
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUniversally applicable, quantitative PCR method utilizing fluorescent nucleobase analogs†
Hyo Yong Kim‡
a,
Taihua Li‡b,
Cheulhee Jung a,
Rongzhan Fua,
Dae-Yeon Choc,
Ki Soo Park*d and
Hyun Gyu Park*a
a,
Rongzhan Fua,
Dae-Yeon Choc,
Ki Soo Park*d and
Hyun Gyu Park*a
aDepartment of Chemical and Biomolecular Engineering (BK21 Program), KAIST, 291 Daehak-ro, Yuseong-gu, Daejeon 305-701, Republic of Korea. E-mail: hgpark@kaist.ac.kr; Fax: +82-42-350-3910; Tel: +82-42-350-3932
bCollege of Biology and the Environment, Co-Innovation Centre for Sustainable Forestry in Southern China, Nanjing Forestry University, 159 Longpan road, Nanjing, Jiangsu 210-037, China
cLabgenomics Clinical Research Institute, Labgenomics Co. Ltd., Yong-In, 449-795, Republic of Korea
dDepartment of Biological Engineering, College of Engineering, Konkuk University, Seoul 05029, Republic of Korea. E-mail: kskonkuk@gmail.com; Fax: +82-2-450-3742; Tel: +82-2-450-3742
First published on 7th November 2018
Abstract
We herein describe a novel quantitative PCR (qPCR) method, which operates in both signal-off and on manners, by utilizing a unique property of fluorescent nucleobase analogs. The first, signal-off method is developed by designing the primers to contain pyrrolo-dC (PdC), one of the most common fluorescent nucleobase analogs. The specially designed single-stranded primer is extended to form double-stranded DNA during PCR and the fluorescence signal from the PdCs incorporated in the primer is accordingly reduced due to its conformation-dependent fluorescence properties. In addition, the second, signal-on method is devised by designing the primers to contain 5′-overhang sequences complementary to the PdC-incorporated DNA probes. At the initial phase, the PdC-incorporated DNA probes are hybridized to the 5′-overhang sequences of the primer, exhibiting the significantly quenched fluorescence signal, but are detached by either hydrolysis or strand displacement reaction during PCR, leading to the highly enhanced fluorescence signal. This method is more advanced than the first one since it produces signal-on fluorescence response and permits the use of a single PdC-incorporated DNA probe for the detection of multiple target nucleic acids, remarkably decreasing the assay cost. With these novel qPCR methods, we successfully quantified target nucleic acids derived from sexually transmitted disease (STD) pathogens with high accuracy. Importantly, the proposed strategies overcome the major drawbacks in the current SYBR Green and TaqMan probe-based qPCR methods such as low specificity and high assay cost.
1. Introduction
Quantitative PCR (qPCR) has been extensively utilized as the most powerful tool for accurate analysis of target nucleic acids in the biological and medical communities.1–4 In principle, qPCR is achieved by measuring the fluorescence signal, which is proportionally increased or decreased as the target nucleic acids are amplified during PCR, thereby enabling the simultaneous amplification and detection. In addition, based on the linear correlation between the initial amount of target nucleic acids and the threshold cycle (Ct) at which the fluorescence signal exceeds a certain threshold signal intensity, the concentration of target nucleic acids is accurately and quantitatively determined, which has not been accomplished in the conventional end-point PCR.5–7With these merits, numerous qPCR methods have been reported, most of which rely on either intercalating fluorescent dyes or sequence-specific fluorescent probes.8–17 The representative example of the intercalating fluorescent dyes is SYBR Green, which emits the high fluorescence signal after it binds to double-stranded DNA. Although this method is universally applied with low assay cost, it has a critical limitation that it is not sequence-specific and thus is susceptible to the false positive signals originating from either primer-dimers or non-specific amplifications. On the other hand, sequence-specific fluorescent probes including TaqMan probe, molecular beacons, FRET probes, Scorpions primer, and universal template (UT) probe enable the accurate quantification of target nucleic acids with high sequence specificity.9,12,14,16,18–21 However, most of them require expensive dual modification with fluorophore and quencher, thereby increasing the total assay cost and limiting the wide-spread use.22,23
To eliminate the drawbacks in the current qPCR methods, interesting alternative strategies have been suggested. Particularly, there have been a lot of efforts to reduce the assay cost by designing new sequence specific fluorescent probes that do not require the quencher modification. For instance, Seidel et al. utilized guanine, which possesses the highest quenching capability among the four natural nucleobases, as the substitute for the expensive quenchers.24 In another study, Nazarenko et al. found that the fluorescence properties of the internally conjugated fluorophores can be altered depending on the structural change from the single- to double-stranded form, which was exploited to monitor PCR process.25 Overall, these strategies offered the promising results with the decreased assay cost, however the signal change was not large enough to achieve the high detection sensitivity and accuracy. Therefore, there still has been high demands for the development of a new qPCR method that can be universally applied with the high sequence specificity.
As a signaling component which can overcome the unsolved drawbacks in the previous efforts, fluorescent nucleobase analogs have gained the special attention due to their high quantum yields and structure-dependent fluorescence properties.26–31 More importantly, it not only eliminates fluorophore or quencher modification, but also enables the high detection sensitivity. However, some limitations exist for the fluorescent nucleobase analogs, which include the spectral overlap of 2-aminopurine with those of biomolecules, and the incompatibility of 3-methylisoxanthopterin with DNA polymerases. These points must be carefully considered for the selection of proper fluorescent nucleobase analogs and development of sensing technologies with high reliability and sensitivity.32–35
By taking these into consideration, we herein devised novel qPCR methods by employing pyrrolo-dC (PdC), one of the most common fluorescent nucleobase analogs.36–38 The PdC intrinsically possesses high fluorescence signal in a single-stranded form, but shows significantly quenched one in a double-stranded form after being hybridized to a complementary DNA. In addition, its small size causes the negligible steric hindrance, which enables the efficient extension reaction catalyzed by DNA polymerase.39–43 With these novel features of PdC, we rationally designed PdC-incorporated DNA primers and probes, which were successfully utilized to quantify the target nucleic acids derived from sexually transmitted disease (STD) pathogens with high accuracy.
2. Experimental section
2.1 Materials
All the oligonucleotides utilized in this study were purchased from Genotech Co. (Daejeon, Korea) and Integrated DNA Technologies (IDT, IA, USA). The fluorescent nucleobase analogs, PdCs, were incorporated into the reverse primer for Chlamydia trachomatis (CT) and the universal fluorescent base (UFB) probe that is complementary to the 5′-overhang sequences of the forward primer for Mycoplasma hominis (MH). The specific sequence information of the oligonucleotides is listed in Table S1.† The two different types of DNA polymerases, i-StarMAX™ II DNA polymerase and Vent® (exo-) DNA polymerase, were purchased from iNtRON Biotechnology (Gyeonggi-do, Korea) and New England Biolabs (MA, USA), respectively.2.2 DNA extraction
Urine samples (30 mL) obtained by prostatic massage of male patients infected by CT and MH were centrifuged at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 2 min, washed twice with 1 mL of 1× phosphate buffered saline (PBS; 0.2 M sodium phosphate, 1.5 M sodium chloride, pH 7.4), and resuspended in 400 mL of 1× PBS for DNA extraction. Genomic DNA was extracted from these samples using the AccuPrep Genomic DNA Extraction Kit (Bioneer, Korea) according to the manufacturer's protocol, and stored at −20 °C before use. Copy numbers of the genomic DNA were calculated by measuring the absorbance at 260 nm using the ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, USA).
000g for 2 min, washed twice with 1 mL of 1× phosphate buffered saline (PBS; 0.2 M sodium phosphate, 1.5 M sodium chloride, pH 7.4), and resuspended in 400 mL of 1× PBS for DNA extraction. Genomic DNA was extracted from these samples using the AccuPrep Genomic DNA Extraction Kit (Bioneer, Korea) according to the manufacturer's protocol, and stored at −20 °C before use. Copy numbers of the genomic DNA were calculated by measuring the absorbance at 260 nm using the ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, USA).
2.3 PdC-based signal-off qPCR
The PCR amplification of the specific gene of CT was performed by using DNA Engine (PTC-200) Peltier Thermal Cycler (Bio-Rad, CA, USA) in 60 μL solution containing 1 μL of target genomic DNA, 0.25 μM of each primer, 0.1 mM of each dNTP, and 1.5 U of i-StarMAX™ II DNA polymerase in 1× PCR reaction buffer (10 mM Tris–HCl, 50 mM KCl, and 1.5 mM MgCl2, pH 9). The PCR protocol was programmed at 94 °C for 5 min, followed by 40 cycles of 94 °C for 20 s, 52 °C for 30 s, and 72 °C for 20 s. The fluorescence signal was recorded at the end of every annealing step using an RF-5301PC spectrofluorophotometer (Shimadzu, Japan) with the excitation and emission wavelength at 350 nm and 450 nm, respectively.2.4 PdC-based signal-on qPCR
The PCR amplification of the specific gene of MH was performed by using DNA Engine (PTC-200) Peltier Thermal Cycler (Bio-Rad, CA, USA) in 60 μL solution containing 1 μL of target genomic DNA, 0.25 μM of each primer, 0.1 mM of each dNTP, and 1.4 U of i-StarMAX™ II DNA polymerase or Vent® (exo-) DNA polymerase in corresponding 1× PCR reaction buffer containing 10 mM Tris–HCl (pH 9), 50 mM KCl, and 1.5 mM MgCl2 for i-StarMAX™ II DNA polymerase and 20 mM Tris–HCl (pH 8.8), 10 mM (NH4)2SO4, 10 mM KCl, 2 mM MgCl2, and 0.1% Triton X-100 for Vent® (exo-) DNA polymerase. The PCR protocol was programmed at 94 °C for 5 min, followed by 40 cycles of 94 °C for 20 s, 52 °C for 30 s, and 72 °C for 30 s. The fluorescence signal was recorded at the end of every extension step using an RF-5301PC spectrofluorophotometer (Shimadzu, Japan) with the excitation and emission wavelength at 350 nm and 450 nm, respectively.2.5 TaqMan probe-based qPCR
The TaqMan probe-based qPCR for the detection of specific genes of CT and MH was performed by using CFX96™ (Bio-Rad, CA, USA) in 20 μL solution containing 1 μL of target genomic DNA, 0.25 μM of each primer, 0.25 μM of TaqMan probe in 1× master mix (Finnzymes, Finland). The PCR protocol for the amplification of CT and MH gene was programmed at 95 °C for 15 min, followed by 50 cycles of 95 °C for 15 s and 60 °C for 60 s. The fluorescence signal was recorded at the end of every extension step.3. Results and discussion
3.1 PdC-based qPCR strategies
The basic principle of the developed qPCR method is illustrated in Fig. 1, which utilizes the forward and PdC-incorporated reverse primer to amplify target nucleic acids. At the initial phase, the PdCs in the single-stranded reverse primer emits high fluorescence signal. However, as the PdC-incorporated reverse primer anneals to the template DNA, the fluorescence signal from PdC is significantly quenched. As a result, the fluorescence signal is decreased in proportion to the amount of DNA products amplified during PCR.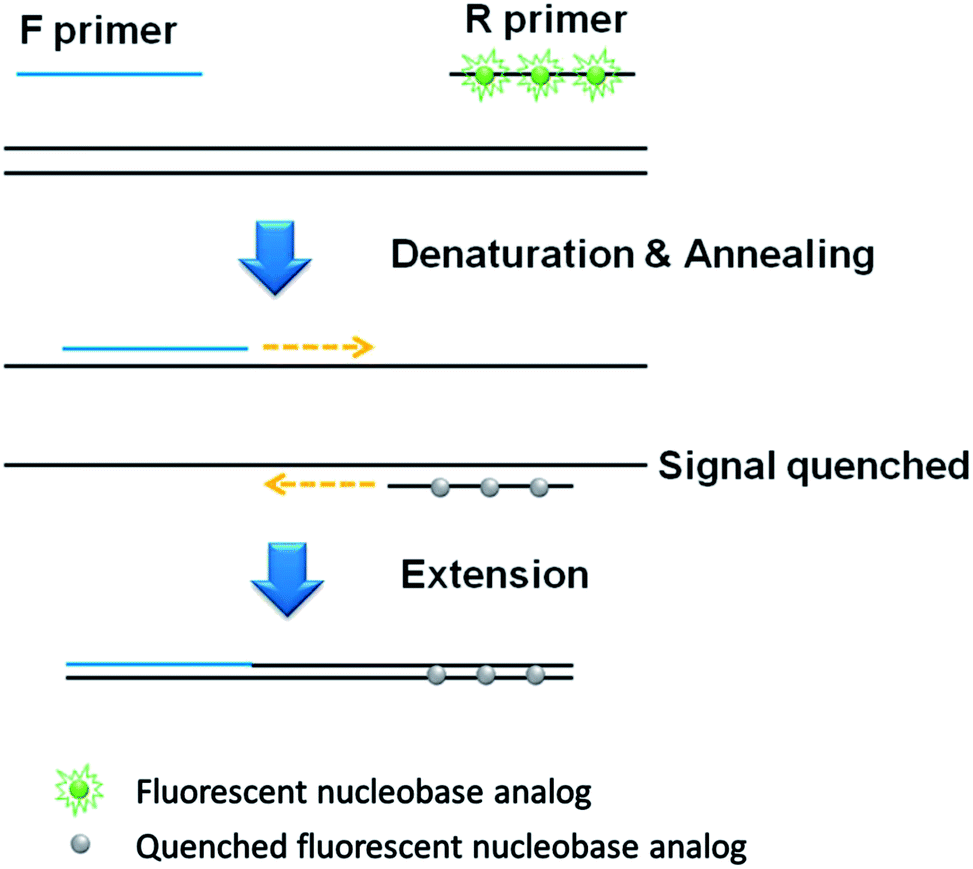 | ||
| Fig. 1 Schematic illustration of the signal-off qPCR strategy utilizing PdC-incorporated primer. | ||
In addition, an advanced qPCR method, which generates the signal-on fluorescence response, is developed by rationally designing universal fluorescent base (UFB) probes that enable the detection of various target nucleic acids at a low cost (Fig. 2). In contrast to the first signal-off strategy using PdC-incorporated primer, the signal-on strategy utilizes UFB probe that initially anneals to the 5′-overhang sequences of the forward primer, exhibiting low fluorescence signal. In the extension step during the PCR process, the bound UFB probe is either hydrolyzed or displaced by the activity of Taq DNA polymerase or Vent® (exo-) DNA polymerase, respectively, leading to the liberation of the PdCs.
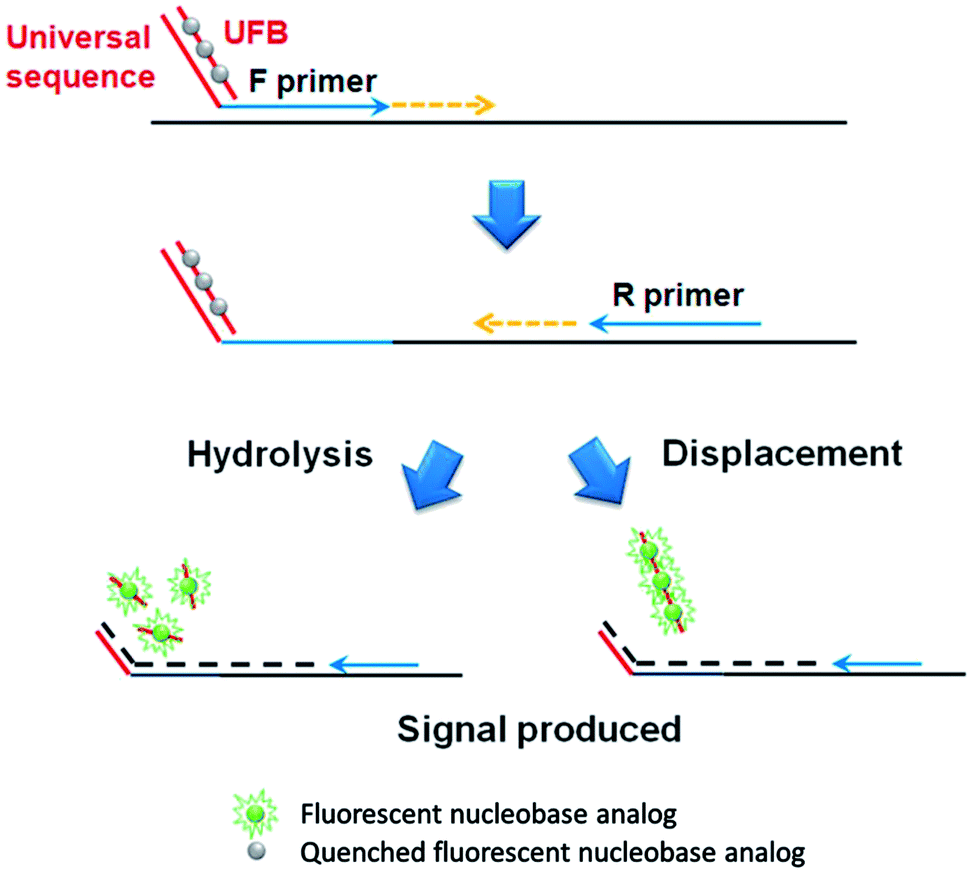 | ||
| Fig. 2 Schematic illustration of the signal-on qPCR strategy utilizing UFB probe. | ||
3.2 Investigation of hybridization-induced fluorescence signal change from PdCs
First, we measured the hybridization-induced fluorescence signal change by varying the number of PdCs. As shown in Fig. S1,† the fluorescence signal from the PdC is remarkably diminished when the PdC-incorporated DNA is hybridized to the complementary DNA. Importantly, as the number of PdC was increased, the more significant fluorescence signal change was observed upon hybridization. However, when more than three PdCs were incorporated, the fluorescence signal change was not significantly increased. Based on these results, three PdCs were incorporated into DNA sequences, which was utilized for further experiments.3.3 Signal-off qPCR strategy utilizing PdC-incorporated primer
Next, we checked the utility of the PdC-incorporated DNA for the real-time monitoring of PCR. As a model target, Chlamydia trachomatis (CT), the most common bacterium causing sexually transmitted diseases (STDs) was selected and the specific DNA primers were designed. As envisioned in the design of the new strategy, the fluorescence signal of PdC was decreased as the number of PCR cycle was increased (Fig. 3). In addition, the standard curve between threshold cycle (Ct) and logarithm of the initial copy number of target nucleic acids was obtained with the great square regression coefficient (R2) of 0.9968. Importantly, the results were well matched with that of TaqMan probe-based assay, which indicates that the PdC-based qPCR method proposed in this study is well suited for the quantitative analysis of target nucleic acids (Fig. S2†).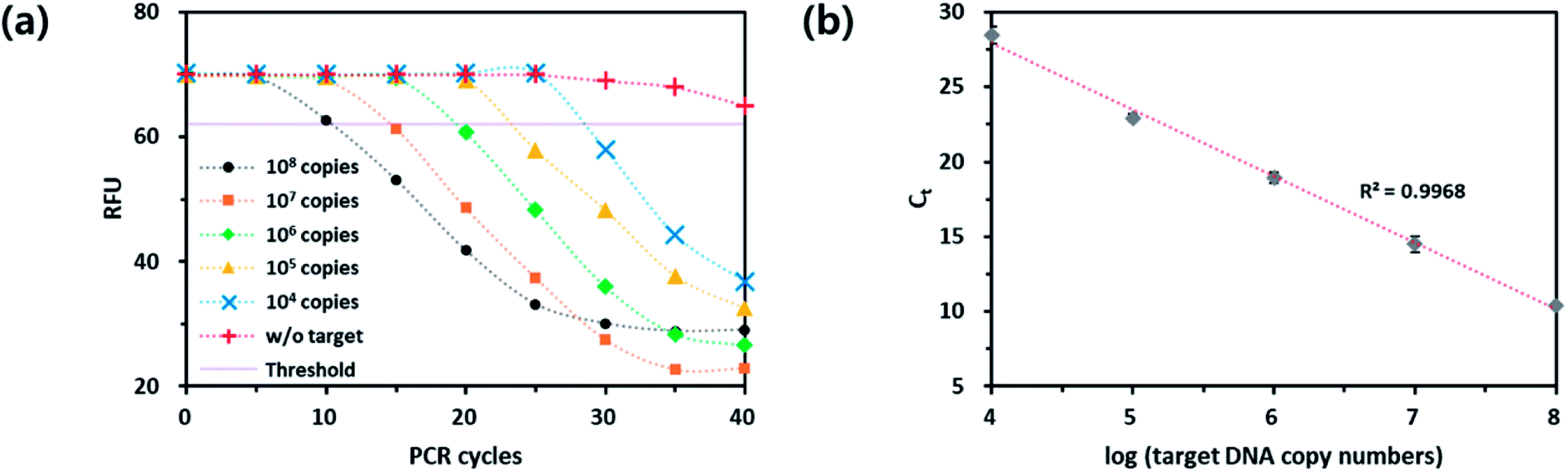 | ||
| Fig. 3 Quantitative analysis of target nucleic acids from CT using the PdC-based signal-off qPCR method. (a) Real-time fluorescence signals from the reaction solutions with varying initial copy numbers of target nucleic acids. (b) Linear relationship between Ct and logarithm of initial copy number of target nucleic acids (104 to 108 copies). | ||
3.4 Signal-on qPCR strategy utilizing UFB probe
In the same manner to the signal-off strategy, the applicability of the UFB probe-based qPCR method was demonstrated through the quantitative analysis of the target genomic DNA from Mycoplasma hominis (MH), another common bacterium causing STDs such as pelvic inflammatory disease (PID), urethritis, and cervicitis. As shown in Fig. 4(a), the fluorescence signal was increased as the PCR proceeded and an excellent linear relationship was observed between Ct and logarithm of initial copy number of the target nucleic acids (R2 = 0.9922). It should be noted that the direct comparison of fluorescence intensities in both modes is difficult because the reaction buffer conditions and DNA polymerase activities are different in both cases. Importantly, the results were well matched with that of TaqMan probe-based assay (Fig. S3†). Overall, the PdC-based qPCR methods are reproducible and could be utilized for the quantitative analysis of target nucleic acids while overcoming the disadvantages of current strategies. It should be noted that the PdC-based qPCR method based on either signal-on or signal-off fluorescence response is highly advantageous compared to current qPCR methods that relies on TaqMan probe because it does not require the expensive quencher moieties, which significantly reduces the assay cost, and it can be universally applied to the detection of various target nucleic acids.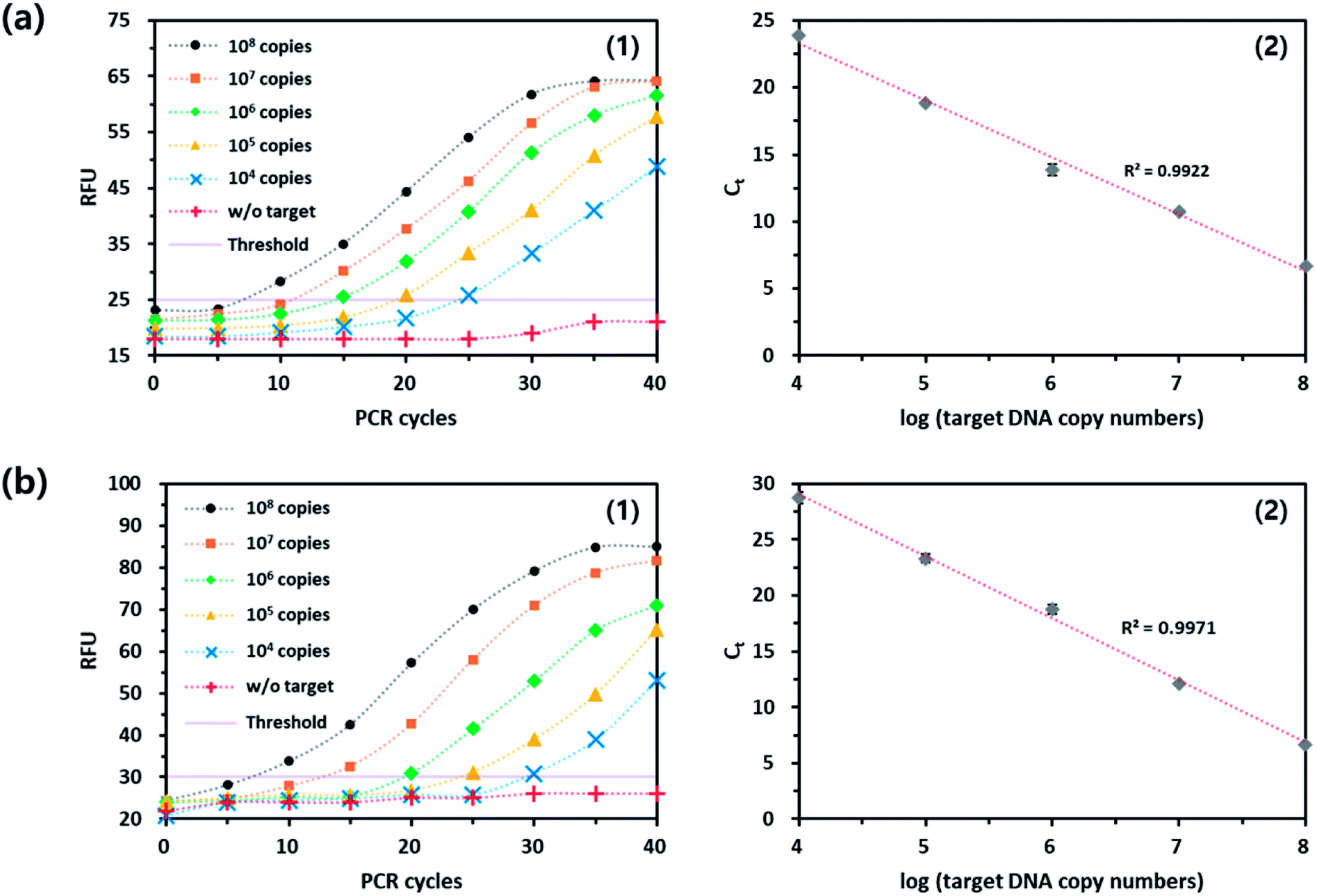 | ||
| Fig. 4 Quantitative analysis of target nucleic acids from MH using PdC-based signal-on qPCR method in (a) hydrolysis or (b) strand displacement mode. (1) Real-time fluorescence signals from the reaction solutions with varying initial copy numbers of target nucleic acids. (2) Linear relationship between Ct and logarithm of initial copy number of target nucleic acids (104 to 108 copies). | ||
4. Conclusions
Herein, we devised new qPCR methods, which operate in signal-off and on manners, based on the unique fluorescence properties of PdC. By rationally designing the PdC-incorporated DNA primers and probes, target nucleic acids extracted from STD pathogens were successfully determined with high accuracy. Importantly, the developed system possesses the high sequence specificity and cost-effectiveness which has not been accomplished by the current qPCR techniques utilizing TaqMan probe, molecular beacon, FRET probe, Scorpions primer, and UT probe. To the best of our knowledge, this is the first report to develop qPCR methods utilizing fluorescent nucleobase analogs, which will pave the way for the accurate and quantitative detection of target nucleic acids in the clinical diagnostics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by BioNano Health-Guard Research Center funded by the Ministry of Science and ICT (MSIT) of Korea as Global Frontier Project (Grant number H-GUARD_2013M3A6B2078964). This research was also supported by the Mid-career Researcher Support Program of the National Research Foundation (NRF) funded by the Ministry of Science, ICT & Future planning (MSIP) of Korea (NRF-2018R1A2A1A05022355).References
- R. K. Saiki, S. Scharf, F. Faloona, K. B. Mullis, G. T. Horn, H. A. Erlich and N. Arnheim, Science, 1985, 230, 1350 CrossRef CAS PubMed.
- R. Higuchi, G. Dollinger, P. S. Walsh and R. Griffith, Nat. Biotechnol., 1992, 10, 413 CrossRef CAS.
- R. Higuchi, C. Fockler, G. Dollinger and R. Watson, Nat. Biotechnol., 1993, 11, 1026 CrossRef CAS.
- B. Y. Won, S. Shin, S. Baek, Y. L. Jung, T. Li, S. C. Shin, D. Y. Cho, S. B. Lee and H. G. Park, Analyst, 2011, 136, 1573 RSC.
- M. Kubista, J. M. Andrade, M. Bengtsson, A. Forootan, J. Jonák, K. Lind, R. Sindelka, R. Sjöback, B. Sjögreen, L. Strömbom, A. Ståhlberg and N. Zoric, Mol. Aspects Med., 2006, 27, 95 CrossRef CAS PubMed.
- A. Ståhlberg, N. Zoric, P. Åman and M. Kubista, Expert Rev. Mol. Diagn., 2005, 5, 221 CrossRef PubMed.
- M. J. Espy, J. R. Uhl, L. M. Sloan, S. P. Buckwalter, M. F. Jones, E. A. Vetter, J. D. C. Yao, N. L. Wengenack, J. E. Rosenblatt, F. R. Cockerill Iii and T. F. Smith, Clin. Microbiol. Rev., 2006, 19, 165 CrossRef CAS PubMed.
- S. Giglio, P. T. Monis and C. P. Saint, Nucleic Acids Res., 2003, 31, e136 CrossRef PubMed.
- L. G. Lee, C. R. Connell and W. Bloch, Nucleic Acids Res., 1993, 21, 3761 CrossRef CAS PubMed.
- U. E. M. Gibson, C. A. Heid and P. M. Williams, Genome Res., 1996, 6, 995 CrossRef CAS PubMed.
- C. A. Heid, J. Stevens, K. J. Livak and P. M. Williams, Genome Res., 1996, 6, 986 CrossRef CAS PubMed.
- S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303 CrossRef CAS PubMed.
- C. T. Wittwer, M. G. Herrmann, A. A. Moss and R. P. Rasmussen, BioTechniques, 1997, 22, 130 CrossRef CAS PubMed.
- A. S. Piatek, S. Tyagi, A. C. Pol, A. Telenti, L. P. Miller, F. R. Kramer and D. Alland, Nat. Biotechnol., 1998, 16, 359 CrossRef CAS PubMed.
- S. Tyagi, D. P. Bratu and F. R. Kramer, Nat. Biotechnol., 1998, 16, 49 CrossRef CAS PubMed.
- X. Chen and P. Y. Kwok, Genet. Anal.: Biomol. Eng., 1999, 14, 157 CrossRef CAS.
- J. Cheng, Y. Zhang and Q. Li, Nucleic Acids Res., 2004, 32, e61 CrossRef PubMed.
- Y. Zhang, D. Zhang, W. Li, J. Chen, Y. Peng and W. Cao, Nucleic Acids Res., 2003, 31, e123 CrossRef PubMed.
- X. Li, Y. Huang, Y. Guan, M. Zhao and Y. Li, Anal. Chem., 2006, 78, 7886 CrossRef CAS PubMed.
- L. Yang, W. Liang, L. Jiang, W. Li, W. Cao, Z. A. Wilson and D. Zhang, BMC Mol. Biol., 2008, 9, 54 CrossRef PubMed.
- D. Whitcombe, J. Theaker, S. P. Guy, T. Brown and S. Little, Nat. Biotechnol., 1999, 17, 804 CrossRef CAS PubMed.
- P. R. Selvin, Methods Enzymol., 1995, 246, 300 CAS.
- I. Nazarenko, B. Lowe, M. Darfler, P. Ikonomi, D. Schuster and A. Rashtchian, Nucleic Acids Res., 2002, 30, e37 CrossRef PubMed.
- C. A. M. Seidel, A. Schulz and M. H. M. Sauer, J. Phys. Chem., 1996, 100, 5541 CrossRef CAS.
- I. Nazarenko, R. Pires, B. Lowe, M. Obaidy and A. Rashtchian, Nucleic Acids Res., 2002, 30, 2089 CrossRef CAS PubMed.
- M. Li, Y. Sato, S. Nishizawa, T. Seino, K. Nakamura and N. Teramae, J. Am. Chem. Soc., 2009, 131, 2448 CrossRef CAS PubMed.
- K. S. Park, J. Y. Lee and H. G. Park, Chem. Commun., 2012, 48, 4549 RSC.
- A. Taylor, A. Joseph, R. Okyere, S. Gogichaishvili, K. Musier-Forsyth and B. Kankia, Biophys. Chem., 2013, 171, 1 CrossRef CAS PubMed.
- C. Y. Lee, K. S. Park and H. G. Park, Chem. Commun., 2015, 51, 13744 RSC.
- W. Zhou, J. Ding and J. Liu, Biosens. Bioelectron., 2017, 87, 171 CrossRef CAS PubMed.
- C. Y. Lee, K. S. Park and H. G. Park, Biosens. Bioelectron., 2017, 98, 210 CrossRef CAS PubMed.
- G. M. Palmer, P. J. Keely, T. M. Breslin and N. Ramanujam, Photochem. Photobiol., 2003, 78, 462 CrossRef CAS PubMed.
- M. E. Hawkins, Methods Enzymol., 2008, 450, 201 CAS.
- K. Datta, N. P. Johnson, G. Villani, A. H. Marcus and P. H. Von Hippel, Nucleic Acids Res., 2012, 40, 1191 CrossRef CAS PubMed.
- S. Gogichaishvili, J. Johnson, D. Gvarjaladze, L. Lomidze and B. Kankia, Biopolymers, 2014, 101, 583 CrossRef CAS PubMed.
- M. E. Hawkins, Cell Biochem. Biophys., 2001, 34, 257 CrossRef CAS PubMed.
- M. J. Rist and J. P. Marino, Curr. Org. Chem., 2002, 6, 775 CrossRef CAS.
- T. Li, R. Fu and H. G. Park, Chem. Commun., 2010, 46, 3271 RSC.
- C. Liu and C. T. Martin, J. Mol. Biol., 2001, 308, 465 CrossRef CAS PubMed.
- C. Liu and C. T. Martin, J. Biol. Chem., 2002, 277, 2725 CrossRef CAS PubMed.
- C. M. Zhang, C. Liu, T. Christian, H. Gamper, J. Rozenski, D. Pan, J. B. Randolph, E. Wickstrom, B. S. Cooperman and Y. M. Hou, RNA, 2008, 14, 2245 CrossRef CAS PubMed.
- R. F. Borkman and S. Lerman, Exp. Eye Res., 1978, 26, 705 CrossRef CAS PubMed.
- T. Li, K. S. Jeon, Y. D. Suh and M. G. Kim, Chem. Commun., 2011, 47, 9098 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06675b |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2018 |