DOI:
10.1039/C8RA06039H
(Paper)
RSC Adv., 2018,
8, 29295-29300
A highly selective fluorescence “turn-on” sensor for Ca2+ based on diarylethene with a triazozoyl hydrazine unit†
Received
16th July 2018
, Accepted 10th August 2018
First published on 17th August 2018
Abstract
A new photochromic diarylethene derivative with a triazozoyl hydrazine unit has been designed and synthesized. Its photochromism and photoswitchable fluorescence behaviors were studied systematically by the stimuli of lights and chemical substances in acetonitrile solution. With the addition of Ca2+, the emission intensity enhanced 6.7 fold, accompanied by an obvious fluorescent color change from dark to light blue. The complexation between the derivative and Ca2+ is reversible with the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry, which was verified by Job's plot and MS. The limit of detection (LOD) for Ca2+ was determined to be 2.49 × 10−8 mol L−1. Based on this unimolecular platform, a logic circuit was designed with fluorescence intensity at 482 nm as the output and the combined stimuli of UV/vis and Ca2+/EDTA as four inputs.
:1 stoichiometry, which was verified by Job's plot and MS. The limit of detection (LOD) for Ca2+ was determined to be 2.49 × 10−8 mol L−1. Based on this unimolecular platform, a logic circuit was designed with fluorescence intensity at 482 nm as the output and the combined stimuli of UV/vis and Ca2+/EDTA as four inputs.
Introduction
Calcium, the fifth most abundant element in the earth's crust, plays a very important role in many environmental and biological processes.1–4 Meantime, Ca2+ is also a pivotal secondary messenger inside cells,5–7 and visualization of Ca2+ intracellular dynamics has generated considerable biological knowledge.8,9 Changes of Ca2+ concentration are related to immune responses and physiological responses to obesity.10–17 High concentrations of Ca2+ ions will reduce the permeability of neuron membranes to sodium ions, thus reducing the excitability, resulting in low tension of smooth muscle.18 Therefore, the effective and selective detection of Ca2+ ions is of great significance to medicine, environmental science and biochemistry.
Up to the present, there are a lot of traditional methods to detect various ions, such as atomic absorption spectrometry (AAS),19 inductively coupled plasma mass spectrometry (ICP-MS),20 voltammetry,21 ion-selective membrane,22 and liquid chromatography-mass spectrometry.23 However, these methods all require high cost and complex instruments, and it is inconvenient to monitor the site quickly in different environments. Compared to these, a fluorescence probe is an effective tool to detect target ions due to its simplicity, easy implementation, high sensitivity and low detection limit.24–31 To date, a number of fluorescent probes based on coumarin,32 rhodamine,33,34 nanoparticles,35–37 polymeric phenols,38,39 for the detection of Ca2+ have been reported. However, the Ca2+ selectivity of some probes is usually interfered by Mg2+ due to the similarly chemical behaviors of Ca2+ and Mg2+. What's more, few of the reported detection capabilities have fluorescent “on–off” mode for the detection of Ca2+ or its fluorescence enhancement rate is very small. Hence, developing novel fluorescence probes with higher sensitivity and selectivity for Ca2+ is of the utmost importance at present.
Among the reported fluorescence probes, diarylethene derivatives are the most promising candidates, due to their excellent thermal stability, remarkable fatigue resistance, drug resistance and rapid response.40–42 Furthermore, the identified ions could induce the diarylethene molecular to undergo polystable conversion,43 and these properties make it possible for the application in the filed of multi-addressable switching. Although some of processes have been made in diarylethenes based on ion recognition,44–50 the sensors for Ca2+ ions based on diarylethenes have rarely been reported.51,52
In this article, a new Ca2+ fluorescent sensor (1o) based on diarylethene and the triazozoyl hydrazine unit was designed and synthesized. The structure of 1o was characterized by 1H NMR, 13C NMR, and HRMS, and the data were shown in ESI (Fig. S1–S3).† Its photochromism and fluorescent properties induced by lights and chemical species were also systematically discussed. The synthesis and photochromism of 1o are shown in Scheme 1.
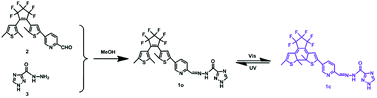 |
| | Scheme 1 The synthetic route and photochromism of 1o. | |
Experimental
General methods
Unless otherwise mentioned, all the reagents for the synthesis of the target compound were acquired from commercial suppliers and were used without further purification. All cations were added in the form of metal nitrates except for K+, Sn2+ and Hg2+ (all of their counter ions were chloride ions). Metal ions solutions (0.1 mol L−1) were prepared by dissolving their respective metal salts in deionized water. Necessary dilutions were made according to each experimental set up. NMR spectra were recorded on a Bruker AV400 spectrometer with deuterium generation of methanol (MeOD-d4) and dimethylsulfoxide (DMSO-d6) as solvents and tetramethylsilane (TMS) as an internal standard. Mass spectra were obtained using a Bruker Amazon SL ion trap mass spectrometer (ESI). The melting point was measured on a WRS-1B melting point apparatus. Absorption spectra were measured on an Agilent 8454 UV/vis spectrometer. Fluorescence spectra were recorded using a Hitachi F-4600 spectrophotometer. Photoirradiation was performed with an MUL-165 UV lamp and a MVL-210 visible lamp. Fluorescence quantum yield was measured with an Absolute PL Quantum Yield Spectrometer QYC11347-11.
Synthesis of 1o
Diarylethene 1o was synthesized via the route shown in Scheme 1. Precursor 2 was synthesized according to the method reported in literature.53 Then compound 2 (0.098 g, 0.2 mmol) was dissolved in 5.0 mL absolute methanol, followed by the addition of 1H-[1,2,4]triazole-3-carboxylic acid hydrazide (0.025 g, 0.2 mmol). The mixture was stirred for 12 h at room temperature in order to complete this reaction. After that, the solution was put into a refrigerator overnight. The crude product was washed with anhydrous methanol (5.0 mL × 3) and dried to give the bluish solid compound 1o (0.081 g, yield: 68%) with the mp of 488–490 K. 1H NMR (400 MHz, DMSO-d6), δ (ppm): 1.86 (s, 3H), 1.95 (s, 3H), 2.41 (s, 3H), 6.84 (s, 1H), 7.72 (s, 1H), 8.00 (d, 1H, J = 7.8 Hz), 8.15 (d, 1H, J = 7.6 Hz), 8.60 (s, 1H), 8.81 (s, 1H), 8.90 (s, 1H), 12.35 (s, 1H), 14.70 (s, 1H). 13C NMR (100 MHz, CH3OD-d4), δ (ppm): 11.6, 11.8, 12.1, 119.8, 122.7, 123.0, 123.3, 125.0, 125.3, 128.1, 129.0, 132.0, 132.4, 136.3, 137.1, 138.6, 142.0, 144.0, 147.4, 150.7. HRMS: m/z = 597.0943 [M + H+]+. Calcd 597.0968.
Results and discussion
Photochromism and fluorescent properties of 1o
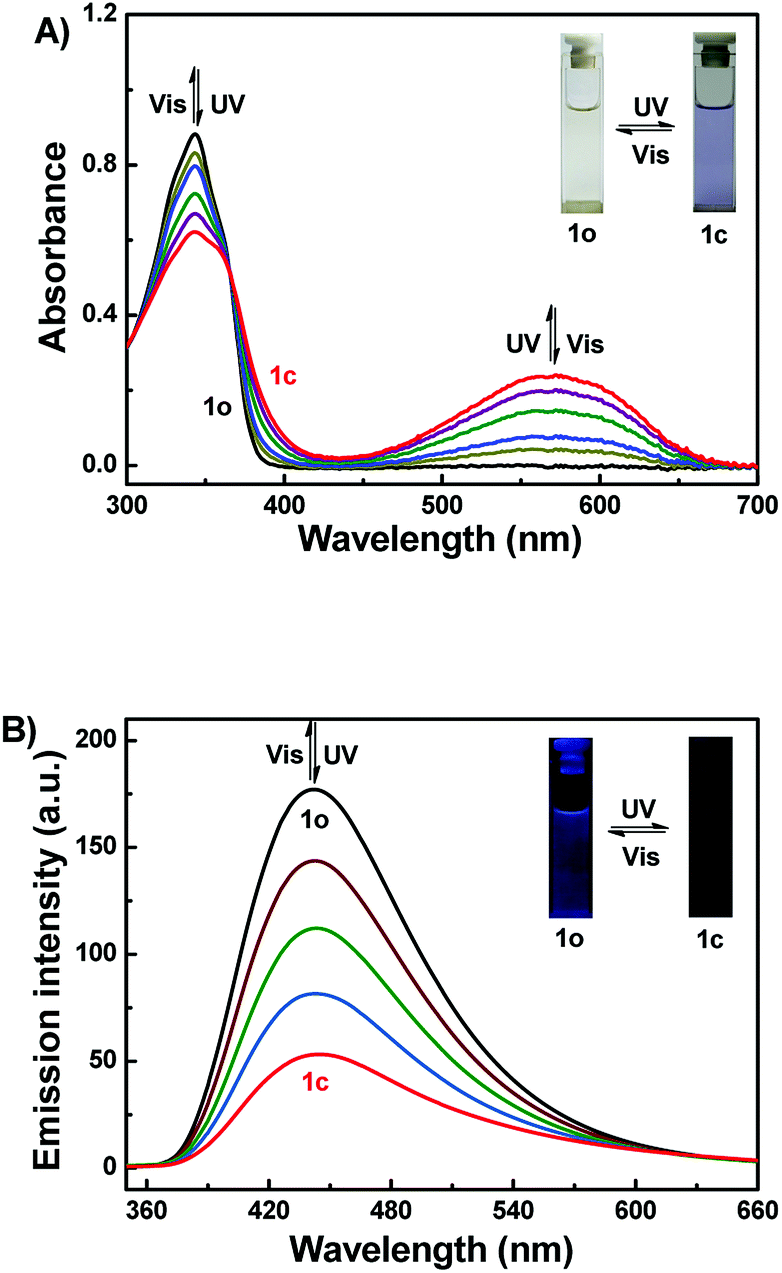
The photochromic properties of 1o were studied in acetonitrile (2.0 × 10−5 mol L−1) at room temperature as shown in Fig. 1A. The absorption maximum of 1o was observed at 343 nm (ε = 4.4 × 104 mol−1 L cm−1). Subsequently, upon irradiation with 297 nm light, a new broad absorption band centered at 572 nm (ε = 1.2 × 104 mol−1 L cm−1) emerged because of the formation of the closed-ring isomer 1c with larger π-electron delocalization in the molecule.54 In the photostationary state (PSS), a clear isosbestic point was observed at 365 nm, accompanied by a distinct color change from colorless to purple, which supported the reversible two-component photochromic reaction.55 Conversely, upon irradiation with visible light (λ > 500 nm), the colored solution of 1c was bleached entirely, and its absorption spectrum recovered to that of the open-ring isomer 1o. The quantum yields of cyclization and cycloreversion were determined to be 0.24 and 0.022, with 1,2-bis(2-methyl-5-phenyl-3-thienyl)perfluorocyclopentene as a reference.56 Additionally, the photochromic cyclization/cycloreversion kinetics were studied in acetonitrile solution (2.0 × 10−5 mol L−1) at room temperature. As described in Fig. S4A,† the relationships between the absorbance and exposure time have good linearity upon irradiation with 297 nm light, suggesting that the cyclization processes of 1o belong to the zeroth order reaction. The reaction rate constant (ko–c) of 1o was determined to be 1.51 × 10−3 s−1. Similarly, the relationship between −log(Abs) and exposure time also has perfect linearity, indicating that the cycloreversion process belong to the first order reaction. The reaction rate constant (kc–o) was determined to be 4.39 × 10−2 s−1 (Fig. S4B†). Furthermore, the fatigue resistance of 1o was also studied by alternating UV and visible lights at room temperature (Fig. S5†). The results indicated that the coloration–decoloration cycles between 1o and 1c could be repeated for 10 times with 15% degradation.
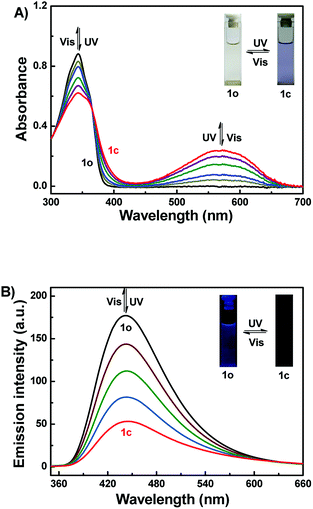 |
| | Fig. 1 Changes in the absorption (A) and fluorescence (B) spectra of 1o upon irradiation with UV/vis lights in acetonitrile (2.0 × 10−5 mol L−1) (λex = 340 nm). | |
Fig. 1B showed the fluorescence spectral changes of 1o upon photoirradiation in acetonitrile solution (2.0 × 10−5 mol L−1). The original state of 1o displayed weak fluorescence at 443 nm when excited at 340 nm, and the absolute fluorescence quantum yield was determined to be 0.005. On irradiation with 297 nm light, its emission intensity at 443 nm decreased gradually due to the generation of non-fluorescent isomer 1c. When the PSS was reached, the emission intensity of 1o was decreased significantly by ca. 70%, accompanied by the fluorescence color changed from dark purple to dark. Back irradiation with the proper wavelength of visible light (λ > 500 nm) regenerated the open-ring isomer 1o and recovered the original state.
Fluorescence response to metal ions
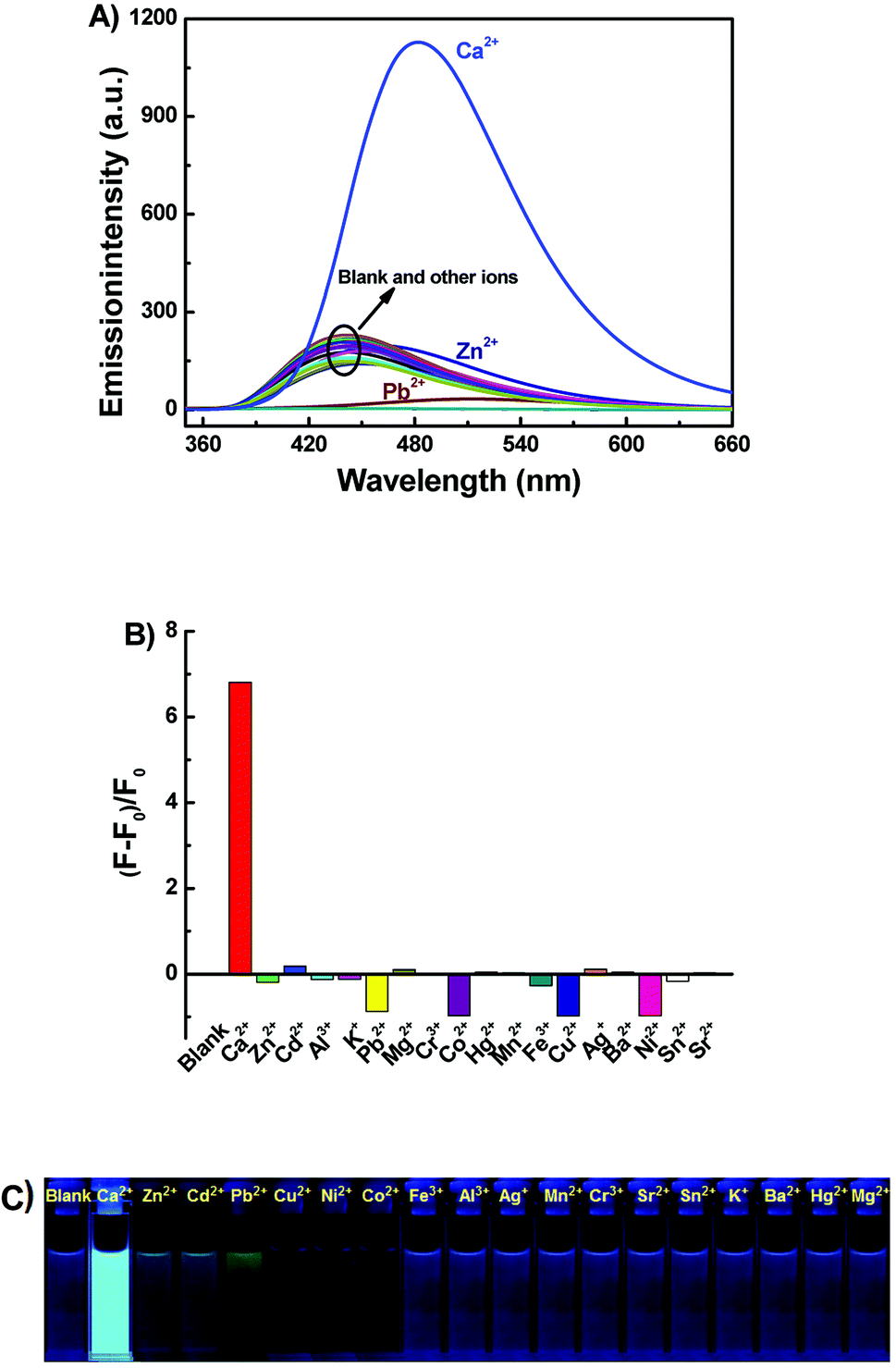
Under the same experimental conditions, the fluorescence responses of 1o toward various metal ions (5 equiv. 0.1 mol L−1) were investigated in acetonitrile such as Al3+, Cu2+, Sn2+, Zn2+, K+, Ag+, Ni2+, Ba2+, Mg2+, Mn2+, Cd2+, Sr2+, Hg2+, Co2+, Cr3+, Fe3+, Pb2+ and Ca2+. As can be seen in Fig. 2, when Ca2+ was added, the fluorescence intensity of 1o was enhanced 6.7 fold as compared with the emission intensity of 1o and the emission peak red shifted from 443 nm to 482 nm, accompanied by the fluorescent color change from dark purple to light blue. Furthermore, the fluorescence intensity of 1o quenched with the addition of Cu2+, Co2+, Ni2+. Moreover, upon addition of other metal ions, including Hg2+, Mg2+, Ca2+, Ba2+, Cr3+, Al3+, Mn2+, Sr2+, Pb2+, Fe3+, and K+, the fluorescence spectra of 1o showed inconspicuous changes. All the results indicated that the excellent capability of 1o for distinguishing Ca2+ from other metals ions. Therefore, the diarylethene 1o could be used as a selective fluorescence sensor for Ca2+ in acetonitrile.
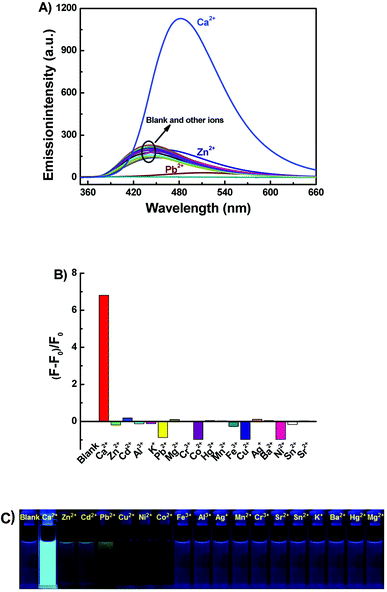 |
| | Fig. 2 Upon addition various metal ions to 1o (2.0 × 10−5 mol L−1) in acetonitrile: (A) fluorescence emission spectral changes (λex = 340 nm); (B) emission intensity changes; (C) fluorescent photos. | |
Fluorescence studies of 1o toward Ca2+
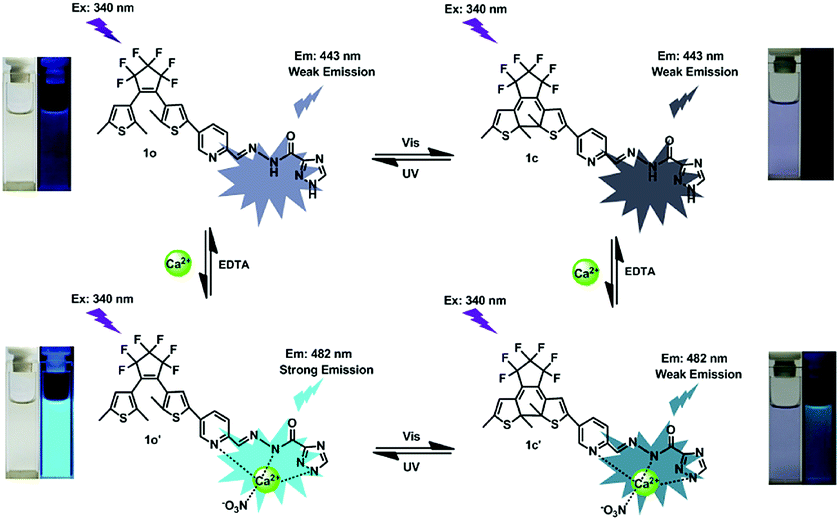
To further assess the responsive nature of 1o induced by Ca2+, the fluorescence spectral responses of 1o toward Ca2+ in acetonitrile were investigated in Fig. 3A. The results turned out that sensor 1o exhibited a very weak emission with a low quantum yield (Φ = 0.005) at 340 nm excitation. With the gradual addition of Ca2+, the emission intensity dramatically increased by 6.7 fold, accompanied by a red shift of 39 nm from 443 nm to 482 nm. Then the fluorescence intensity achieved its maximum until the amount of Ca2+ reached 2.2 equivalents of 1o (Fig. S4†), and the absolute quantum yield of fluorescence was determined to be 0.03, which is 6 fold of 1o. Meanwhile, the fluorescent color changed from dark purple to light blue, which was coincident with the changes in the fluorescence spectra. The weak fluorescence of the initial state 1o was put down to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N isomerization, which has long been known as the dominant decay process.57,58 However, a stable chelate 1o–Ca2+ was formed with the existence of Ca2+. The isomerization of CN bond was inhibited, which enhanced the rigidity of the molecule, thus leading to the chelation enhanced fluorescence (CHEF) effect.59 Furthermore, the reversibility experiment was established by adding 6.0 equiv. EDTA (0.1 mol L−1) to 1o–Ca2+ (1o′) solution which possibly deprives Ca2+ away from the binding position. Eventually, the fluorescence spectrum of 1o′ was brought back to that of 1o, suggesting that the complexation–decomplexation reaction between 1o and Ca2+ was reversible.
N isomerization, which has long been known as the dominant decay process.57,58 However, a stable chelate 1o–Ca2+ was formed with the existence of Ca2+. The isomerization of CN bond was inhibited, which enhanced the rigidity of the molecule, thus leading to the chelation enhanced fluorescence (CHEF) effect.59 Furthermore, the reversibility experiment was established by adding 6.0 equiv. EDTA (0.1 mol L−1) to 1o–Ca2+ (1o′) solution which possibly deprives Ca2+ away from the binding position. Eventually, the fluorescence spectrum of 1o′ was brought back to that of 1o, suggesting that the complexation–decomplexation reaction between 1o and Ca2+ was reversible.
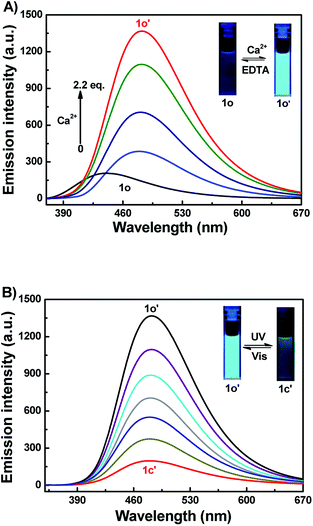 |
| | Fig. 3 (A) Fluorescence spectra changes of 1o (2.0 × 10−5 mol L−1) in acetonitrile induced by Ca2+ (0–2.2 equiv.); (B) fluorescence spectra changes of 1o′ (2.0 × 10−5 mol L−1 in acetonitrile), upon irradiation with UV-vis light (λex = 340 nm). | |
Complexation mechanism of 1o with Ca2+
To further investigate the coordination mode of 1o and Ca2+, Job's plot analysis was performed by using the emission intensity at 482 nm for Ca2+ as a function of molar fraction of 1o according to the reported method.60 As shown in Fig. 4, the maximum value was achieved when the molar fraction of [Ca2+]/([1o] + [Ca2+]) was about 0.5, suggesting that 1o was bound to Ca2+ with a 1:1 stoichiometry in acetonitrile. Based on these results and fluorescence titration data, the association constant (Ka) of 1o with Ca2+ was calculated from the slope and intercept of these linear plots to be 8.86 × 103 L mol−1 with a good linear relationship (R = 0.990) (Fig. S6†). According to the method reported in previous literature,61 the limit of detection (LOD) of 1o toward Ca2+ was determined to be 2.49 × 10−8 mol L−1 (Fig. S7†). Therefore, 1o could serve as a fluorescent sensor for detection of Ca2+ with high selectivity and sensitivity in acetonitrile.
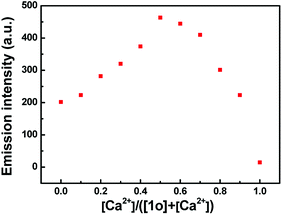 |
| | Fig. 4 Job's plot showing the 1:1 complex of 1o and Ca2+ in acetonitrile (2.0 × 10−5 mol L−1). | |
1H NMR titration experiments were also carried out in DMSO-d6 to further study the binding mode of 1o and Ca2+. As shown in Fig. 5, the signals of the Ha (imino proton) at 12.48 ppm and the Hb (–NH– proton on the triazole) at 14.70 ppm were found in the 1H NMR spectrum of 1o. With the addition of Ca2+, the imino proton (Ha) disappeared completely, indicating the coordinate bond between imino N and Ca2+ was formed. Meanwhile, the –NH– proton (Hb) on the triazole was also disappeared finally, indicating that the formation of coordinate bond between the N atom on the triazole and Ca2+. On the other hand, the signal of Hc on CHN decreased gradually, and the Hc displayed a downfield shift of 0.01 ppm from 8.81 ppm to 8.82 ppm ultimately, showing that the formation of the coordinate bond between the N atom on the Schiff base and Ca2+. All of the results suggested that the imino N, the N on the Schiff base unit, and the N on the triazole ring are the most reasonable binding sites. Furthermore, the HRMS analysis was also carried out to confirm the interaction between 1o–Ca2+. The testing sample was prepared by adding excessive Ca2+ to 1o in acetonitrile, and the result displayed that the signal located at m/z = 698.0362 was consistent with the ensemble [1o + Ca2+ + NO3−]+ (m/z calcd: 698.0392) (Fig. S8†). These results further proved that 1o and Ca2+ formed a 1:1 complex. Based on these facts, the most likely binding mode was shown in Scheme 2. Furthermore, the affection of pH to the sensor was also investigated. According to the methods in our previous work,62 the fluorescence intensity changes of 1o and 1o′ over different pH values in CH3CN:H2O (9:1, v/v) were shown in Fig. S11.† The results demonstrated that the optimal pH region for sensor 1o and 1o′ was 6.0–9.0.
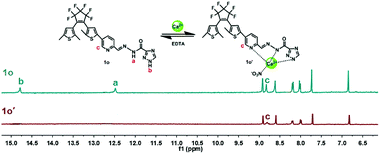 |
| | Fig. 5 Changes in 1H NMR of 1o and 1o′ in DMSO-d6 (inset shows the proposed binding mode of 1o′ complex). | |
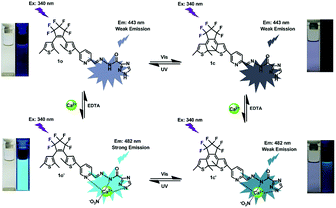 |
| | Scheme 2 Dual-controlled fluorescent switching behaviors of 1o induced by Ca2+/EDTA and UV/vis light. | |
Application in logic circuit and practical sample
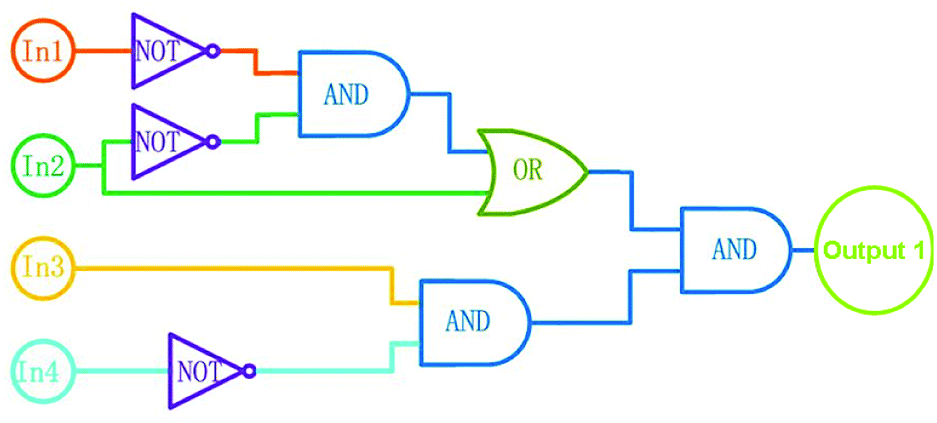
According to the properties described above, the fluorescence emission of 1o could be effectively modulated by either UV-vis lights or chemical reagents stimuli in acetonitrile, and the photoswitching behaviors of 1o are shown in Scheme 2. On the basis of these facts, a logic circuit was constructed by using UV (In1), vis (In2), Ca2+ (In3), and EDTA (In4) as four input signals, and the emission intensity at 482 nm as the output signal (Fig. 6). The emission intensity of 1o at 443 nm was considered as the initial value. When the emission intensity at 482 nm was 6.7 fold larger than the initial value, the output signal could serve as ‘on’ with a Boolean value of ‘1’. Otherwise, it serves as ‘off’ with a Boolean value of ‘0’. For instance, when the input strings were ‘1, 1, 1, and 0’, the corresponding signals to In1, In2, In3, and In4 are ‘on, on, on, and off’. Under this condition, 1o was converted to 1o′ with the stimuli of Ca2+, and the emission intensity increased dramatically. As a result, the output digital was ‘1’. Similarly, others stimuli would cause different on–off fluorescence switching. All of the possible logic strings were listed in the combinational logic circuit as shown in Table 1.
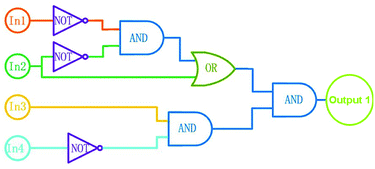 |
| | Fig. 6 Combinational logic circuits equivalent to the truth table given in Table 1: In1 (UV light), In2 (visible light), In3 (Ca2+), In4 (EDTA). | |
Table 1 Truth table for all the possible strings of four binary-input data and the corresponding output digit
| Inputs |
Outputa (λem = 482 nm) |
| In1 (UV) |
In2 (Vis) |
In3 (Ca2+) |
In4 (EDTA) |
| At 482 nm, the emission intensity 6.7 fold of the original value is defined as 1, otherwise defined as 0. |
| 0 |
0 |
0 |
0 |
0 |
| 1 |
0 |
0 |
0 |
0 |
| 0 |
1 |
0 |
0 |
0 |
| 0 |
0 |
1 |
0 |
1 |
| 0 |
0 |
0 |
1 |
0 |
| 1 |
1 |
0 |
0 |
0 |
| 1 |
0 |
1 |
0 |
0 |
| 1 |
0 |
0 |
1 |
0 |
| 0 |
1 |
1 |
0 |
1 |
| 0 |
1 |
0 |
1 |
0 |
| 0 |
0 |
1 |
1 |
0 |
| 1 |
1 |
1 |
0 |
1 |
| 1 |
1 |
0 |
1 |
0 |
| 1 |
0 |
1 |
1 |
0 |
| 0 |
1 |
1 |
1 |
0 |
| 1 |
1 |
1 |
1 |
0 |
The application of 1o to the practical samples was also performed. The Ca2+ content in real water samples from the Ganjiang River in Nanchang, Jiangxi province were determined. Table 2 displayed the results measured with 1o after adding a moderate amount of Ca2+. The recovery was ranged from 96.8% to 105%. These results indicated that 1o could be used for detecting Ca2+ in practical samples with higher accuracy, and has certain practical value.
Table 2 Application in practical samples detection for Ca2+
| Sample |
Ca2+ added (μM) |
Ca2+ determined (μM) |
Recovery (%) |
| 1 |
0.40 |
0.42 |
105 |
| 2 |
0.80 |
0.81 |
101 |
| 3 |
1.20 |
1.18 |
98.3 |
| 4 |
1.60 |
1.55 |
96.8 |
| 5 |
2.00 |
1.97 |
98.5 |
Conclusions
In conclusion, a novel fluorescent sensor based on a diarylethene derivative with triazozoyl hydrazine unit was developed. The sensor exhibited high selectivity toward Ca2+ over other metal ions, and the detection limit for Ca2+ could be as low as 2.49 × 10−8 mol L−1. Furthermore, a logic circuit was designed and constructed with the emission intensity at 482 nm as output signal and the UV/vis lights, Ca2+/EDTA as input signals. The application results indicated that the sensor could be used for the detection of Ca2+ in practical samples. All these results will be helpful for the design and construction of new sensors for Ca2+ with high selectivity and sensitivity in the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful for the financial support from the “5511” Science and Technology Innovation Talent Project of Jiangxi (2016BCB18015), the key project of Natural Science Foundation of Jiangxi Province (20171ACB20025), the Project of the Science Funds of the Education Office of Jiangxi (GJJ160773), the Young Talents Project of Jiangxi Science and Technology Normal University (2015QNBJRC004), the Project of Jiangxi Science and Technology Normal University Advantage Sci-Tech Innovative Team (2015CXTD002).
Notes and references
- M. Landoni, B. F. Cerino and N. Haman, et al., J. Agric. Food Chem., 2013, 61, 4622–4630 CrossRef PubMed.
- S. M. Bawin and W. R. Adey, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 1999–2003 CrossRef.
- S. V. Dorozhkin and M. Epple, Angew. Chem., Int. Ed., 2002, 41, 3130–3146 CrossRef PubMed.
- S. Orrenius, B. Zhivotovsky and P. Nicotera, Nat. Rev. Mol. Cell Biol., 2003, 4, 552 CrossRef PubMed.
- D. E. Clapham, Cell, 2007, 131, 1047–1058 CrossRef PubMed.
- E. Arunkumar, A. Ajayaghosh and J. Daub, J. Am. Chem. Soc., 2005, 127, 3156–3164 CrossRef PubMed.
- T. Alizadeh, A. N. Shamkhali and Y. Hanifehpour, et al., New J. Chem., 2016, 40, 8479–8487 RSC.
- M. Raju, R. R. Nair and I. H. Raval, et al., Analyst, 2015, 140, 7799–7809 RSC.
- Y. Nakagawa, Y. Watanabe and Y. Igarashi, et al., Bioorg. Med. Chem. Lett., 2015, 25, 2963–2966 CrossRef PubMed.
- A. T. Harootunian, J. P. Kao and S. Paranjape, et al., Science, 1991, 251, 75–78 CrossRef PubMed.
- A. Minta, J. P. Kao and R. Y. Tsien, J. Biol. Chem., 1989, 264, 8171–8178 Search PubMed.
- G. Grynkiewicz, M. Poenie and R. Y. Tsien, J. Biol. Chem., 1985, 260, 3440–3450 Search PubMed.
- K. V. Kuchibhotla, C. R. Lattarulo and B. T. Hyman, et al., Science, 2009, 323, 1211–1215 CrossRef PubMed.
- I. Micu, A. Ridsdale and L. Zhang, et al., Nat. Med., 2007, 13, 874 CrossRef PubMed.
- D. Skokos, G. Shakhar and R. Varma, et al., Nat. Immunol., 2007, 8, 835 CrossRef PubMed.
- J. Chambers, R. S. Ames and D. Bergsma, et al., Nature, 1999, 400, 261 CrossRef PubMed.
- C. M. Armstrong and G. Cota, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 4154–4157 CrossRef.
- M. Frankowski, A. Zioła-Frankowska and J. Siepak, Talanta, 2010, 80, 2120–2126 CrossRef PubMed.
- A. Sanz-Medel, A. B. S. Cabezuelo and R. Milačič, et al., Coord. Chem. Rev., 2002, 228, 373–383 CrossRef.
- R. N. Goyal, V. K. Gupta and S. Chatterjee, Biosens. Bioelectron., 2009, 24, 3562–3568 CrossRef PubMed.
- V. K. Gupta, A. K. Singh and S. Mehtab, et al., Anal. Chim. Acta, 2006, 566, 5–10 CrossRef.
- V. K. Gupta, R. Jain and S. Sharma, et al., Int. J. Electrochem. Sci., 2012, 7, 569–587 Search PubMed.
- H. N. Kim, W. X. Ren and J. S. Kim, et al., Chem. Soc. Rev., 2012, 41, 3210–3244 RSC.
- Z. Shi, Y. Tu and S. Z. Pu, RSC Adv., 2018, 8, 6727–6732 RSC.
- Y. L. Pak, K. M. K. Swamy and J. Yoon, Sensors, 2015, 15, 24374–24396 CrossRef PubMed.
- S. Pan, H. Tang and Z. Song, et al., Chin. J. Chem., 2017, 35, 1263–1269 CrossRef.
- M. Morimoto, Y. Takagi and K. Hioki, et al., Dyes Pigm., 2018, 153, 144–149 CrossRef.
- N. S. Wang, R. J. Wang and Y. Y. Tu, et al., Spectrochim. Acta, Part A, 2018, 196, 303–310 CrossRef PubMed.
- N. Tetsuya, Y. Miyasaka and Y. Yokoyama, Chem. Commun., 2018, 54, 3207–3210 RSC.
- C. Yu, L. Chen and J. Zhang, et al., Talanta, 2011, 85, 1627–1633 CrossRef PubMed.
- C. Yu, J. Zhang and J. Li, et al., Microchim. Acta, 2011, 174, 247–255 CrossRef.
- L. Cao, C. Jia and Y. Huang, et al., Tetrahedron Lett., 2014, 55, 4062–4066 CrossRef.
- X. Yu, P. Zhang and Q. Liu, et al., Mater. Sci. Eng., C, 2014, 39, 73–77 CrossRef PubMed.
- M. S. Eom, W. Jang and Y. S. Lee, et al., Chem. Commun., 2012, 48, 5566–5568 RSC.
- J. Zhang, Y. Wang and X. Xu, et al., Analyst, 2011, 136, 3865–3868 RSC.
- A. Ajayaghosh, E. Arunkumar and J. Daub, Angew. Chem., 2002, 114, 1844–1847 CrossRef.
- S. K. Sharma, S. Kumar and R. Tyagi, et al., Microchem. J., 2008, 90, 89–92 CrossRef.
- R. Sahli, N. Raouafi and E. Maisonhaute, et al., Electrochim. Acta, 2012, 63, 228–231 CrossRef.
- S. Z. Pu, T. S. Yang and J. K. Xu, et al., Tetrahedron, 2005, 61, 6623–6629 CrossRef.
- S. Z. Pu, J. K. Xu and L. Shen, et al., Tetrahedron Lett., 2005, 46, 871–875 CrossRef.
- R. J. Wang, P. P. Ren and S. Z. Pu, et al., J. Photochem. Photobiol., A, 2014, 294, 44–53 CrossRef.
- H. Tian, B. Qin and R. Yao, et al., Adv. Mater., 2003, 15, 2104–2107 CrossRef.
- Q. Zou, X. Li and J. Zhang, et al., Chem. Commun., 2012, 48, 2095–2097 RSC.
- A. Bianco, S. Perissinotto and M. Garbugli, et al., Laser Photonics Rev., 2011, 5, 711–736 CrossRef.
- Q. Zou, J. Jin and B. Xu, et al., Tetrahedron, 2011, 67, 915–921 CrossRef.
- Z. Zhou, H. Yang and M. Shi, et al., ChemPhysChem, 2007, 8, 1289–1292 CrossRef PubMed.
- Z. Zhou, S. Xiao and J. Xu, et al., Org. Lett., 2006, 8, 3911–3914 CrossRef PubMed.
- Z. Li, C. Zhang and Y. Ren, et al., Org. Lett., 2011, 13, 6022–6025 CrossRef PubMed.
- S. Z. Pu, D. Jiang and W. Liu, et al., J. Mater. Chem., 2012, 22, 3517–3526 RSC.
- R. Wang, X. Dong and G. Liu, et al., Luminescence, 2015, 30, 1290–1296 CrossRef PubMed.
- S. Q. Cui, Z. Y. Tian and S. Z. Pu, et al., RSC Adv., 2016, 6, 19957–19963 RSC.
- S. Z. Pu, Z. P. Tong and G. Liu, et al., J. Mater. Chem. C, 2013, 1, 4726–4739 RSC.
- M. Irie, Chem.
Rev., 2000, 100, 1685–1716 CrossRef PubMed.
- Z. X. Li, L. Y. Liao and W. Sun, et al., J. Phys. Chem. C, 2008, 112, 5190–5196 CrossRef.
- S. Z. Pu, T. Yang and J. Xu, et al., Tetrahedron, 2005, 61, 6623–6629 CrossRef.
- M. Irie, T. Lifka, S. Kobatake and N. Kato, J. Am. Chem. Soc., 2000, 122, 4871–4876 CrossRef.
- W. K. Dong, X. L. Li and L. Wang, et al., Sens. Actuators, B, 2016, 229, 370–378 CrossRef.
- E. T. Feng, Y. Y. Tu and C. B. Fan, et al., RSC Adv., 2017, 7, 50188–50194 RSC.
- Y. M. Xue, R. J. Wang and C. H. Zheng, et al., Tetrahedron Lett., 2016, 57, 1877–1881 CrossRef.
- W. Liu, L. Xu and R. Sheng, et al., Org. Lett., 2007, 9, 3829–3832 CrossRef PubMed.
- S. Guo, G. Liu and C. Fan, et al., Sens. Actuators, B, 2018, 266, 603–613 CrossRef.
- Z. Wang, S. Q. Cui, S. Y. Qiu and S. Z. Pu, Spectrochim. Acta, A, 2018, 205, 21–28 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06039h |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Shouyu Qiu and
Shouzhi Pu*
*,
Shouyu Qiu and
Shouzhi Pu*