
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGold nanorods or nanospheres? Role of particle shape on tuning the shape memory effect of semicrystalline poly(ε-caprolactone) networks†
Haikun Xu and
Bridgette Maria Budhlall *
*
Department of Plastics Engineering and Nanomanufacturing Center, University of Massachusetts, Lowell, MA 01854, USA. E-mail: Bridgette_Budhlall@uml.edu
First published on 17th August 2018
Abstract
In this study, modified poly(ε-caprolactone) (PCL) tri-block copolymers were successfully synthesized through ring opening polymerization. The nanocomposite films containing either colloidal gold nanorods (AuNRs) or gold nanospheres (AuNSs) in the polymer matrix were fabricated without chemical modification to realize light-responsive shape memory behaviors. The localized surface plasmon resonance of AuNPs was utilized by irradiating the selective wavelength of light to create a photothermal effect. The polymer microstructures were investigated by NMR, and the thermal properties of the polymer networks were studied by TGA and DSC. The addition of AuNPs did not change the melting temperature, (Tm) of the SMP. The AuNRs were fairly well dispersed within the PCL matrix as observed using SEM-EDAX analysis and as indicated from the uniform shape memory transitions of the SMP/AuNR nanocomposites. The shape memory behavior was quantitatively analyzed by cyclic thermomechanical tests using DMA. The laser-triggered shape memory properties of the nanocomposites were analyzed and the shape recovery process from a rectangular shaped film to a helical coil was demonstrated. The speed of SMP recovery was found to be dependent on the geometry and concentration of the AuNPs in the nanocomposite, as well as on the laser wavelength and intensity.
1. Introduction
In the last decade, shape memory polymers (SMPs) have attracted a lot of research interest in the field of multi-functional smart materials.1–5 This category of SMPs can change from a temporary shape to a permanent shape when exposed to external stimuli (heat,1,6 light,2 pH,7,8 magnetic field9). They can be programmed to exhibit multiple temporary shapes and the shape change process is reversible.10 Reversible mesomorphism, where a polymer changes its phase and gas permeability in response to external stimuli such as heat, has also been recently demonstrated.11As polymers have significantly higher deformation and compliance than alloys, the SMPs can undergo transformations between very complex temporary and permanent shapes for many potential applications.12 The concept of an intravascular thrombectomy device that can change from a rod to a corkscrew was demonstrated by Small et al. to remove the blood clots using a laser-activated SMP.13 Light-induced SMPs seem to be promising functional materials for biomedical devices since remote control and actuation can be realized by selecting the appropriate wavelength of light.
Although a variety of light-induced SMPs have been developed,2,14–17 there are several drawbacks. First of all, light-induced SMPs mostly rely on the reversible photoisomerization reaction ([2 + 2] cycloaddition reaction) of the photoresponsive cinnamoyl groups so they only respond to UV light.18,19 Second, good biodegradability is often required for biomedical devices, however most light-induced SMPs are non-biodegradable.
To address these issues, thermally-induced SMPs that can be indirectly heated by near infrared (NIR) radiation provide an alternative solution for remotely light-triggered implantable biomedical devices. When used as biomedical implants, light in the NIR spectrum as the stimulus is preferred because they can have the maximal depth of penetration in the biological tissues with less photo-damage and actuate the shape memory effect while UV-vis light cannot serve this purpose efficiently.20
To achieve the light-triggering feature, many researchers have recently focused on incorporating various types of organic and inorganic particles into thermally-induced SMPs.21–27 The photothermal effect of these nanoparticles has the ability to heat the polymer either thoroughly or partially in a specific region upon selective exposure to light, and to control the shape recovery process in a step-manner by turning the light off and on without changing the environmental temperature.21
Another advantage of using inorganic particles to absorb light is that all the favorable features of thermally-induced SMPs can be retained and molecular design and modifications are convenient (e.g. large fixity ratio, controllable degradability).28 Furthermore, photothermal heating allows remote actuation, is more targeted and spatially sensitive than induction heating of magnetic nanoparticles,29 which necessitates a strong (large) magnetic field in close proximity to the SMP.
To date, of these inorganic nanoparticles, gold nanoparticles (AuNPs) have been studied as non-toxic in human cells and the absorption wavelengths of gold nanorods (AuNRs) fall into the NIR region.30 In 2009, Burdick et al. reported an amorphous and biodegradable SMP and AuNR composite system that can be heated above the SMP's transition temperature to initiate the shape recovery by IR exposure, and investigated the cytotoxicity effect of the AuNRs.31 This study however, did not focus on the effects of AuNPs on the mechanical, thermal and shape-memory properties of the polymeric systems. In fact, the AuNPs could have different effects on various polymeric systems.
Poly(caprolactone) (PCL) copolymers have been recently reported to exhibit two-way shape memory behavior that is reversible under stress-free conditions.32 Nanoparticles have also been incorporated into PCL SMP. Recently, it was shown that by incorporating cellulose nanocrystals into a melt-recyclable PCL-based polymer, the extent of the crosslinking in the networks could be adjusted, and thus the SMP properties.33
In our previous study, crosslinking with cinnamoyl functional groups was exploited as a mechanism to tune the transition temperatures of a semicrystalline PCL polymer with thermally-responsive shape-memory behavior.34 It is our hypothesis that by introducing AuNPs into these modified-PCL copolymers with weak hydrolyzable bonds, which can cleave under physiological conditions, light-triggered shape-memory properties can be realized in a controlled manner in a biodegradable polymeric composite system.
Herein, we report on a new SMP/AuNP composite film in which either colloidal gold nanospheres (AuNSs) or AuNRs were incorporated at different loading ratios. Since the localized surface plasmon resonance (SPR) absorption wavelengths of the AuNPs can be easily tuned in a wide range of light spectra (500–1400 nm) by changing their sizes or shapes,35 the wavelength of light can be selected to trigger the shape memory response of a polymer/AuNP hybrid composite. In this research, we demonstrate the shape recovery from a flat film to a helical coil by exposure to an either a 800 nm IR laser or a 527 nm green laser at different laser intensities, where AuNPs act as light absorbers in the thermally-responsive SMPs to convert the photon energy to heat.
2. Results and discussion
2.1 Synthesis of poly(ε-caprolactone) shape polymer and characterization of its microstructure
The synthesis of the cinnamate-modified ε-caprolactone (CCL) monomer, was achieved in three steps. Improvements were made to each step to increase the yield and accommodate large-scale syntheses as shown in Scheme 1. 1H NMR spectra for each step in the synthesis of the monomer is shown in the ESI Fig. S1–S3.† First, 1,4-cyclohexanediol reacted with 1 equivalent of cinnamoyl chloride in dichloromethane to yield Step I monomer-intermediate in 55% yield. This new approach to the reaction in Step I resulted in higher yield than previously reported (25%),34 and greatly improved the overall efficiency of the monomer synthesis.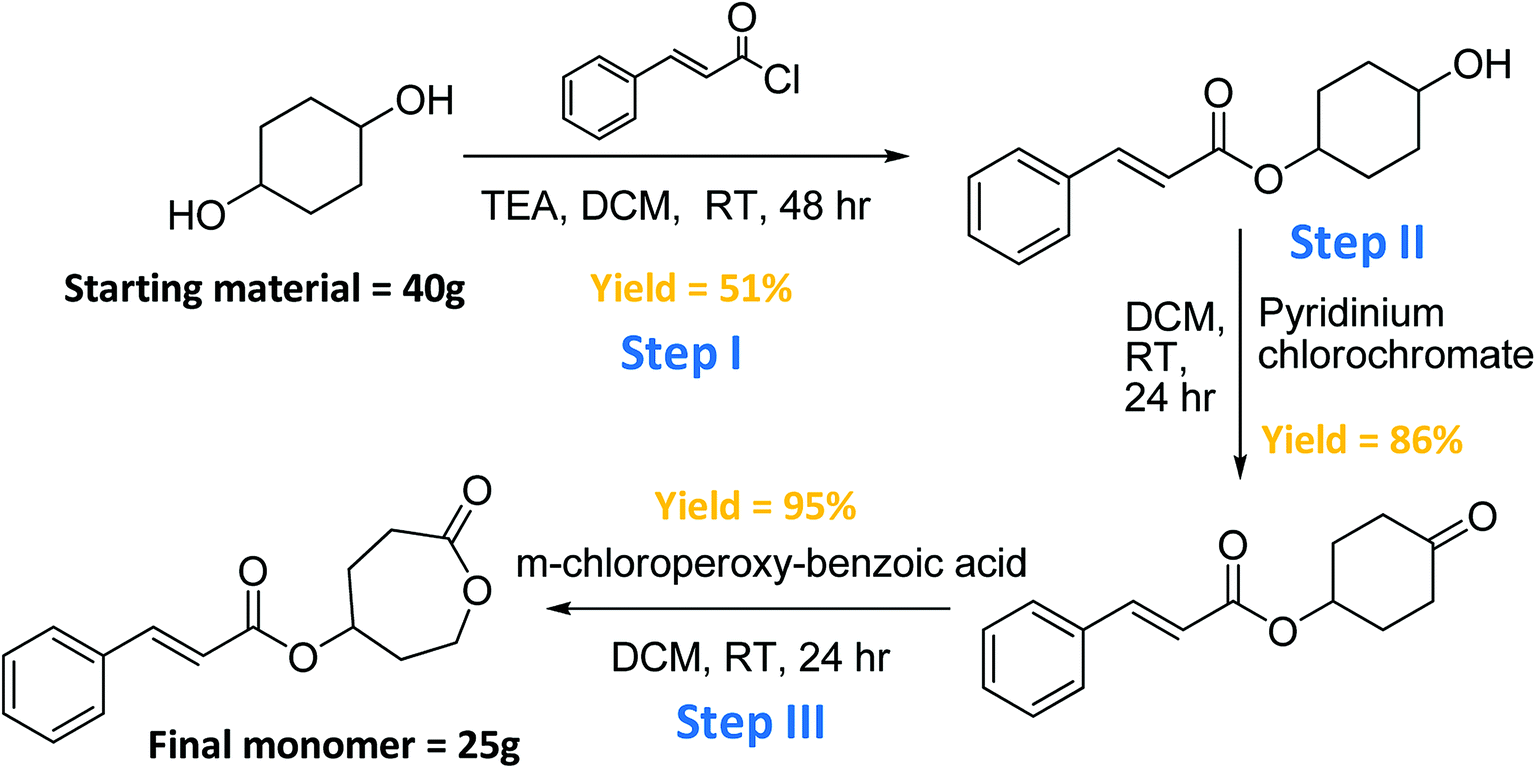 | ||
| Scheme 1 Synthesis route for novel 4-(cinnamoyloxy)-ε-caprolactone monomer. | ||
In Fig. S1,† individual doublet peaks a at 3.6 ppm, and d at 5.0 ppm in the 1H NMR spectrum confirmed the mono-incorporation of the cinnamate group on the di-functional alcohol. The alcohol was then isolated and oxidized to form a ketone (Step II monomer-intermediate) by use of pyridinium chlorochromate (PCC) giving 86% yield. In Fig. S2,† the disappearance of the peak a around 3.5–4.0 ppm 1H NMR spectrum indicated the oxidization of the mono-functional hydroxyl group to ketone.
Finally, the cyclohexanone was converted to a 7-membered lactone (final monomer) in 95% yield by a Baeyer–Villiger oxidation using m-chloroperoxybenzoic acid (m-CPBA). In Fig. S3,† the two peaks b at 2.5–3.0 ppm and the two peaks c at 4.0–4.5 ppm confirmed the successful synthesis of the lactone in the 7-membered ring.
The PCL triblock copolymer was synthesized by sequential copolymerization of CCL and the ε-caprolactone (CL) is depicted in Scheme 2. The 1H NMR spectrum of the PCL modified tri-block copolymer is illustrated in the ESI Fig. S4.† The peaks in the spectrum were assigned to the protons shown in the chemical formula, with peak c as the confirmation of the attachment of cinnamoyl moieties on the polymer backbone. Sequential copolymerization of the CL with CCL was accomplished by ring-opening polymerization reaction using Sn(Oct)2 as the catalyst and tri(ethylene glycol) as the initiator in toluene. The feed ratios of the two monomers were 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with 0.01 equiv. of both initiator and catalyst. The product was washed a couple of times by methanol before vacuum dried to ensure the purity of the polymer.
:1 with 0.01 equiv. of both initiator and catalyst. The product was washed a couple of times by methanol before vacuum dried to ensure the purity of the polymer.
 | ||
| Scheme 2 Ring opening polymerization of the PCL modified triblock copolymer using a step-addition approach. | ||
It has been observed that any residue monomers in the polymer could influence all the mechanical, thermal, and even shape-memory properties of the polymer. Also the yield of the polymer and the molar content of CCL in the copolymer calculated from the NMR spectra would be affected. The molar content of CCL (or CCL:CL ratio) in the copolymers was determined by comparing the integrated signals of the hydrogens (c peak in NMR spectrum) in the CCL blocks and those (h peak in NMR spectrum) in the CL blocks.
2.2 Molecular weight analysis via GPC and thermal transitions by DSC
The molecular weight analysis, CCL:CL ratio and the melting point of the PCL modified tri-block copolymers were reported in Table 1. These two batches of copolymers were based on a homo-block copolymer architecture. The melting points of the copolymers were obtained from DSC analysis. In general, the anionic ROP resulted in copolymers with relatively high Mw and low polydispersity. ROP is very sensitive to moisture and oxygen however, so the reaction environment was tightly controlled by using an inert atmosphere.
| SM polymer | Mn (g mol−1) | Mw (g mol−1) | PDI | Initiator & monomer | CCL:CL ratio (%) |
Tm (°C) |
|---|---|---|---|---|---|---|
| 1 | 5402 | 10330 |
1.91 | TEG | 30 | 51 |
| (M1: CL) | ||||||
| (M2: CCL) | ||||||
| 2 | 19057 |
23818 |
1.25 | TEG | 22 | 53 |
| (M1: CL) | ||||||
| (M2: CCL) |
As seen in Table 1, SMP2 has a higher Mw and is closer to 1.0 polydispersity, whereas SMP1 has a lower Mw and higher polydispersity. This could indicate that ROP of SMP1 was conducted in a less than ideal inert atmosphere, or the monomers were not as pure. On the other hand, the incorporation of the CCL causes a decrease of the Tm because the CCL blocks not only are composed of the amorphous portions of the triblock copolymers (lowering the crystalline portions of the copolymers), but they also disturb the crystallization process of the PCL block. Thus, an increase of the CCL block in the copolymer will lower the Tm.
SMP1 has about 30% CCL incorporation and 51 °C melting point, while SMP2 has about 22% CCL incorporation and 53 °C melting point. Since the synthesis method can be used to tune the Mw, it is a viable method to also tune the Tm of the polymer, which serves as the switching temperature for the shape-memory polymer.
It is also worth noting that both batches of triblock copolymers gave good shape memory properties. It was reported in our previous study that reasonably high Mw and CCL incorporation ratios were required in the copolymers in order to achieve good shape memory properties.34 The reason for this is that higher CL block lengths promote higher degrees of crystallinity in the copolymer, and higher CCL ratios lead to higher crosslinking ratios in the polymer network.
Since the CL crystalline segments act as the switching domain, (determines the temporary shape) and the crosslinked network acts as the elastic domain (determines the permanent shape), our hypothesis is that a higher CCL incorporation will lead to better shape memory properties. Indeed, this hypothesis was tested and all the results presented in the following sections were those of the photo-crosslinked SMP2 copolymers, which gave better shape memory properties.
2.3 Effect of gold nanoparticles on the thermal transitions of the polymer nanocomposites
The AuNRs in two concentrations in acetone (0.1 and 0.2 wt%) and the AuNSs at 0.2 wt% in acetone were added to SMP2 to prepare separate nanocomposites, whose thermal stabilities were characterized via TGA. The TGA curves for SMP, SMP/AuNRs and SMP/AuNSs composites are shown in ESI in Fig. S5, S6, and S7,† respectively. It was observed from the TGA curves that both the SMP and SMP/AuNPs composites remained thermally stable up to ∼270 °C. The 1% onset weight loss of SMP/AuNS (0.2 wt%) composite occurred at 268 °C, with the maximal degradation rate at 359 °C. The 1% onset weight loss of SMP/AuNR (0.2 wt%) composite occurred at 268 °C, with the maximal degradation rate at 356 °C. The incorporation of AuNPs in our SMP did not have any effect on the thermal stability of the SMP.The thermal transitions of the nanocomposites were characterized via DSC. The DSC curves for SMP, SMP/AuNR (0.1 wt% and 0.2 wt%), and SMP/AuNS (0.2 wt%) nanocomposites are illustrated in Fig. 1. The melting temperature (Tm) indicates the presence of the crystalline domains of PCL blocks in the copolymers. As the switching domains, PCL crystalline domains are important to determine the temporary shape of the shape memory polymer.
 | ||
| Fig. 1 DSC curves for SMP, SMP/AuNS (0.1 and 0.2 wt%), and SMP/AuNR (0.2 wt%) nanocomposite. | ||
Previously, Zhang H. et al. concluded that AuNPs act as an impurity for the polymers and consequently result in a depression of Tm.21 The theoretical reasons for this are that the increased entropy (ΔSm) of the composite when AuNPs were introduced into the mixture would result in a decrease in Tm while the enthalpy (ΔHm) remained constant, based on the thermodynamic equation: 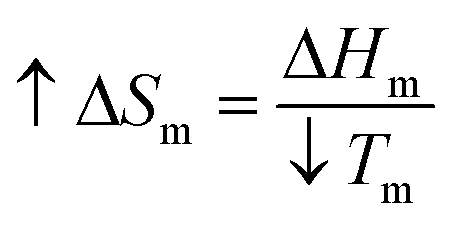 .
.
In the current study however, the melting transitions of the SMP with and without AuNPs were found to be the same at ∼55 °C. That is, the incorporation of AuNPs in the SMP had little to no effect on the Tm of the polymer. Neither the amount of the AuNPs nor the shape of the particles had an influence on the melting point of the SMP. The heat of fusions (ΔH) calculated from the DSC curves (via TA Universal Analysis software) for SMP, SMP/AuNSs, and SMP/AuNRs were 41, 41 and 43 J g−1, respectively. Therefore, the heat of fusions (ΔH) also provided evidence that the presence of AuNPs in the polymer did not lead to a decrease in the degree of crystallinity.
These results are in contrast to that reported previously for thiol-functionalized gold nanoshell/PCL nanocomposites, where it was found that both the Tm and the degree of crystallinity decreased simultaneously with increasing gold nanoshells loading.23 Presumably because the surface functionalized gold nanoshells caused a reduction of polymer chains around the NP due to an entropically driven repulsion of polymer from the surface of the NP, resulting in synchronized decrease in the crystallinity and Tm of the PCL.
2.4 Thermomechanical characterization using dynamic mechanical analysis
The effect of AuNPs on the tensile thermo-mechanical properties of the SMP were studied by DMA. 0.2 wt% AuNSs and AuNRs were separately incorporated into SMPs and the results are shown in Fig. 2. The incorporation of AuNPs not only had little effect on the Tm of the SMP, but also had no influence on the glass transition temperature (Tg) of the SMP. The Tm results obtained from DMA were also consistent with the DSC results at ∼55 °C, and the Tg was approx. −24 °C.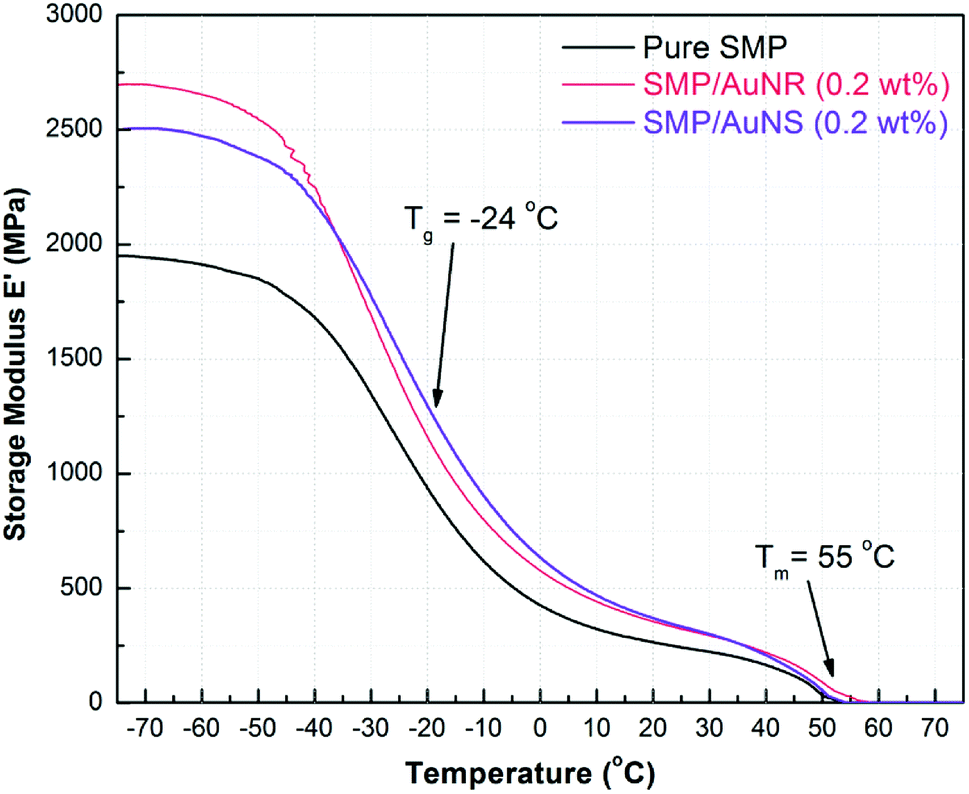 | ||
| Fig. 2 Tensile storage modulus (E′) as a function of temperature for SMP, SMP/AuNS (0.2 wt%), and SMP/AuNR (0.2 wt%) nanocomposites. The glass transitions and the melting transitions were approx. −24 °C and 55 °C, respectively for all the SMP and SMP/AuNP composites. | ||
The storage modulus of SMP was ∼2000 MPa at −70 °C and decreased to ∼240 MPa before reaching a plateau once the SMP was heated above its Tg. The plateau seen above Tg is attributed to the existence of PCL crystalline portions in the copolymer, even though the copolymers were crosslinked. During the plateau, the amorphous segments became mobile; however the crystalline domains were able to maintain the shape of the polymer. Above Tm, the copolymer started to melt and the modulus dropped to zero.
The storage moduli of SMP/AuNS and SMP/AuNR composites were higher than that of the SMP in all the temperature ranges until the polymers melted. In other words, the AuNPs, act as an inorganic filler, strengthening the SMP and enhancing the overall tensile properties of the SMP. The storage moduli of both the SMP/AuNS and SMP/AuNR composites were 2500 to 2700 MPa below the Tg and were ∼330 MPa at the plateau temperatures (including at room temperature).
2.5 Morphology of the nanocomposite films via electron microscopy
Both transmission and scanning electron microscopy, TEM and SEM, respectively, were used to determine the dispersion or aggregation of AuNRs in the SMP film containing 0.2 wt% AuNRs. The PCL polymer matrix was very challenging to cut into ultrathin and wrinkle-free sections even at cryo-conditions. As such, the resolution of the TEM images of SMP/AuNRs shown in the ESI Fig. S8† is only fair, due to the relatively thick and wrinkled sections obtained during sample preparation. Thus, the entire distribution profile of the AuNRs may not be visible in the TEM photographs. However, based on the observation of a uniform shape memory transition in the SMP/AuNR nanocomposites during the laser actuation experiments (as described in the following section), it is expected that the majority of AuNRs were reasonably distributed in the nanocomposite.Indeed, on careful observations of the TEM images, the majority of AuNRs were found to be fairly well dispersed in the polymer matrix (albeit at the low dilution of 0.2 wt%), and appeared as isolated nanorods, while some clustering did occur. This clustering did not adversely affect the light-actuation response of the nanocomposite films. Furthermore, there were no observed changes in the shape, length or aspect ratio of the AuNRs in the nanocomposites.
This finding is significant as AuNRs absorb and scatter light due to the excitation of the transverse and longitudinal surface plasmons that characterize these nanoparticles.36 Therefore, any changes in the geometry of the AuNRs would change the wavelength of light absorbed, which in-turn would adversely affect the speed and/or the extent of shape memory recovery at a wavelength previously identified as the plasmon resonance for that particular geometry.
The TEM images, shown in Fig. S8,† indicated that the length, Lr and aspect ratios (ν = Lr/Wr) of the Au NRs were found to be similar to that reported by the manufacturer at Lr ∼ 40 nm and 4, respectively, both before and after they were incorporated into the SMP films.
SEM with energy-dispersive X-ray spectroscopy (SEM-EDAX) analysis was used to complement the TEM studies. It verified the presence, distribution and the relative abundance of AuNRs in the SMP/AuNR nanocomposite film. From the SEM-EDAX image of the film shown in Fig. S9,† all the AuNRs were located below the surface of the film and not protruding from the surface.
Moreover, the EDS spectrum taken at the cluster of a rod-shaped structure (marked by  ), confirmed the presence of AuNRs, as well as a many individual AuNRs (highlighted in the image). Overall, the SEM image provided additional evidence that the majority of AuNRs were fairly well dispersed in the polymer matrix.
), confirmed the presence of AuNRs, as well as a many individual AuNRs (highlighted in the image). Overall, the SEM image provided additional evidence that the majority of AuNRs were fairly well dispersed in the polymer matrix.
2.6 Effect of gold nanoparticles on thermo-responsive shape memory properties
The shape memory programming and recovery process of the nanocomposites is depicted in Scheme 3. As shown in the three dimensional (3D) graph of (6) consecutive shape memory cycles of the SMP (Fig. 3), in each cycle the nanocomposites underwent five different stages (loading, cooling, unloading, heating, and recovery). In the loading stage, at 60 °C the polymer was deformed to ∼10% strain under 16 kPa stress. In the cooling stage, the polymer was cooled from 60 °C to 0 °C under the constant loading to fix the applied strain. The strain remained constant until a sharp decrease was observed, due to the crystallization phase of the polymer during cooling, which leads to a contraction of the polymer.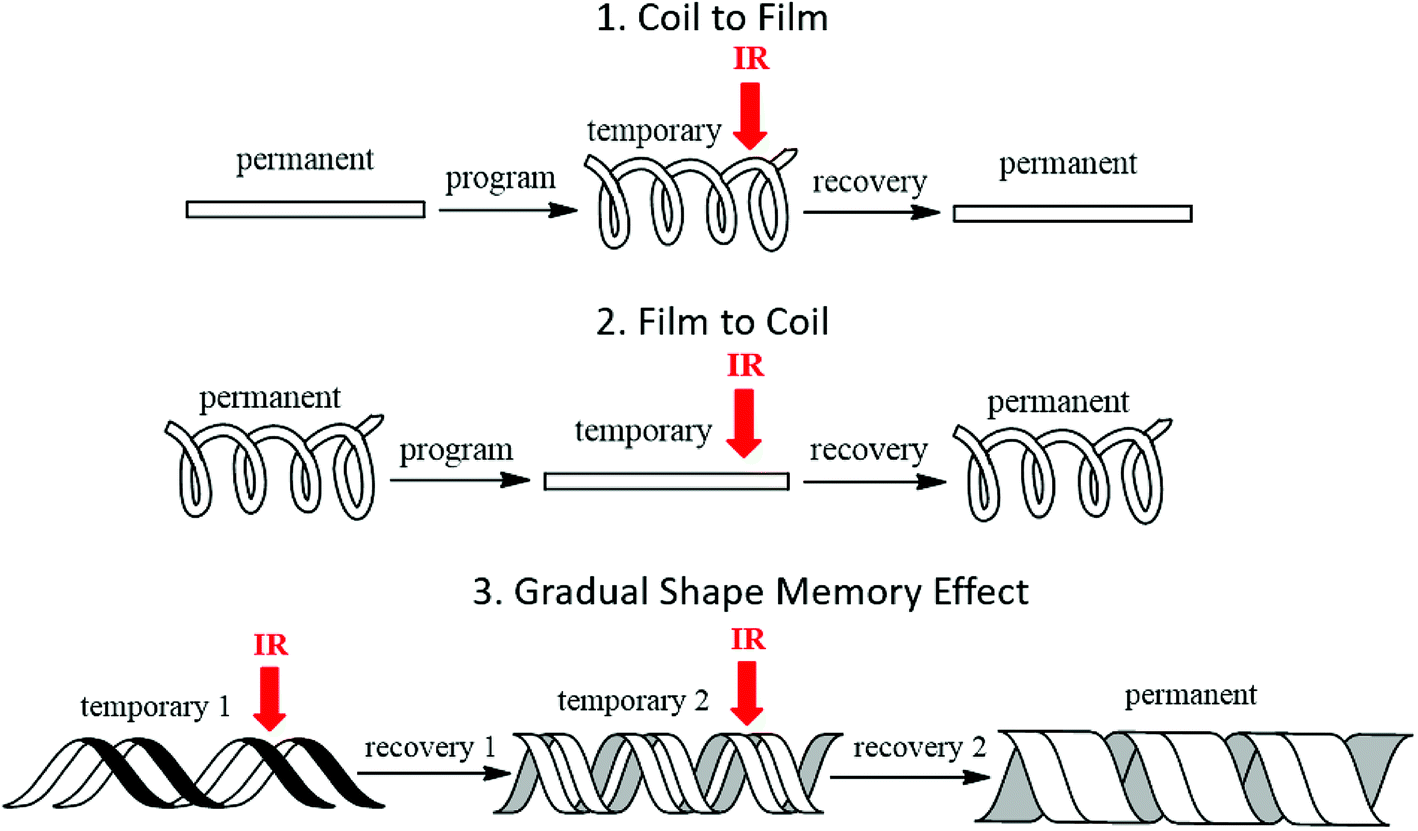 | ||
| Scheme 3 Illustration of proof of concept: (1) coil to film; (2) film to coil; (3) gradual shape recovery by IR laser triggered shape recovery process of the AuNR/SMP nanocomposites. | ||
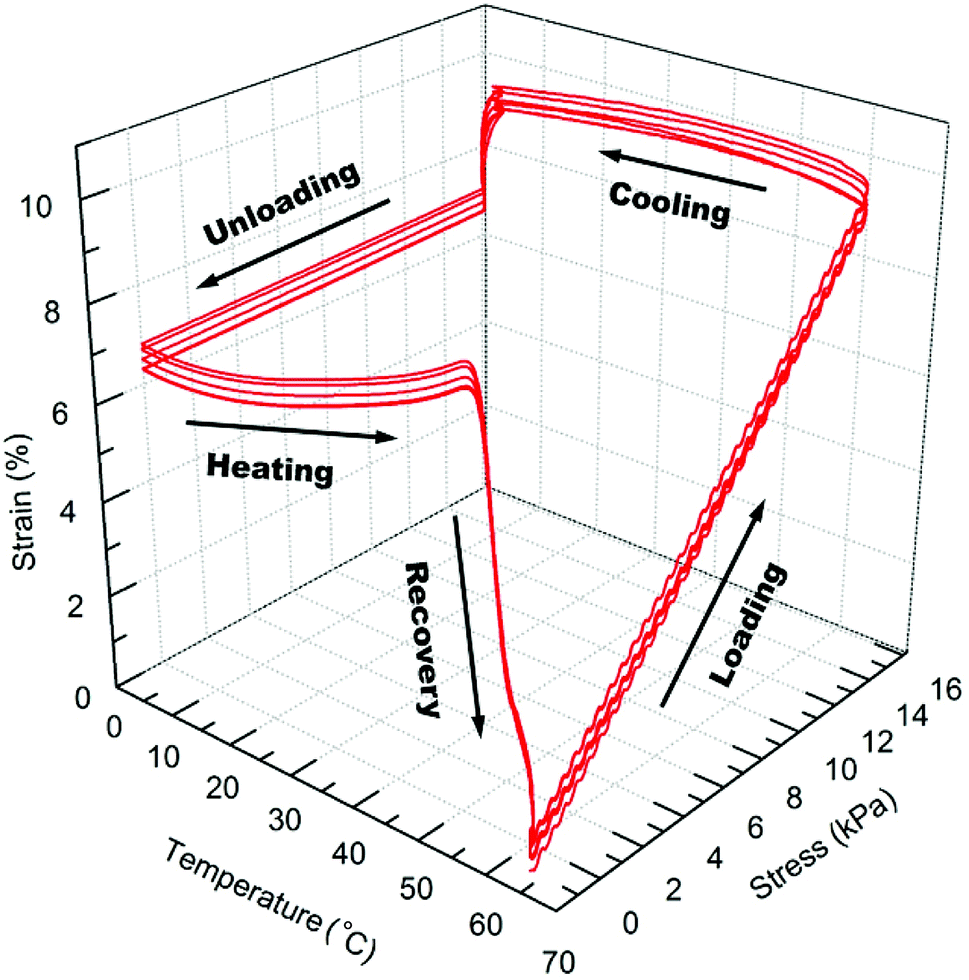 | ||
| Fig. 3 3D graph of 6 thermomechanical shape memory cycles for SMP without AuNPs. | ||
In the unloading stage, the polymer stayed in the fixed strain at 0 °C while the stress was released to the pre-load force (0.001 N). In the heating stage, the polymer slowly expanded upon the crystallization temperature of the PCL domains, and finally during recovery stage, the polymer recovered to the initial strain at pre-load force upon the switching temperature (the melting transition of the PCL domains).
As summarized in Table 2, starting from the first cycle, each shape memory cycle is highly reproducible, with a slight increase of the creep after the previous cycle. The increased creep can be explained as the interconnected crosslinked polymer network starts to become loose after each heating cycle, losing the ability to maintain the permanent shape of the polymer. The reachable shape-fixity rate (Rf) and the shape recovery rate (Rr) were ∼96% and 99%, respectively. However, the instantaneous recovery only reached about 80% at Tr (60 °C), while the remaining recovery requires ∼20 min of isothermal heating due to the decreased driving force for recovery.
In Fig. 4 and 5, the 3D graphs of (6) consecutive shape memory cycles of the SMP/AuNS and SMP/AuNR composites respectively, were shown. Both the SMP/AuNS and SMP/AuNR composites demonstrated similar behaviors as the pure polymers. As shown in Tables 3 and 4, both the composites reached ∼99% Rf and 99% Rr. However, the shape memory cycles only become reproducible from the second cycles while the first cycles differ from the rest. In this regard, the thermal history of the composite samples had a significant influence on the shape memory properties.
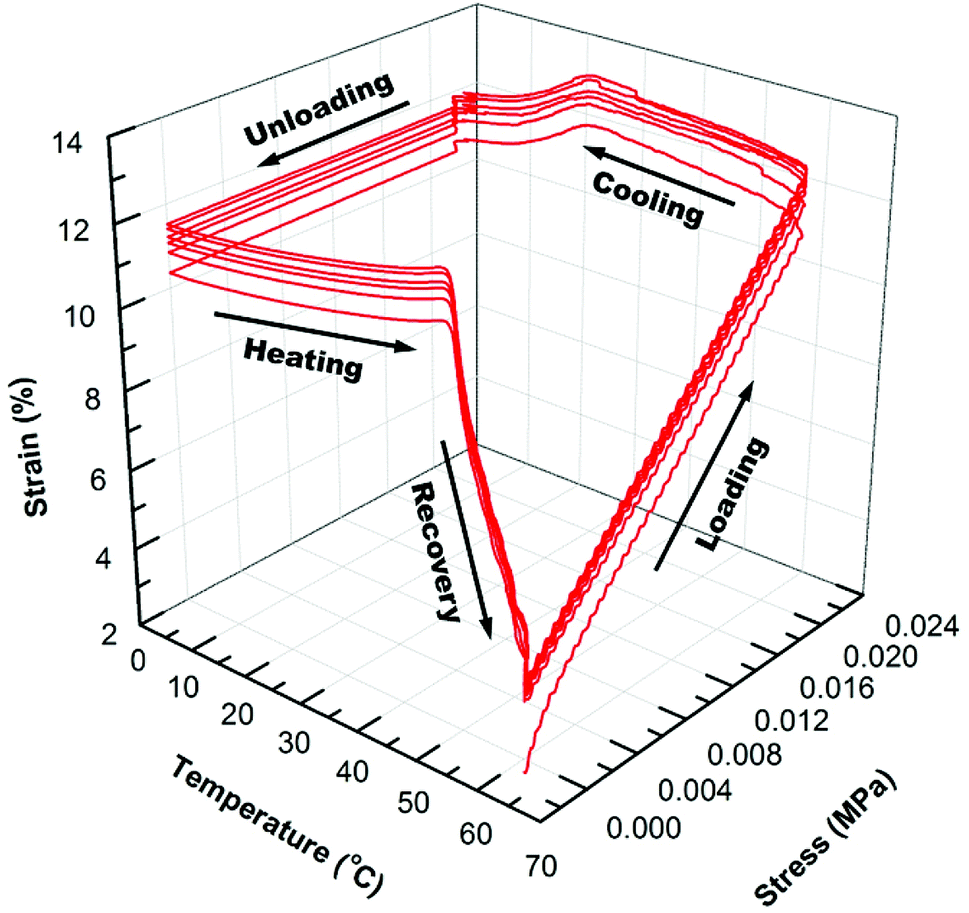 | ||
| Fig. 4 3D graph of 6 shape memory cycles for SMP/AuNS (0.2 wt%) nanocomposite. | ||
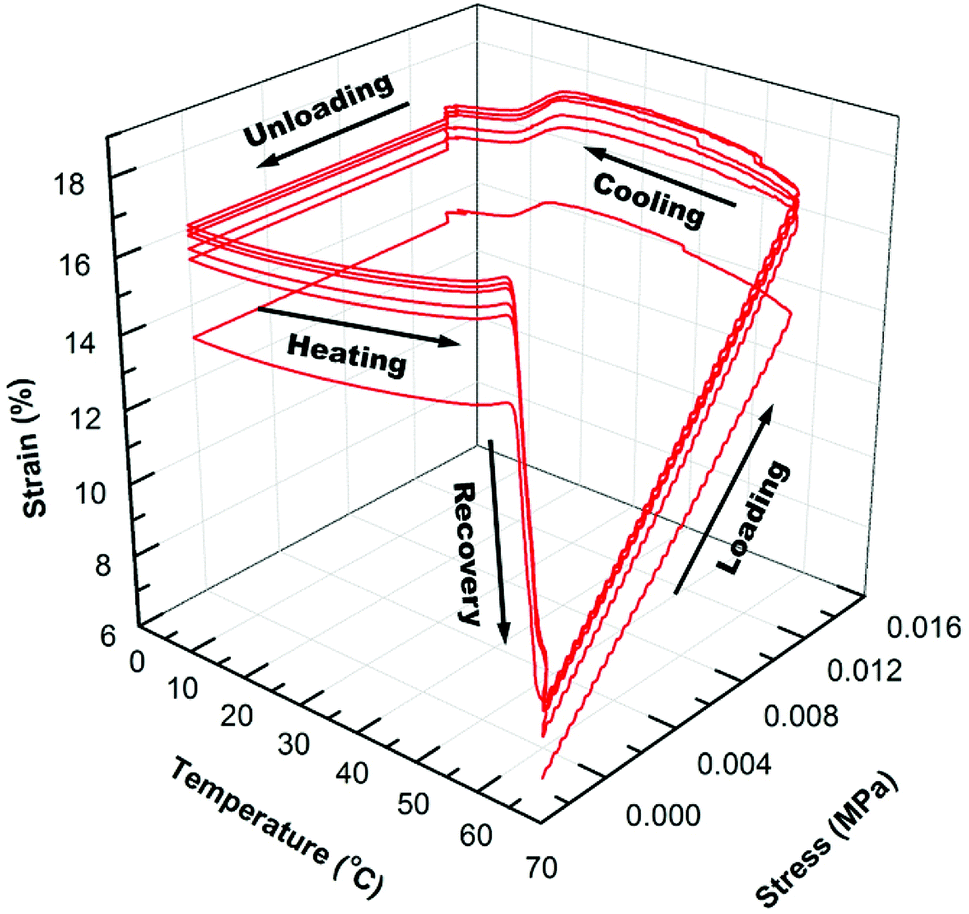 | ||
| Fig. 5 3D graph of 6 shape memory cycles for SMP/AuNR (0.2 wt%) nanocomposite. | ||
Due to the presence of AuNPs in the polymer and prior processing, the polymer re-orients themselves on the molecular level after the first cycles, removing the thermal history. Also, the SMP/AuNS and SMP/AuNR composites achieved an instantaneous recovery as high as 85% Rrins and 89% Rrins, respectively. Therefore, the incorporation of AuNPs enhanced the overall shape memory properties of the SMP.
2.7 Effect of gold nanoparticles on IR light-responsive shape memory properties
A visual demonstration of shape recovery tests was video-recorded at a rate of 60 fps. The shape recovery process from a stent coil to a strip was used to study the shape recovery speed. Three samples were tested under each condition, and the reported time for the onset of recovery and the total recovery time was the average of three repeated tests. N/A indicates the polymer did not recover to its original shape.The laser-activated shape memory properties of the SMP/AuNP composites were studied. The kinetic analysis of the shape recovery process from a rectangular shaped film to a helical coil was summarized in Table 5. To study the effect of gold loading in the polymer matrix on the light responsiveness, the SMP/AuNR composite films with 0.1 wt% and 0.2 wt% AuNRs was compared. With 0.2 wt% gold loading, the SMP/AuNR composite started its recovery process after 2 s laser exposure and completed its recovery in 15 s when irradiated vertically from the top using 800 nm NIR laser with a 990 mW cm−2 intensity. However, with 0.1 wt% gold loading, the composite responded much slower under the same radiation conditions and could not recover to its original shape.
| SMP/gold nanoparticle | Gold loading (wt%) | SPR peak wavelength (nm) | Laser wavelength (nm) | Laser intensity (mW cm−2) | Onset of recovery (s) | Total recovery time (s) |
|---|---|---|---|---|---|---|
| AuNR | 0.10 | 808 | 800 | 990 | 12 | N/A |
| AuNR | 0.20 | 808 | 800 | 990 | 2 | 15 |
| AuNR | 0.20 | 808 | 800 | 130 | 10 | 180 |
| AuNS | 0.20 | 520 | 527 | 990 | 6 | 45 |
| AuNS | 0.20 | 520 | 527 | 130 | 20 | 190 |
To study the effect of laser intensity on the shape recovery speed, both SMP/AuNR and SMP/AuNS composite films were irradiated with 990 mW cm−2 and 130 mW cm−2 laser intensities at their respective on-resonance frequencies. The SMP/AuNR (0.2 wt%) composite films responded to the NIR laser at least 3 times slower at 130 mW cm−2 intensity than at 990 mW cm−2 intensity, while the SMP/AuNS (0.2 wt%) samples showed similar results under green laser radiation. The total recovery time at lower laser intensities were also much longer than that at higher intensities. The AuNRs act as NIR absorbers and the AuNSs act as the green light absorbers. The photothermal effect, whereby heat is generated from light absorption was significantly lower at lower laser intensities. This heat can also be dissipated during the course of total shape recovery.
To elucidate the role of nanoparticle shape on the surface plasmon induced photothermal effect, the shape recovery efficiencies of the SMP nanocomposites with AuNRs and AuNSs were investigated. With the same gold loading and laser intensity, SMP/AuNR samples had a significantly faster response to on-resonance laser actuation than SMP/AuNS samples. These results provide evidence that the AuNRs had higher light absorbing efficiency at the on-resonance frequencies facilitating a faster photo-thermal effect and thus a faster shape recovery process. It is important to note that, fast light activation of the nanocomposites was achieved at a very low (0.2 wt%) gold loading. These results were expected as it has been reported that the geometry-dependent plasmonic heating of AuNPs results in highly non-uniform temperature distribution even at the nanoscale,37 which would influence the polymer chain mobility and speed of recovery of the permanent shape.
While the Femtosecond laser was used for our experiment, such high power laser is not needed for actuation. A lower power laser can be used when AuNP is loaded at higher weight ratios in the SMP. Even though the SPR wavelengths of the AuNRs and AuNSs had slight discrepancies with the laser wavelengths, the differences were considered negligible, and on-resonance actuation was achieved.
Several control experiments were conducted by shining either a NIR laser or a green laser on the SMP samples with either AuNS or AuNR. The SMP/AuNS showed no response to the NIR laser, while the SMP/AuNR showed only small shape recovery with the green laser. These results were expected as there would be no SPR in the SMP/AuNS using the NIR laser due to the mis-match in the wavelengths.
It is known however, that AuNR exhibits both a longitudinal and transverse plasmon resonance, where the longitudinal plasmon resonance depends on the aspect ratio of the AuNR.35 In the current study, AuNR with an aspect ratio of ∼4 was used. These AuNR will exhibit SPR at a wavelength of 808 nm, which closely matches that of the NIR laser wavelength at 800 mn. In contrast, the wavelength of the green laser (520 nm) does not exactly match the transverse plasmon resonance of the AuNR (548 nm). It was reported that the especially large cross-sectional absorption area of AuNR at the longitudinal resonance gives a greater photothermal conversion effiency than that at the transverse resonance,22 thereby explaining the significant difference in shape memory recovery observed in the on-resonance response of the AuNR using the NIR laser and the off-resonance response (or lack thereof) using the green laser, respectively.
Another control experiment was conducted by irradiating the SMP without AuNP with either NIR or green laser. The SMP samples had no response to the laser light but subsequently recovered its original shape when heated above Tm using a water bath. Therefore, only AuNPs were responsible for the heat generation in the SMP when irradiated by the appropriate wavelength of light.
The shape recovery process of a SMP/AuNR nanocomposite sample from a rectangular-shaped film to a helical coil activated by an 800 nm NIR laser is shown in Fig. 6. The film gradually recovered to the helical shape in a course of 20 s. Since the light source is a defined spot, the change in film shape can be controlled spatially at a localized spot, or undergo a discontinuous recovery in a step-wise manner by turning the laser off and on.
 | ||
| Fig. 6 Visual demonstration of 800 nm NIR laser actuation of the SMP/AuNR nanocomposites from a series of photographs. After the SMP/AuNR nanocomposite helical coil was prepared as the permanent shape, it was softened at 60 °C to program the temporary shape as a flat film. When radiated by the 800 nm NIR laser, the film recovered to its permanent helical coil shape in 20 s. This shape recovery process is also seen in a Movie video (M1) in the ESI.† | ||
3. Conclusions
Herein, we described the synthesis of a PCL triblock copolymer through ROP of the cinnamate-modified caprolactone monomers in higher yield than was previously reported. AuNSs and AuNRs were incorporated in the synthesized copolymers to obtain the SMP/AuNS or SMP/AuNR nanocomposites and the corresponding nanocomposite films were successfully fabricated and photo-crosslinked.No changes in the shape, length or aspect ratio of the AuNRs were observed in the TEM images. Moreover, both TEM and SEM-EDAX analysis showed that the majority of AuNRs were fairly well dispersed in the polymer matrix and appear as isolated nanorods, while some clustering did occur. This clustering did not adversely affect the light-actuation response of the nanocomposite films, and the thermal and mechanical studies showed that the incorporation of AuNSs or AuNRs had no influence on the thermal properties, but reinforced the overall tensile properties of the polymer.
The shape memory properties of the pure SMP and SMP/AuNP nanocomposites were quantified by DMA and the AuNPs also enhanced the shape memory properties of the polymer and reached ∼99% Rf and 99% Rr. Furthermore, the SMP/AuNS and SMP/AuNR nanocomposites were triggered by a green laser and a NIR laser, respectively. The kinetics of the light actuation were investigated at different laser intensities, and the AuNRs had much higher light absorbing efficiencies at the on-resonance frequencies and resulted faster shape recovery process.
This work demonstrated the idea to achieve light-triggered shape memory properties of a thermally induced SMP using the surface plasmon induced photo-thermal effect of AuNPs. By simply varying the shape of the AuNPs, the light absorption wavelengths could be tuned in a wide range without compromising the chemical composition of the polymers, therefore all the preferable properties and features of the polymers could be preserved.
Lastly, the visualization of laser-triggered shape change process of the nanocomposites from a rectangular shaped film to a helical coil was realized in less than 2 s suggesting these SMP/AuNP nanocomposites may have potential biomedical applications as NIR light-triggered vascular stents.
4. Experimental section
4.1 Materials
1,4-Cyclo-hexanediol, cinnamoyl chloride, pyridinium chlorochromate (PCC), triethylamine (TEA), 3-chloroperoxybenzoic acid (m-CPBA), ε-caprolactone, tin(II) 2-ethylhexanoate (Sn(Oct)2) and anhydrous toluene were purchased from Alfa Aesar and used as received. All other reagents and solvents were purchased from Aldrich and used without further purification. Organic spherical gold nanoparticles (AuNS) 20 nm in diameter and a with maximal absorption at 520 nm along with organic gold nanorods (AuNR) 10 nm in diameter and 41 nm in length (aspect ratio ∼4) and with maximal absorption at 808 nm were purchased from Nanopartz and used as received.4.2 Synthesis of monomers
The synthesis of the cinnamate-modified ε-caprolactone monomer was previously reported.34 To be brief, 1,4-cyclohexanediol was reacted with cinnamoyl chloride in dichloromethane to yield the mono-substituted alcohol, which was further oxidized by pyridinium chlorochromate. The cinnamate-modified ε-caprolactone was then obtained through a Baeyer–Villiger reaction. Modifications were made in each step as detailed below to increase the yield and accommodate large-scale syntheses (Scheme 1).:1 molar equivalent of 1,4-cyclohexanediol and cinnamoyl chloride was used here in order to improve the yield of the mono-substituted product and minimize the formation of di-substituted by-product, compared to the previously reported method by Amit Garle, et al.34 After 24 h of reaction at room temperature, DI water was added, and the mixture was washed with dilute HCl (1 M) until pH reached 7. Then the water layer was extracted with DCM twice and the organic layer was washed twice with DI water and once with brine before it was dried over anhydrous MgSO4, filtered and concentrated. The crude product was purified using silica gel column chromatography (hexane/ethyl acetate as eluent with the gradient of 10, 20, 30, 40 and 50 vol% of EtOAc). The product was isolated as a yellow viscous liquid. Yield: 42.9 g, 51%. 1H NMR (500 MHz, CDCL3, δ): 1.72 (m, 4H, CH2), 2.06 (m, 4H, CH2), 3.75 (m, 1H, CH), 4.99 (m, 1H, CH), 6.43 (d, 1H, CH), 7.36 (m, 3H, CH), 7.53 (m, 2H, CH), 7.67 (d, 1H, CH). 13C NMR (500 MHz, CDCl3, δ): 28 (C2), 31 (C2), 69 (C–O), 72 (C–O), 118 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 128 (C2), 129 (C2), 131 (–C), 135 (–C), 145 (CC), 167 (O–CO).C), 128 (C2), 129 (C2), 131 (–C), 134 (–C), 145 (CC), 166 (O–CO), 210 (CO).C), 128 (C2), 129 (C2), 131 (–C), 134 (–C), 146 (CC), 166 (O–CO), 175 (O–CO).
C), 128 (C2), 129 (C2), 131 (–C), 135 (–C), 145 (CC), 167 (O–CO).C), 128 (C2), 129 (C2), 131 (–C), 134 (–C), 145 (CC), 166 (O–CO), 210 (CO).C), 128 (C2), 129 (C2), 131 (–C), 134 (–C), 146 (CC), 166 (O–CO), 175 (O–CO).4.3 Ring opening polymerization (ROP) of the monomer
The PCL triblock copolymer was synthesized by two different approaches. The sequential copolymerization of the cinnamate-modified ε-caprolactone monomer (CCL) and the ε-caprolactone (CL) was depicted in Scheme 2 and carried out as follows. The ε-caprolactone (5.7 g, 50 mmol), the TEG as the initiator (75 mg, 0.5 mmol) and Sn(Oct)2 as the catalyst (203 mg, 0.5 mmol) were added into a 3-neck round bottom flask which was carefully evacuated and purged with argon gas (3 times). Then anhydrous toluene (200 mL) was added into the flask via a septum. The reaction system was heated to 120 °C for refluxing. After 24 h of reaction, the previously dried cinnamate-modified ε-caprolactone monomer (13.1 g, 50 mmol) in anhydrous toluene (50 mL) was added to the reaction mixture under inert gas protection. After another 48 h, the reaction was terminated by acetic acid (0.5 M) in methanol. The mixture was concentrated and then transferred in cold methanol to precipitate out the polymer. The final product was washed by methanol (3 times) and dried under vacuum to give a white power. Yield: 9.2 g, ∼50%.4.4 Preparation of polymer film networks
The polymer films were prepared using a Carver Laboratory Press (Model C, serial no. 33000-778) in a mold made of Teflon coated aluminum sheet. The compression molding was performed at 70 °C with a minimum of 10 MPa pressure, and the films were cooled under pressure at room temperature. The thickness of the films was measured to be ∼200 μm. The polymer films were then photo-crosslinked by UV exposure (Dymax Bluewave 200 UV spot lamp, 40 mW cm−2 intensity) under a light guide from a distance of 10–20 cm. The films were UV radiated for a minimum of 30 min on each side to ensure adequate crosslinking.4.5 Preparation of polymer film
After the polymer films were prepared by the compression molding as described above, they were rolled onto a cylindrical steel rod while they were softened at 48 °C. After cooling at room temperature, the polymer stents were exposed to UV light (Dymax Bluewave 200 UV spot lamp, 40 mW cm−2 intensity) under a light guide with a distance of 10 cm while the steel rod was rotating at 6 rpm powered by a motor (Fig. S10, ESI†).4.6 Preparation of AuNP-SMP composite films
After the polymer was fully dissolved in acetone in a vial, the organic AuNP in acetone was added into the polymer solution using a syringe and needle. The mixture was sonicated for ∼5 min for complete dissolution to achieve uniform dispersion of AuNP in the SMP matrix (Fig. S11, ESI†). Three samples were prepared with AuNS and AuNR at different loading ratios (Table S1, ESI†). The mixture solution was poured into a Teflon dish and dried in the fume hood to form a thin layer of composite film, which was further dried in the vacuum oven for at least another 6 h. The SMP/AuNP composite powder was collected and fabricated to a film using the Carver Lab Press as previously described (Fig. S12, ESI†).4.7 Characterization techniques
1H NMR and 13C NMR spectra for the structural analysis were recorded with a Bruker 500 MHz spectrometer. The molecular weight and polydispersity index (Mw/Mn) of the polymer was analyzed by gel permeation chromatography (GPC) on a Waters system, using tetrahydrofuran (THF) (1.0 mL min−1) as the eluent and polystyrene of known molecular weight as the calibration standards. The thermal properties of the polymer were investigated by thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC). TGA was performed on TA Instrument Q50 by heating the samples from room temperature to 800 °C at a heating rate of 10 °C min−1 under nitrogen atmosphere. DSC was performed TA Instrument Q200 with heat–cool–heat cycles in the temperature range of −70 to 80 °C with the heating and cooling rate of 10°C min−1 under nitrogen purging. The second heating cycle was used to analyze the calorimetric data to determine the melting transition (Tm) and the heat of fusion (ΔH).Transmission electron microscopy (TEM) images of the SMP/AuNR nanocomposites were examined to indicate the dispersion and distribution of the AuNRs in the polymer matrix. TEM samples were prepared by cryo-microtomy via a Leica UC6 cryo-ultramicrotome. 50 nm thick sections of the specimen embedded in EpoFix cold-setting embedding resin were cut using the cryo-ultramicrotome. Glass knives and the sample block were held at −100 °C during sectioning. The sections were transferred to 3 mm diameter 200 mesh Cu TEM grids. The prepared grids were air dried on a filter paper for 1 hour prior to TEM imaging. A Philips EM400 transmission electron microscope was used to image the thin sections using a 100 kV electron beam at various magnifications.
The block face created from the cryo-sectioning of the embedded samples were also investigated using field emission scanning electron microscopy (FE-SEM). The surface of the specimens were carbon coated using a Denton Vacuum Desk II carbon evaporator. The freshly carbon coated samples were submitted to SEM imaging immediately after coating. A JEOL 7401F field emission scanning electron microscope was use to image the micotomed embedded block face of the samples. A 10 kV electron beam was used to capture secondary electron images at various magnification and to excite X-ray fluorescence to determine the presence of the Au nanorods using the EDAX Energy Dispersive Spectroscopy (EDS) attachment on the FE-SEM.
The thermo-mechanical properties of the SMP and SMP/AuNP composites were studied by the dynamic mechanical analyzer (DMA) (TA Instruments, Model Q800) in the tensile mode with a pre-load force of 0.001 N, oscillation strain of 0.1%, force tracking of 110%, and an oscillation frequency of 1 Hz. The graphs of the tensile storage modulus (E′) and the loss modulus (E′′) versus temperature were obtained.
4.8 Shape memory property analysis
The shape memory properties of the polymer films and the composites were examined by the cyclic thermo-mechanical test38 as follows. DMA (TA Instruments, Model Q800) was performed in the controlled-force mode with a preload force of 0.001 N. The previously prepared rectangular film sample (10 mm × 5 mm × 0.3 mm) was equilibrated at 60 °C and elongated by applying a force of 0.024 N at a rate of 0.005 N min−1. To fix the temporary shape, the sample was cooled to 0 °C (fixation temperature, Tf) at a rate of 3 °C min−1 while the force was maintained, then the force was unloaded to 0.001 N at a rate of 0.005 N min−1. To recover the original shape, the sample was heated to 60 °C (recovery temperature, Tr) a rate of 3 °C min−1, and kept isothermal for 20 min to fully restore the permanent shape. The shape memory properties were quantified using shape fixity rate, Rf, and the shape recovery rate, Rr:
 | (1) |
 | (2) |
4.9 Light-triggered shape memory effect
The laser exposure experiment was conducted using a NIR laser with ∼800 nm wavelength and a green laser with ∼527 nm wavelength (Quantronix Corp., Model TITAN 1.0 FS). Within the capacity of the instrument, the NIR laser-triggering properties of AuNR/SMP nanocomposite and the green laser-triggering properties of AuNS/SMP nanocomposite were both investigated at two different laser power densities (130 mW and ∼1 W). The shape recovery process of the nanocomposite samples from a strip to a stent coil was video recorded to study the light responsiveness and shape recovery speed of the nanocomposites. The shape recovery process of the nanocomposite samples was depicted in Scheme 3.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the National Science Foundation (Award # DUE-1044363) for research funding, the Plastics Engineering Department for a teaching assistantship, the Nanomanufacturing Center for facilities support and Wendy Gavin and Jean Vangsness for help with characterizing the SMP/AuNP films using GPC, NMR and DMA respectively. Prof. Mengyan Shen and Prof. Jayant Kumar are gratefully acknowledged for use of their laser instruments for our experiments. The authors would also like to thank Prof. Ramaswamy Nagarajan for the use of his thermogravimetric characterization equipment and Dr Earl Ada for characterizing the SMP/AuNR films using TEM and SEM-EDAX.References
- A. Lendlein, Science, 2002, 296, 1673–1676 CrossRef PubMed.
- A. Lendlein, H. Jiang, O. Jünger and R. Langer, Nature, 2005, 434, 879–882 CrossRef PubMed.
- M. Behl and A. Lendlein, Mater. Today, 2007, 10, 20–28 CrossRef.
- M. Behl, M. Y. Razzaq and A. Lendlein, Adv. Mater., 2010, 22, 3388–3410 CrossRef PubMed.
- Z. Yang, Q. Wang and T. Wang, J. Mater. Chem. A, 2017, 5, 13823–13833 RSC.
- Y. Zhang, Y. Li and W. Liu, Adv. Funct. Mater., 2015, 25, 471–480 CrossRef.
- X.-J. Han, Z.-Q. Dong, M.-M. Fan, Y. Liu, J.-H. li, Y.-F. Wang, Q.-J. Yuan, B.-J. Li and S. Zhang, Macromol. Rapid Commun., 2012, 33, 1055–1060 CrossRef PubMed.
- Y. Hu, C.-H. Lu, W. Guo, M. A. Aleman-Garcia, J. Ren and I. Willner, Adv. Funct. Mater., 2015, 25, 6867–6874 CrossRef.
- R. Mohr, K. Kratz, T. Weigel, M. Lucka-Gabor, M. Moneke and A. Lendlein, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3540–3545 CrossRef PubMed.
- T. Xie, Nature, 2010, 464, 267–270 CrossRef PubMed.
- S. Hassan, R. Anandakathir, M. J. Sobkowicz and B. M. Budhlall, Polym. Chem., 2016, 7, 1452–1460 RSC.
- Q. Chai, Y. Huang, T. L. Kirley and N. Ayres, Polym. Chem., 2017, 8, 5039–5048 RSC.
- W. Small IV, T. S. Wilson, W. J. Benett, J. M. Loge and D. J. Maitland, Opt. Express, 2005, 13, 8204 CrossRef PubMed.
- H. Y. Jiang, S. Kelch and A. Lendlein, Adv. Mater., 2006, 18, 1471–1475 CrossRef.
- E. Havens, E. A. Snyder and T. H. Tong, Proc. SPIE, 2005, 5762, 48–55 CrossRef.
- E. A. Snyder and T. H. Tong, MRS Online Proc. Libr., 2005, 872, J18.6 CrossRef.
- L. Wu, C. Jin and X. Sun, Biomacromolecules, 2011, 12, 235–241 CrossRef PubMed.
- L. Wang, X. Yang, H. Chen, T. Gong, W. Li, G. Yang and S. Zhou, ACS Appl. Mater. Interfaces, 2013, 5, 10520–10528 CrossRef PubMed.
- D. Tunc, C. Le Coz, M. Alexandre, P. Desbois, P. Lecomte and S. Carlotti, Macromolecules, 2014, 47, 8247–8254 CrossRef.
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316–317 CrossRef PubMed.
- H. Zhang, H. Xia and Y. Zhao, J. Mater. Chem., 2012, 22, 845 RSC.
- Z. Xiao, Q. Wu, S. Luo, C. Zhang, J. Baur, R. Justice and T. Liu, Part. Part. Syst. Charact., 2013, 30, 338–345 CrossRef.
- D. M. Le, M. A. Tycon, C. J. Fecko and V. S. Ashby, J. Appl. Polym. Sci., 2013, 130, 4551–4557 Search PubMed.
- X. Shen, C. Viney, C. Wang and J. Q. Lu, Adv. Funct. Mater., 2014, 24, 77–85 CrossRef.
- H. Zhang and Y. Zhao, ACS Appl. Mater. Interfaces, 2013, 5, 13069–13075 CrossRef PubMed.
- H. Lu, Y. Yao, W. M. Huang, J. Leng and D. Hui, Composites, Part B, 2014, 62, 256–261 CrossRef.
- Z. Yang, Q. Wang and T. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 21691–21699 CrossRef PubMed.
- D. Habault, H. Zhang and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7244 RSC.
- U. N. Kumar, K. Kratz, M. Heuchel, M. Behl and A. Lendlein, Adv. Mater., 2011, 23, 4157–4162 CrossRef PubMed.
- E. E. Connor, J. Mwamuka, A. Gole, C. J. Murphy and M. D. Wyatt, Small, 2005, 1, 325–327 CrossRef PubMed.
- K. C. Hribar, R. B. Metter, J. L. Ifkovits, T. Troxler and J. A. Burdick, Small, 2009, 5, 1830–1834 CrossRef PubMed.
- K. Wang, Y.-G. Jia and X. X. Zhu, Macromolecules, 2017, 50, 8570–8579 CrossRef.
- F. Pilate, Z.-B. Wen, F. Khelifa, Y. Hui, S. Delpierre, L. Dan, R. Mincheva, P. Dubois, K.-K. Yang and J.-M. Raquez, RSC Adv., 2018, 8, 27119–27130 RSC.
- A. Garle, S. Kong, U. Ojha and B. M. Budhlall, ACS Appl. Mater. Interfaces, 2012, 4, 645–657 CrossRef PubMed.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 4212–4217 CrossRef.
- D. Wang, M. J. A. Hore, X. Ye, C. Zheng, C. B. Murray and R. J. Composto, Soft Matter, 2014, 10, 3404–3413 RSC.
- G. Baffou, R. Quidant and F. J. García de Abajo, ACS Nano, 2010, 4, 709–716 CrossRef PubMed.
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06715e |
| This journal is © The Royal Society of Chemistry 2018 |