
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantitative syntheses of permethylated closo-1,10-R2C2B8Me8 (R = H, Me) carboranes. Egg-shaped hydrocarbons on the Frontier between inorganic and organic chemistry†
Mario Bakardjieva,
Oleg L. Toka,
Aleš Růžička b,
Zdeňka Růžičkováb,
Josef Holuba,
Drahomír Hnyka,
Jindřich Fanfrlíkc and
Bohumil Štíbr*a
b,
Zdeňka Růžičkováb,
Josef Holuba,
Drahomír Hnyka,
Jindřich Fanfrlíkc and
Bohumil Štíbr*a
aInstitute of Inorganic Chemistry, Academy of Sciences of the Czech Republic, Husinec-Řež 1001, Czech Republic. E-mail: stibrb@seznam.cz
bDepartment of General and Inorganic Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 532 10 Pardubice, Czech Republic
cInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nám. 2, 166 10 Prague 6, Czech Republic
First published on 14th November 2018
Abstract
Electrophilic methylation of the closo-1,10-R2C2B8H8 (1) (R = H or Me) dicarbaboranes at higher temperatures or thermal rearrangement of the 1,6-R2C2B8Me8 (3) compounds at 400–500 °C generated the B-permethylated derivatives closo-1,10-R2C2B8Me8 (2) in quantitative (>95%) yields. The compounds exhibit extreme air stability as a consequence of a rigid, egg shaped hydrocarbon structures incorporating inner 1,10-C2B8 carborane core.
Introduction
Methods for cage substitution on the cage of closo-1,10-C2B8H10 (1a) generally parallel those employed for the larger C2B10H12 icosahedral carboranes.1 The CH hydrogens in 1a are sufficiently protonic in character to undergo lithiation with butyllithium in ethereal solvents, generating the mono- and dilithio derivatives.2 The C-lithiated species afford the main entry to alkyl, aryl, carboxyl, silyl, and other C-substituted derivatives via treatment with appropriate reagents. For example, exo-polyhedral metal complexes,3,4 and the silyl-linked mixed-carboranes5 have also been prepared by this route along with numerous C-metallated compounds containing main-group metals.1 The only B-substitution processes so far reported are, however, the direct reaction between 1a and Cl2 affording the B-perchloro species 1,10-H2C2B8Cl8 (ref. 2) and those leading to a series of halo derivatives 1,10-H2C2B8H7-2-X.6 The successful B-methylation experiments in the 12-vertex carborane series,7–9 together with those achieved by our group in the B-methylation of closo-1,6-C2B8H10,10 arachno-6,9-C2B8H14,11 prompted us to extend boron-methylation strategy to the most stable members of the 10-vertex closo series, closo-1,10-R2C2B8H10 (1) (where R = H or Me), which have been now relatively easily available.12 In this article we would like to present electrophilic reactions with methylation agents leading to quantitative permethylation of B-vertexes under the formation of rigid, hydrocarbon–boron structures of egg shape that exhibit outstandingly high stability.Results and discussion
The electrophilic CH3I/AlCl3 methylation of carborane closo-1,10-H2C2B8H8 (1a) (Scheme 1) led on heating at 115 °C for 15 h to exclusive formation of the B-permethylated dicarbaborane closo-1,10-H2C2B8Me8 (2a) in practically quantitative yield (>95%). The scheme also shows that the CH3OTf/HOTf (Tf = SO2CH3) methylation proceeded excellently at 165 °C for 48 h, giving again a quantitative yield of 2a (>95%). It is, however, interesting that the CH3I/AlCl3 methylation (115 °C, 15 h) of closo-1,10-Me2C2B8H8 (1b) has completely failed, while the CH3OTf/HOTf methylation of 1b proceeded smoothly at 175 °C for 48 h, giving again a quantitative yield of the decamethylated closo-1,10-Me2C2B8Me8 (2b). This finding is in accord with that observed in the comparable 12-vertex closo-1,12-Me2C2B10Me10 series (reflux, 20 h, 91% yield).9 As also observed for the latter species, an attempt at methylating the CH1,10 vertexes in 2a via the Li2+ salt as in Scheme 1 has failed, too. The difficulty in forcing this reaction to completion must be due to the relative lack of reactivity of the CH vertices present in 2a, which is exacerbated by the steric protection afforded to each CH vertex by the methyl groups of the four surrounding BMe, vertices.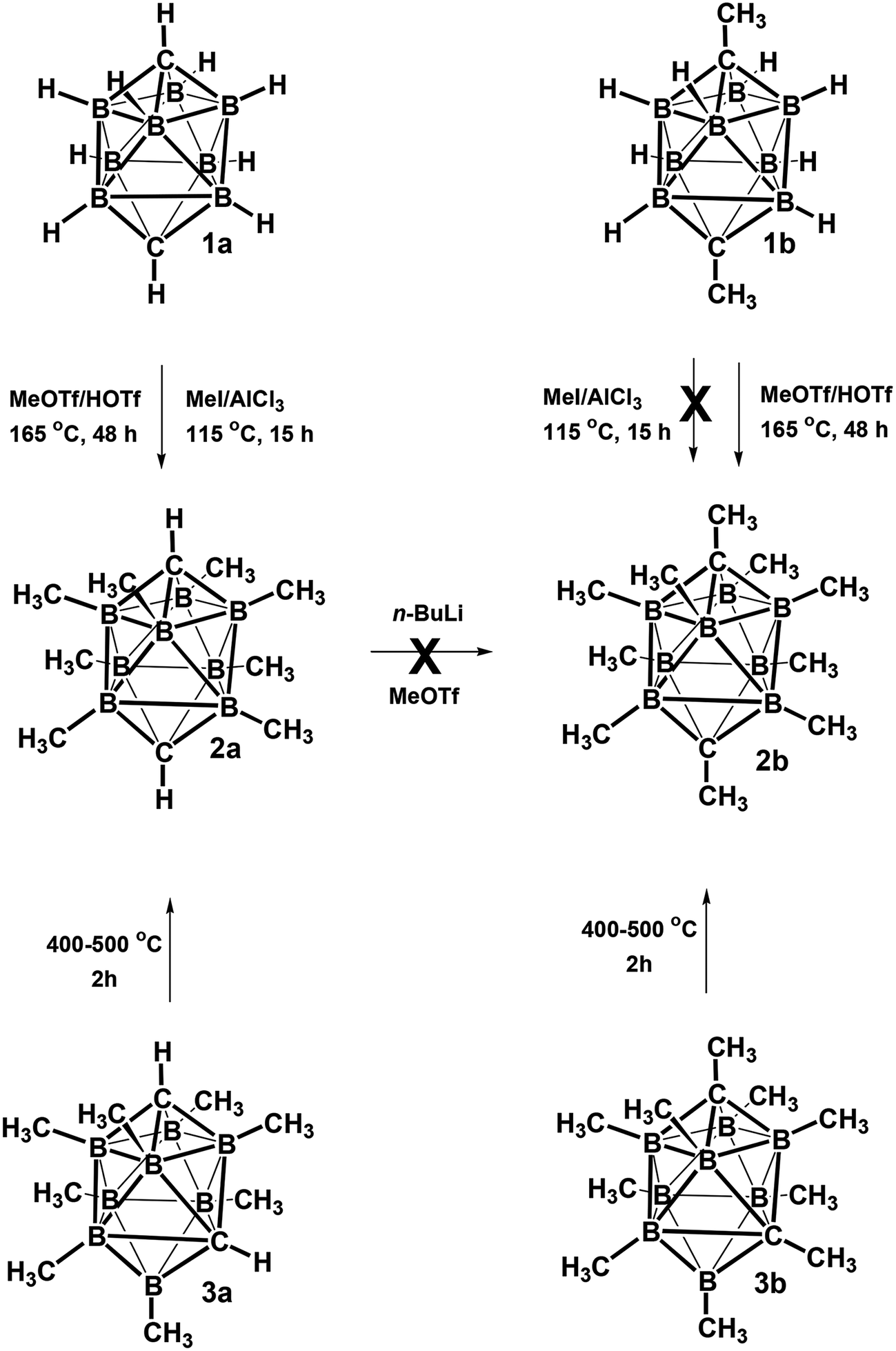 | ||
| Scheme 1 Quantitative syntheses of permethylated derivatives of the closo-1,10-R2C2B8H8 family. | ||
Another straightforward route leading to quantitative formation of permethylated dicarbaboranes 2 consists in thermal isomerisation of closo-1,6-R2C2B8Me8 (3) carboranes (R = H 3a or Me 3b)10 by heating at 400–500 °C in a sealed tube for 2 h. A similar thermal isomerisation principle could be also applied to the substituted derivatives, for example to closo-1,6-H2C2B8Me7-8-OTf (4a),10 which underwent cage rearrangement to afford 97% of the closo-1,10-H2C2B8Me7-2-OTf (5a) upon a similar heating, as shown in Scheme 2.
 | ||
| Scheme 2 Quantitative synthesis of the permethylated derivative closo-1,10-R2C2B8Me7-2-OTf. | ||
The structures of derivatives 2a and 5a were determined by an X-ray diffraction study (see Fig. 1 and 2). Both carboranes adopt the expected bicapped Archimedean antiprismatic geometry with two unsubstituted apical CH1,10 vertexes along with eight or seven BMe groups, respectively. The structure 5a unambiguously confirms the B2-substitution with the O-SO2-CF3 group and permethylation in all other B-positions of the cluster. Unfortunately, the crystals of the permethylated 2b were not found suitable for crystallographic studies and the 2a/2b pair was therefore geometry optimized at the MP2/TZVP level (Fig. 3 and 4).
 | ||
| Fig. 1 ORTEP (30% probability level) representation of the molecular structure of closo-1,10-H2C2B8Me8 (2a). Selected bond lengths (Å): C1–B2 1.600(4), C1–B4 1.595(4), B3–B2 1.870(4), B2–B3a 1.821(4), mean B–CH3 1.639(4); angles (°): B5–B2–B3 89.99(18), C1–B2–B3a 107.2(2), B2–B3a-B3 61.70(17), B5–C1–B2 71.39(17). | ||
 | ||
| Fig. 2 ORTEP (30% probability level) representation of the molecular structure of closo-1,10-H2C2B8Me7-2-OTf (5a). Selected bond lengths (Å): C1–B2 1.584(2), C1–B4 1.598(3), B2–B3 1.851(3), B2–B6 1.806(3), mean B–CH3 1.578(3), B2–O 1.453(2), mean C10–B 1.601(3), angles (°): B5–B2–B3 91.96(11), C1–B2–B6 109.72(13), B2–B6–B3 61.03(10), B2–C1–B5 70.64(12). | ||
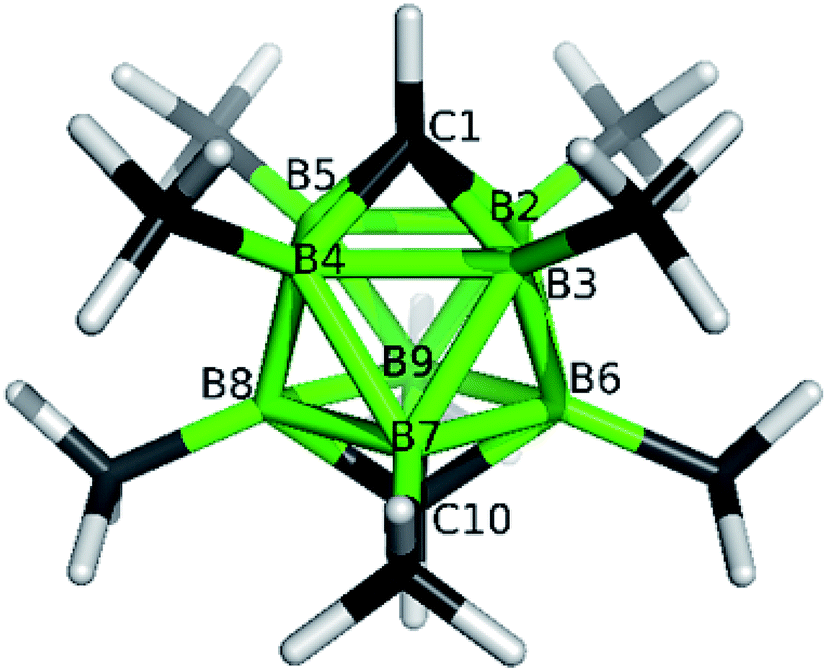 | ||
| Fig. 3 Molecular model for closo-1,10-H2C2B8Me8 (2a) as obtained from the MP2/TZVP optimization. The selected internal coordinates are (distances in Å, angles in °): C1–B2 1.602, C1⋯C10 3.329, B2–B3 1.866, B2–B6 1.818, B6–B7 1.866. | ||
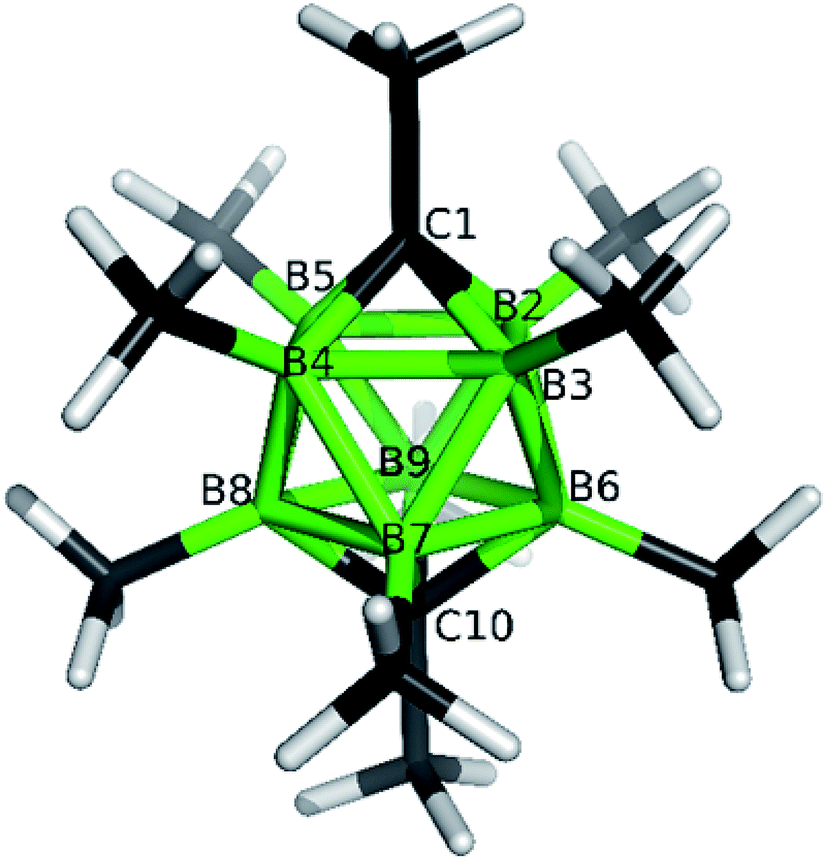 | ||
| Fig. 4 Molecular model for closo-1,10-Me2C2B8Me8 (2b) as obtained from the MP2/TZVP optimization. The selected internal coordinates are (distances in Å, angles in °): C1–B2 1.607, C1⋯C10 3.367, B2–B3 1.856, B2–B6 1.812, B6–B7 1.858. | ||
The optimization revealed that the comparable B–B, C–B, and B–Me bonding vectors are very similar to those found crystallographically for 2a and 5a. The computation has also led to a good agreement between theoretical and experimental δ(11B) values for 2a and 2b (max. deviation less than 3 ppm), for individual values see ESI.†
The HF/cc-pVTZ calculations of the electrostatic potential (ESP) surface show that the parent 1a has hydridic hydrogen atoms, which can form dihydrogen bonds,13 the hydridic B-bound hydrogens in 2a and 2b are now replaced by methyl groups of amphiphilic character.14 The hydrogen atoms of the methyl groups have positive ESP surface and the exo-skeletal carbon atoms have negative ESP surface (see Fig. 5). From the viewpoint of electron transmission, Me groups behave as weak electron acceptors, when compared to toluene, xylene, hexamethylbenzene etc. This is in accord with the 1,6-isomers of the same molecular shape10 and the same also applies to CMe methyl groups; the lower electron density at C1,10 in 2b in relation to 2a (see C1⋯C10 body diagonals) may perhaps prove this concept. The electron transmission thus follows the established electronegativity concept (C in CH3 is more electronegative than C (cage)) reflecting the fact that exo-skeletal substituents are bound via classical 2-centre 2-electron bonds.15 This agrees with the concept elaborated by Viñas et al. on a hexamethylated closo-H2C2B10H4Me6 system.16 Conceivably, the whole 11B NMR spectra of 2a and 2b are significantly paramagnetically deshielded to high frequencies (i.e. downfield shifted) with respect to those of parent compounds, for the computed 11B NMR spectra see ESI.†
 | ||
| Fig. 5 Computed (HF/cc-pVTZ) electrostatic potential (ESP) surface for (a) 1a, (b) 2a and (c) 2b. The color range of the ESP in kcal mol−1. | ||
The constitution of all compounds isolated in this study is also in agreement with the results of multinuclear 11B, [11B–11B]-COSY,17 1H, and 13C NMR measurements that led to complete assignments of individual cage BMe, BX, CH, and CMe units (for hardcopies of the NMR measurements, see ESI, Fig. S1–S13†). The multinuclear (11B, 1H and 13C) NMR spectra for all compounds isolated in this work are compared and depicted in Fig. 6, a general feature of the persubstituted compounds being the identity of the 11B and 11B–{1H}-decoupled NMR spectra that exhibit only singlet resonances.
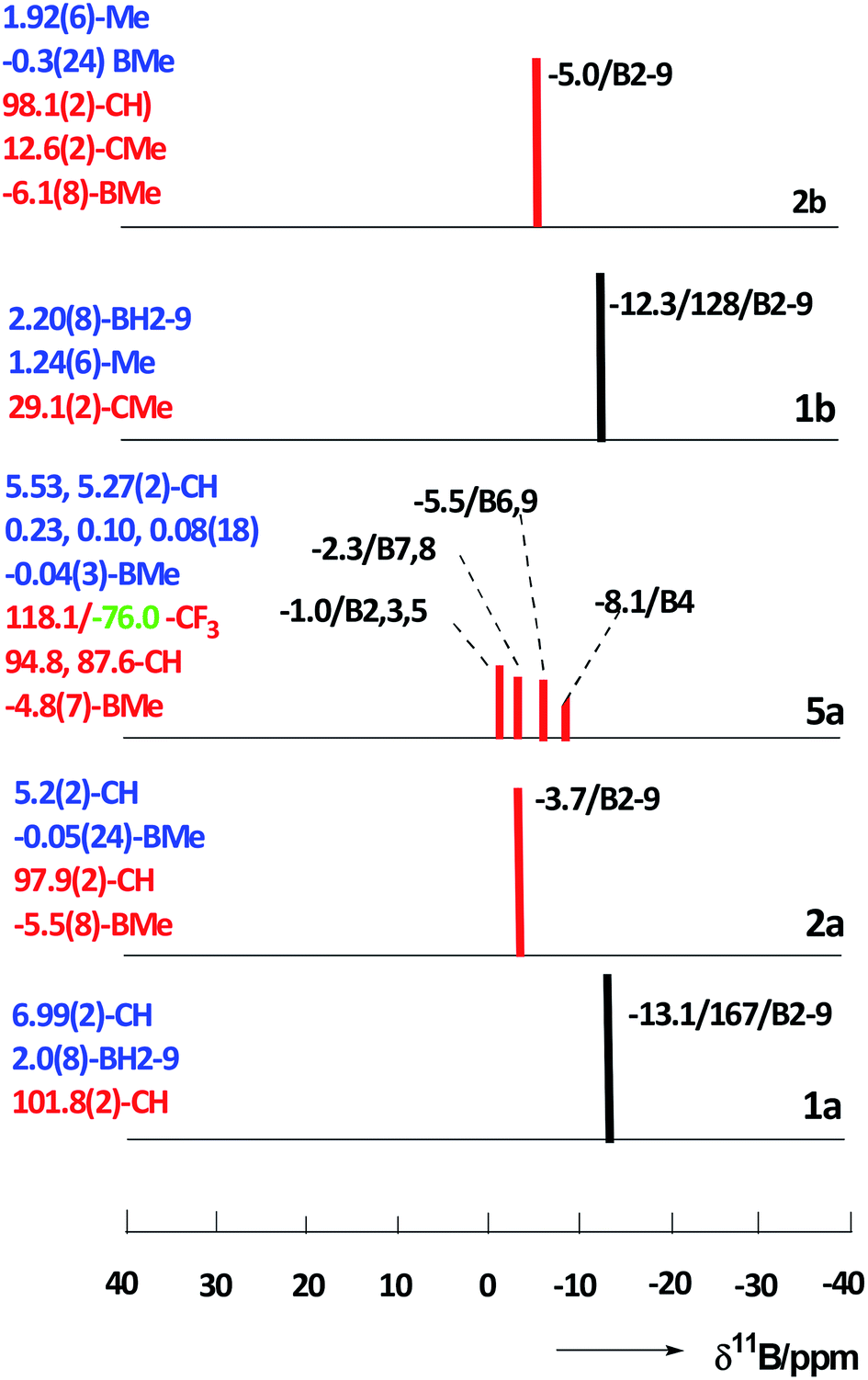 | ||
| Fig. 6 Stick diagrams comparing the 11B NMR chemical shifts and relative intensities for closo compounds 1,10-H2C2B8H8 (1a), 1,10-H2C2B8Me8 (2a), 1,10-H2C2B8Me7-2-OTf (5a), 1,10-Me2C2B8H8 (1b), and 1,10-Me2C2B8Me8 (2b) (red sticks = singlets, black = doublets). The data for individual cluster positions are ordered as: δ(11B)/1JBH (where applicable)/assignment. 1H (blue text) and [13C,19F] (red and green text) NMR resonances (δ in ppm/TMS) are summarized on the left side of each diagram with relative intensities other than 1 in parentheses. | ||
The 11B NMR spectra of the D4h-symmetry derivatives 2a and 2b (Fig. 3, S7 and S10†), exhibit only one singlet due to equivalency of all B-positions; the corresponding 1H spectra (Fig. 6, S8 and S10†) consist of two sharp 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 or 1:4 singlets for 2a and 2b, respectively, attributed to CH1,10 (or Me1,10) and BMe resonances. The 13C–{1H} NMR spectra of carboranes of structure 2 (Fig. 6, S9 and S11†) show one lower-field C1,10 singlet along with a very broad (JC–B coupling) high-field BMe resonance of relative areas 1:4, whereby that of 2b shows an additional C–Me signal. For NMR spectra of the key permethylated derivatives 2a and 2b, see Fig. 7.
:12 or 1:4 singlets for 2a and 2b, respectively, attributed to CH1,10 (or Me1,10) and BMe resonances. The 13C–{1H} NMR spectra of carboranes of structure 2 (Fig. 6, S9 and S11†) show one lower-field C1,10 singlet along with a very broad (JC–B coupling) high-field BMe resonance of relative areas 1:4, whereby that of 2b shows an additional C–Me signal. For NMR spectra of the key permethylated derivatives 2a and 2b, see Fig. 7.
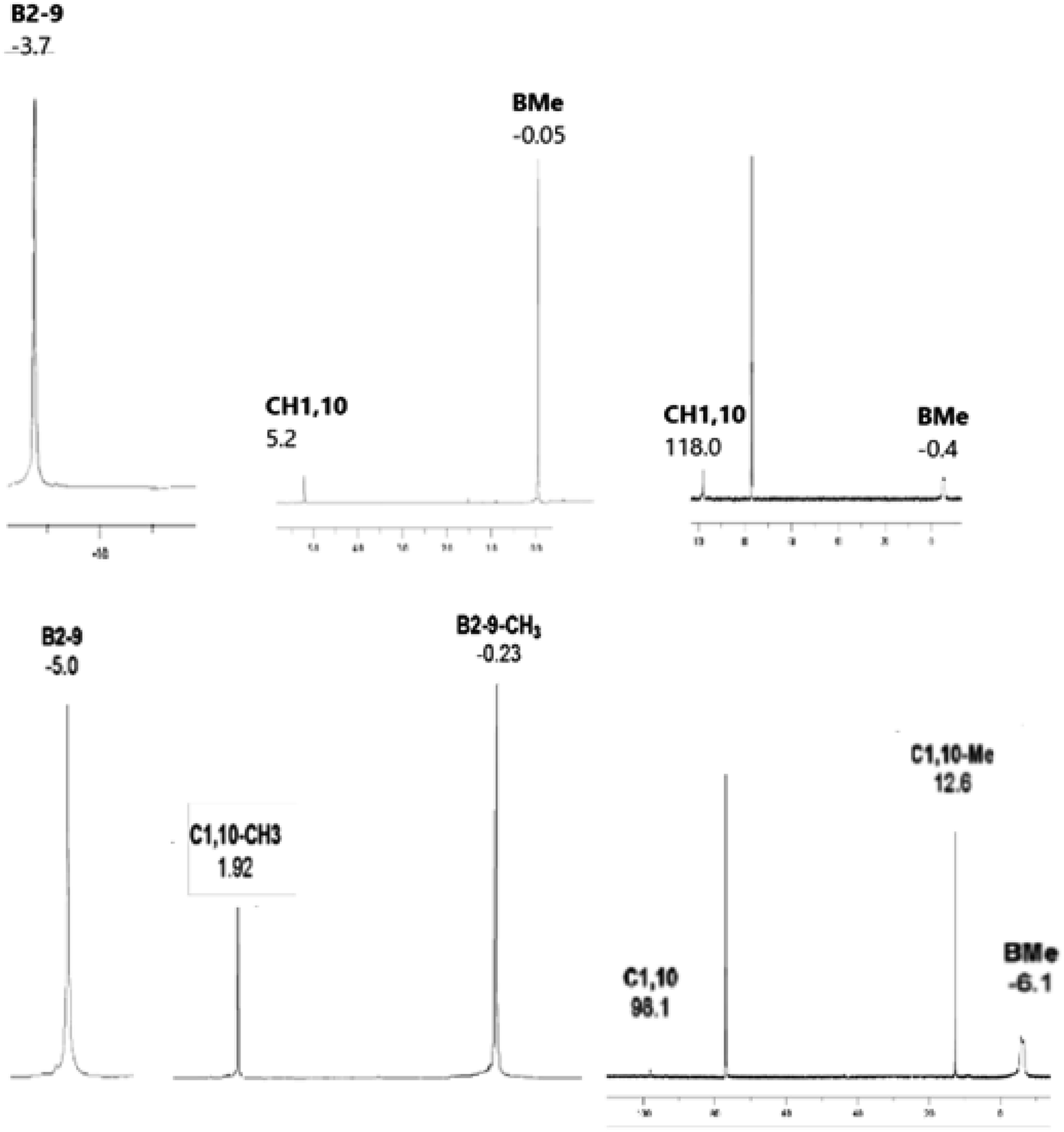 | ||
| Fig. 7 192.6 MHz 11B, 600 MHz 1H, and 150.9 MHz 13C NMR spectra of closo-1,10-H2C2B8Me8 (2a) (top) and closo-1,10-Me2C2B8Me8 (2b) (bottom). | ||
On the other hand, the 11B NMR spectrum of the Cs-symmetry compound 5a (Fig. 3 and S12†) displays 3:2:2:1 patterns of singlets with one coincidental overlap. The 1H spectrum of 5a (Fig. 6 and S13†) shows two different CH signals and four well resolved 2:2:2:1 patterns of BMe resonances in the high-field; the 13C–{1H} NMR spectrum contains two different low-field CH resonances together with a typical low-field CF3 quartet, apart from a broad, high-field BMe signal of intensity 7 (see Fig. S13†).
The 19F NMR spectrum of 5a shows one singlet resonance at −76.0 ppm, as expected.
Experimental
Materials and methods
All the reactions were carried out under argon atmosphere. Dichloromethane and hexane were dried over CaH2 and freshly distilled before use. Other conventional chemicals were of reagent or analytical grade and were used as purchased. NMR spectroscopy was performed at 400 and 600 Mz (for 1H), inclusive of standard [11B–11B]-COSY17 experiments (all theoretical cross-peaks were observed) leading to complete assignments of all resonances to individual cage B-vertexes. Chemical shifts are given in ppm to high-frequency (low field) of Ξ = 32.083971 MHz (nominally F3B·OEt2 in CDCl3) for 11B (quoted ± 0.5 ppm), Ξ = 25.144 MHz for 13C (quoted ± 0.5 ppm), and Ξ = 100 MHz for 1H (quoted ± 0.05 ppm), Ξ is defined as in ref. 18 and the solvent resonances were used as internal secondary standards. The starting carboranes of structures 1 and 3 were prepared according to the reported methods.10,12Dimethylation of closo-1,10-C2B8H10 (1a) on carbon vertices
A solution of 1a (120 mg, 1 mmol) in dry Et2O (ca. 10–20 ml) was cooled to −78 °C and then treated dropwise with 2.5 M LiBu (solution in hexane) (1 ml, 2.5 mmol) under stirring. The off-white slurry of the Li+ salt was stirred for additional 1 h prior to addition of methyl triflate (MeOTf, m.w. 164.1) drop by drop, (410 mg, 2.5 mmol) under cooling down in an dry-ice bath. The mixture was then left stirring for additional 2 h at room temperature. After adding 5 % hydrochloric acid (10 ml) under repeated cooling and shaking, the Et2O layer was separated and evaporated to provide a crude product closo-1,10-Me2C2B8H8 (1b) in practically a quantitative yield, as assessed by NMR spectroscopy (see Fig. S4–S6†).closo-1,10-R2C2B8Me8 (2) (where R = H 2a or Me 2b)
Thermal rearrangement of closo-1,6-R2C2B8Me7-8-OTf (4a)
Compound 4a (74 mg, 0.2 mmol) was heated in a sealed ampoule at 400–500 °C (heating gun) for 2 hours. The resulting product was identified as closo-1,10-R2C2B8Me7-2-OTf (5a) and isolated on sublimation (bath temperature ∼ 150 °C) in essentially quantitative yield. For 5a MS (ESI−): m/z (max.) calcd 366.20, found 366.19; for C10H23B8O3SF3 (m.w. 366.84) calcd 32.74 %C, 6.32 %H; found 32.28 %C, 6.15 %H. For NMR spectra, see Fig. 6, S13 and S14.†Computational details
Magnetic shielding was calculated using the GIAO-MP2 method incorporated into Gaussian09 (ref. 19) utilizing the IGLO-II basis with the MP2/TZVP geometry and frozen core electrons. Electrostatic potentials were computed at the HF/cc-pVDZ level using Gaussian09 and Molekel4.3 (ref. 20) programs. It has recently been shown that this basis set size is sufficient for these purposes.21X-ray crystallography
The X-ray data for the derivatives 2a and 5a (colourless crystals by slow evaporation of a hexane solution) were collected at 150(2) K with a Bruker D8-Venture diffractometer equipped with Mo (Mo/Kα radiation; λ = 0.71073 Å) microfocus X-ray (IμS) source, photon CMOS detector and Oxford Cryosystems cooling device was used for data collection. The frames were integrated with the Bruker SAINT software package using a narrow-frame algorithm. Data were corrected for absorption effects using the Multi-Scan method (SADABS). Obtained data were treated by XT-version 2014/5 and SHELXL-2014/7 software implemented in APEX3 v2016.5-0 (Bruker AXS) system.22 Hydrogen atoms were mostly localized on a difference Fourier map, however to ensure uniformity of treatment of crystal, all hydrogen were recalculated into idealized positions (riding model) and assigned temperature factors Hiso(H) = 1.2Ueq (pivot atom) or of 1.5Ueq (methyl). H atoms in methyl groups were placed with C–H distances of 0.96 while the hydrogen atoms of the C–H in the carborane cage were assigned according to the maxima on the difference Fourier map. Rint = ∑∣Fo2 − Fo,mean2∣/∑Fo2, S = [∑(w(Fo2 − Fc2)2)/(Ndiffrs – Nparams)]1/2 for all data, R(F) = ∑∣∣Fo∣ − ∣Fc∣∣/∑∣Fo∣for observed data, wR(F2) = [∑(w(Fo2 − Fc2)2)/(∑w(Fo2)2)]1/2 for all data. Crystallographic data for structural analysis have been deposited with the Cambridge Crystallographic Data Centre, CCDC no. 1573271 and 1573272 for 2a and 5a, respectively. Crystallographic data for 2a: C10H26B8, M = 232.79, orthorhombic, Pbcn a = 8.5140(13), b = 15.098(3), c = 12.670(2) Å, β = 90°, Z = 4, V = 1628.6(5) Å3, Dc = 0.949 g cm−3, μ = 0.045 mm−1, Tmin/Tmax = 0.6416/0.7456; −10 ≤ h ≤ 11, −19 ≤ k ≤ 19, −15 ≤ l ≤ 16; 14313 reflections measured (θmax = 27.55°), 1875 independent (Rint = 0.0566), 136 with I > 2σ(I), 91 parameters, S = 1.078, R1(obs. data) = 0.0977, wR2(all data) = 0.2866; max., min. residual electron density = 0.792, −0.319 eÅ−3. Crystallographic data for 5a: C10H23B8F3O3S, M = 366.82, monoclinic, P21/c, a = 13.0608(9), b = 9.8980(6), c = 16.0303(9) Å, β = 111.202(2) °, Z = 4, V = 1932.1(2) Å3, Dc = 1.261 g cm−3, μ = 0.200 mm−1, Tmin/Tmax = 0.6787/0.7456; −16 ≤ h ≤ 16, −12 ≤ k ≤ 12, −20 ≤ l ≤ 19; 47313 reflections measured (θmax = 27.54°), 4445 independent (Rint = 0.0300), 3971 with I > 2σ(I), 241 parameters, S = 1.049, R1(obs. data) = 0.0502, wR2(all data) = 0.1431; max., min. residual electron density = 0.729, −0.628 eÅ−3.
Conclusions
There are not too many reactions in the area of carborane chemistry that proceed quantitatively.1 To these rare cases belong syntheses leading to permethylated derivatives of closo-1,10-R2C2B8H8 (1) reported this work. It was shown that all B-positions in structures 1 can be furnished with methyl substituents, via electrophilic reactions with MeOTf or MeI reagents. In quantitative yields proceed also the 1,6- → 1,10-carbon rearrangement reactions of the isomeric compounds closo-1,6-R2C2B8Me8 (3). Moreover, the permethylated compounds, such as 2a and 2b, can be, in fact, envisaged as egg (or ellipsoid) shaped hydrocarbons (see Fig. 5) of remarkable air stability due to the protective sheath of the surrounding methyl groups. For example, the persubstituted 2a can be stored in air for at least a month without any noticeable change, while the unprotected 1a is decomposed in air within a couple of hours, especially in a solution. The less stability of the unprotected intermediate-sized carborane 1a derives from its non-icosahedral constitution (though it exhibits features of 3D aromaticity23). The quantitative yields and relative easiness of the synthesis predestinate these persubstituted derivatives for using in designed syntheses in specific areas of carborane chemistry as multipurpose reagents, for example in cluster-insertion/expansion or cage-degradation processes. Apart from this, such compounds are expected to exhibit extreme hydrophobicity, which can be made use of in various directions of chemical or biochemical research. Relevant experiments aimed at extension of permethylation chemistry are therefore in progress in our laboratories.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Grant Agency of the Czech Republic (project no. 16-01618S).Notes and references
- For review, see: R. N. Grimes, Carboranes, Academic Press, 3rd edn, 2016 Search PubMed.
- See, for example: P. M. Garrett, J. C. Smart and M. F. Hawthorne, J. Am. Chem. Soc., 1969, 91, 4707–4710 CrossRef CAS.
- D. A. Owen, J. C. Smart, P. M. Garrett and M. F. Hawthorne, J. Am. Chem. Soc., 1971, 93, 1362–1368 CrossRef.
- T. J. Wedge, A. Herzog, R. Huertas, M. W. Lee, C. B. Knobler and M. F. Hawthorne, Organometallics, 2004, 23, 482–489 CrossRef CAS.
- T. D. Getman, P. M. Garrett, C. B. Knobler, M. F. Hawthorne, K. Thorne and J. D. MacKenzie, Organometallics, 1992, 11, 2723–2725 CrossRef CAS.
- M. Bakardjiev, B. Štíbr, J. Holub, Z. Padělková and A. Růžička, Organometallics, 2015, 34, 450–454 CrossRef CAS.
- For review, see: J. J. Rockwell, A. Herzog, T. Peymann, C. B. Knobler and M. F. Hawthorne, Curr. Sci., 2000, 78, 405 CAS.
- For review, see: (a) S. Körbe, P. J. Schreiber and J. Michl, Chem. Rev., 2006, 106, 5208–5249 CrossRef PubMed; (b) C. Douvris and J. Michl, Chem. Rev., 2013, 113, PR179–PR233 CrossRef PubMed.
- W. Jiang, C. B. Knobler, M. D. Mortimer and M. F. Hawthorne, Angew. Chem., Int. Ed., 1995, 34, 1332 CrossRef CAS.
- M. Bakardjiev, O. L. Tok, A. Růžička, Z. Růžičková, J. Holub, Z. Špalt and B. Štíbr, Dalton Trans., 2018, 47, 11070–11076 RSC.
- M. Bakardjiev, B. Štíbr, J. Holub, O. L. Tok, P. Švec, Z. Růžičková and A. Růžička, Inorg. Chem., 2016, 55, 7068–7074 CrossRef CAS PubMed.
- B. Štíbr, J. Plešek and S. Heřmánek, Collect. Czech. Chem. Commun., 1973, 38, 338 CrossRef.
- J. Fanfrlík, M. Lepšík, D. Hořínek, Z. Havlas and P. Hobza, ChemPhysChem, 2006, 7, 1100 CrossRef PubMed.
- (a) C. Esterhuysen, A. Hesselmann and T. Clark, ChemPhysChem, 2017, 18, 1 CrossRef PubMed; (b) J. Fanfrlík, A. Pecina, J. Řezáč, R. Sedlák, D. Hnyk, M. Lepšík and P. Hobza, Phys. Chem. Chem. Phys., 2017, 19, 18194 RSC.
- In contrast, in the closo-1,10-C2B8 skeleton the cage carbon atoms participate in five multicenter bondings of 3c2e and 4c2e types, which results in in their positive charges as opposed to classical electronegativity complex. See P. Melichar, D. Hnyk and J. Fanfrlík, A systematic examination of classical and multi-center bonding in heteroborane clusters, Phys. Chem. Chem. Phys., 2018, 20, 4666–4675 RSC Positive charges of cage carbons in closo systems were also verified experimentally: D. Hnyk, V. Všetečka, L. Drož and O. Exner, Charge Distribution Within 1,2-Dicarba-closo-dodecaborane: Dipole Moments of Its Phenyl Derivatives, Collect. Czech. Chem. Commun., 2001, 66, 1375–1379 CrossRef CAS.
- F. Teixidor, G. Barberà, A. Vaca, R. Kivekäs, R. Sillanpää, J. Oliva and C. Viñas, J. Am. Chem. Soc., 2005, 127, 10158–10159 CrossRef CAS PubMed.
- W. C. Hutton, T. L. Venable and R. N. Grimes, J. Am. Chem. Soc., 1984, 106, 29–37 CrossRef.
- W. A. McFarlane, Proc. R. Soc. London, Ser. A, 1968, 306, 175–199 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- (a) P. Flükiger, H. P. Lüthi, S. Portmann and J. Weber, MOLEKEL 4.3, Swiss Center for Scientific Computing, Manno (Switzerland), 2000 Search PubMed; (b) S. Portmann and H. P. Lüthi, MOLEKEL: CHIMIA, 2007, Vol. 28, p. 555 Search PubMed.
- K. E. Riley, K.-A. Tran, P. Lane, J. S. Murray and P. Politzer, J Comput Sci, 2016, 17, 273 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, A71, 3–8 Search PubMed.
- (a) J. Poater, M. Sola, C. Viñas and F. Teixidor, Angew. Chem., Int. Ed., 2014, 53, 12191–12195 CrossRef CAS PubMed; (b) J. Poater, M. Sola, C. Viñas and F. Teixidor, Chem.–Eur. J., 2016, 22, 7437–7443 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR data and computed 11B NMR, cartesian geometries. CCDC 1573271 and 1573272. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra06640j |
| This journal is © The Royal Society of Chemistry 2018 |