3L) were prepared according to our reported procedure.38 Elemental analyses were performed with a Carlo-Erba 1106 elemental analyzer. IR spectra (KBr pellets) were recorded in the range 400–4000 cm−1 on a Nicolet NEXUS 470 FT-IR spectrophotometer (Fig. S1†). Thermal analysis curves were scanned from 30 to 800 °C under air on a STA 409 PC thermal analyzer (Fig. S2†). X-ray diffraction (PXRD) patterns of the samples were recorded on a RIGAKU-DMAX2500 X-ray diffractometer with Cu Kα radiation (Fig. S3–S4†). The luminescence spectra for the solid and suspension samples were determined at room temperature on a Hitachi F-4500 fluorophotometer with a xenon arc lamp as light source. The fluorescence lifetime τ was determined by a FLS980 fluorescence spectrometer. The emission quantum yield measurements were carried out on solid samples using an integrating sphere on an Edinburgh FLS980 fluorescence spectrometer. Solid-state circular dichroism (CD) spectra (KBr pellets) were recorded at room temperature on a MOS-450 spectrometer. Inductively coupled plasma (ICP) spectroscopy was detected on a Thermo ICAP 6000 DUO spectrometer. On the basis of international CIE standards, the Commission International de I'Eclairage (CIE) color coordinates were calculated. The general colour rendering index (CRI) were designated by the symbol Ra, which is the average value of R1 to R8.39,40 The numbers in parentheses indicate the Munsell colour system.41 The correlated colour temperature (CCT) values were obtained based on the corresponding CIE colour coordinates.Synthesis of compounds
All compounds including the mixed-lanthanide ones were synthesized in a similar procedure except for the different starting lanthanide salts. The synthesis of {[Gd2(HL)2(H2O)4]·2Cl·5H2O}n (1) is detailedly introduced as a representative: a mixture of H3L (0.0134 g, 0.05 mmol), GdCl3·6H2O (0.0183 g, 0.05 mmol), deionized H2O (0.5 mL) and acetonitrile (2 mL) was sealed in a 25 mL Teflon-lined stainless autoclave and heated at 120 °C for 24 h. After the mixture was cooled to room temperature at a rate of 5 °C h−1, colorless block crystals were obtained, washed with distilled water, and dried in air, resulting in 62% yield (based on Gd). Anal. calcd for C24H40Cl2Gd2N2O21 (%): C, 26.74; H, 3.74; N, 2.60. Found: C, 26.58; H, 3.75; N, 2.58. IR (KBr, cm−1): 3439 (m), 2960 (w), 1641 (s), 1553 (s), 1534 (s), 1451 (s), 1395 (s), 1249 (w), 1116 (w), 816 (w) 780 (s), 732 (s).{[Eu2(HL)2(H2O)4]·2Cl·5H2O}n (2). Yield 62% (based on Gd). Anal. calcd for C24H40Cl2Eu2N2O21 (%): C, 27.01; H, 3.78; N, 2.62. Found: C, 26.90; H, 3.81; N, 2.65. IR (KBr, cm−1): 3428 (m), 2981 (w), 1640 (s), 1552 (s), 1534 (s), 1449 (s), 1393 (s), 1249 (w), 1112 (w), 815 (w) 781 (s), 731 (s).
{[Tb2(HL)2(H2O)4]·2Cl·5H2O}n (3). Yield 64% (based on Tb). Anal. calcd for C24H40Cl2N2O21Tb2 (%): C, 26.66; H, 3.73; N, 2.59. Found: C, 26.44; H, 3.75; N, 2.61. IR (KBr, cm−1): 3443 (m), 2952 (w), 1643 (s), 1548 (s), 1539 (s), 1457 (s), 1396 (s), 1250 (w), 1114 (w), 816 (w) 780 (s), 733 (s).
{[Dy2(HL)2(H2O)4]·2Cl·5H2O}n (4). Yield 65% (based on Dy). Anal. calcd for C24H40Cl2Dy2N2O21 (%): C, 26.48; H, 3.70; N, 2.57. Found: C, 26.37; H, 3.73; N, 2.56. IR (KBr, cm−1): 3430 (m), 2952 (w), 1646 (s), 1550 (s), 1540 (s), 1456 (s), 1397 (s), 1249 (w), 1111 (w), 818 (w) 782 (s), 732 (s).
As for the isostructural mixed-lanthanide analogues, Eu3+/Tb3+ or Eu3+/Gd3+/Tb3+ codoped compounds adopt the same methods as those mentioned above just by adding the corresponding LnCl3·6H2O (Ln3+ = Gd3+, Eu3+ or Tb3+) as the starting materials in different stoichiometric ratios. Their structure and purity were confirmed by PXRD (Fig. S4†), IR spectra (Fig. S1†), and elemental analyses (ICP) (Table S1†).
Luminescence sensing experiments
A ground sample (10 mg) of the Tb-HMOF was dispersed in an aqueous solution (3 mL) of M(NO3)x (Mx+ = Na+, K+, Ag+, Cd2+, Ba2+, Mg2+, Mn2+, Ni2+, Zn2+, Cu2+, Co2+, Hg2+, Pb2+, Fe3+, Al3+, or Cr3+, 1 × 10−3 mol L−1) which was ultrasonicated for 30 min at room temperature. The resulted mixtures were used for luminescent measurements.X-ray structure determination
On a Bruker APEX-II CCD diffractometer, single-crystal X-ray data of 1–4 were collected at 293(2) K using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). Absorption corrections were applied by using the multiscan program SADABS.42 The structures were solved by direct methods and refined on F2 full-matrix least-squares using the SHELXTL program package.43,44 All of the non-hydrogen atoms were refined with anisotropic displacement parameters during the final cycles. The H atoms attached to C were generated geometrically while the H atoms attached to O and N were located from different Fourier maps and treated as idealized contributions. Unluckily, the H atoms of some water molecules were not added owing to not suitable Fourier maps being picked up. Notably, the precision of the C–C bonds of 2–4 is slightly low, which perhaps results from the weak diffractions owing to the bad quality and small sizes of the single crystals and/or from the effects of heavy Ln atoms whose strong diffraction signals affect the accurate detection of lighter atoms C. Further attempts to get suitable single crystals by different methods as well as the recollecting crystal data at a lower temperature were of fail. The crystal data were summarized in Table 1, and the selected bond distances were given in Table S2.†| Compounds | 1 | 2 | 3 | 4 |
| Formula | C24H40Cl2Gd2N2O21 | C24H40Cl2Eu2N2O21 | C24H40Cl2Tb2N2O21 | C24H40Cl2Dy2N2O21 |
| Temp (K) | 293(2) | 293(2) | 293(2) | 293(2) |
| Formula weight | 1077.98 | 1067.40 | 1081.32 | 1088.48 |
| Crystal system | Triclinic | Triclinic | Triclinic | Triclinic |
| Space group | P1 | P1 | P1 | P1 |
| a (Å) | 9.6682(6) | 9.6686(6) | 9.6763(4) | 9.6947(4) |
| b (Å) | 10.0302(6) | 10.0202(4) | 10.0338(4) | 10.0533(5) |
| c (Å) | 10.9110(7) | 10.8727(4) | 10.9853(5) | 11.0032(7) |
| α/° | 104.893(2) | 104.824(3) | 105.072(3) | 105.039(5) |
| β/° | 93.675(2) | 93.710(5) | 93.691(3) | 93.823(4) |
| γ/° | 110.003(2) | 109.569(5) | 110.107(4) | 110.052(4) |
| V (Å3) | 947.30(10) | 946.31(8) | 953.10(7) | 958.48(9) |
| Z, ρcalcd (g cm−3) | 1, 1.890 | 1, 1.873 | 1, 1.884 | 1, 1.886 |
| GOF | 1.061 | 1.052 | 1.060 | 1.051 |
| Flack parameter | 0.195(8) | 0.20(3) | 0.25(2) | 0.22(2) |
| R1, wR2 (I > 2 σ(I)) | 0.0164, 0.0427 | 0.0603, 0.1298 | 0.0415, 0.0804 | 0.0467, 0.0840 |
| Largest diff. peak and hole | 0.668, −0.517 | 3.370, −2.031 | 0.994, −0.778 | 1.159, −0.985 |
Results and discussion
Synthesis and general characterization of compounds
The isostructural Ln-HMOFs including the mixed-lanthanide species were solvothermally synthesized at 120 °C using the corresponding LnCl3·6H2O and the chiral ligand H3L in a mixed solvent of deionized H2O and acetonitrile. The crystalline products are air-stable and insoluble in water or any common organic solvents.The chemical formulas of single-lanthanide compounds 1–4 have been confirmed by satisfactory elemental analysis and single-crystal X-ray diffraction. In the IR spectra of 1–4, the strong and broad absorption bands in a range of 3400–3500 cm−1 indicates the presence of νN–H and the νO–H stretching frequencies of amino groups and water molecules, respectively. Four complexes exhibit strong characteristic absorptions around 1534–1646 cm−1 for νas(–COO−) and 1393–1457 cm−1 for νs(–COO−), respectively. However, no strong characteristic absorptions around 1700 cm−1 for (–COOH) were observed, being in agreement with the single-crystal X-ray diffraction analysis results that all carboxyl groups of ligand H3L in compounds 1–4 were deprotonated. The accurate molar ratios of the individual lanthanide elements in the mixed-lanthanide compounds have been determined by inductively coupled lasma (ICP) spectroscopy (Table S1†), being well consistent with the corresponding ratios in the starting mixture. The phase purities and isostructuralism of all the as-synthesized Ln-HMOFs have been verified by powder X-ray diffraction (PXRD) measurements. Clearly, the experimental PXRD patterns of single-lanthanide compounds 1–4 are very similar to each other and match well with those simulated, indicating the phase purities and isostructuralism of those polycrystalline samples (Fig. S3†). Meanwhile, the observed PXRD patterns of all the bi- and tri-metallic doped samples are in conformity to the simulated pattern of 1 (Fig. S4†), demonstrating that they are isostructural with single-lanthanide compounds 1–4. Single-lanthanide compounds 1–4 all experience a very similar three-stepped thermal decomposition behavior revealed by TGA (Fig. S2†). The first weight loss corresponds to the removal of five lattice water molecules (calcd. 8.36%, obsed. 7.70% for 1; calcd. 8.44%, obsed. 7.60% for 2; calcd. 8.33%, obsed. 8.23% for 3; calcd. 8.28%, obsed. 7.65% for 4) before 130, 135, 150, and 155 °C, respectively. The second weight loss occurs in a temperature range of 155–230 °C, being attributable to the release of four coordinated water molecules (calcd. 6.68%, obsed. 6.60% for 1; calcd. 6.75%, obsed. 6.62% for 2; calcd. 6.66%, obsed. 6.10% for 3; calcd. 6.62%, obsed. 6.07% for 4). The removal of the organic components (calcd. 51.33%, obsed. 48.65% for 1; calcd. 51.84%, obsed. 48.02% for 2; calcd. 51.17%, obsed. 48.84% for 3; calcd. 50.83%, obsed. 49.45% for 4) occurred in a temperature range of 300–700 °C. The final residue for each complex corresponds to the formation of the respective lanthanide oxide. In addition, the thermal decomposition behavior of the trimetallic doped sample has been also investigated as a representative of the mixed-lanthanide species, and the result shows that its thermal decomposition behavior (weight loss and decomposition temperature) is surprisingly similar to that of all single-lanthanide compounds (Fig. S2†). The TGA results show that these single- and mixed-lanthanide frameworks have higher thermal stabilities being suitable for potential applications in solid-state luminescence materials, and again confirm the number of lattice and coordinated water molecules in their chemical formulas.
Crystal structure descriptions
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) in their structural models, this can be due to the pseudotranslation symmetry of heavy Ln atoms.45,46 Their chirality was further confirmed by the solid-state CD spectra (vide infra). As a representative case, only the crystal structure of 1 is herein discussed in detailed. The asymmetric unit of 1 consists of two (HL)2− ligands, two independent Gd3+ ions, four coordinated water molecules, five lattice water molecules, and two free Cl−. Gd1 and Gd2 are nine-coordinated by seven O atoms from two chelating carboxylates and three bridging carboxylates of five different (HL)2− ligands and two O atoms from water molecules, respectively, forming distorted single-capped square antiprism (C4v) polyhedron configurations (Fig. 1a). The Gd–O bond lengths vary from 2.310(8) Å to 2.925(8) Å, which corresponds to those reported for other lanthanide–oxygen donor complexes,47 and the O–Gd–O angles range from 47.4(2) to 148.2(3)°. Every H3L ligand in 1, with its three carboxyls being completely deprotonated and meanwhile its amino group being protonated, exists in a zwitterionic form of (HL)2− as observed in its Pb-HMOF.38 Interestingly, the three carboxylate groups of (HL)2− adopt three different coordination modes, namely, chelating fashion μ1 − η1:η1, chelating and bridging fashion μ2 − η2:η1, and bridging fashion μ2 − η1:η1, unlike to those found in its transition metal frameworks.38 Consequently, ligand (HL)2− acts as a κ7-linker to connect five Gd3+ ions.
in their structural models, this can be due to the pseudotranslation symmetry of heavy Ln atoms.45,46 Their chirality was further confirmed by the solid-state CD spectra (vide infra). As a representative case, only the crystal structure of 1 is herein discussed in detailed. The asymmetric unit of 1 consists of two (HL)2− ligands, two independent Gd3+ ions, four coordinated water molecules, five lattice water molecules, and two free Cl−. Gd1 and Gd2 are nine-coordinated by seven O atoms from two chelating carboxylates and three bridging carboxylates of five different (HL)2− ligands and two O atoms from water molecules, respectively, forming distorted single-capped square antiprism (C4v) polyhedron configurations (Fig. 1a). The Gd–O bond lengths vary from 2.310(8) Å to 2.925(8) Å, which corresponds to those reported for other lanthanide–oxygen donor complexes,47 and the O–Gd–O angles range from 47.4(2) to 148.2(3)°. Every H3L ligand in 1, with its three carboxyls being completely deprotonated and meanwhile its amino group being protonated, exists in a zwitterionic form of (HL)2− as observed in its Pb-HMOF.38 Interestingly, the three carboxylate groups of (HL)2− adopt three different coordination modes, namely, chelating fashion μ1 − η1:η1, chelating and bridging fashion μ2 − η2:η1, and bridging fashion μ2 − η1:η1, unlike to those found in its transition metal frameworks.38 Consequently, ligand (HL)2− acts as a κ7-linker to connect five Gd3+ ions.
 | ||
| Fig. 1 (a) Coordination geometries of Gd3+ in 1; (b) binuclear cluster [Gd2(COO)4] unit; (c) wave-like homochiral layer structure of 1 based on binuclear cluster units [Gd2(COO)4] and zwitterionic (HL)2− linkers. Symmetry code: (A) x, 1 + y, z; (B) −1 + x, y, z; (C) 1 + x, 1 + y, z. | ||
Notably, the two independent Gd3+ ions are connected by four carboxylate groups from four (HL)2− ligands to form a binuclear cluster [Gd2(COO)4] (Fig. 1b), which is connected by (HL)2− to produce a wave-like homochiral layer paralleling to ab plane (Fig. 1c). In the layer, each (HL)2− ligand bonds three [Gd2(COO)4] units. Topologically, the (HL)2− ligand and [Gd2(COO)4] unit can be considered as 3- and 6-connected nodes, respectively. Thus, the whole framework of 1 can be described as a (3,6)-connected kgd net with point (Schläfli) symbol of (43)2(46.66.83) [Fig. 2]. Those layers stack up along c axis with their chiral aminopropionate groups arraying up and down. Moreover, every protonated amino group of (HL)2− ligand hydrogen-bonds two independent chloride anions (Cl1 and Cl2) which are further hydrogen-bonded by another amino group from the adjacent layer, and in such a way it results in an interesting 3D porous homochiral pillared-layer supramolecular framework with lattice water molecules in apertures when regarded the hydrogen-bonded (N2Cl2) rings as pillars (Fig. 3).
 | ||
| Fig. 2 Schematic representation of 2D (3,6)-connected kgd topology of 1. | ||
 | ||
| Fig. 3 View of interesting 3D porous homochiral pillared-layer supramolecular framework of 1, regarding the hydrogen-bonded (N2Cl2) rings as pillars (N2⋯Cl1 = 3.209(10) Å, N2⋯Cl2 = 3.071(9) Å, N1D⋯Cl1 = 3.129(9) Å, N1D⋯Cl2 = 3.210(10) Å; lattice water molecules in apertures were omitted for clarity). Symmetry code: (D) −1 + x, y, 1 + z. | ||
It should be mentioned that with the radium decrease of Ln atoms the κ7-linker (HL)2− in 1 and 2 becomes the κ6-linker (HL)2− in 3 and 4 owing to the chelating and bridging fashion μ2 − η2:η1 of the carboxylate becoming the bridging fashion μ2 − η1:η1 (Scheme 1, Fig. S5–S7†). As a result, both Tb3+ in 3 and Dy3+ in 4 adopt eight-coordinated slightly distorted square antiprism (C4v) polyhedron configurations (Fig. S6–S7†). In their binuclear clusters [Tb2(COO)4] and [Dy2(COO)4], the bridging model between the two metal centers is slightly different from that found in the binuclear clusters [Gd2(COO)4] and [Eu2(COO)4] (Fig. 1 and S5–S7†).
Solid-state circular dichroism (CD) studies
To further demonstrate their homochirality, solid-state CD spectra of 1–4 have been measured with KBr pellets (Fig. 4). In their CD spectra, obvious chiroptical signals have been observed in the ultra band of 200–400 nm, which perhaps originate in the π → π* transitions of (HL)2− ligand. For 1, positive Cotton effects center at 203 and 224 nm, and negative Cotton effects center at 218 and 295 nm. For 2, positive Cotton effects center at 205, 223 and 240 nm, and negative Cotton effects center at 217 and 234 nm. The CD spectrum of 3 exhibits positive Cotton effects centered at 203 and 240 nm, and negative Cotton effects centered at 220 and 235 nm. As for 4, it displays positive Cotton effects centered at 203, 242 and 270 nm, and negative Cotton effects centered at 219 and 249 nm. From the above results, the inherent chirality of H3L ligand was transmitted to the resulting Ln-HMOFs. | ||
| Fig. 4 Solid-state CD spectra of compounds 1–4. | ||
Photoluminescence properties
The lanthanide MOFs usually show excellent luminescent properties. Hence, the photoluminescence of complexes 1–4 and their lifetimes were investigated at room temperature. The excitation spectra for free ligand H3L and complexes 1–4 are given in Fig. S8–S9.† Upon excitation at 310 nm, the free ligand H3L exhibits a broad fluorescent emission centered at 369 nm (Fig. 5a), maybe attributing to intraligand π* → π charge transfer. Complex 1 presents a broad blue emission centered at 443 nm (λex = 365 nm) [Fig. 5b], also assignable to the intraligand charge transition of (HL)2− ligand. Compared with the emission of free ligand H3L, the emission peak of 1 red-shifts by 74 nm, indicating obvious influence upon the HOMO and LUMO levels of the coordinated ligand (HL)2− as well as the energy transitions between them owing to the coordination of Gd3+ ions. This phenomenon is similar to the previously reported.48 The CIE color coordinate is (0.198, 0.214) for 1. Upon excitation at 395 nm, 2 emits the bright characteristic red color of Eu3+ ions with the CIE color coordinate of (0.587, 0.316). The emission peaks at 593, 615, 653 and 701 nm are assigned to 5D0 → 7FJ (J = 1, 2, 3, 4) transitions of Eu3+ ions, respectively, without the emission peak corresponding to 5D0 → 7F0 transition being observed (Fig. 5c). The electric dipole transition 5D0 → 7F2 dominates the red color emission while the 5D0 → 7F1 transition is insensitive to the site symmetry as a magnetic dipole. For 2 the intensity ratio of I(5D0 → 7F2)/I(5D0 → 7F1) is about 5.28, indicating that the Eu3+ ions locate at a low symmetric coordination environment without an inversion center.34,37 This result is in agreement with the crystal structural analysis result that every Eu3+ ion adopts a distorted noncentral single-capped square antiprism (C4v) polyhedron configurations (Fig. S6†). Upon excitation at 352 nm, complex 3 exhibits characteristic Tb3+ emissions at 490, 545, 586 and 622 nm, which can be attributed to 5D4 → 7FJ (J = 6, 5, 4, 3) transitions, respectively (Fig. 5d). The most intense transition of 5D4 → 7F5 leads to strong green luminescence of the crystal sample, which makes 3 become a promising green emitting phosphors with the CIE color coordinate of (0.278, 0.541). | ||
| Fig. 5 Solid-state emission spectra of free ligand H3L (a), 1 (b), 2 (c), and 3 (d) at room temperature. The insets show the corresponding luminescence pictures under UV-light irradiation at 365 nm. | ||
It is worth noting that the ligand-based emission in the fluorescence spectra of compounds 2 and 3 was not observed, indicating effective energy transfer from ligand (HL)2− to the lanthanide centers via “antenna effect”. The luminescence decay curves of 1–3 were obtained in the solid state at room temperature (Fig. S10–S12†). Complexes 1–3 display the single-exponential luminescent decay with the lifetimes of 9.88, 352.95 and 826.26 μs, respectively, and the quantum yields Φoverall are 3.9% for complex 2 and 5.2% for complex 3, respectively. When excited at 365 nm at room temperature, compound 4 displays blue photoluminescence mainly originating from the intraligand charge transition of (HL)2− ligand (Fig. S9†).
Tuning of luminescent color for bimetallic doped Eu/Tb-HMOFs
The isostructuralism of the resulting Ln-HMOFs 1–4 gives a chance to synthesize heterometallic frameworks by codoping. A series of bimetallic doped Eu/Tb-HMOFs [(EuxTb1−x)2(HL)2(H2O)4]·2Cl·5H2O were successfully obtained by adjusting different molar ratios of Eu3+ and Tb3+ reactants, and the molar ratio of Eu3+ and Tb3+ ions was determined by means of ICP spectroscopy (Table S1†).As shown in Fig. 6a, the bimetallic doped Eu/Tb-HMOFs exhibit the dual emissions of Eu3+ and Tb3+ ions in their luminescent spectra when excited at 370 nm. With the increase of Eu3+/Tb3+ molar ratios, the emission intensity of Tb3+ at 545 nm (5D4 → 7F5) decreases monotonically, while that of Eu3+ at 615 nm (5D0 → 7F2) increases stepwise to reach a maximum when the mole fraction of Eu3+ is 29.23 mol%, and then decreases slightly due to the concentration quenching effect.17,49 The observation suggests the enhanced probability of energy transfer from Tb3+ to Eu3+ with the increase of Eu3+ concentration.15,17,19,49 Furthermore, the fluorescence decay of the 5D4 → 7F5 transition of Tb3+ and the 5D0 → 7F2 transition of Eu3+ in the bimetallic doped samples of [(EuxTb1−x)2(HL)2(H2O)4]·2Cl·5H2O (x = 14.88–38.51 mol%) was also investigated. The results show that with the increase of Eu3+ doping, the obtained effective lifetime of Tb3+ shortens while that of Eu3+ basically increases (Table S3 and Fig. S13†), which indicates that the concentration of doped Eu3+ significantly changes the fluorescent dynamics of Tb3+ and further confirm an efficient energy transfer from Tb3+ to Eu3+. Notably, the lifetime of Eu3+ also reaches a maximum when the mole fraction of Eu3+ is 29.23 mol%, and then shortens slightly with the further increase of Eu3+ doping, reflecting the above mentioned concentration quenching effect. In addition, the observed energy transfer between Tb3+ and Eu3+ also confirms that the as-synthesized bimetallic doped compounds are single-phase coordination polymers where Eu3+ has been successfully incorporated into the lattice of 3 because the energy transfer could not occur in a mixture of separated phases.
 | ||
| Fig. 6 (a) Solid-state emission spectra of the bimetallic Eu/Tb-HMOFs [(EuxTb1−x)2(HL)2(H2O)4]·2Cl·5H2O when excited at 370 nm (x = 10.08–79.01 mol%); (b) the CIE chromaticity diagram for Eu-HMOF (2), Tb-HMOF (3), and the bimetallic doped Eu/Tb-HMOFs. The inset shows the luminescence picture of the bimetallic doped Eu/Tb-HMOFs under UV-light irradiation at 365 nm. | ||
Interestingly, the bimetallic doped Eu/Tb-HMOFs present a wide range of visible emission colors such as green, yellow, orange, orange-red, and red under UV-light irradiation at 365 nm (Fig. 6b), indicating that the luminescent colors of the bimetallic doped Eu/Tb-HMOFs [(EuxTb1−x)2(HL)2(H2O)4]·2Cl·5H2O can be systematically modulated through adjusting different mole fractions of Tb3+ and Eu3+. At the same time, with the change of the concentration of the doping Eu3+ in the heterometallic frameworks, the corresponding CIE chromaticity coordinates change from (0.325, 0.468) to (0.470, 0.287) (Fig. 6b and Table S4†).
WLED for trimetallic doped Eu/Gd/Tb-HMOF
Being considered as fourth-generation light sources, white-light-emitting diodes (WLEDs) have greatly attracted current interest owing extensive applications in displays and lighting. Isostructural compounds 1, 2 and 3, as observed above, exhibit blue, red and green primary colors, respectively, which is expected that white light emission could be obtained by the doping of Eu3+ and Tb3+ ions into the Gd3+ compound 1. Indeed, the emission output of Eu/Gd/Tb-HMOFs can be controlled precisely through the compositional adjustment of the three metal ions. By optimizing the molar ratio of Eu3+, Gd3+ and Tb3+ ions, the trimetallic doped Eu/Gd/Tb-HMOF [(Eu0.1388Gd0.6108Tb0.2504)2(HL)2(H2O)4]·2Cl·5H2O was synthesized successfully. Upon excitation at 370 nm, this three-component compound simultaneously shows the triple emissions originating from the emissions of Eu3+, Tb3+and the coordinated ligand (HL)2−, respectively (Fig. 7a), and its CIE color coordinate is (0.285, 0.298). Furthermore, we have systematically studied the subtle effects of excitation wavelength changes upon the luminescent color of the trimetallic doped Eu/Gd/Tb-HMOF (Fig. 7b and c). As shown in Fig. 6b, when the excitation wavelength is controlled from 310 to 370 nm, the emission color of the trimetallic doped Eu/Gd/Tb-HMOF changes from yellow to white (CIE coordinates in Table S5†). The white emission of the trimetallic doped Eu/Gd/Tb-HMOF has been obtained upon excitation at 355 nm [the CIE color coordinate is (0.317, 0.331); the average lifetime values for Tb3+and Eu3+ ions are 753.59 and 473.03 μs, respectively (Fig. S14†), and the overall quantum yield is about 4.2%,], which is very close to that of pure white light (0.333, 0.333) according to 1931 CIE coordinate diagram. The colour rendering index (CRI) and corresponding colour temperature (CCT) are 82 and 6261 K, respectively, which satisfy the necessary conditions for high quality white light-emitting materials (CRI above 80 and CCT in the range of 2500–6500 K). Although the quantum yield value is smaller than that of several recent trimetallic doped 3D Eu/Gd/Tb-MOFs,15,19 studies on 2D Ln-HMOFs as multicolor luminescence materials are fewer at present.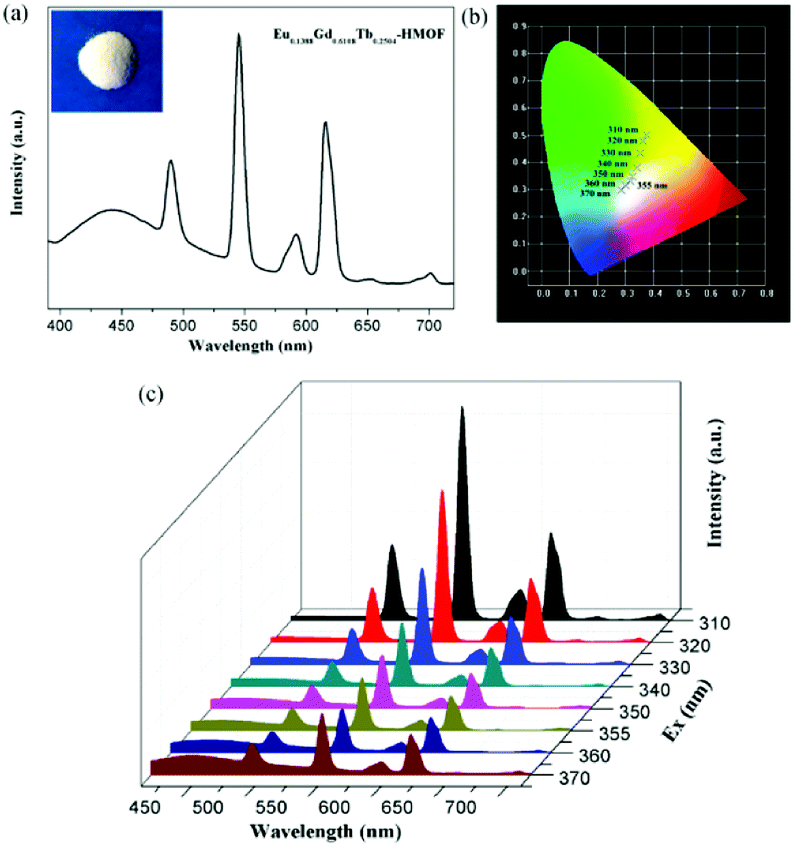 | ||
| Fig. 7 (a) Solid-state emission spectra of the trimetallic doped Eu/Gd/Tb-HMOF [(Eu0.1388Gd0.6108Tb0.2504)2(HL)2(H2O)4]·2Cl·5H2O when excited at 370 nm (the inset shows the corresponding luminescence picture under UV-light irradiation at 365 nm); (b) the CIE-1931 chromaticity diagram for the trimetallic doped Eu/Gd/Tb-HMOF with excitation wavelengths varying from 310 to 370 nm; (c) solid-state emission spectra of the trimetallic doped Eu/Gd/Tb-HMOF with excitation wavelengths varying from 310 to 370 nm. | ||
Luminescent sensing of metal ions
To research the potential of Tb-HMOF for sensing of metal ions, the ground samples of the as-synthesized Tb-HMOF were dispersed in aqueous solutions of M(NO3)x (Mx+ = Na+, K+, Ag+, Cd2+, Ba2+, Mg2+, Mn2+, Ni2+, Zn2+, Cu2+, Co2+, Hg2+, Pb2+, Fe3+, Al3+, and Cr3+), and handled with ultrasonication treatment for 30 min to form a metal-ion-incorporated Tb-HMOF suspension whose fluorescent measurements were carried out at room temperature upon excited at λex = 352 nm. As shown in Fig. 8, the incorporated various metal ions exhibit different influences upon the luminescence of Tb3+. For the incorporated Na+, K+, Cd2+, Ba2+, Mg2+, Mn2+, Ni2+, Zn2+, Co2+, Hg2+, or Pb2+, the effect on the luminescence intensity at 545 nm of Tb-HMOF is negligible. However, for the incorporated Ag+, Cu2+, Cr3+, Al3+, or Fe3+, they display different degrees of quenching on the fluorescence of Tb3+ with obviously distinguished spectra: Ag+, Al3+ or Cu2+ has a certain quenching effect on the luminescence intensity, whereas Cr3+ or Fe3+ has a rapidly quenching effect on the luminescence intensity, and especially for Fe3+ it can efficiently and completely quench the emission of Tb3+. In particular, accompanied by the quenching effect on the fluorescence of Tb3+, the incorporated Al3+ can largely enhance the ligand-related emission (Fig. 8a). These results suggest that the fluorescence of Tb-HMOF is very sensible to Fe3+, Cr3+, and Al3+ ions, and perhaps has potential application in ion detection. | ||
| Fig. 8 (a) Emission spectra of Tb-HMOF (10 mg) dispersed in various nitrate salts aqueous solutions (3 mL, 10−3 M). (b) Cation selectivity of the Tb-HMOF (I0/I) in H2O. | ||
To further understand the fluorescent sensing behaviours of Tb-HMOF to Fe3+, Cr3+ and Al3+, concentration-dependent luminescence measurements were carried out. The as-synthesized Tb-HMOF samples were ground and immersed in different concentrations of Fe3+, Al3+, or Cr3+, and then their luminescence spectra were recorded. As shown in Fig. 9 and S15,† the emission intensity of Tb-HMOF suspension is rapidly quenched with increasing concentration Fe3+ or Cr3+. When the concentration reaches 5 mM for Fe3+ or 30 mM for Cr3+, the luminescence of Tb3+ is almost completely quenched. Comparatively, the fluorescent quenching effect of Fe3+ on the Tb-HMOF is more quick and thorough than that of Cr3+. For the Al3+-incorporated sample, the luminescence intensity of Tb3+ decreases slowly and the emission of the ligand-related emission centered at 425 nm increases gradually with increasing concentration Al3+. When the Al3+ concentration reaches 10 mM, clear quenching of the luminescence of Tb3+ can be seen (Fig. 10). As the concentration of Al3+ increases to 100 mM, the luminescence of Tb3+ is nearly completely quenched and only the emission of the ligand can be observed. Further with the increase of Al3+ concentration, Tb-HMOF dissolves gradually, and finally forms a clear aqueous solution as the Al3+ concentration reaches 0.1 M. This phenomenon has been reported in previous literature.50
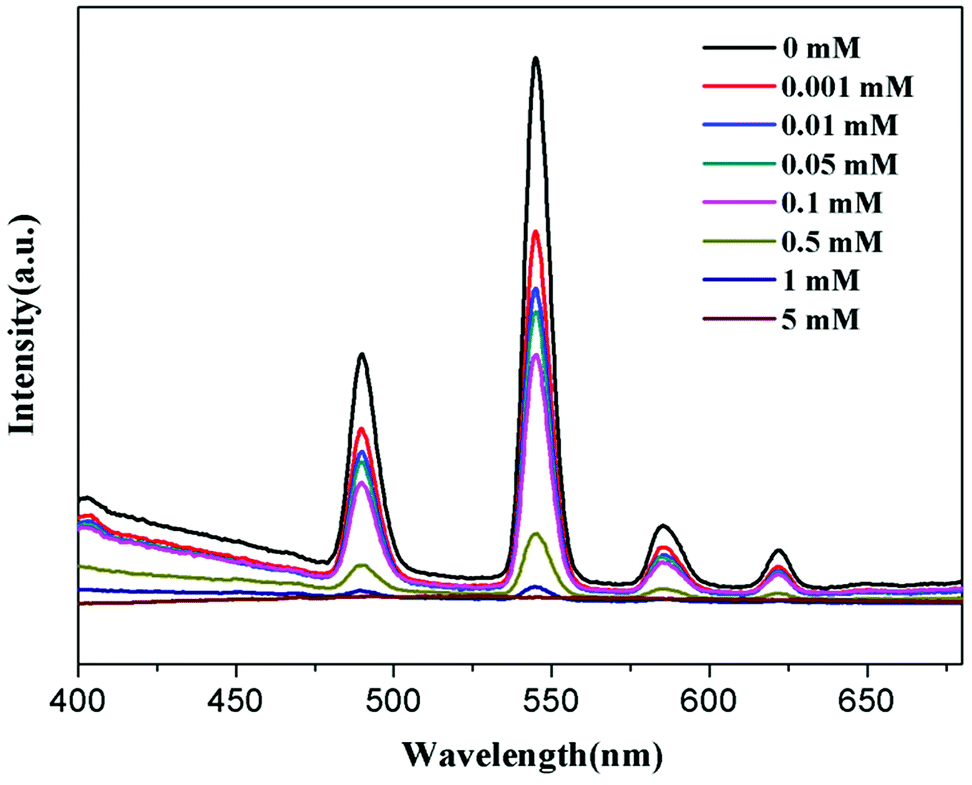 | ||
| Fig. 9 Emission spectra of Tb-HMOF in aqueous solutions of various concentrations of Fe3+ under excitation at 352 nm. | ||
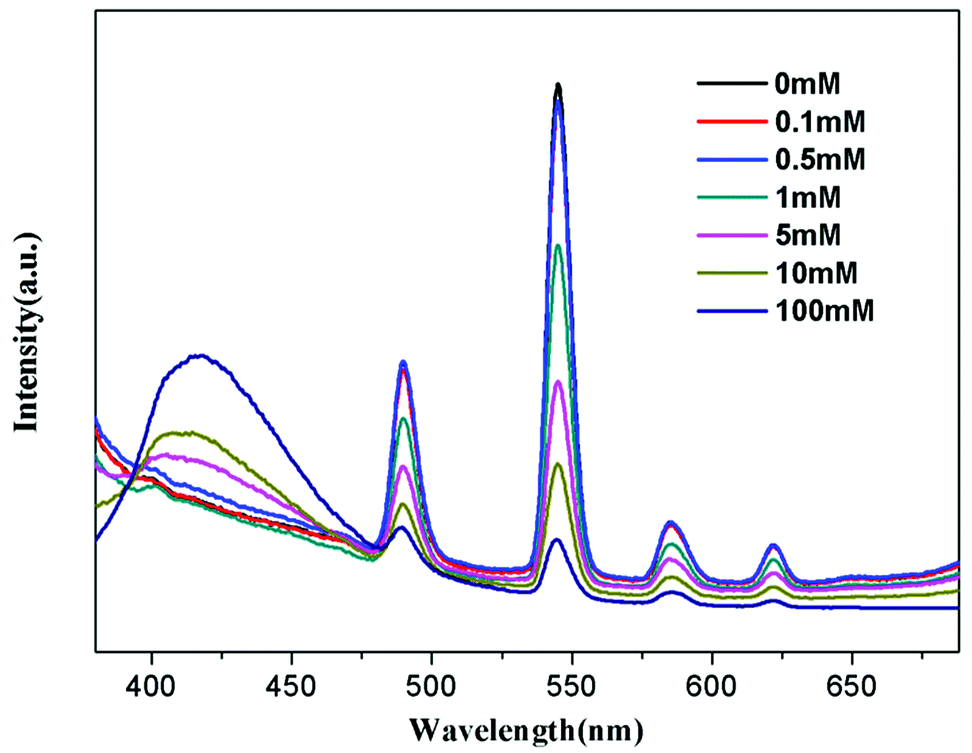 | ||
| Fig. 10 Emission spectra of Tb-HMOF in aqueous solutions of various concentrations of Al3+ under excitation at 352 nm. | ||
Conclusions
In summary, we have successfully synthesized a series of novel Ln-HMOFs based on chiral ligand (S)-5-(((1-carboxyethyl)amino)methyl)isophthalic acid (H3L). All complexes are isostructural with (3,6)-connected homochiral frameworks whose phase purities and chirality are further confirmed by PXRD and CD studies, respectively. The luminescent investigation of 1–4 suggests that compounds 2 and 3 display the strong characteristic emissions of the corresponding Eu3+ and Tb3+ ions, while 1 and 4 exhibit blue emissions arising from ligand (HL)2−. By doped different Ln3+ ions into single-phase Ln-HMOFs, the bimetallic Eu/Tb-HMOFs [(EuxTb1−x)2(HL)2(H2O)4]·2Cl·5H2O and trimetallic Eu/Gd/Tb-HMOF [(Eu0.1388Gd0.6108Tb0.2504)2(HL)2(H2O)4]·2Cl·5H2O have been successfully fabricated. The emission colors of bimetallic Eu/Tb-HMOFs can be tuned among green, yellow, orange, orange-red, and red by adjusting the doping concentration of Eu3+ ions into the Tb-HMOF. Very important, the trimetallic doped Eu/Gd/Tb-HMOF [(Eu0.1388Gd0.6108Tb0.2504)2(HL)2(H2O)4]·2Cl·5H2O features white light emission upon excitation at 355 nm, whose emission can also be switched between different colors when excited with different ultraviolet light. Furthermore, the fluorescence response of Tb-HMOF to various usual metal ions, and especially fluorescent sensing behaviours to Fe3+, Cr3+ and Al3+ has been preliminarily investigated.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (21271157), and the Foundation and Research in Cutting-Edge Technologies in the Project of Henan Province (122300410092).Notes and references
- M. Roushan, X. Zhang and J. Li, Angew. Chem., Int. Ed., 2012, 51, 436–439 CrossRef CAS PubMed.
- Y. C. Liao, C. H. Lin and S. L. Wang, J. Am. Chem. Soc., 2005, 127, 9986–9987 CrossRef CAS PubMed.
- P. C. P. Lima, F. A. Almeida Paz, R. A. S. Ferreira, V. De Zea Bermudez and L. S. D. Carlos, Chem. Mater., 2009, 21, 5099–5111 CrossRef CAS.
- Y. S. Zhao, H. B. Fu, F. Q. Hu, A. D. Peng, W. S. Yang and J. N. Yao, Adv. Mater., 2008, 20, 79–83 CrossRef CAS.
- G. He, D. Guo, C. He, X. Zhang, X. Zhao and C. Duan, Angew. Chem., Int. Ed., 2009, 48, 6132–6135 CrossRef CAS PubMed.
- Q. Ju, D. Tu, Y. Liu, R. Li, H. Zhu, J. Chen, Z. Chen, M. Huang and X. Chen, J. Am. Chem. Soc., 2012, 134, 1323–1330 CrossRef CAS PubMed.
- J. Rocha, L. D. Carlos, F. A. A. Paz and D. Ananias, Chem. Soc. Rev., 2011, 40, 926–940 RSC.
- A. Schoedel, M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2016, 116, 12466–12535 CrossRef CAS PubMed.
- J. C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- L. D. Carlos, R. A. S. Ferreira, V. De Zea Bermudez, B. Julian-Lopez and P. Escribano, Chem. Soc. Rev., 2011, 40, 536–549 RSC.
- F. F. Chen, Z. Q. Chen, Z. Q. Bian and C. H. Huang, Coord. Chem. Rev., 2010, 254, 991–1010 CrossRef CAS.
- J. Heine and K. Muller-Buschbaum, Chem. Soc. Rev., 2013, 42, 9232–9242 RSC.
- H. Jeong, B. Lee, S. Byeon, H. Jeong, B. Lee and S. Byeon, ACS Appl. Mater. Interfaces, 2016, 8, 10946–10953 CrossRef CAS PubMed.
- L. Armelao, D. Belli Dell Amico, L. Bellucci, G. Bottaro, L. Labella, F. Marchetti and S. Samaritani, Inorg. Chem., 2016, 55, 939–947 CrossRef CAS PubMed.
- Q. Tang, S. X. Liu, Y. W. Liu, D. F. He, J. Miao, X. Q. Wang, Y. J. Ji and Z. P. Zheng, Inorg. Chem., 2014, 53, 289–293 CrossRef CAS PubMed.
- X. Yang, X. Lin, Y. Zhao, Y. S. Zhao and D. Yan, Angew. Chem., Int. Ed., 2017, 56, 7853–7857 CrossRef CAS PubMed.
- M. L. Ma, C. Ji and S. Q. Zang, Dalton Trans., 2013, 42, 10579–10586 RSC.
- J. Rong, W. Zhang and J. Bai, RSC Adv., 2016, 6, 103714–103723 RSC.
- Y. W. Zhao, F. Q. Zhang and X. M. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 24123–24130 CrossRef CAS PubMed.
- H. Xu, C. S. Cao, X. M. Kang and B. Zhao, Dalton Trans., 2016, 45, 18003–18017 RSC.
- K. X. Shang, J. Sun, D. C. Hu, X. Q. Yao, L. H. Zhi, C. D. Si and J. C. Liu, Cryst. Growth Des., 2018, 18, 2112–2120 CrossRef CAS.
- Y. Y. An, L. P. Lu, S. S. Feng and M. L. Zhu, CrystEngComm, 2018, 20, 2043–2052 RSC.
- Y. Yang, F. L. Jiang, C. P. Liu, L. Chen, Y. L. Gai, J. Pang, K. Z. Su, X. Y. Wan and M. C. Hong, Cryst. Growth Des., 2016, 16, 2266–2276 CrossRef CAS.
- J. Rong, W. Zhang and J. Bai, CrystEngComm, 2016, 18, 7728–7736 RSC.
- K. Kim, J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh and Y. J. Jeon, Nature, 2000, 404, 982–986 CrossRef PubMed.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed.
- Y. Liu, W. M. Xuan and Y. Cui, Adv. Mater., 2010, 22, 4112–4235 CrossRef CAS PubMed.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- K. K. Bisht, B. Parmar, Y. Rachuri, A. C. Kathalikattil and E. Suresh, CrystEngComm, 2015, 17, 5341 RSC.
- Z. G. Gu, C. Zhan, J. Zhang and X. Bu, Chem. Soc. Rev., 2016, 45, 3122–3144 RSC.
- K. Mo, Y. H. Yang and Y. Cui, J. Am. Chem. Soc., 2014, 136, 1746–1749 CrossRef CAS PubMed.
- Z. G. Gu, W. Q. Fu, M. Liu and J. Zhang, Chem. Commun., 2017, 53, 1470–1473 RSC.
- Z. X. Xu, Y. X. Tan, H. R. Fu, J. Liu and J. Zhang, Inorg. Chem., 2014, 53, 12199–12204 CrossRef CAS PubMed.
- Q. Yue, J. Yang, G. H. Li, G. D. Li and J. S. Chen, Inorg. Chem., 2006, 45, 4431–4439 CrossRef CAS PubMed.
- C. H. Gao, L. Zhang, G. F. Hou, D. S. Ma, W. H. Jiang and Y. H. Yu, Inorg. Chem. Commun., 2017, 78, 70–73 CrossRef CAS.
- L. Sun, G. F. Hou, W. H. Jiang, Q. Huang, Y. H. Yu and J. S. Gao, Polyhedron, 2017, 129, 55–59 CrossRef CAS.
- T. Liu, Y. H. Yu, H. Z. Zhang, W. H. Jiang, J. S. Gao and G. F. Hou, Cryst. Growth Des., 2017, 17, 1788–1795 CrossRef CAS.
- X. Wang, K. Zhang, L. Lv, R. Chen, W. Wang and B. Wu, Cryst. Growth Des., 2018, 18, 1799–1808 CrossRef CAS.
- H. S. Jang, H. Yang, S. W. Kim, J. Y. Han, S. G. Lee and D. Y. Jeon, Adv. Mater., 2008, 20, 2696–2702 CrossRef CAS PubMed.
- Y. Lu and B. Yan, Chem. Commun., 2014, 50, 15443–15446 RSC.
- G. N. Wyszecki and W. S. Stiles, Color Science: Concepts and Methods, Quantitative Data, and Formulae, John Wiley & Sons, New York, Wiley classics library edn, 2000 Search PubMed.
- G. M. Sheldrick, SADABS, Program for Empirical Absorption Correction of AreaDetector Data, University of Götingen, Götingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXS-97, Program for X-ray Crystal Structure Determination, University of Göttingen: Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXS-97, Program for X-ray Crystal Structure Refinement, University of Göttingen: Göttingen, Germany, 1997 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- B. Wu, S. Wang, R. Wang, J. Xu, D. Yuan and H. Hou, Cryst. Growth Des., 2013, 13, 518–525 CrossRef CAS.
- J. P. Costes, T. Yamaguchi, M. Kojima and L. Vendier, Inorg. Chem., 2009, 48, 5555–5561 CrossRef CAS PubMed.
- Z. J. Li, X. Y. Li, Y. T. Yan, L. Hou, W. Y. Zhang and Y. Y. Wang, Cryst. Growth Des., 2018, 18, 2031–2039 CrossRef CAS.
- K. Liu, H. You, Y. Zheng, G. Jia, Y. Song, Y. Huang, M. Yang, J. Jia, N. Guo and H. Zhang, J. Mater. Chem., 2010, 20, 3272–3279 RSC.
- L. H. Cao, F. Shi, W. M. Zhang and S. Q. Zang, Chem.–Eur. J., 2015, 21, 15705–15712 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and tables, TGA, PXRD, IR spectra, excitation and emission spectra, decay curves (PDF)s. CCDC 1852703–1852706 for 1–4. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra06793g |
| This journal is © The Royal Society of Chemistry 2018 |