
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of kealiiquinone: the regio-controlled strategy for accessing its 1-methyl-4-arylbenzimidazolone core†
Velayudham Ramadoss a,
Angel J. Alonso-Castrob,
Nimsi Campos-Xolalpac,
Rafael Ortiz-Alvaradod,
Berenice Yahuaca-Juárezd and
César R. Solorio-Alvarado*a
a,
Angel J. Alonso-Castrob,
Nimsi Campos-Xolalpac,
Rafael Ortiz-Alvaradod,
Berenice Yahuaca-Juárezd and
César R. Solorio-Alvarado*a
aUniversidad de Guanajuato, Departamento de Química, División de Ciencias Naturales y Exactas, Campus Guanajuato, Cerro de la Venada S/N, 36040, Guanajuato, Gto., Mexico. E-mail: csolorio@ugto.mx
bUniversidad de Guanajuato, Departamento de Farmacia, División de Ciencias Naturales y Exactas, Noria Alta S/N, 36050, Guanajuato, Gto., Mexico
cUniversidad Metropolitana, Unidad Xochimilco, Calzada del Hueso 1100, Coyoacán 04960, México D.F., Mexico
dUniversidad Michoacana de San Nicolás de Hidalgo, Facultad de Químico Farmacobiología, Tzintzuntzan 173, col. Matamoros 58240, Morelia, Michoacán, Mexico
First published on 31st August 2018
Abstract
A practical, concise and straightforward total synthesis of kealiiquinone 1, a naphtho[2,3-d]imidazole alkaloid obtained from the Micronesian marine sponge Leucetta sp. was accomplished. The squaric acid chemistry to construct the 1,4-quinoid ring and the regioselective N-methylation through a benzo[c][1,2,5]selenadiazolium heterocycle are the key features in this report. The full details of the representative approaches involving the different attempted synthetic strategies are also presented. Finally a successful total synthesis of this complex secondary metabolite is described.
Introduction
Natural secondary metabolites obtained from marine sponges have been an extensive source of pharmacologically active new drugs.1 Representative examples obtained from the sponge Leucetta chagosensis include clathridine, naamidines A, G, H,2 and (−)-spiroleucettadine.3 In 1990 Scheuer and Clardy isolated two new imidazole alkaloids from the Leucetta sp. sponge, the pyronaamidine and kealiiquinone 1 (Fig. 1).4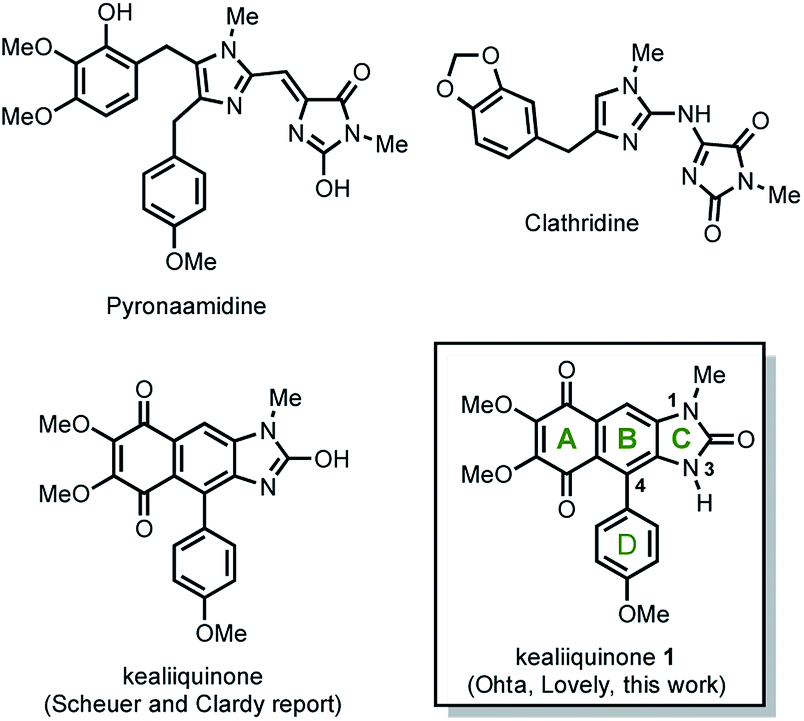 | ||
| Fig. 1 Representative imidazole alkaloids isolated from the Leucetta sp. sponge. | ||
Kealiiquinone 1, is a complex natural compound, which contains a 1,4-quinoid ring (A ring), fused to a regio-differentiated N-methyl-4-arylbenzimidazolone with the aryl and the methyl groups at opposite hemispheres of the alkaloid. Due to its attractive molecular architecture and to its modest biological activity, two total syntheses have been developed to date (Scheme 1).
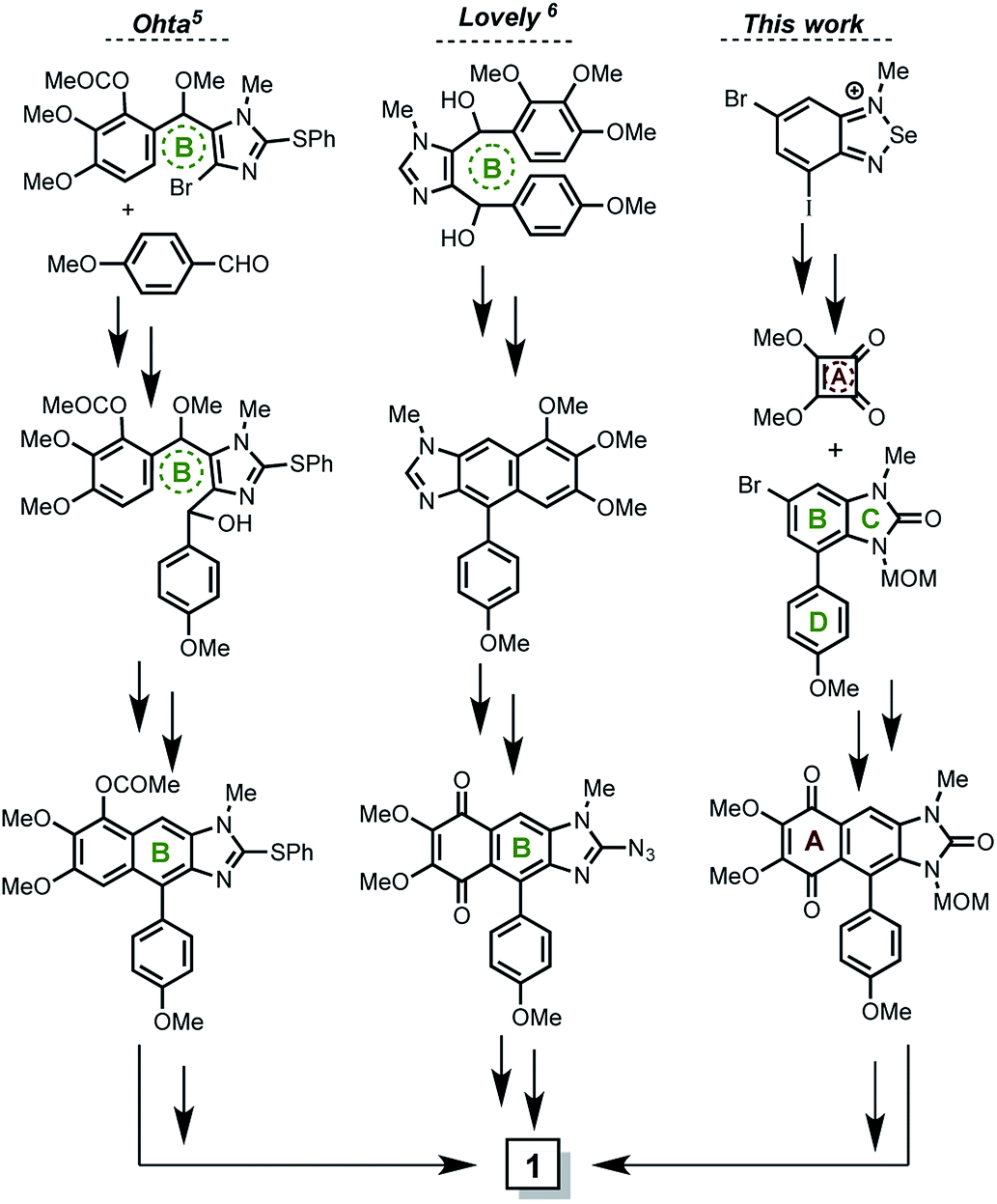 | ||
| Scheme 1 Described total synthesis of kealiiquinone to date. | ||
The first total synthesis of kealiiquinone was described by Ohta.5 This strategy was mainly based upon the central ring (B ring, Scheme 1) formation in acid media via a Friedel–Crafts-type condensation. The procedure is overall efficient, however a functionalized imidazole as building block as well as the use of several protecting groups (four of them) could be pointed as limitants to scale up the process. The second total synthesis of kealiiquinone was described by Lovely.6 In this approach, the same two main features about the use of a functionalized imidazole, as well as the Friedel–Crafts-type condensation also in acidic media for equally constructing the central B ring of the alkaloid are found. Notwithstanding great similarities can be identified, the synthesis was completed but several (four of them) organic redox fluctuations (oxidation–reduction–oxidation) are found. Additionally the biological assays of activity were carried out in a successful way.7
A particular and well-identified synthetic challenge in the total synthesis of keliiquinone is the regioselective N-methylation at the first position nitrogen of the imidazolone ring (see Fig. 1), this is the key objective of our work to complete the total synthesis of 1. To highlight the importance of the aforementioned molecular regio-differentiation, is worth to mention the efforts to address this problem, mainly by Lovely group.8 In the same context, our group recently described the total synthesis of the 3-methylkealiiquinone,9 in route to the total synthesis of the natural alkaloid 1.
In regards to previous reports, our procedure overcomes the necessity for using imidazole as building block as described by Ohta and Lovely. This is an advantage in our strategy since it allowed us to scale up in a straightforward fashion the starting materials. Also a metallation–sulphinilation–oxidation sequence at the first imidazole carbon was necessary for the final imidazolone construction in the Ohta route, which did not occur in ours. On the other hand, at least four sequences implying REDOX fluctuations were presented in the Lovely kealiiquinone synthesis, while in our route only two of them were used and correspond to the protecting groups used. Additionally as we described9 our strategy is modular and is possible to introduce different functional groups in the 1,4-quinone as well as in the aryl pendant rings. Finally is important to mention that the regiodifferentiation to get selective methylation in final natural alkaloid include several steps. In our synthesis the use of protecting groups is necessary. However we use only two protecting groups instead of four as in the Ohta protocol.
Considering all of these reports, herein we describe the full details about representative strategies attempted to prepare 1. Also we present our developed route, which was totally focused in the regioselective N-methylation of the imidazole ring and finally allowed us to accomplish the total synthesis of kealiiquinone 1 by late stage construction of the 1,4-quinoid ring (A ring).
Results
The first attempted strategy to prepare 1 is following outlined (Scheme 2).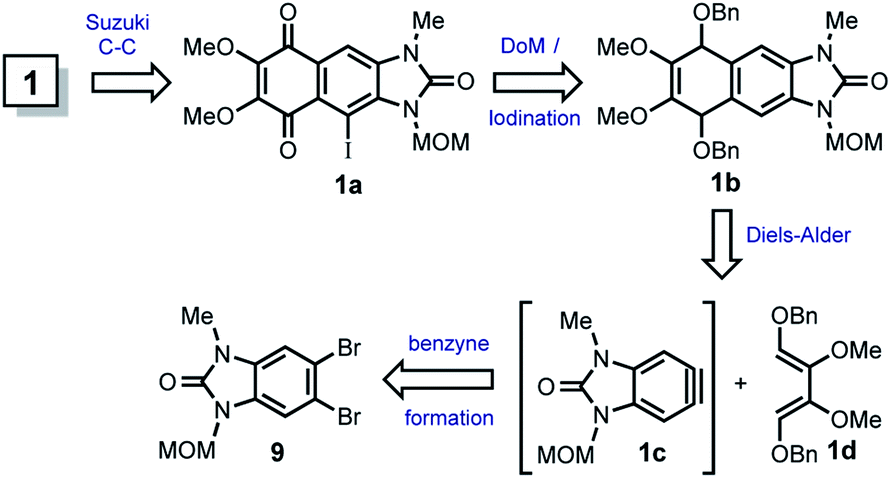 | ||
| Scheme 2 First strategy towards the total synthesis of kealiiquinone 1. | ||
In this route, kealiiquinone 1 can be obtained from the iodoquinone 1a by MOM removal and cross-coupling reaction with the p-anisyl boronic acid. The 1,4-quinone 1a could be obtained by oxidation of the Diels–Alder adduct 1b, where the MOM group may act as protecting-directing group for the directed ortho-metalation (DoM)/iodination.10 The compound 1b is the result of the Diels–Alder reaction between diene 1d and benzyne 1c, which comes from 9. Thus, different routes intended to prepare 9 are following summarized (Scheme 3).
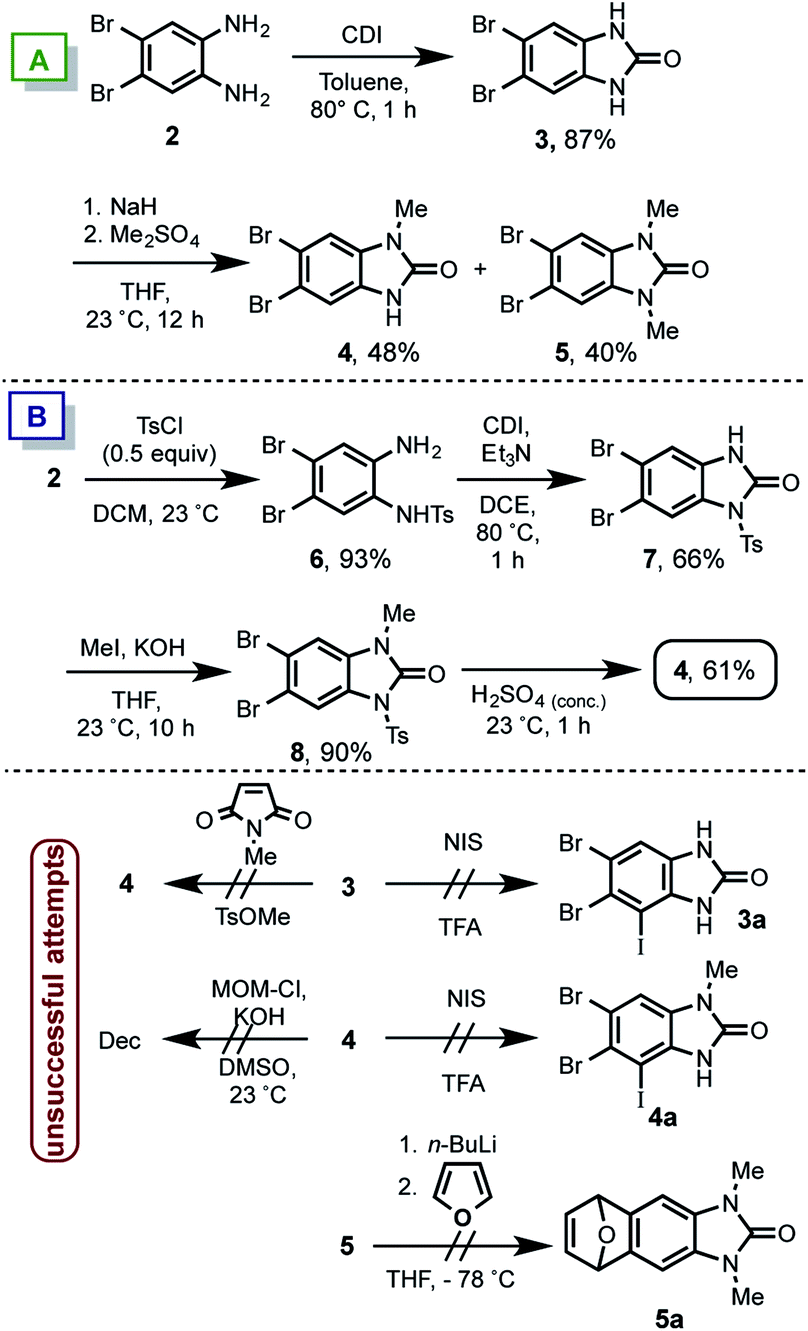 | ||
| Scheme 3 Synthesis of benzimidazolones 4 and 5 for preparing 9. | ||
We prepared the known symmetric aniline 2,11 that reacted with CDI in toluene at 80 °C affording the benzimidazolone 3. Then, by using dimethylsulfate, the single methylation for accessing to the non-symmetric benzimidazolone 4 was not observed. Instead a mixture of 4 and 5 in almost (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) ratio was obtained (Scheme 3A). Accordingly to the required nitrogens differentiation in 4, a different route was attempted (Scheme 3B). Therein the mono-tosylation of 2, was achieved giving rise to 6 in 93% of yield. The subsequent benzimidazolone formation by using CDI in DCE yielded 7 in 66%, which was methylated in presence of methyliodide to furnish 8 in 90% of yield. Finally the treatment with sulphuric acid lead to 4 in 61%. This derivative shows the desired non-symmetric mono-methylated benzimidazolone.
:1) ratio was obtained (Scheme 3A). Accordingly to the required nitrogens differentiation in 4, a different route was attempted (Scheme 3B). Therein the mono-tosylation of 2, was achieved giving rise to 6 in 93% of yield. The subsequent benzimidazolone formation by using CDI in DCE yielded 7 in 66%, which was methylated in presence of methyliodide to furnish 8 in 90% of yield. Finally the treatment with sulphuric acid lead to 4 in 61%. This derivative shows the desired non-symmetric mono-methylated benzimidazolone.
Several other trials (Scheme 3C) to obtain 4 such as the use of N-methylsuccinimide or methyltosylate as methylating reagent on 3 resulted in no reaction. On the other hand accordingly with the 1b structure (see Scheme 2), a MOM group is necessary as protecting-directing group for the DoM/iodination sequence. Thereby the incorporation of MOM was tried in 4, unfortunately the decomposition of starting material was found after different attempts. Upon the failure to attach a single MOM group in 4, it was decided to install the iodine atom of the iodoquinone 1a. Different trials by using NIS/TFA in 3 or 4 to get 3a/4a respectively only showed the low reactivity of the starting material and as consequence no reaction was observed. Even tough we did not succeed in the compound 9 synthesis, it was tested the benzyne formation since is the key step in the proposed route. Thus, compound 5 was mixed with n-BuLi at −78 °C in presence of furane. Several conditions changing base equivalents, time and temperatures were assayed, however we were unable to get any Diels–Alder adduct 5a (see ESI†). Presumably due to an electron-rich dienophile formation in a direct electron demand Diels–Alder reaction.
In this point, we decided to change substantially the general strategy towards the total synthesis of 1. A very important aspect was not to complete the total synthesis by using a Friedel–Crafts strategy as key step, since Ohta and Lovely previously described it. Based upon our expertise,9 we focused in the construction of the quinone ring as the last step of the route, we planned the use of the squaric acid chemistry12 for building up it. Accordingly, the new synthetic strategy is described (Scheme 4).
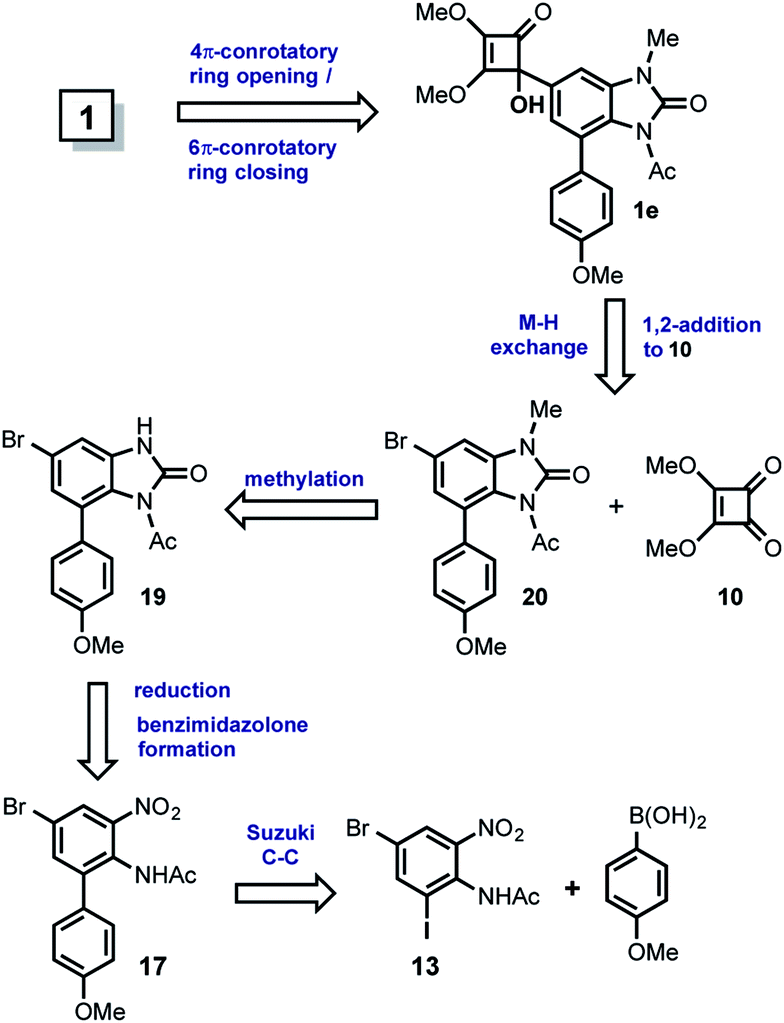 | ||
| Scheme 4 Second strategy towards the total synthesis of kealiiquinone 1. | ||
In this approach, kealiiquinone 1 comes from the tertiary alcohol 1e via the thermally-promoted electrocyclic sequence 4π-conrotatory ring opening/6π-conrotatory ring closure. The alcohol 1e can be the result of the 1,2-addition between the dimethylsquarate 10 and the organolithium generated form 20 by metal–halogen (M–H) exchange. Compound 20 is obtained by the methylation of the bromobenzimidazolone 19 which can be prepared by the nitro group reduction concomitant phosgene treatment of 17. Finally this ortho-nitroarylaniline can be synthesized by a Suzuki cross-coupling reaction within p-anisylboronic acid and the regio-differentiated bromoiodoacetyl-nitroaniline 13. The most relevant aspect in this alternative is the regioselective proposal for introducing the desired methyl group at first position nitrogen of the natural compound. This regiochemistry is based upon the different oxidation states at the adjacent nitrogens (Scheme 5).
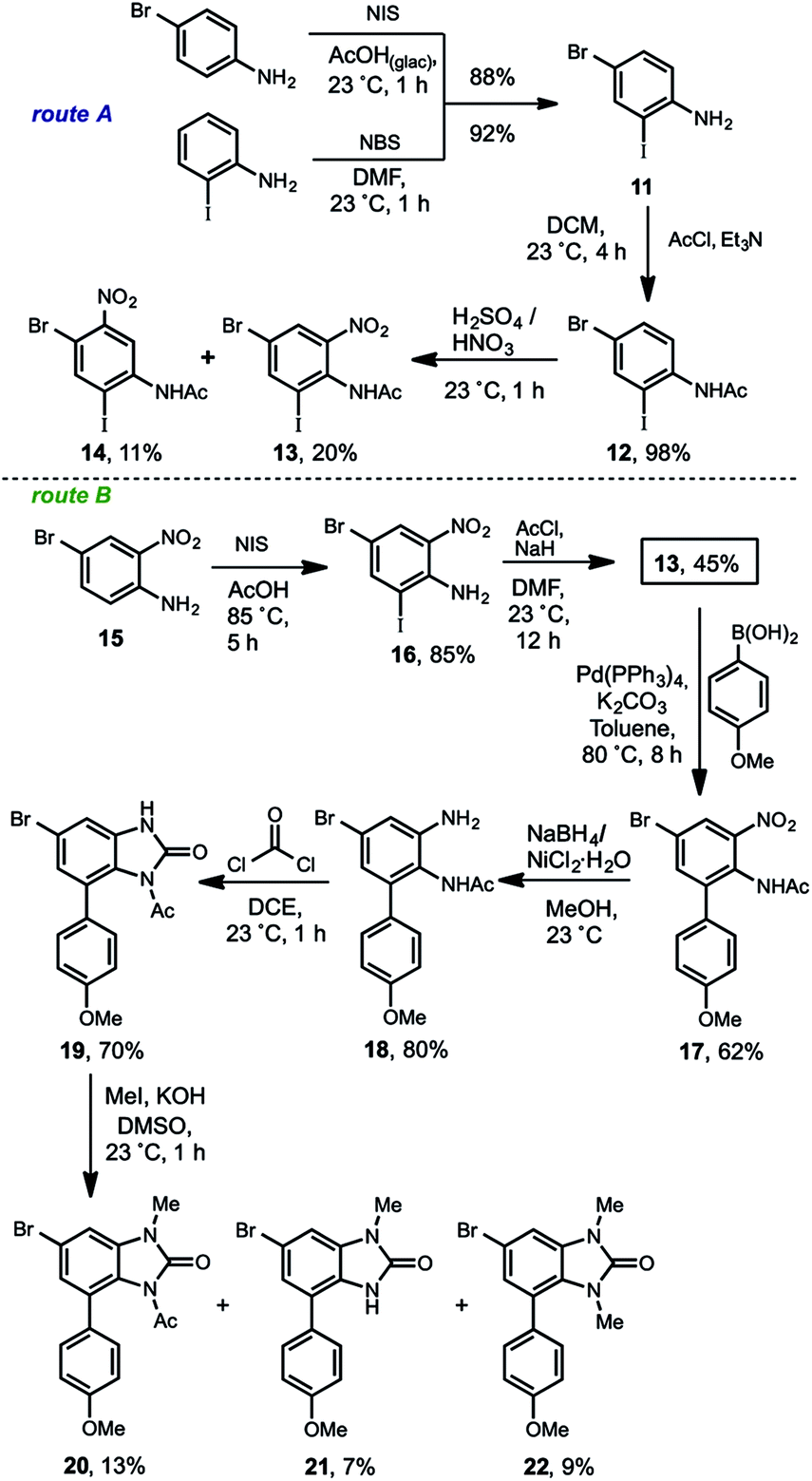 | ||
| Scheme 5 Synthesis of the regio-differentiated arylbenzimidazolone 20. | ||
The synthesis of the arylbenzimidazolone 20 was developed by two convergent routes (route A and B), which merged in 13. In the route A, two commercially available compounds were used for starting. In one side para-bromoaniline was iodinated and on the other side the ortho-iodoaniline was brominated in 88% and 92% of yield respectively, to get 11. After an extensive experimental analysis, we determined this as a good point for the acetyl group introduction, which produced 12 almost quantitatively. Next, the sulfonitric mixture gave rise to a low-yielding and non-selective nitration that produced the regioisomers 13 (20%) and 14 (11%). Other attempts such as nitration of 11 to get 1613 for the regioselective introduction of the nitro group ortho to the amino, resulted in no reaction. Regarding the non-selective nitration, although the regioisomeric mixture of 13 and 14 is chromatographically separable, the strong acid medium promoted the deacetylation of 12 giving poor yields for each compound, thus resulting in a no viable alternative. In consequence, another route (route B) with the initially adjacent installed nitrogens but in different oxidation states was envisioned. Thereby the known compound 15,14 was synthesized from o-nitroaniline. The iodination of 15 gave rise to 16 in 85% of yield, which was acetylated in a modest 45% to furnish the convergence compound 13. This moderate yield is more evident if the acetylation reactions of 16 to 13 with 11 to 12 are compared. The bulkiness of the iodine atom in 16 as well as of the nitro group, severely restricted the functionalization of the amino group due to a bis-ortho-substitution effect.15 This steric effect was observed during all of our work development including the fails for some basic-resistant groups (TIPS, MOM, Piv) we tried to introduce. Nevertheless a moderate overall yield at this stage was obtained, we decided to continue with the route to validate it. The following Suzuki cross-coupling reaction between 13 and the p-anisyl boronic acid afforded the arylnitroaniline 17 in 62% of yield. The non-acidic Hassanloie16 conditions for chemoselective nickel-catalyzed nitro group reduction, gave rise to 18 in 80% without acetyl removal. This compound was subjected to benzimidazolone formation by phosgene treatment to furnish 19 in 70% of yield. Finally, the methylation of 19 in basic medium was carried out, unfortunately a very complex reaction mixture was observed. Several optimization assays were done, however in the best of our results a very low-yielding mixture consisting of the desired compound 20 (13%), methyl-deacetylated benzimidazolone 21 (7%) and the bismethylated derivative 22 (9%) were isolated. This result evidenced the extremely poor resistance of the acetyl group to basic conditions17 and strongly limits the scalability. Therefore the M–H exchange over 20 (see Scheme 4) was not carried out.
After this set of reactions we preliminarily concluded two relevant points: (1) the haloaniline 16 was identified as a very important building block due to the properly functionalization for further orthogonal reactions. In 16, there is a nitro group, which was reduced and regioselective methylated, an iodine atom for the chemoselective Suzuki cross-coupling and a bromine for organolithium generation via M–H exchange. (2) Even tough this second strategy failed, it could be useful with a more basic-resistant group instead of an acetyl. However was not possible by this route.
Thus, an alternative procedure based on 16 to introduce regioselectively a basic-resistant group was started (Scheme 6).
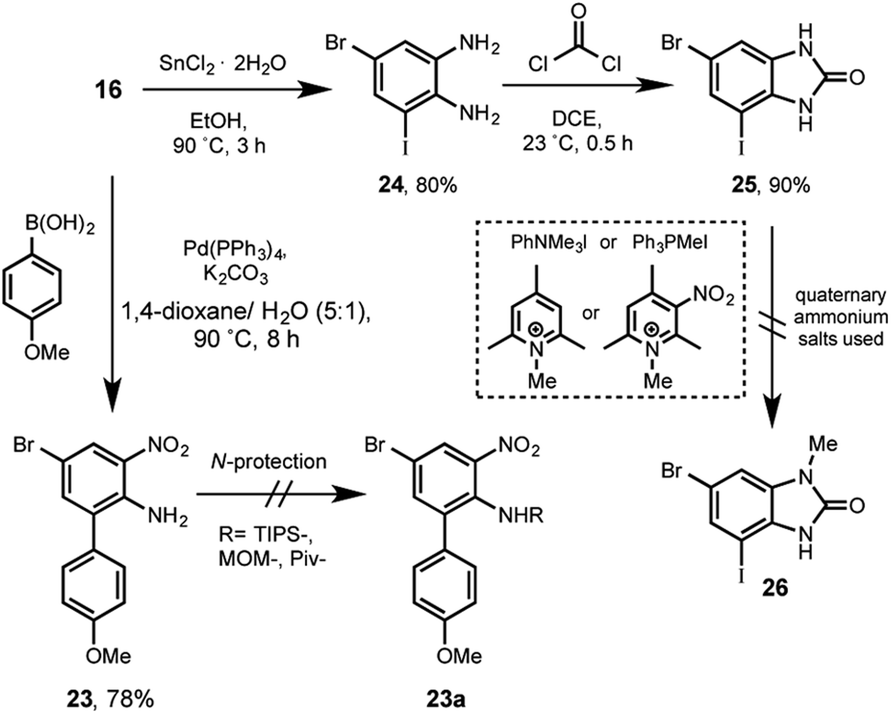 | ||
| Scheme 6 Attempts to synthesize the aniline 23a and benzimidazolone 26. | ||
In this strategy the aniline 16 participated in a Suzuki cross-coupling reaction with p-anisylboronic acid, giving rise to the arylnitroaniline 23 in 78% of yield. Next, after too many attempts to functionalized the amino group with basic-resistant groups such as TIPS, MOM or Piv (see ESI†), we systematically failed. Presumably due to the aforementioned bis-ortho-substitution steric effect.
Previous to rule out this sequence, an additional alternative was considered. For instance, 16 was reduced in presence of tin(II) chloride affording 24 in 80%. The following reaction with phosgene allowed the formation of 25 in excellent 90%. In this stage the regioselective methylation was rationalized by using a bulky methylating reagent, in order to direct the reaction at first position nitrogen. Different quaternary ammonium salts such as triphenylanilinium, methyltriphenylphosphonium, N-methylcollidine or the N-methylnitrocollidine were tested. All of them did not show any reaction to yield the regioselective-methylated derivative 26.
Frustratingly we can preliminarily summarize that after three different strategies and more than three hundred experiments, we were unable to establish a successful route for the total synthesis of kealiiquinone 1. Therefore a deep analysis of the developed work revealed potential compounds such as 25 (Scheme 6), which is an excellent chemo-differentiated building block for orthogonal Suzuki cross-coupling and M–H exchange reactions. Also we identified the compound 21 (Scheme 5). This resulted a very attractive derivative, since it contains the desired methyl group regioselectively installed, the p-anisyl group in the correct position, the third-position nitrogen available for a protecting group introduction and finally a bromine atom for the organolithium generation by M–H exchange.
On the other side after an extensive bibliographic search18 it was found an excellent opportunity in the selenium chemistry described by Milata19 for the regioselective methylation of 2-aminoanilines that can give the desired regiodifferentiated methylbenzimidazolone after the reaction with phosgene. The compounds 16, 25 and 21 were considered in this approach that finally allowed us the total synthesis of kealiiquinone 1. The overall strategy is illustrated in the following retrosynthetic analysis (Scheme 7).
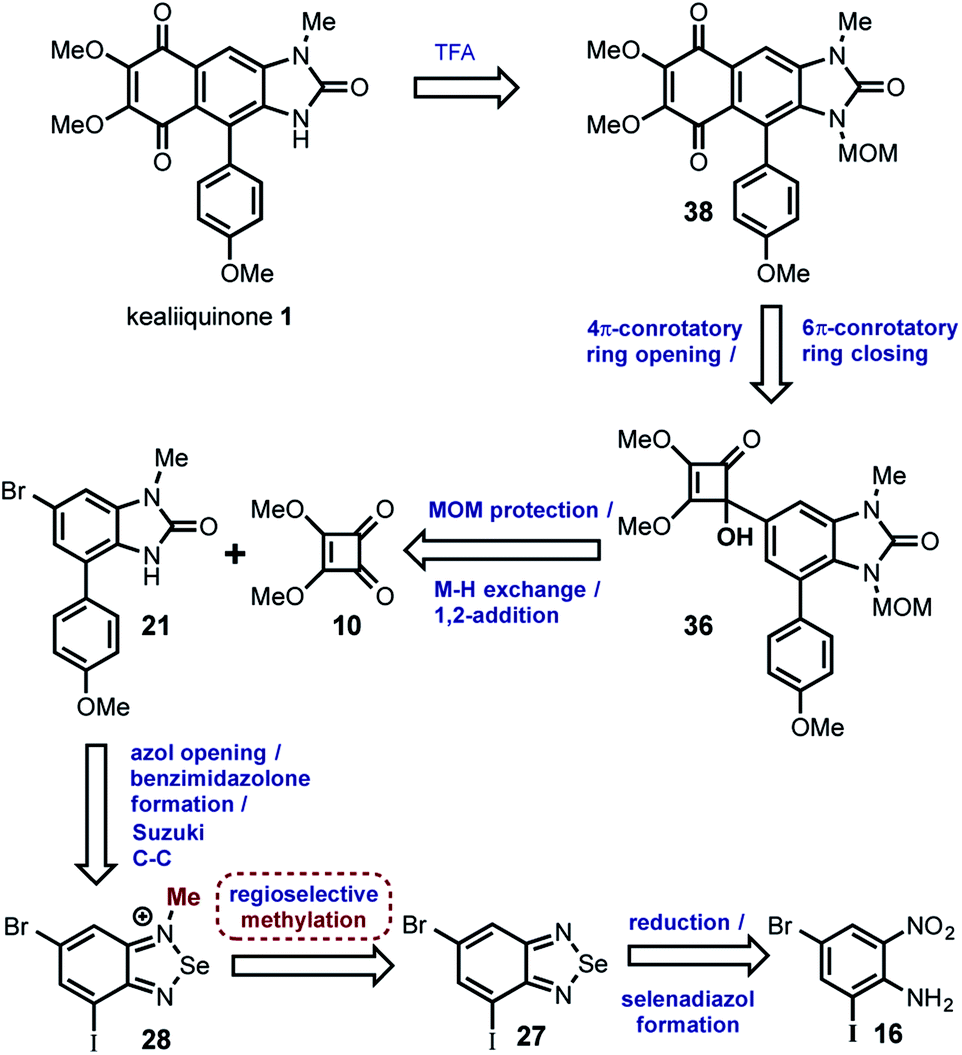 | ||
| Scheme 7 Successful retrosynthetic analysis of the kealiiquinone 1. | ||
In this strategy the naturally occurring kealiiquinone 1, was obtained from 38 after MOM-deprotection in acidic medium. This protected 1,4-quinone is the result of the electrocyclic 6π-ring closure carried out in the thermal ring expansion of 36. This tertiary alcohol comes from the 1,2-addition of the organolithium generated from 21 to the dimethylsquarate 10. The mentioned arylbenzimidazolone was obtained from the slenadiazolium 28 after the regioselective methylation of the corresponding selenadiazol 27 that is obtained form the halonitroaniline 16.
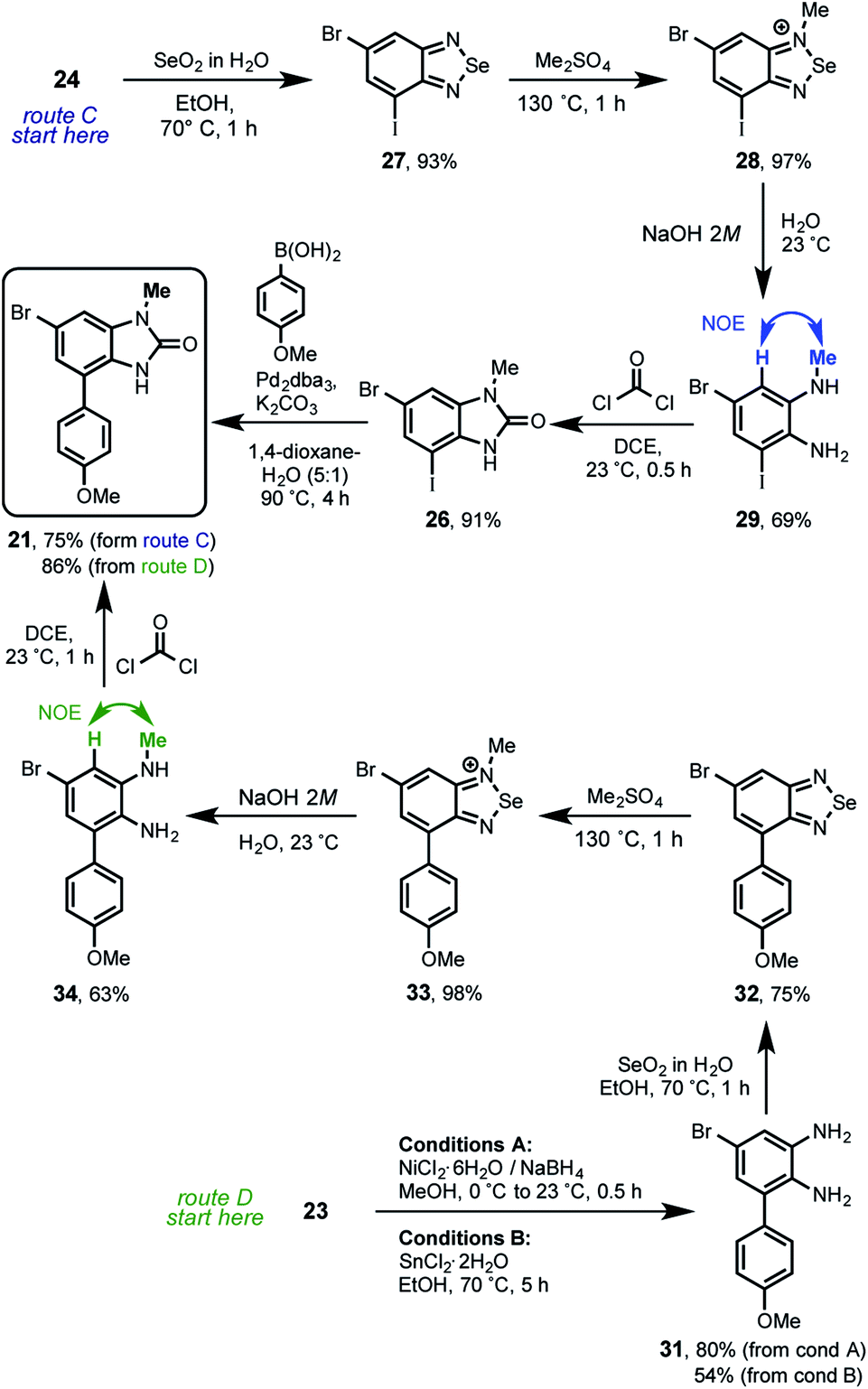
From this analysis it was determined that compound 21 is key to complete the total synthesis of kealiiquinone 1. Thereby two routes (C and D) were developed allowing for accessing in good yield as well as in a regio-controlled fashion to the 1-methyl-4-arylbenzimidazolone core of kealiiquinone (Scheme 8).
 | ||
| Scheme 8 Synthetic route for accessing to the 1-methyl-4-arylbenzimidazolone 21 of the kealiiquinone. | ||
The route C started with the reaction between the 2-aminoaniline 24 and selenium dioxide, to produce the benzo[c][1,2,5]selenadiazol 27 in excellent 93% of yield. In agreement with the Milata19 reports, these derivatives are methylated by heating in dimethyl sulfate as solvent. Based upon our previous strategies results (see Scheme 5), it was rationalized that the steric hindrance provided by the iodine atom, would direct the methylation to the nitrogen at the first position. To our delight we found the regioselective methylation in such expected nitrogen, giving rise to the 1-methylbenzo[c]selenadiazol-1-ium 28 nearly to quantitative yield. Is worth to mention that these two last steps proceeded without any purification. Next, the ring opening of 28 in basic-medium yielded the aniline 29 in 69%. The NOESY experimentation for this compound unequivocally confirmed the required regioselective methylation (see ESI†). The route continued with the benzimidazolone formation by phosgene treatment of 29 furnishing the 1-methyl-6-bromo-4-iodobenzimidazol-2-one 26 in 91% of yield. This compound was used for the MOM-group introduction trials, however it was found only complex reaction mixtures after exhaustive attempts (see ESI†). Therefore it was introduced in a different synthetic stage. The route C concludes with the Suzuki cross-coupling within 26 and p-anisylboronic acid leading to 21 in 75%. On the other hand, the route D started with the reduction of nitroaniline 26 by using two reaction conditions. The first are the Hassanloie-reducing16 conditions using sodium borohydride in presence of nickel chloride to get 31 in 80%. As alternative, the usage of tin(II) chloride in ethanol yielded the nitro group reduction in 54%. Similarly the reaction with selenium dioxide of this 2-aminoarylaniline furnished the structurally related 4-arylbenzo-[c][1,2,5]selenadiazol 32 in 75% of yield. Under the same rationale, the regioselective methylation took place by heating 32 at 130 °C in dimethyl sulfate, giving rise to the 4-anisylbenzo[c][1,2,5]selenadiazolium 33 in excellent 98% of yield. The following ring opening in basic medium provided 34 in 63%. The regioselective methylation of this route was unequivocally confirmed by NOESY experimentation in this compound (see ESI†). Afterwards the treatment of 34 with phosgene convergently produced 21 in 75%.
After carry out this group of experiments, gratifyingly we were able to establish a regio-controlled strategy for accessing to the 1-methyl-4-arylbenzimidazolone core of kealiiquinone.
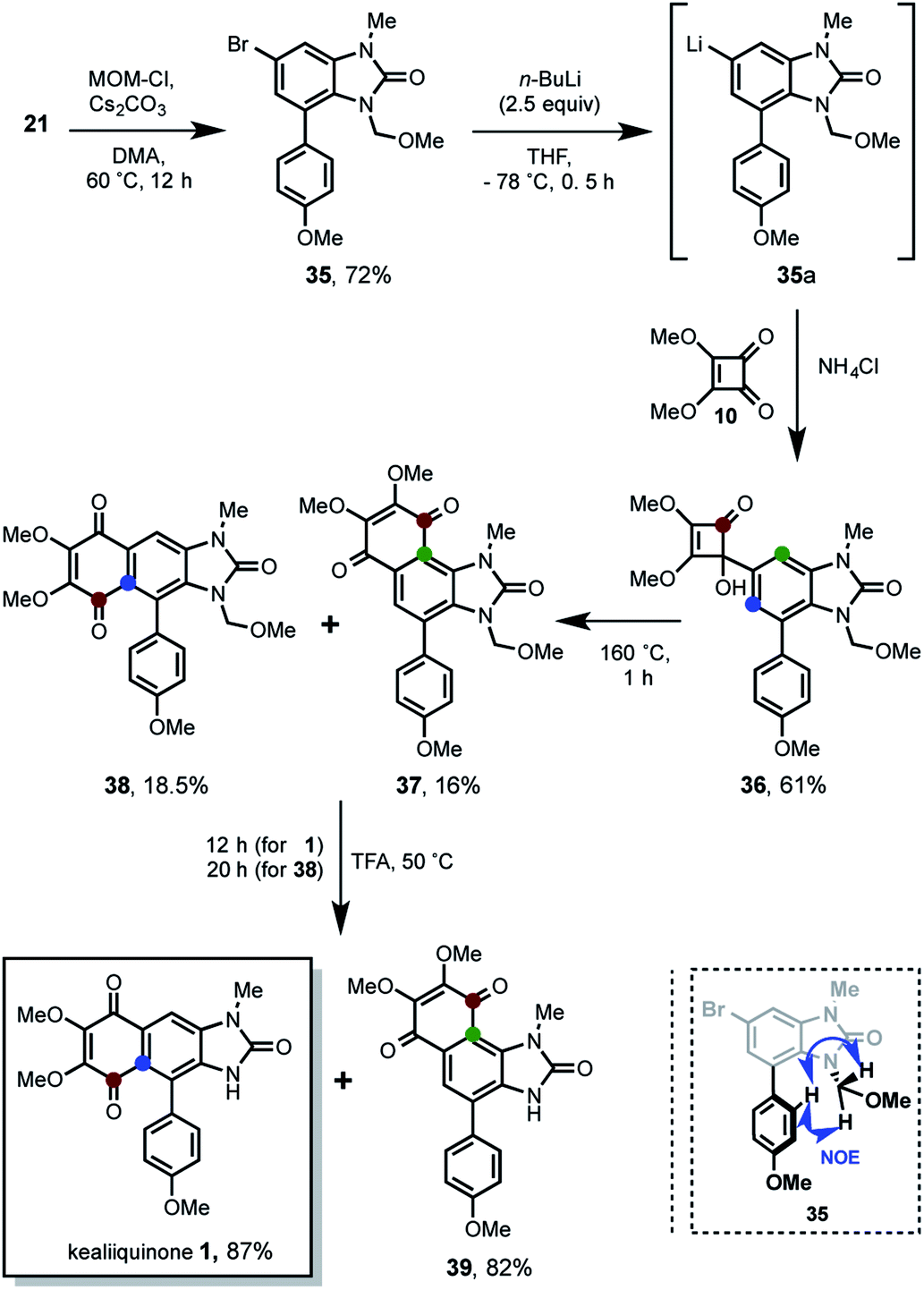
Finally, the total synthesis of the natural alkaloid was accomplished (Scheme 9).
 | ||
| Scheme 9 Completion of the total synthesis of kealiiquinone 1. | ||
After the successful preparation of the regio-differentiated 1-methyl-4-p-anisylbromobenzimidazolone 21, the challenge to address is the introduction of a basic-resistant protecting group, which tolerates the M–H exchange reaction conditions. We focused our attention to the MOM group, since is a strong basic-resistant group even used for DoM reactions,9 can be deprotected in relative milder acid medium and has moderate steric hindrance. This former aspect is very important because our previous experimentation (see Schemes 6 and 8) showed great difficulties to introduce big groups in the third position of benzimidazolone. We assumed was due to the bis-ortho-substitution effect. Thus, after the fail to introduce de MOM group in 26 (see Scheme 8) such functionalization was attempted in 21. In this way, 35 was obtained in 72% of yield with the desired MOM-protection at nitrogen. In view that benzimidazolone is bidentated at nitrogen and oxygen, the regioselectivity in the nitrogen MOM-functionalization was confirmed by NOESY experiments (see ESI† and down of Scheme 9). Then the M–H exchange generated in situ the organolithium 35a, which reacted with the dimethylsquarate 10 yielding 61% of the tertiary alcohol 36. The following heating of 36 at 160 °C along 1 h, promoted the thermal 4π-conrotatory ring opening/6π-conrotatory ring closure for the π-homologation system. This transformation take place through the accepted ketene intermediates,20 which have been described for this chemistry.12 The reaction furnished a (1:1) regioisomeric mixture of the 1,4-quinones 37 and 38 in 16% and 18.5% of yield respectively. For simplicity we refer to them as angular and linear compounds. This expected result showed the desired linear 3-MOM-kealiiquinone 38 as well as an angular 3-MOM-kealiiquinone 37 which posses a novel naphtho[1,2-d]imidazole nucleus. Is important to highlight that even though the yields are moderately-low, they are the result of a two consecutives reactions (hydroquinone formation/oxidation to 1,4-quinone). Therefore this two-step one-pot reaction represents a ca. 40% and 43% of yield respectively for each transformation calculated in linear manner. Finally the MOM group-removal by heating at 50 °C in trifluoroacetic acid lead to the completion of the total synthesis of kealiiquinone 1 in 87% of yield after 12 h. From the angular derivative was also obtained in 82% of yield along of 20 h of reaction.
The spectroscopic data were compared and they match with those reported by Ohta and Lovely (Tables 1 and 2).
| Kealiiquinone (1) | |||
|---|---|---|---|
| 1H NMR acquired in DMSO-d6 | |||
| Naturala | Ohtab | Lovelyc | This work |
| a The spectroscopic data were obtained from ref. 4.b The spectroscopic data were obtained from ref. 5.c The spectroscopic data were obtained from ref. 6. | |||
| 3.58 (s, 3H) | 3.39 (s, 3H) | 3.40 (s, 3H) | 3.39 (s, 3H) |
| 3.78 (s, 3H) | 3.82 (s, 3H) | 3.83 (s, 3H) | 3.82 (s, 3H) |
| 3.83 (s, 3H) | 3.85 (s, 3H) | 3.85 (s, 3H) | 3.85 (s, 3H) |
| 3.92 (s, 3H) | 3.94 (s, 3H) | 3.94 (s, 3H) | 3.94 (s, 3H) |
| 6.88 (d, 2H) | 6.98 (d, 2H) | 6.98 (d, 2H) | 6.98 (d, 2H) |
| 7.12 (d, 2H) | 7.13 (d, 2H) | 7.13 (d, 2H) | 7.13 (d, 2H) |
| 7.69 (s, 1H) | 7.68 (s, 1H) | 7.68 (s, 1H) | 7.68 (s, 1H) |
| — | 11.03 (bs, 1H) | 11.02 (bs, 1H) | 11.03 (bs, 1H) |
| Kealiiquinone (1) | |||
|---|---|---|---|
| 13C NMR obtained in DMSO-d6 | |||
| Naturala | Ohtab | Lovelyc | This work |
| a The spectroscopic data were obtained from ref. 4.b The spectroscopic data were obtained from ref. 5.c The spectroscopic data were obtained from ref. 6. | |||
| 182.4 | 181.3 | 181.3 | 181.3 |
| 181.8 | 181.1 | 181.1 | 181.1 |
| 159.0 | 158.5 | 158.5 | 158.5 |
| 158.3 | 154.8 | 154.8 | 154.8 |
| 148.2 | 147.8 | 147.8 | 147.8 |
| 147.8 | 134.0 | 145.2 | 145.2 |
| 146.0 | 132.6 | 134.0 | 134.0 |
| 137.9 | 129.9 | 132.7 | 132.7 |
| 131.1 | 127.7 | 129.9 | 129.9 |
| 130.6 | 126.5 | 127.7 | 127.7 |
| 129.4 | 126.5 | 126.4 | 126.4 |
| 124.1 | 123.5 | 123.5 | 123.5 |
| 122.9 | 122.6 | 122.6 | 122.6 |
| 113.3 | 113.9 | 113.9 | 113.9 |
| 105.1 | 104.6 | 104.5 | 104.5 |
| 61.0 | 60.8 | 60.8 | 60.8 |
| 61.0 | 60.8 | 60.8 | 60.8 |
| 55.4 | 55.0 | 55.0 | 55.0 |
| 29.2 | 26.8 | 26.8 | 26.8 |
The Table 1 shows a very good correlation among the spectroscopic data for the 1H NMR of kealiiquinone reported by Ohta and Lovely and our synthetic alkaloid. Certainly the naturally occurring compound we obtained is in the carbonyl tautomer form at the benzimidazolone ring more than an enol-form such as was isolated by Scheuer and Clardy.4 Previously, this observation has been pointed out.5,6
On the other hand the spectroscopic data obtained for the 13C NMR of our synthetic kealiiquinone also matched with the reports of Ohta and Lovely (Table 2).
We found an excellent correlation in the comparison of the 13C NMR spectroscopic data. Also we found a better correlation with Ohta and Lovely reports than form original isolation report. Surely due to the thermodynamically more stable ketonic-form of kealiiquinone instead to its enolic-form as mentioned by Lovely.6
Therefore kealiiquinone 1, was successfully synthesized.
Conclusions
In summary, we have developed a new, convergent and modular total synthesis of the naturally occurring marine alkaloid kealiiquinone 1. Our procedure was completely focused in the regioselective methylation at the first nitrogen position of its arylbenzimidazolone core. Accordingly, several experimentations we described, progressively guided us to the final successful strategy through which we were able to synthesized in a regio-controlled manner the 1-methyl-4-(p-anisyl)benzimidazolone fragment as key building block of 1. This was a big challenge addressed via a selenadiazolium intermediate formation. On the other hand unless of the thermal ring expansion, which is the only step showing moderately-low yield, the rest of this route proceeds in good to excellent yields. Our strategy required only two protecting groups whose procedures for introduction and removal took place under mild and efficient conditions. The use of MOM as basic-resistant protecting group resulted in an excellent choice since we demonstrated that bulkier analogues such as TIPS or Piv were not possible to introduce owing to a bis-ortho-substitution effect. Finally as expected, in the thermal ring expansion result, it was found a non-described angular analogue of kealiiquinone that contains an attractive naphtho[1,2-d]imidazole moiety. The complete characterization for the final naturally occurring alkaloid kealiiquinone 1, concisely matched with the previous reports confirming the ketonic-form in its imidazolone ring.Experimental section
General information
All moisture and oxygen sensitive reactions were carried out in flame-dried round bottom flasks under an inert atmosphere of nitrogen. Unless otherwise specified, all commercial materials were used as received without further purification. Anhydrous solvents were purchased from Sigma Aldrich in SureSeal® bottles. Column chromatography was performed using silica gel of size 100–200 and 230–400 mesh (Sigma Aldrich). Thin layer chromatography was performed with TLC Silica gel 60 F256 plates, and visualization was effected with short wavelength UV light (254 nm). Compounds were characterized using 1H NMR, 13C NMR. (Copies of 1H NMR and 13C NMR spectra are provided for all the compounds.) Data of known compounds were compared with existing literature characterization data and the references are given. 1H and 13C NMR spectra were recorded with 500 MHz and Bruker advance 400 MHz instruments using deuterated solvents purchased from Sigma Aldrich like CDCl3. 1H spectra were referenced with tetramethyl silane (TMS, 0.0 ppm) or chloroform (CDCl3, 7.26 ppm) and are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant (Hz), and integration. Chemical shifts of the 13C NMR spectra were measured relative to CDCl3 (δ = 77.16 ppm). All the starting materials were synthesized according to reported procedures in the literature. High resolution mass (HRMS) analysis was obtained using MAXIS IMPACT BRUKER. Chemical nomenclature was generated using Chemdraw. Infrared (IR) spectra were recorded using Perkin-Elmer system 2000 FT-IR spectrometer. Melting points of solids were measured using Fisher-Johns melting point apparatus.Scheme 1 to prepare 4,5-dibromobenzene-1,2-diamine (2)
Sequence followed in Scheme 3
Sequence followed in Scheme 5
4-Bromo-2-iodoaniline (11).
Procedure A. 4-Bromoaniline (0.5 g, 2.941 mmol) was charged in 100 mL two necked round bottom flask and dissolved in 10 mL of glacial acetic acid. To the reaction mixture was added N-iodosuccinimide (0.650 g, 2.941 mmol, 1 equiv.) portion-wise and stirred at 23 °C for 1 h. The reaction mixture was neutralized by addition saturated NaHCO3 solution (50 mL) and extracted with EtOAc (50 mL). The resulted crude was purified by column chromatography (8% EtOAc/hexane) to furnish 11 (0.5 g, 88%) as light yellowish solid.
Procedure B. A 100 mL round bottom flask was charged with 2-iodoaniline (4.5 g, 20.6422 mmol) and was dissolved in 25 mL of dry dimethylformamide. To this reaction mixture was added N-bromosuccinimide (3.6 g, 20.6422 mmol, 1 equiv.) portion-wise and stirred at 23 °C for 1 h. The resulted reaction mixture was evaporated and extracted with EtOAC (100 mL) and water (200 mL). The organic layers were evaporated under reduced pressure and the resulted crude was purified by column chromatography (10% EtOAc/hexane) to give 4-bromo-2-iodoaniline 11 (5.6 g, 92%) as light yellowish solid. Mp 69–71 °C. Rf = 0.6 (24% EtOAC/hexane). IR (neat, cm−1): 3383, 3288, 3176, 2923, 2853, 1866, 1738, 1621, 1557, 1471, 1384, 1283, 1251, 1081, 1020, 870, 830, 807. 1H NMR (CDCl3, 500 MHz): 7.73 (d, J = 2.2 Hz, 1H), 7.22 (dd, J = 8.5, 2.2 Hz, 1H), 6.62 (d, J = 8.5 Hz, 1H), 4.05 (s, 2H). 13C NMR (CDCl3, 126 MHz); δ 146.1, 140.5, 132.2, 115.7, 110.0, 84.2, 77.2. HRMS (ESI+): m/z calcd for C6H6BrIN [M + H]+ = 297.8728, found 297.8726.
N-(4-Bromo-2-iodophenyl)acetamide (12). A flame dried 100 mL round bottom flask was charged with 4-bromo-2-iodoaniline 11 (5.5 g, 18.5810 mmol) and dissolved in 25 mL of dry dichloromethane. To the reaction mixture was added triethylamine (2.6 mL, 18.581 mmol, 1 equiv.) at room temperature followed by the dropwise addition of acetyl chloride (1.3 mL 18.581 mmol, 1 equiv.). The reaction mixture was stirred at room temperature for 5 h and then the solvent evaporated under the reduced pressure. The resulted crude was purified by column chromatography (15% EtOAC/hexane) to get the N-(4-bromo-2-iodophenyl)acetamide 12 (6.2 g, 98% yield) as pale white solid. Mp 210–212 °C. Rf = 0.5 (40% EtOAC/hexane). IR (neat, ν/cm−1): 3275, 3068, 2928, 1892, 1737, 1656, 1562, 1514, 1459, 1368, 1282, 1245, 1086, 1031, 1006, 870, 816, 724. 1H NMR (CDCl3, 500 MHz): δ 8.12 (d, J = 8.3 Hz, 1H), 7.90 (d, J = 1.9 Hz, 1H), 7.45 (dd, J = 8.8, 2.1 Hz, 1H), 7.38 (s, 1H), 2.23 (s, 3H). 13C NMR (CDCl3, 126 MHz): δ 168.3, 140.6, 137.6, 132.4, 122.9, 117.5, 77.12, 24.9. HRMS (ESI+): m/z calcd for C8H8BrINO [M + H]+ = 339.8834, found 339.8817.
Nitration on N-(4-bromo-2-iodophenyl)acetamide (12). To a solution of N-(4-bromo-2-iodophenyl)acetamide 12 (4.0 g, 11.8343 mmol, 1 equiv.) in 20 mL of concentrated sulfuric acid, was added sulfonitric mixture (4 mL) at 0 °C. The reaction mixture was stirred at 23 °C for 3 h. The reaction mixture was quenched by saturated NaHCO3 solution (100 mL) and extracted with EtOAc (200 mL) and washed with brine (100 mL). The organic layers were dried over Na2SO4, concentrated in vacuum and the resulted crude was purified by column chromatography to afford the two regioisomeric compounds 13 (0.6 g, 20%) as light yellow solid and the compound 14 (0.5 g, 11%) as pale white solid.
Spectra data for N-(4-bromo-2-iodo-6-nitrophenyl)acetamide (13). Mp 182–184 °C. Rf = 0.4 (40% EtOAC/hexane). IR (neat, ν/cm−1): 3264, 2923, 2853, 1666, 1560, 1522, 1506, 1446, 1366, 1325, 1281, 1123, 1062, 1014, 968, 887, 882. 1H NMR (CDCl3, 500 MHz): δ 8.87 (s, 1H), 8.15 (s, 1H), 7.52 (s, 1H), 2.28 (s, 3H). 13C NMR (CDCl3, 126 MHz): δ 168.4, 150.3, 143.5, 138.6, 116.9, 108.6, 93.3, 25.0. HRMS (ESI+): m/z calcd for C8H7BrIN2O3 [M + H]+ = 384.8685, found 384.8679.
Spectra data for N-(4-bromo-2-iodo-6-nitrophenyl)acetamide (14). Mp 179–181 °C. Rf = 0.3 (40% EtOAC/hexane) IR (neat, ν/cm−1): 3257, 3083, 3083, 2992, 1730, 1706, 1663, 1552, 1503, 1439, 1367, 1334, 1276, 1247, 1110, 1034, 1009, 870, 738. 1H NMR (CDCl3, 500 MHz): δ 8.61 (s, 1H), 7.99 (s, 1H), 7.35 (s, 1H), 2.25 (s, 3H).13C NMR (CDCl3, 126 MHz): δ 168.4, 150.3, 143.5, 138.8, 117.00, 108.5, 93.4, 25.0. HRMS (ESI+): m/z calcd for C8H7BrIN2O3 [M + H]+ = 384.8685, found 384.8662.
4-Bromo-2-iodo-6-nitroaniline (16). A 500 mL two-necked round-bottom flask, was charged with a solution of 4-bromo-2-nitroaniline 15 (2.0 g, 9.216 mmol, 1 equiv.) in glacial AcOH (25 mL) and heated to 80 °C. N-iodosuccinimide (4.0 g, 18.416 mol, 2 equiv.) was added portion-wise and stirred for 2 h. The reaction mixture was poured into ice-cooled water (50 mL) and immediately an orange colour solid was formed. The resulted solid was filtered-off, washed with DCM (50 mL) and dried under vacuo. The aqueous portion was neutralized with saturated NaHCO3 (50 mL) solution, followed by extraction with EtOAc (100 mL). The combined organic fractions were washed with brine (50 mL), dried with Na2SO4, and concentrated under reduced pressure to afford additional amount of such solid. The orange solid obtained from filtration and extraction process were collected to furnish 16 (2.2 g, 85%) that was used without further purification for next step. Mp 135–137 °C. Rf = 0.5 (20% EtOAc/hexane). IR (neat, ν/cm−1): 3572, 3453, 3101, 1541, 1435, 1346, 1241, 1070, 872, 671. 1H NMR (500 MHz, CDCl3): δ 8.31 (d, J = 2.3 Hz, 1H), 8.01 (d, J = 2.3 Hz, 1H), 6.67 (bs, 2H). 13C NMR (126 MHz, CDCl3): δ 147.5, 143.3, 131.9, 129.4, 108.1, 88.0. HRMS (ESI): m/z calcd for C6H4BrIN2O2 [M + H]+ = 342.8501, found 342.8504.
N-(4-Bromo-2-iodo-6-nitrophenyl)acetamide (13). A dried 100 mL round bottom flask was charged with 4-bromo-6-iodo-2-nitroaniline 16 (1.0 g, 2.9321 mmol) and dissolved in 25 mL of dry dimethylformamide. To the reaction mixture was added sodium hydride (0.281 g, 11.7.30 mmol, 4 equiv.) at 0 °C followed by the dropwise addition of acetyl chloride (4.1 mL, 58.642 mmol, 20 equiv.). The reaction mixture was stirred at room temperature for 12 h and then the solvent evaporated under the reduced pressure. The resulted crude was purified by column chromatography (20% EtOAC/hexane) to get the N-(4-bromo-2-iodo-6-nitrophenyl)acetamide 13 (0.380 g, 45% yield) as pale white solid.
N-(5-Bromo-4′-methoxy-3-nitro-[1,1′-biphenyl]-2-yl)acetamide (17). To a solution of N-(4-bromo-2-iodo-6-nitrophenyl)acetamide 13 (0.5 g, 1.305 mmol) in toluene (30 mL) and water (3 mL) was added 4-methoxyphenyl boronic acid (0.44 g, 2.872 mmol, 2.2 equiv.), sodium carbonate (0.34 g, 3.262 mmol, 2.5 equiv.) and Pd(PPh3)4 (0.12 g, 0.1044 mmol, 8 mol%). The reaction mixture was purged with N2 and stirred at 80 °C for 8 h. The reaction was quenched by addition of water and extracted with EtOAc. The organic layers were washed with brine (100 mL), dried with Na2SO4, and concentrated under reduced pressure. The resulted crude was purified by column chromatography (25% EtOAc/hexane) to yield N-(5-bromo-4′-methoxy-3-nitro-[1,1′-biphenyl]-2-yl)acetamide 17 (0.297 g, 62%) as yellowish solid. Mp 172–174 °C. Rf = 0.4 (40% EtOAc/hexane). IR (neat, ν/cm−1): 3319, 3236, 3083, 2997, 2951, 2926, 2854, 2832, 1739, 1673, 1608, 1511, 1455, 1362, 1334, 1243, 1181, 1030, 966, 883, 836, 814, 717. 1H NMR (500 MHz, CDCl3): δ 8.69 (s, 1H), 7.45 (s, 2H), 7.25 (d, J = 10.2 Hz, 3H), 7.09 (s, 1H), 7.02 (d, J = 11.3 Hz, 2H), 3.87 (s, 3H), 2.03 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 168.2, 160.1, 135.1, 134.4, 132.2, 130.3, 127.9, 125.7, 124.0, 119.1, 114.9, 55.6, 24.9. HRMS (ESI+): m/z calcd for C15H14BrN2O4 [M + H]+ = 365.0317, found 365.0119.
N-(3-Amino-5-bromo-4′-methoxy-[1,1′-biphenyl]-2yl)acetamide (18). N-(5-Bromo-4′-methoxy-3-nitro-[1,1′-biphenyl]-2-yl)acetamide 17 (0.2 g, 0.5494 mmol, 1 equiv.) was charged in a 250 mL two-necked round-bottom flask and dissolved in MeOH (30 mL). To the resulting solution was added NaBH4 (0.208 g, 5.4945 mmol, 10 equiv.) portion wise within 5 minutes at 0 °C and followed by NiCl2·6H20 (0.021 g, 0.1648 mmol, 0.3 equiv.). The reaction mixture was stirred at 23 °C for 0.5 h and was poured into ice-cold water. The resulting mixture was extracted with EtOAc (200 mL) and water (200 mL). The organic layers were dried over Na2SO4 and concentrated under reduced pressure. The crude was purified by column chromatography (60% EtOAc/hexane) to furnish N-(3-amino-5-bromo-4′-methoxy-[1,1′-biphenyl]-2-yl)acetamide 18 (0.144 g, 80%) as yellowish solid. Mp 185–187 °C. Rf = 0.3 (100% EtOAc). IR (neat, ν/cm−1): 3481, 3360, 3320, 3233, 2950, 2928, 2832, 1674, 1645, 1608, 1511, 1425, 1364, 1291, 1242, 1178, 1033, 928, 828, 815, 805, 735, 718. 1H NMR (500 MHz, CDCl3): δ 7.21 (d, J = 8.7 Hz, 2H), 6.95 (d, J = 8.7 Hz, 2H), 6.92 (d, J = 2.1 Hz, 1H), 6.86 (d, J = 2.1 Hz, 1H), 6.63 (s, 1H), 4.13 (s, 2H), 3.85 (s, 3H), 2.05 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 169.6, 159.6, 144.5, 140.1, 130.2, 123.2, 121.1, 120.5, 119.4, 114.2, 55.5, 23.3. HRMS (ESI+): m/z calcd for C15H16BrN2O2 [M + H]+ = 335.0395, found 335.0374.
1-Acetyl-5-bromo-7-(4-methoxyphenyl)-1,3-dihydro-2H-benzo[d]imidazol-2-one (19). To a 100 mL two-neck round-bottom flask, was added N-(3-amino-5-bromo-4′-methoxy-[1,1′-biphenyl]-2-yl)acetamide 18 (0.140 g, 0.4191 mmol) and dissolved in 15 mL of DCE at room temperature. Then, a solution of phosgene 15 wt% in toluene (0.8 mL, 3.3528 mmol, 8 equiv.) was added drop wise to the reaction mixture and stirred for 1 h. The reaction mixture was poured into ice-cooled water (30 mL) and immediately a white solid was formed. The resulting solid was filtered-off and dried under vacuum to give 19 (0.105 g, 70%) as an off-white solid. The compound was used without purification for next step. Mp 194–196 °C. Rf = 0.3 (40% EtOAc/hexane). IR (neat) ν/cm−1 = 2938, 2837, 2274, 1714, 1667, 1611, 1515, 1466, 1442, 1367, 1383, 1388,1309, 1287, 1246, 1143, 1045, 1020, 995, 840, 762. 1H NMR (500 MHz, DMSO-d6): δ 11.57 (s, 1H), 7.24 (d, J = 7.5 Hz, 2H), 7.13 (s, 1H), 7.12 (s, 1H), 6.92 (d, J = 8.8 Hz, 2H), 3.78 (s, 3H), 2.50 (s, 3H).13C NMR (126 MHz, DMSO-d6): δ 168.6, 158.5, 152.9, 132.3, 131.2, 130.3, 128.1, 125.5, 123.22, 116.4, 113.6, 110.1, 55.0, 39.5, 25.5. HRMS (ESI+): m/z calcd for C16H14.BrN2O3 [M + H]+ = 361.0188, found 361.0176.
Methylation of N-(3-amino-5-bromo-4′-methoxy-[1,1′-biphenyl]-2-yl)acetamide (19). A dried 50 mL two-neck flask was charged with dry DMSO (10 mL), powdered KOH (0.14 g, 2.55 mol, 4 equiv.) and was stirred for 10 minutes at room temperature. Then 6-bromo-4-iodo-1,3-dihydro-2H-benzo[d]imidazol-2-one 19 (0.230 g, 0.638 mol, 1 equiv.) was added in portions to the reaction mixture followed by the drop wise addition of MeI (0.25 mL, 3.833 mmol, 6 equiv.) over a period of 5 minutes. The mixture was stirred at room temperature for 1 h and quenched by the addition of H2O (20 mL). The crude was extracted with EtOAc (2 × 25 mL). The combined organic layers were washed with brine (100 mL), dried over Na2SO4 and finally concentrated under reduced pressure. The resulted crude was purified by column chromatography (20% EtOAc/hexane) to furnish 20 (32 mg, 13%) as pale solid, 21 (16 mg, 7%) as white solid and 22 (20 mg, 9%) as white solid.
Spectral data for 3-acetyl-6-bromo-4-(4-methoxyphenyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (20). The following compound was obtained by column chromatography purification with a 20% EtOAc/hexane gradient. Mp 229–231 °C. Rf = 0.6 (40% EtOAc/hexane). IR (neat, ν/cm−1): 3035, 2976, 2939, 2832, 1738, 1722, 1605, 1595, 1517, 1450, 1364, 1272, 1251, 1185, 1174, 1024, 994, 827. 1H NMR (500 MHz, CDCl3): δ 7.47 (d, J = 1.8 Hz, 1H), 7.25 (d, J = 8.6 Hz, 2H), 7.19 (d, J = 1.8 Hz, 1H), 6.93 (d, J = 8.7 Hz, 2H), 3.78 (s, 3H), 3.35 (s, 3H), 2.49 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 168.4, 158.5, 152.4, 133.8, 131.2, 130.2, 128.2, 125.9, 121.9, 116.8, 113.7, 109.7, 55.1, 27.3, 25.5. HRMS (ESI+): m/z calcd for C17H16BrN2O3 [M + H]+ = 375.0344, found 375.0341.
Spectral data for 6-bromo-4-(4-methoxyphenyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (21). The following compound was obtained by column chromatography purification with a 30% EtOAc/hexane gradient as white solid. Mp 225–227 °C. Rf = 0.4 (40% EtOAc/hexane). IR (neat, ν/cm−1): 3137, 3048, 2953, 2927, 2833, 1703, 1608, 1578, 1517, 1450, 1380, 1339, 1291, 1247, 1178, 962, 823, 767. 1H NMR (500 MHz, CDCl3): δ 8.72 (s, 1H), 7.43 (d, J = 8.7 Hz, 2H), 7.24 (d, J = 1.8 Hz, 1H), 7.06 (d, J = 1.4 Hz, 1H), 7.02 (d, J = 8.7 Hz, 2H), 3.87 (s, 3H), 3.40 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 159.9, 155.0, 132.7, 129.2, 128.5, 125.1, 124.3, 124.2, 114.9, 114.7, 109.5, 55.5, 27.2. HRMS (ESI+): m/z calcd for C15H14BrN2O2 [M + H]+ = 333.0239, found 333.0218.
Spectral data for 6-bromo-4-(4-methoxyphenyl)-1,3-dimethyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (22). The following compound was obtained by column chromatography purification with a 25% EtOAc/hexane gradient as white solid. Mp 154–156 °C. Rf = 0.55 (40% EtOAc/hexane). IR (neat, ν/cm−1) = 3071, 2945, 1696,1607, 1481, 1450, 1388, 1241, 1176, 1122, 1025, 832, 741, 692. 1H NMR (500 MHz, CDCl3): δ 7.27 (d, J = 8.6 Hz, 2H), 7.09 (d, J = 1.9 Hz, 1H), 7.08 (d, J = 1.9 Hz, 1H), 6.96 (d, J = 8.6 Hz, 2H), 3.87 (s, 3H), 3.42 (s, 3H), 2.97 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 159.6, 155.1, 131.8, 130.9, 129.2, 126.6, 126.5, 126.1, 113.5, 113.2, 109.4, 55.4, 30.4, 27.4. HRMS (ESI+): m/z calcd for C16H16BrN2O2 [M + H]+ = 347.0395, found 347.0396.
Sequence followed in Scheme 6
Sequence followed in Scheme 8
6-Bromo-4-(4-methoxyphenyl)-1-methyl-1,3-dihydro-2H-benzo-6-bromo-4-iodo-benzo[c][1,2,5]selenadiazole (27). To a solution of 5-bromo-3-iodobenzene-1,2-diamine 24 (3.8 g, 12.218 mmol) in 60 mL of dry ethanol, was added SeO2 (1.9 g, 18.3279 mmol, 1.5 equiv.) in water. The reaction mixture was stirred at 70 °C for 1 h, then the resulted solid was filtered-off and dried under vacuum. The yellowish solid was obtained from filtration process was collected to furnish 6-bromo-4-iodobenzo[c][1,2,5]selenadiazole 27 (4.4 g, 93%) that was used without further purification for next step. Mp 168–170 °C. Rf = 0.8 (40% EtOAc/hexane). IR (neat) ν/cm−1: 3295, 3229, 2955, 2915, 2850, 1738, 1729, 1571, 1469, 1392, 1245, 1195, 1178, 1048, 991, 940, 850, 740, 719. 1H NMR (500 MHz, CDCl3): δ 8.11 (d, J = 1.7 Hz, 1H), 8.03 (d, J = 1.6 Hz, 1H). 13C NMR (126 MHz, CDCl3): δ 158.8, 157.6, 141.9, 125.7, 125.5, 91.1. HRMS (ESI+): m/z calcd for C6H3BrIN2Se [M + H]+ = 388.7690, found 388.7676.
6-Bromo-4-iodo-1-methylbenzo[c][1,2,5]selenadiazol-1-ium (28). A 100 mL two-necked round bottom flask was charged 6-bromo-4-iodobenzo[c][1,2,5]selenadiazole 27 (4.3 g, 0.011 mmol, 1 equiv.) and dimethyl sulphate (22.0 mL, 0.2216 mmol, 20 equiv.). The reaction mixture was heated at 130 °C for 1 h. To the resulted reaction mixture was added diethyl ether at room temperature to get an orange solid. This solid was filtered-off and dried well. The orange solid obtained from filtration process was collected to furnish 6-bromo-4-iodo-1-methylbenzo[c][1,2,5]selenadiazol-1-ium 28 (4.5 g, 97%) as orange solid. Mp: 224–226 °C. Rf = 0.8 (40% EtOAc/hexane). IR (neat) ν/cm−1: 3071, 3002, 2863, 2573, 2230, 1572, 1510, 1478, 1441, 1478, 1392, 1311, 1284, 1202, 1157, 1013, 845, 749. 1H NMR (500 MHz, DMSO-d6): δ 8.55 (d, J = 1.6 Hz, 1H), 8.39 (d, J = 1.6 Hz, 1H), 4.46 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 154.6, 150.2, 141.3, 132.5, 119.1, 97.8, 52.7, HRMS (ESI+): m/z calcd for C7H5BrIN2Se+ [M + H]+ = 402.7841, found 402.7837.
5-Bromo-3-iodo-N1-methylbenzene-1,2-diamine (29). A 50 mL two-necked round bottom flask was charged with 25 mL of 2 M NaOH solution. To this solution was added 6-bromo-4-iodo-1-methylbenzo[c][1,2,5]selenadiazol-1-ium 28 (4.5 g, 11.1940 mmol) portion wise at room temperature. The reaction mixture was stirred at 23 °C vigorously for 1 h. The reaction mixture was extracted with EtOAc (100 mL) and the organic layers were collected, dried over Na2SO4, concentrated under reduced pressure. The resulted crude was purified by column chromatography (15% EtOAc/hexane) to afford 5-bromo-3-iodo-N1-methylbenzene-1,2-diamine 29 (3.2 g, 69%) as red solid. Mp 61–63 °C. Rf = 0.6 (40% EtOAc/hexane). IR (neat, ν/cm−1): 3363, 3346, 3079, 2975, 2931, 2883, 2847, 2794, 2553, 2457, 1578, 1505, 1470, 1445, 1430, 1399, 1274, 1224, 1058, 833, 822, 774, 674. 1H NMR (500 MHz, CDCl3): δ 7.25 (d, J = 2.1 Hz, 1H), 6.69 (d, J = 2.1 Hz, 1H), 3.57 (bs, 3H), 2.82 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 139.7, 134.3, 129.0, 113.7, 113.3, 86.4, 31.1. HRMS (ESI+): m/z calcd for C7H9BrN2 [M + H]+ = 326.8994, found 326.8998. The regioselectivity of this methylation reaction was confirmed by NOESY experiments (see spectra file).
6-Bromo-4-iodo-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (26). In a 250 mL two-neck round-bottom flask, was added 5-bromo-3-iodo-N1-methylbenzene-1,2-diamine 29 (3.2 g, 9.815 mmol) and dissolved in 50 mL of DCE at room temperature. Then, a solution of phosgene 15 wt% in toluene (8.5 mL, 78.527 mmol, 8 equiv.) was added drop-wise to the reaction mixture and stirred for 1 h. The reaction mixture was poured into ice-cooled water (200 mL) and immediately a white solid was formed. The resulting solid was filtered-off and dried under vacuum to give 26 (2.7 g, 91%) as an off-white solid. The compound was used without purification for next step. Mp 136–138 °C. Rf = 0.5 (60% EtOAc/hexane). IR (neat, ν/cm−1): 3376, 3113, 3032, 2919, 2850, 2258, 1687, 1616, 1586, 1485, 1440, 1376, 1307, 1240, 1178, 1105, 1074, 959, 879, 826, 736, 718, 670. 1H NMR (500 MHz, CDCl3): δ 8.42 (s, 1H), 7.51 (d, J = 1.6 Hz, 1H), 7.07 (d, J = 1.0 Hz, 1H), 3.37 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 153.9, 131.7, 131.6, 130.2, 114.8, 110.9, 73.0, 27.5. HRMS (ESI+): m/z calcd for C8H7BrIN2O [M + H]+ = 352.8786, found 352.8784.
6-Bromo-4-(4-methoxyphenyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (21). To a solution of 6-bromo-4-iodo-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one 26 (2.5 g, 7.207 mmol) in 1,4-dioxane (60 mL) and water (5 mL) were added p-anisyl boronic acid (1.35 g, 8.649 mmol, 1.2 equiv.), potassium carbonate (2.48 g, 18.019 mmol, 2.5 equiv.) and Pd2(dba)3 (0.68 g, 0.7207 mmol, 1 mol%). The reaction mixture was purged with N2 and stirred at 90 °C for 6 h. The reaction was quenched by addition of water and extracted with EtOAc (200 mL). The organic layers were washed with brine (100 mL), dried with Na2SO4, and concentrated under reduced pressure. The resulted crude was purified by column chromatography (30% EtOAc/hexane) to afford 6-bromo-4-(4-methoxyphenyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one 21 (1.8 g, 75%) as white solid. Mp 225–227 °C. Rf = 0.55 (60% EtOAc/hexane). IR (neat, ν/cm−1): 3137, 3048, 2953, 2927, 2833, 1703, 1608, 1578, 1517, 1450, 1380, 1339, 1291, 1247, 1178, 962, 823, 767. 1H NMR (500 MHz, CDCl3): δ 8.72 (s, 1H), 7.43 (d, J = 8.7 Hz, 2H), 7.24 (d, J = 1.8 Hz, 1H), 7.06 (d, J = 1.4 Hz, 1H), 7.02 (d, J = 8.7 Hz, 2H), 3.87 (s, 3H), 3.80 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 159.9, 155.0, 132.7, 129.2, 128.5, 125.1, 124.2, 114.9, 114.7, 109.5, 55.5, 27.2. HRMS (ESI+): m/z calcd for C15H14BrN2O2 [M + H]+ = 333.0239, found 333.0218.
Sequence followed in Scheme 8
Synthesis of 5-bromo-4′-methoxy-[1,1′-biphenyl]-2,3-diamine (31).
Conditions A. To a 100 mL two-necked round bottom flask was added 5-bromo-4′-methoxy-3-nitro-[1,1′-biphenyl]-2-amine 23 (0.300 g, 0.9345 mmol, 1 equiv.) and dissolved in 15 mL of dry ethanol at 70 °C. To the reaction mixture SnCl2·2H2O (1.05 g, 4.6728 mmol, 5 equiv.) was added portion wise and the reaction mixture was stirred at 70 °C for 5 h. Then the reaction mixture was poured into 50 mL of ice-cold water and extracted with EtOAc (100 mL) followed by washing with brine (20 mL). The resulted organic layers were dried over Na2SO4 and concentrated under reduced pressure. The resulted crude was purified by column chromatography (30% EtOAc/hexane) to afford 5-bromo-4′-methoxy-[1,1′-biphenyl]-2,3-diamine 31 (0.160 g, 50%) as orange solid.
Conditions B. 5-Bromo-4′-methoxy-3-nitro-[1,1′-biphenyl]-2-amine 23 (3.2 g, 9.9688 mmol, 1 equiv.) was charged in a 500 mL two-necked round-bottom flask and was dissolved in MeOH (100 mL). To the reaction mixture was added NaBH4 (3.8 g, 99.688 mmol, 10 equiv.) portion-wise within 5 minutes at 0 °C followed by NiCl2·6H20 (0.720 g, 2.9906 mmol, 0.3 equiv.). The reaction mixture was stirred at 23 °C for 1 h. The reaction mixture was poured into ice-cold water and the resulting mixture was extracted with EtOAc (200 mL) and water (200 mL). The organic layers were dried over Na2SO4 and concentrated under reduced pressure. The crude such obtained was purified by column chromatography (30% EtOAc/hexane) to furnish 5-bromo-4′-methoxy-[1,1′-biphenyl]-2,3-diamine 31 (1.78 g, 54%) as yellowish solid. Mp 137–139 °C. Rf = 0.3 (40% EtOAc/hexane). IR (neat, ν/cm−1): 3407, 3394, 3310, 3224, 3054, 3009, 2961, 2922, 2840, 1607, 1557, 1510, 1463, 1241, 1174, 1024, 830, 778, 699. 1H NMR (500 MHz, CDCl3): δ 7.32 (d, J = 8.7 Hz, 1H), 6.98 (d, J = 8.7 Hz, 1H), 6.83 (d, J = 2.1 Hz, 1H), 6.82 (d, J = 2.1 Hz, 1H), 3.85 (s, 3H), 3.45 (s, 2H). 13C NMR (126 MHz, CDCl3): δ 159.3, 136.3, 131.7, 130.7, 130.6, 130.3, 124.1, 118.1, 114.5, 111.4, 55.5. HRMS (ESI+): m/z calcd for C13H14BrN2O [M + H]+ = 293.0290 found 293.0286.
6-Bromo-4-(4-methoxyphenyl)benzo[c][1,2,5]selenadiazole (32). To a solution of 5-bromo-4′-methoxy-[1,1′-biphenyl]-2,3-diamine 31 (1.75 g, 5.9931 mmol) in 10 mL of dry ethanol, was added SeO2 (0.98 g, 8.9897 mmol, 1.5 equiv.) in water. The reaction mixture was stirred at 70 °C for 1 h, and the resulted solid was filtered-off and dried under vacuum. The yellowish solid was obtained from filtration process and collected to furnish 6-bromo-4-(4-methoxyphenyl)benzo[c][1,2,5]selenadiazole 32 (1.7 g, 75%) which was used without further purification for next step. Mp 188–190 °C. Rf = 0.8 (40% EtOAc/hexane). IR (neat) ν/cm−1: 3072, 2986, 2955, 2930, 2907, 2832, 1877, 1718, 1665, 1606, 1585, 1501, 1463, 1316, 1289, 1235, 1185, 1060, 1035, 952, 884, 829, 797, 763, 745. 1H NMR (500 MHz, CDCl3): δ 8.01 (d, J = 1.8 Hz, 1H), 7.79 (d, J = 8.8 Hz, 2H), 7.57 (d, J = 1.8 Hz, 1H), 7.05 (d, J = 8.8 Hz, 2H), 3.89 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 161.2, 160.2, 158.0, 136.1, 130.81, 130.6, 128.9, 125.9, 123.7, 114.0, 55.4. HRMS (ESI+): m/z calcd for C13H10BrN2OSe [M + H]+ = 368.9142, found 368.9147.
6-Bromo-4-(4-methoxyphenyl)-1-methylbenzo[c][1,2,5]selenadiazol-1-ium (33). A 100 mL two-necked round bottom flask was charged with 6-bromo-4-(4-methoxyphenyl)benzo[c][1,2,5]selenadiazole 32 (1.62 g, 4.4277 mmol) and dimethyl sulphate (5.0 mL, 44.2779 mmol, 10 equiv.). The reaction mixture was heated at 130 °C for 1 h. To the resulted reaction mixture was added diethyl ether to get a yellowish solid. This solid was filtrated-off and dried in vacuum to furnish 6-bromo-4-(4-methoxyphenyl)-1-methylbenzo[c][1,2,5]selenadiazol-1-ium 33 (1.8 g, 98%) that was used without further purification for next step. Mp 208–210 °C. Rf = 0.8 (40% EtOAc/hexane). IR (neat) ν/cm−1: 3059, 2941, 2841, 2540, 1605, 1585, 1492, 1443, 1375, 1287, 1246, 1143, 1045, 1020, 995, 840, 827, 762. 1H NMR (500 MHz, DMSO-d6): δ 8.31 (d, J = 1.4 Hz, 1H), 7.95 (d, J = 1.4 Hz, 1H), 7.89 (d, J = 8.7 Hz, 1H), 7.15 (d, J = 8.7 Hz, 1H), 4.52 (s, 3H), 3.85 (s, 3H). 13C NMR (126 MHz, DMSO-d6): δ 160.5, 153.5, 151.8, 136.9, 133.3, 131.7, 130.1, 126.4, 117.1, 114.2, 55.4, 38.6. HRMS (ESI+): m/z calcd for C13H10BrN2OSe [M]+ = 382.9298, found 382.9296.
5-Bromo-4′-methoxy-N3-methyl-[1,1′-biphenyl]-2,3-diamine (34). A 250 mL two-necked round bottom flask was charged with 50 mL of 2 M NaOH solution. To this solution was added 6-bromo-4-(4-methoxyphenyl)-1-methylbenzo[c][1,2,5]selenadiazol-1-ium 33 (1.7 g, 4.691 mmol) portion wise. The reaction mixture was stirred at 23 °C vigorously for 1 h. The reaction mixture was extracted with EtOAc (100 mL) and the organic layers were collected, dried over Na2SO4 and concentrated under reduced pressure. The resulted crude was purified by column chromatography (15% EtOAc/hexane) to afford 5-bromo-4′-methoxy-N3-methyl-[1,1′-biphenyl]-2,3-diamine 34 (0.9 g, 63%) as red solid. Mp 120–122 °C. Rf = 0.4 (20% EtOAc/hexane). IR (neat, ν/cm−1): 3384, 3310, 3039, 2098, 2934, 2854, 2834, 1608, 1596, 1565, 1513, 1501, 1460, 1472, 1288, 1241, 1176, 1025, 821, 776, 709. 1H NMR (500 MHz, CDCl3): δ 7.33–7.28 (m, 1H), 7.00–6.96 (m, 1H), 6.80 (d, J = 1.5 Hz, 1H), 6.74 (d, J = 2.1 Hz, 1H), 3.86 (d, J = 7.4 Hz, 2H), 3.41 (bs, 3H), 2.84 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 159.2, 140.4, 130.9, 130.7, 130.4, 130.1, 122.4, 114.5, 112.7, 112.7, 55.5, 31.2. HRMS (ESI+): m/z calcd for C14H16BrN2O [M]+ = 307.0446, found 307.0455. The regioselectivity of this methylation reaction was confirmed by NOESY experiments (see spectra file).
6-Bromo-4-(4-methoxyphenyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (21). In a 250 mL two-neck round-bottom flask, was added 5-bromo-4′-methoxy-N3-methyl-[1,1′-biphenyl]-2,3-diamine 34 (0.9 g, 2.931 mmol, 1.0 equiv.) and dissolved in 20 mL of DCE at room temperature. Then, a solution of phosgene 15 wt% in toluene (3.2 mL, 29.31 mmol, 10 equiv.) was added drop wise to the reaction mixture and stirred for 1 h. The reaction mixture was poured into ice-cooled water (300 mL) and extracted with EtOAc (100 mL). The organic layers were dried over Na2SO4 and concentrated under reduced pressure. The crude was purified by column chromatography (30% EtOAc/hexane) to afford 6-bromo-4-(4-methoxyphenyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one 21 (0.82 g, 86%) as a pale white solid. Mp 225–227 °C. Rf = 0.55 (60% EtOAc/hexane). IR (neat, ν/cm−1): 3137, 3048, 2953, 2927, 2833, 1703, 1608, 1578, 1517, 1450, 1380, 1339, 1291, 1247, 1178, 962, 823, 767. 1H NMR (500 MHz, CDCl3): δ 8.72 (s, 1H), 7.43 (d, J = 8.7 Hz, 2H), 7.24 (d, J = 1.8 Hz, 1H), 7.06 (d, J = 1.4 Hz, 1H), 7.02 (d, J = 8.7 Hz, 2H), 3.87 (s, 3H), 3.80 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 159.9, 155.0, 132.7, 129.2, 128.5, 125.1, 124.2, 114.9, 114.7, 109.5, 55.5, 27.2. HRMS (ESI+): m/z calcd for C15H14BrN2O2 [M + H]+ = 333.0239, found 333.0218.
Sequence followed in Scheme 9
Thermolysis of 36. A flame dried 20 mL glass vial was charged with the intermediate 36 (0.14 g). Without the addition of solvent, the vial was placed in a preheated oil bath for 1 hour in which temperature was previously adjusted and fixed at 160 °C. After this period, the vial was removed from the hot bath allowed to reach room temperature and dissolved in DCM (20 mL). The DCM was evaporated under reduced pressure and the crude of reaction such obtained was purified by column chromatography (28% EtOAc/hexane) to afford the angular compound 37 (22 mg, 18.5%) as a yellow solid and the linear isomer 38 (19 mg, 16%) as a yellow solid.
Removal of MOM-group from kealiiquinone analogous
:2) to afford angular isomer of kealiiquinone 39 (6 mg, 82%) as light red solid. Mp > 300 °C. Rf = 0.4 (50% EtOAc/hexane). IR (neat, ν/cm−1): 3159, 2953, 2921, 2851, 1708, 1665, 1646, 1623, 1515, 1460, 1377, 1330, 1249, 1195, 1104, 1059, 1031, 962, 908, 829, 735. 1H NMR (500 MHz, CDCl3): δ 8.16 (s, 1H), 7.93 (s, 1H), 7.46 (d, J = 8.6 Hz, 2H), 7.06 (d, J = 8.6 Hz, 2H), 4.13 (s, 3H), 4.10 (s, 3H), 3.89 (s, 3H), 3.75 (s, 3H). HRMS (ESI): m/z calcd for C21H19N2O6 [M + H]+ = 395.1243, found 395.1238.Kealiiquinone
:2) to afford kealiiquinone 1 (6.5 mg, 87%) as red solid. Mp 291–293 °C. Rf = 0.4 (50% EtOAc/hexane). IR (neat, ν/cm−1): 3183, 2954, 2921, 2852, 1712, 1665, 1648, 1625, 1611, 1516, 1460, 1333, 1288, 1253, 1198, 1186, 1107, 1060, 1031, 961, 830, 747, 724, 670. 1H NMR (400 MHz, DMSO-d6) δ 11.02 (s, 1H), 7.68 (s, 1H), 7.12 (d, J = 8.0 Hz, 2H), 6.97 (d, J = 7.9 Hz, 2H), 3.93 (s, 3H), 3.85 (s, 3H), 3.82 (s, 3H), 3.39 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 181.3, 181.2, 158.6, 154.8, 147.8, 145.2, 134.0, 132.6, 129.9, 127.7, 126.5, 123.5, 122.8, 113.9, 104.6, 60.8, 60.8, 55.1, 26.8. HRMS (ESI+): m/z calcd for C21H19N2O6 [M + H]+ = 395.1243, found 395.1242. The spectroscopic data match for those reported by Ohta5 and Lovely.6Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to CONACyT (CB-2013/220836) for financial support. We acknowledge the facilities of the DCNyE, the Chemistry Department, and the National Laboratory UG-CONACyT (LACAPFEM) of the University of Guanajuato for full characterization. We also thank to CONACyT for fellowships to V. Ramadoss.Notes and references
- N. M. Shady, E. M. El-Hossary, M. A. Fouad, T. A. M. Gulder, M. S. Kamel and U. R. Abdelmohsen, Molecules, 2017, 22, 781 CrossRef PubMed.
- A. R. Carrol, B. F. Bowden and J. C. Coll, Aust. J. Chem., 1993, 46, 1229–1234 CrossRef.
- R. A. Lamb, N. S. Aberle, N. T. Lucas, G. Lessen and B. C. Hawkins, Angew. Chem., Int. Ed., 2017, 56, 14663–14666 CrossRef PubMed.
- R. K. Akee, T. R. Carrol, W. Y. Yoshida, P. J. Scheuer, T. J. Stout and J. Clardy, J. Org. Chem., 1990, 55, 1944–1946 CrossRef.
- I. Kawasaki, N. Taguchi, T. Yamamoto, M. Yamashita and S. Ohta, Tetrahedron Lett., 1995, 36, 8251–8254 CrossRef.
- J. Das, A. Bhan, S. S. Mandal and C. J. Lovely, Bioorg. Med. Chem. Lett., 2013, 23, 6183–6187 CrossRef PubMed.
- P. B. Koswatta, S. Kasiri, J. K. Das, A. Bhan, H. M. Lima, B. García-Barboza, N. N. Khatibi, M. Yousufuddin, S. S. Mandal and C. J. Lovely, Bioorg. Med. Chem. Lett., 2017, 25, 1608–1621 CrossRef PubMed.
- (a) H. M. Lima, R. Sivappa, M. Yousufuddin and C. J. Lovely, Org. Lett., 2012, 14, 2274–2277 CrossRef PubMed; (b) H. M. Lima, R. Sivappa, M. Yousufuddin and C. J. Lovely, J. Org. Chem., 2014, 79, 2481–2490 CrossRef PubMed; (c) J. Das, R. Mukherjee and A. Basak, J. Org. Chem., 2014, 79, 3789–3798 CrossRef PubMed; (d) For the synthesis of isokealiiquinone (methylation at the position 3) S. Nakamura, N. Tsuno, M. Yamashita, I. Kawasaki, S. Ohta and Y. Ohishi, J. Chem. Soc., Perkin Trans. 1, 2001, 429–436 RSC.
- V. Ramados, A. J. Alonso-Castro, N. Xolalpa and C. R. Solorio-Alvarado, J. Org. Chem., 2018 DOI:10.1021/acs.joc.8b01436 , ASAP.
- (a) For excellent revision on DoM reactions see: V. Sniekus, Chem. Rev., 1990, 90, 879–933 CrossRef; (b) For use of the MOM group in DoM reactions see: M. R. Winkle and R. C. Ronald, J. Org. Chem., 1982, 47, 2102–2108 CrossRef; (c) For the sequence DoM/iodination see: C. A. Townsend and L. M. Bloom, Tetrahedron Lett., 1981, 22, 3923–3924 CrossRef.
- J. Shao, J. Chang and C. Chi, Org. Biomol. Chem., 2012, 10, 7045–7052 RSC.
- (a) L. S. Liebeskind, R. W. Fengl, K. R. Wirtz and T. T. Shawe, J. Org. Chem., 1988, 53, 2482–2488 CrossRef; (b) B. M. Trost, O. R. Thiel and H.-C. Tsui, J. Am. Chem. Soc., 2003, 125, 13155–13164 CrossRef PubMed; (c) M. Mohamed, T. P. Goncalves, R. J. Whitby, H. F. Sneddon and D. C. Harrowen, Chem.–Eur. J., 2011, 17, 13698–13705 CrossRef PubMed; (d) B. Yucel, B. Sanil, H. Akbulut, S. Ozbey and A. B. Benniston, Org. Biomol. Chem., 2012, 10, 1775–1784 RSC; (e) L. S. Liebeskind, S. Iyer and C. F. Jewell Jr, J. Org. Chem., 1986, 51, 3065–3067 CrossRef; (f) S. T. Perri and H. W. Moore, J. Am. Chem. Soc., 1990, 112, 1897–1905 CrossRef; (g) A. Enhsen, K. Karabelas, J. M. Heerding and H. W. Moore, J. Org. Chem., 1990, 55, 1177–1185 CrossRef; (h) H. W. Moore and S. T. Perri, J. Org. Chem., 1988, 53, 996–1003 CrossRef; (i) S. T. Perri and H. W. Moore, Tetrahedron Lett., 1987, 28, 4507–4510 CrossRef.
- X. Zhang and X. Zheng and L. S. Chupack, Aryl substituted bicyclic heteroaryl compounds, WO 2017019828, February 2, 2017.
- R. Salmasi, M. Gholizadeh, A. Salimi and J. Garrison, J. Iran. Chem. Soc., 2016, 13, 2019–2028 CrossRef.
- (a) E. Lindstedt, E. Stridfeldt and B. Oloffson, Org. Lett., 2016, 18, 4234–4237 CrossRef PubMed; (b) D. Liu, L. P. Sanow and C. Zhang, Tetrahedron Lett., 2014, 55, 3090–3092 CrossRef; (c) T. F. Woiwode, C. Rose and T. J. Wandless, J. Org. Chem., 1998, 63, 9594–9596 CrossRef.
- The NiCl2-NaBH4 system has been used for different organic transformations: (a) For selective carbonyl reduction in presence of acetyl group see: N. Hassanloie, B. Zeynizadeh, S. Ashuri and F. Hassanloie, Open Cancer Immunol. J., 2014, 59–62 Search PubMed; (b) For desulfurization reactions see: T. G. Back, D. L. Baron and K. Yang, J. Org. Chem., 1992, 58, 2407–2413 CrossRef.
- Is well known the poor basic- or acid-resistance of the acetyl group, however when is bonded to nitrogen its liability decreases substantially..
- (a) E. V. D. Brge and R. Robiette, J. Org. Chem., 2013, 78, 12220–12223 CrossRef PubMed; (b) R. Soundararajan and T. R. Balasubramanian, Tetrahedron Lett., 1984, 25, 5555–5558 CrossRef; (c) Y. Ono, Y. Izawa and Z.-I. Fu, J. Chem. Soc., Chem. Commun., 1995, 9 RSC; (d) E. Boredon, B. Chabaud, A. Gaset, S. Thiebaud-Roux and S. Ouk, Monomethylation of Nitrogenous Heterocycles, US 2004/0024205 A1, February 5, 2004.
- M. Bella and V. Milata, J. Heterocycl. Chem., 2008, 45, 425–427 CrossRef.
- (a) A. D. Allen, J. D. Colomvakos, I. Egle, J. Lusztyk, M. A. McAllister, T. T. Tidwell, B. D. Wagner and D.-C. Zhao, J. Am. Chem. Soc., 1995, 117, 7552–7553 CrossRef; (b) G. Cerioni, R. Janoshchek, Z. Rappaport and T. T. Tidwell, J. Org. Chem., 1996, 61, 6212–6217 CrossRef PubMed.
- J. Shao, J. Chang and C. Chi, Org. Biomol. Chem., 2012, 10, 7045–7052 RSC.
- L. S. Liebeskind, R. W. Fengl, K. R. Wirtz and T. T. Shawe, J. Org. Chem., 1988, 53, 2482–2488 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06676k |
| This journal is © The Royal Society of Chemistry 2018 |